Folate Deficiency Could Restrain Decidual Angiogenesis in Pregnant Mice

Abstract
:1. Introduction
2. Experimental Section
2.1. Animals
2.2. Detection of Serum Folate, Hcy, P4, E2, FSH, LH, and PRL
2.3. Hematoxylin-Eosin (H&E) Staining
2.4. Immunohistochemistry
2.5. Western Blotting
2.6. Real-Time polymerase chain reaction (RT-PCR)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Forward | Reverse |
|---|---|---|
| VEGFA | GTCCAACTTCTGGGCTCTTCT | CCCTCTCCTCTTCCTTCTCTTC |
| VEGFR2 | TGGCAAATACAACCCTTCAGAT | GTCACCAATACCCTTTCCTCAG |
| PLGF | GCCGATAAAGACAGCCAACA | CATTCACAGAGCACATCCTGA |
| β-actin | CCTGAGGCTCTTTTCCAGCC | TAGAGGTCTTTACGGATGTCAACGT |
2.7. Statistical Analysis
3. Results
3.1. Validation of the Folate-Deficient Pregnant Mouse Model
| Normal (n) | Folate-deficient (n) | p | |
|---|---|---|---|
| Serum folate | >20 (10) | 4.83 ± 0.48 (19) | <0.01 |
| Normal (n) | Folate-deficient (n) | p | |
|---|---|---|---|
| E6 | 7.50 ± 0.24 (5) | 11.06 ± 0.40 (5) | <0.01 |
| E7 | 6.75 ± 0.41 (5) | 12.05 ± 0.57 (5) | <0.01 |
| E8 | 8.26 ± 0.32 (5) | 12.46 ± 0.61 (5) | <0.01 |
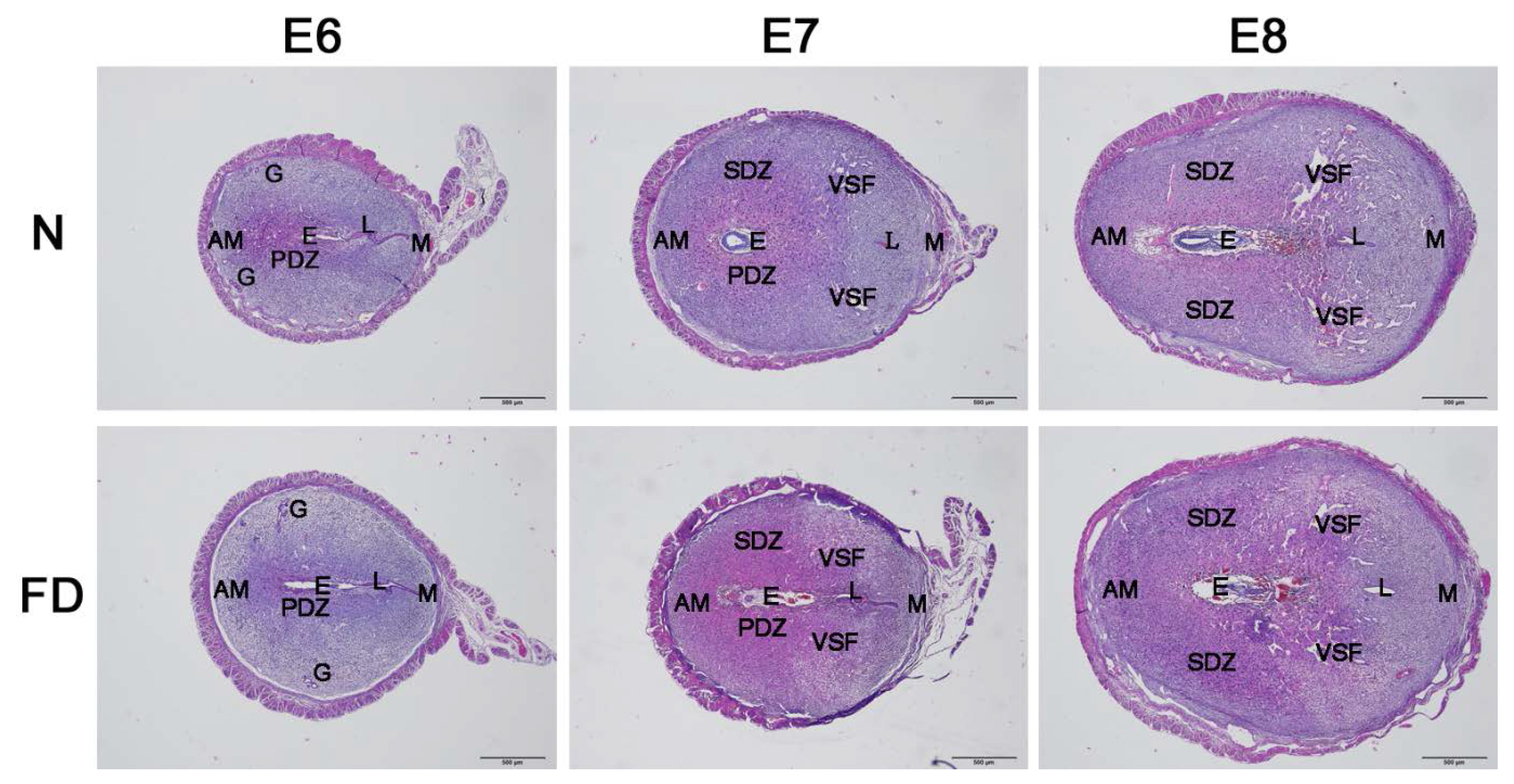
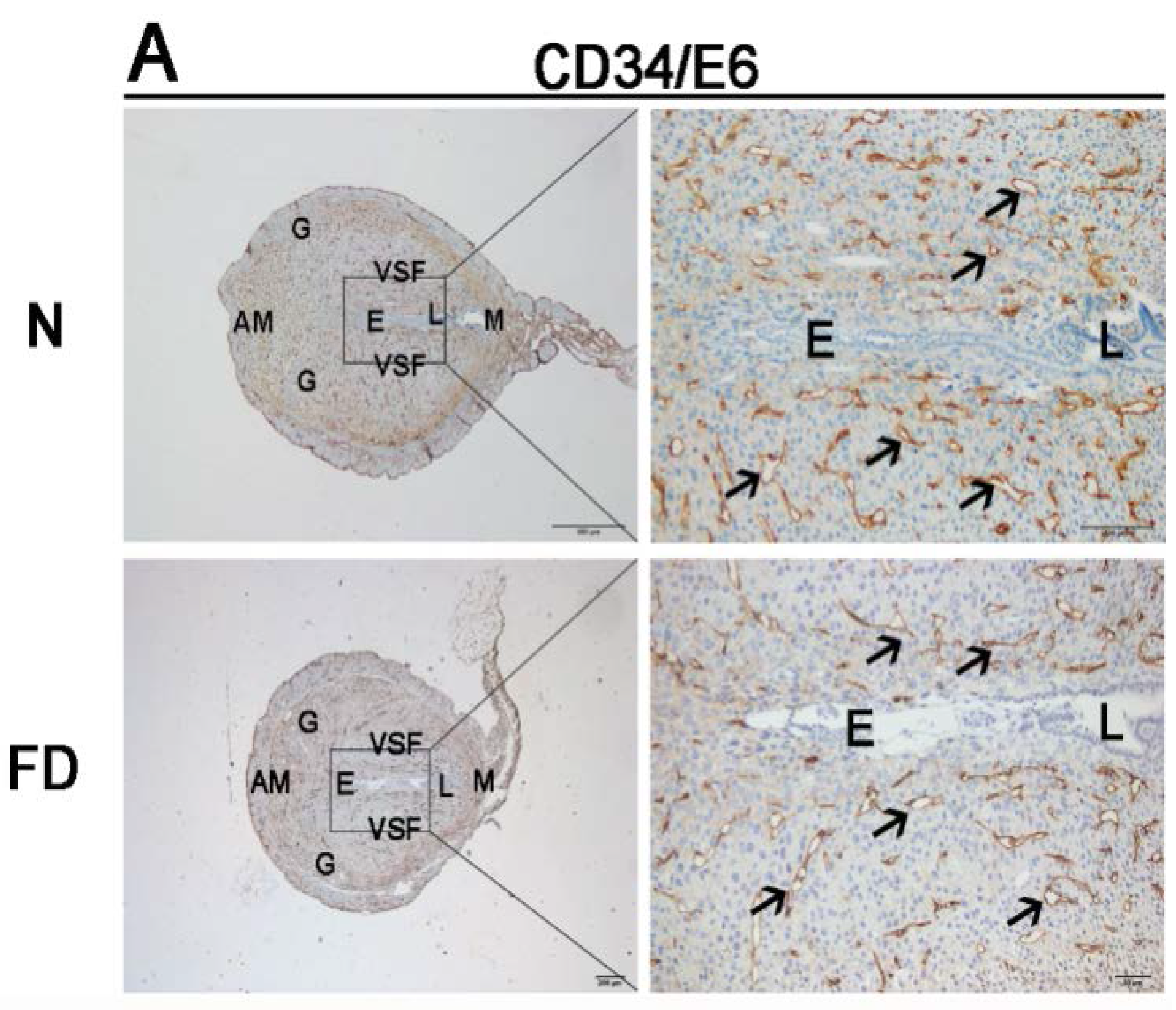
3.2. Decidual Angiogenesis Was Restrained in Folate-Deficient Pregnant Mice


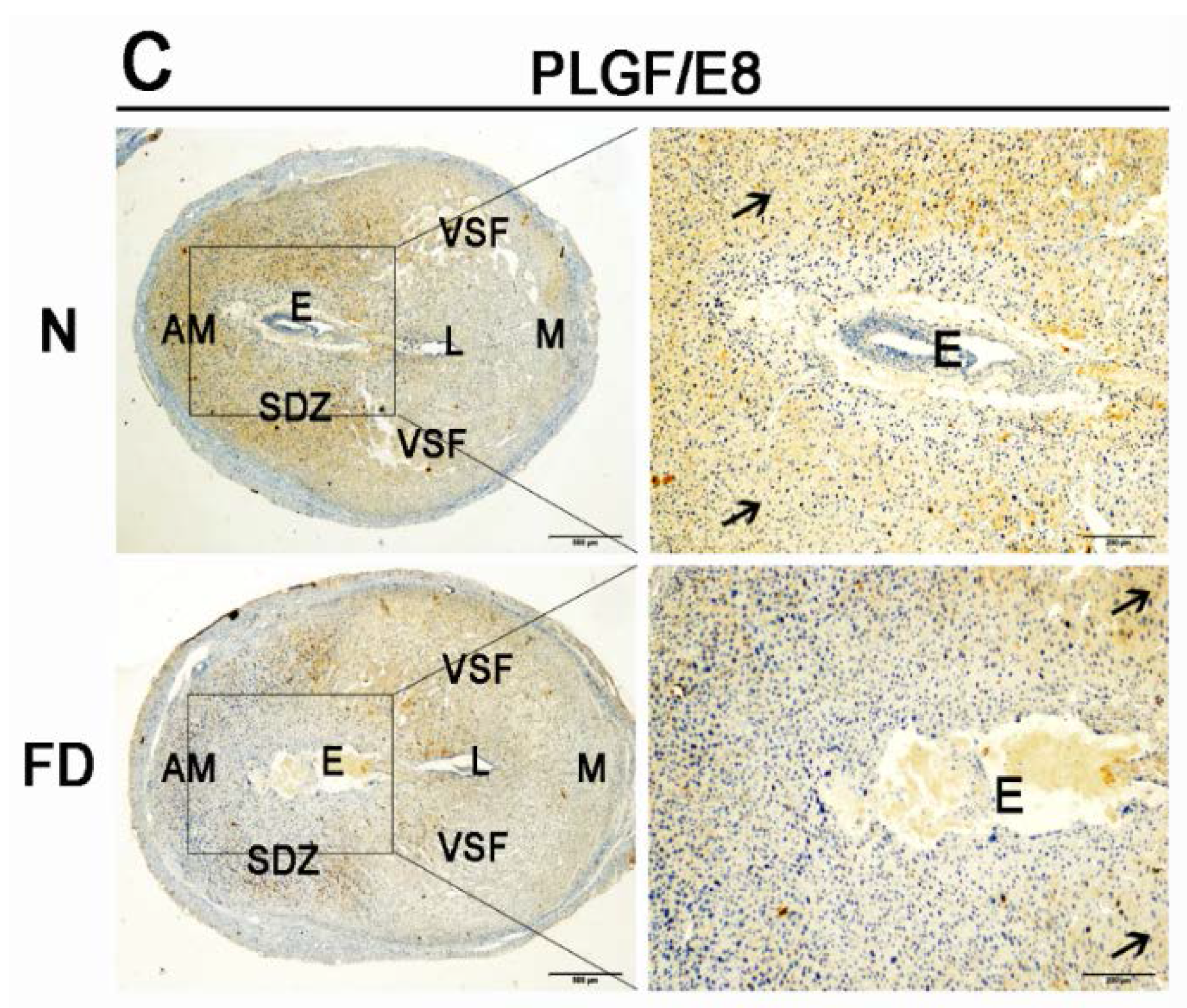
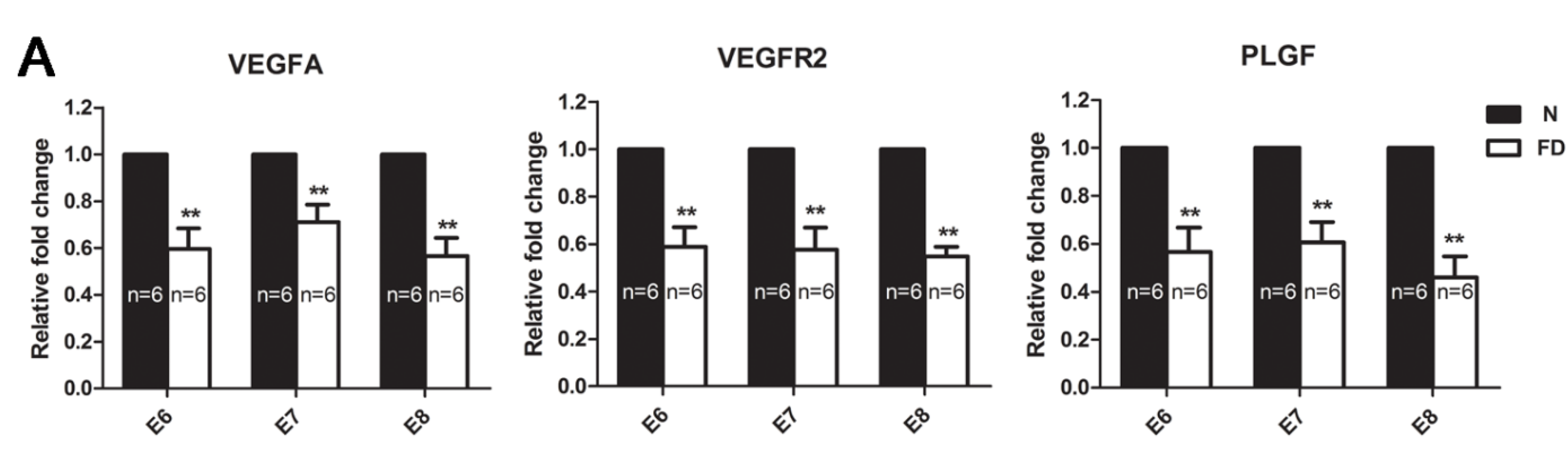
3.3. The Expression of VEGFA, VEGFR2, and PLGF Was Reduced in Folate-Deficient Decidual Tissue

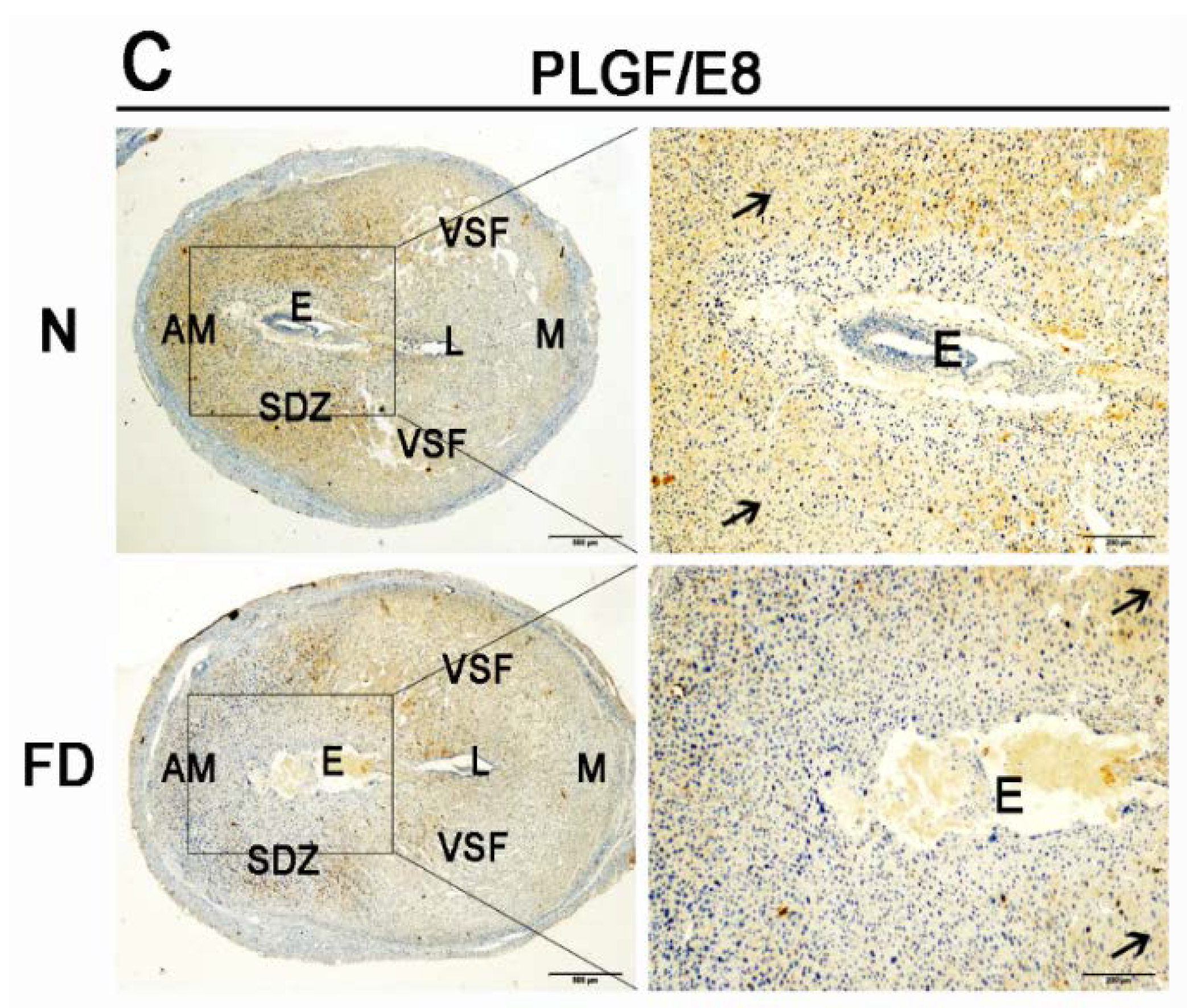
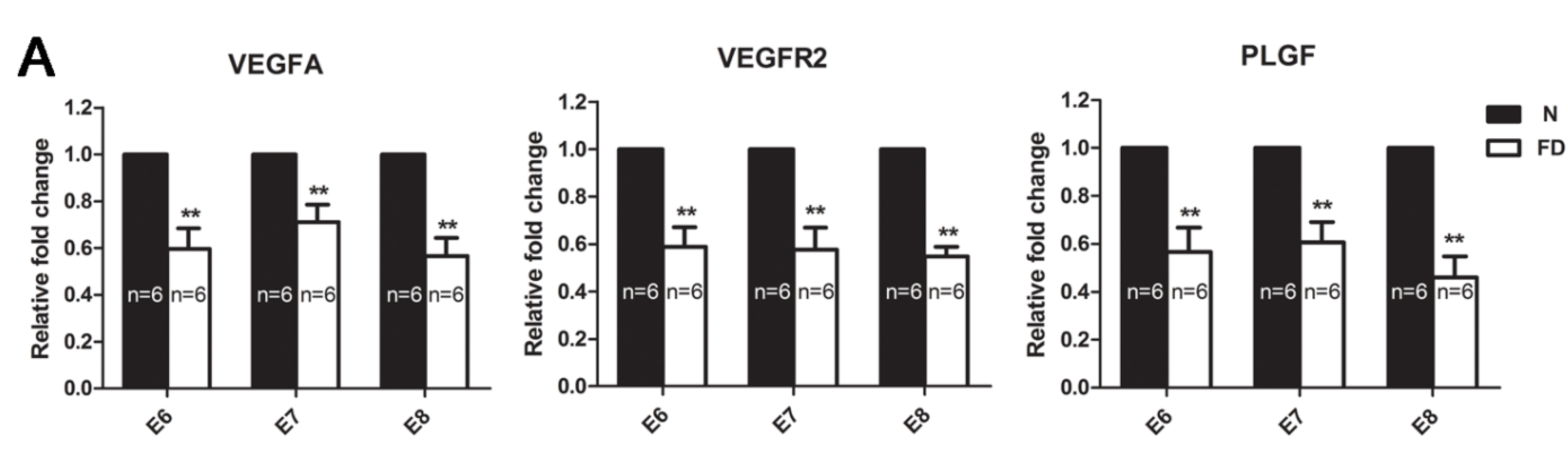
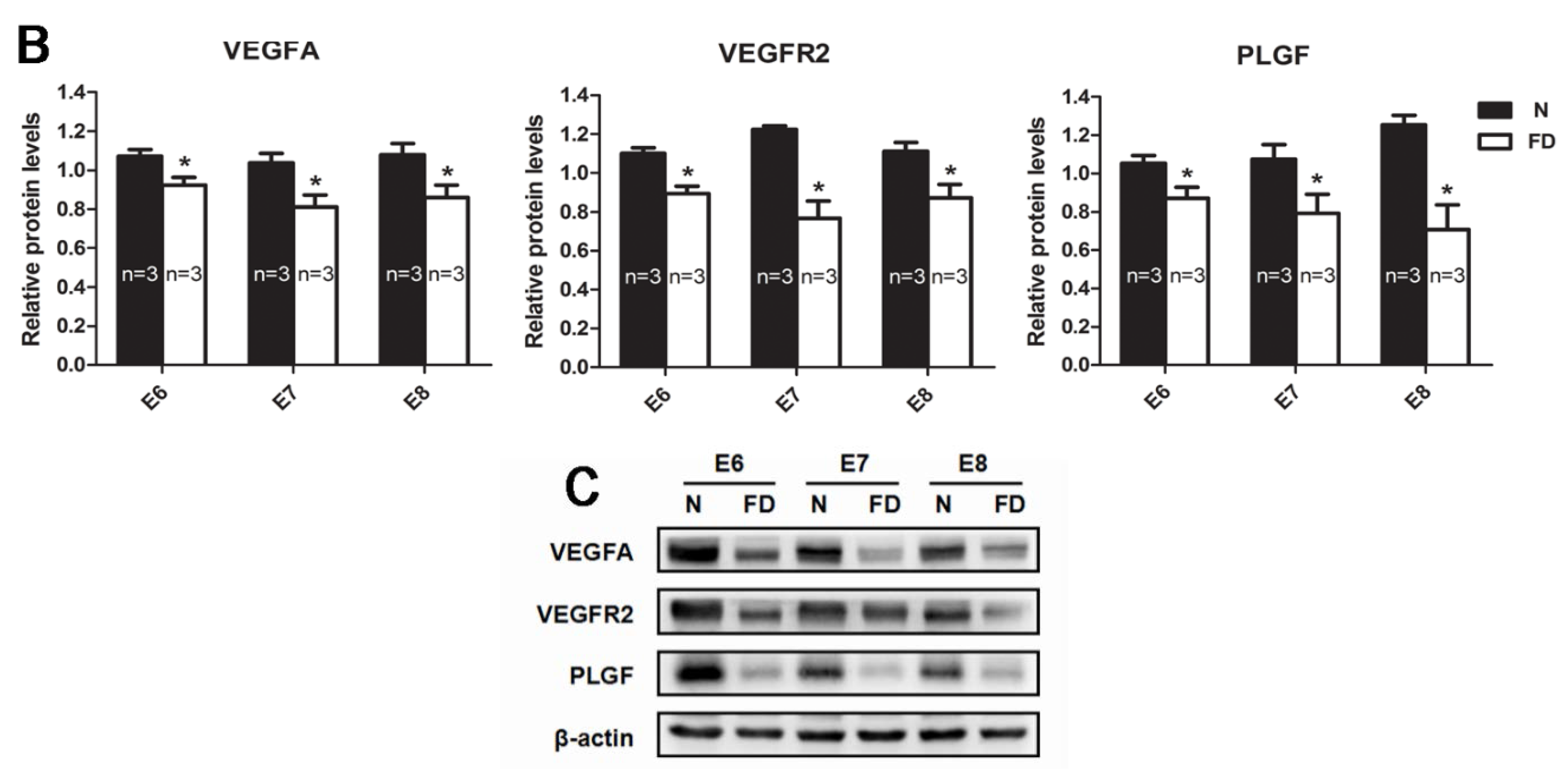
3.4. Folate-Deficient Mice Showed Disordered Reproductive Hormone Levels
| P4 (ng/mL) | E2 (pmol/L) | |||||
|---|---|---|---|---|---|---|
| Normal (n) | Folate-deficient (n) | p | Normal (n) | Folate-deficient (n) | p | |
| E6 | 11.02 ± 0.44 (4) | 8.79 ± 0.17 (4) | 0.009 | 42.39 ± 0.82 (3) | 29.97 ± 0.60 (4) | <0.001 |
| E7 | 10.02 ± 0.21 (4) | 7.56 ± 0.18 (4) | <0.001 | 38.01 ± 0.77 (3) | 26.50 ± 0.65 (4) | <0.001 |
| E8 | 10.24 ± 0.16 (4) | 6.20 ± 0.26 (4) | <0.001 | 38.76 ± 0.71 (3) | 23.26 ± 0.71 (4) | <0.001 |

| FSH (mIU/mL) | LH (mIU/mL) | PRL (ng/mL) | ||||
|---|---|---|---|---|---|---|
| Normal (n) | Folate-deficient (n) | Normal (n) | Folate-deficient (n) | Normal (n) | Folate-deficient (n) | |
| E6 | 47.38 ± 4.02 (3) | 45.50 ± 2.23 (5) | 3.72 ± 0.51 (3) | 2.66 ± 0.15 (5) | 31.74 ± 3.23 (3) | 24.21 ± 1.76 (5) |
| E7 | 53.49 ± 4.43 (3) | 51.07 ± 2.46 (5) | 4.52 ± 0.60 (3) | 3.28 ± 0.20 (5) | 32.94 ± 3.53 (3) | 25.21 ± 1.42 (5) |
| E8 | 48.36 ± 3.62 (3) | 46.45 ± 2.19 (5) | 3.88 ± 0.45 (3) | 2.95 ± 0.21 (5) | 31.46 ± 1.70 (3) | 24.78 ± 1.55 (5) |

4. Discussion
5. Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mathers, C.D.; Loncar, D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med. 2006, 3, e442. [Google Scholar] [CrossRef] [PubMed]
- Castillo, T.S. Services for the care and prevention of birth defects. Reduced report of a World Health Organization and March of Dimes Foundation meeting. Rev. Med. Chile 2007, 135, 806–813. [Google Scholar]
- Zheng, X.Y.; Song, X.M.; Chen, G.; Chen, J.P.; Ji, Y.; Wu, J.L.; Liu, J.F.; Zhang, L.; Fan, X.H. Epidemiology of birth defects in high-prevalence areas of China. Zhonghua Liu Xing Bing Xue Za Zhi 2007, 28, 5–9. [Google Scholar] [PubMed]
- Wills, L. Treatment of “pernicious anaemia of pregnancy” and “tropical anaemia” with special reference to yeast extract as a curative agent. 1931. Natl. Med. J. India 2013, 26, 117–122. [Google Scholar] [PubMed]
- Czeizel, A.E.; Dudas, I. Prevention of the first occurrence of neural-tube defects by periconceptional vitamin supplementation. N. Engl. J. Med. 1992, 327, 1832–1835. [Google Scholar] [CrossRef] [PubMed]
- Ionescu-Ittu, R.; Marelli, A.J.; Mackie, A.S.; Pilote, L. Prevalence of severe congenital heart disease after folic acid fortification of grain products: Time trend analysis in Quebec, Canada. BMJ 2009, 338. [Google Scholar] [CrossRef] [PubMed]
- Tolarova, M.; Harris, J. Reduced recurrence of orofacial clefts after periconceptional supplementation with high-dose folic acid and multivitamins. Teratology 1995, 51, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Frias, M.L.; Perez, B.; Desviat, L.R.; Castro, M.; Leal, F.; Rodriguez, L.; Mansilla, E.; Martinez-Fernandez, M.L.; Bermejo, E.; Rodriguez-Pinilla, E.; et al. Maternal polymorphisms 677C-T and 1298A-C of MTHFR, and 66A-G MTRR genes: Is there any relationship between polymorphisms of the folate pathway, maternal homocysteine levels, and the risk for having a child with Down syndrome? Am. J. Med. Genet. A 2006, 140, 987–997. [Google Scholar] [CrossRef] [PubMed]
- Hibbard, B.M.; Hibbard, E.D.; Hwa, T.S.; Tan, P. Abruptio placentae and defective folate metabolism in Singapore women. Int. J. Obstet. Gynaecol. Br. Commonw. 1969, 76, 1003–1007. [Google Scholar] [CrossRef]
- Mignini, L.E.; Latthe, P.M.; Villar, J.; Kilby, M.D.; Carroli, G.; Khan, K.S. Mapping the theories of preeclampsia: The role of homocysteine. Obstet. Gynecol. 2005, 105, 411–425. [Google Scholar] [CrossRef] [PubMed]
- George, L.; Mills, J.L.; Johansson, A.L.; Nordmark, A.; Olander, B.; Granath, F.; Cnattingius, S. Plasma folate levels and risk of spontaneous abortion. JAMA 2002, 288, 1867–1873. [Google Scholar] [CrossRef] [PubMed]
- Lindblad, B.; Zaman, S.; Malik, A.; Martin, H.; Ekstrom, A.M.; Amu, S.; Holmgren, A.; Norman, M. Folate, vitamin B12, and homocysteine levels in South Asian women with growth-retarded fetuses. Acta Obstet. Gynecol. Scand. 2005, 84, 1055–1061. [Google Scholar] [CrossRef] [PubMed]
- Gao, R.; Ding, Y.; Liu, X.; Chen, X.; Wang, Y.; Long, C.; Li, S.; Guo, L.; He, J. Effect of folate deficiency on promoter methylation and gene expression of Esr1, Cdh1 and Pgr, and its influence on endometrial receptivity and embryo implantation. Hum. Reprod. 2012, 27, 2756–2765. [Google Scholar] [CrossRef] [PubMed]
- Winterhager, E.; Gellhaus, A.; Blois, S.M.; Hill, L.A.; Barr, K.J.; Kidder, G.M. Decidual angiogenesis and placental orientation are altered in mice heterozygous for a dominant loss-of-function Gja1 (connexin43) mutation. Biol. Reprod. 2013, 89, 111. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.; Park, H.J.; Seol, J.W.; Jang, J.Y.; Cho, Y.S.; Kim, K.R.; Choi, Y.; Lydon, J.P.; Demayo, F.J.; Shibuya, M.; et al. VEGF-A regulated by progesterone governs uterine angiogenesis and vascular remodelling during pregnancy. EMBO Mol. Med. 2013, 5, 1415–1430. [Google Scholar] [CrossRef] [PubMed]
- Charnock-Jones, D.S.; Kaufmann, P.; Mayhew, T.M. Aspects of human fetoplacental vasculogenesis and angiogenesis. I. Molecular regulation. Placenta 2004, 25, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Senger, D.R.; Ledbetter, S.R.; Claffey, K.P.; Papadopoulos-Sergiou, A.; Peruzzi, C.A.; Detmar, M. Stimulation of endothelial cell migration by vascular permeability factor/vascular endothelial growth factor through cooperative mechanisms involving the alphavbeta3 integrin, osteopontin, and thrombin. Am. J. Pathol. 1996, 149, 293–305. [Google Scholar] [PubMed]
- Zhao, S.; Yuan, H.; Xie, C.; Xiao, D. Determination of folic acid by capillary electrophoresis with chemiluminescence detection. J. Chromatogr. A. 2006, 1107, 290–293. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.G.; Li, Y.L.; Gao, R.F.; Geng, Y.Q.; Chen, X.M.; Liu, X.Q.; Ding, Y.B.; Mu, X.Y.; Wang, Y.X.; He, J.L. Folate deficiency decreases apoptosis of endometrium decidual cells in pregnant mice via the mitochondrial pathway. Nutrients 2015, 7, 1916–1932. [Google Scholar] [CrossRef] [PubMed]
- Chandana, E.P.; Maeda, Y.; Ueda, A.; Kiyonari, H.; Oshima, N.; Yamamoto, M.; Kondo, S.; Oh, J.; Takahashi, R.; Yoshida, Y.; et al. Involvement of the Reck tumor suppressor protein in maternal and embryonic vascular remodeling in mice. BMC Dev. Biol. 2010, 10, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burri, P.H.; Hlushchuk, R.; Djonov, V. Intussusceptive angiogenesis, its emergence, its characteristics, and its significance. Dev. Dyn. 2004, 231, 474–488. [Google Scholar] [CrossRef] [PubMed]
- Hyder, S.M.; Stancel, G.M. Regulation of angiogenic growth factors in the female reproductive tract by estrogens and progestins. Mol. Endocrinol. 1999, 13, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Willmott, M.; Bartosik, D.B.; Romanoff, E.B. The effect of folic acid on superovulation in the immature rat. J. Endocrinol. 1968, 41, 439–445. [Google Scholar] [CrossRef] [PubMed]
- Mohanty, D.; Das, K.C. Effect of folate deficiency on the reproductive organs of female rhesus monkeys: A cytomorphological and cytokinetic study. J. Nutr. 1982, 112, 1565–1576. [Google Scholar] [PubMed]
- Mooij, P.N.; Wouters, M.G.; Thomas, C.M.; Doesburg, W.H.; Eskes, T.K. Disturbed reproductive performance in extreme folic acid deficient golden hamsters. Eur. J. Obstet. Gynecol. Reprod. Biol. 1992, 43, 71–75. [Google Scholar] [CrossRef]
- Xiao, S.; Hansen, D.K.; Horsley, E.T.; Tang, Y.S.; Khan, R.A.; Stabler, S.P.; Jayaram, H.N.; Antony, A.C. Maternal folate deficiency results in selective upregulation of folate receptors and heterogeneous nuclear ribonucleoprotein-E1 associated with multiple subtle aberrations in fetal tissues. Birth Defects Res. A Clin. Mol. Teratol. 2005, 73, 6–28. [Google Scholar] [CrossRef] [PubMed]
- Abrahamsohn, P.A.; Zorn, T.M. Implantation and decidualization in rodents. J. Exp. Zool. 1993, 266, 603–628. [Google Scholar] [CrossRef] [PubMed]
- Folkman, J.; Klagsbrun, M. Angiogenic factors. Science 1987, 235, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Reynolds, L.P.; Killilea, S.D.; Redmer, D.A. Angiogenesis in the female reproductive system. FASEB J. 1992, 6, 886–892. [Google Scholar] [PubMed]
- Plaisier, M.; Rodrigues, S.; Willems, F.; Koolwijk, P.; van Hinsbergh, V.W.; Helmerhorst, F.M. Different degrees of vascularization and their relationship to the expression of vascular endothelial growth factor, placental growth factor, angiopoietins, and their receptors in first-trimester decidual tissues. Fertil. Steril. 2007, 88, 176–187. [Google Scholar] [CrossRef] [PubMed]
- Cooper, J.C.; Sharkey, A.M.; McLaren, J.; Charnock-Jones, D.S.; Smith, S.K. Localization of vascular endothelial growth factor and its receptor, flt, in human placenta and decidua by immunohistochemistry. J. Reprod. Fertil. 1995, 105, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Douglas, N.C.; Tang, H.; Gomez, R.; Pytowski, B.; Hicklin, D.J.; Sauer, C.M.; Kitajewski, J.; Sauer, M.V.; Zimmermann, R.C. Vascular endothelial growth factor receptor 2 (VEGFR-2) functions to promote uterine decidual angiogenesis during early pregnancy in the mouse. Endocrinology 2009, 150, 3845–3854. [Google Scholar] [CrossRef] [PubMed]
- Torry, D.S.; Wang, H.S.; Wang, T.H.; Caudle, M.R.; Torry, R.J. Preeclampsia is associated with reduced serum levels of placenta growth factor. Am. J. Obstet. Gyneco.l 1998, 179, 1539–1544. [Google Scholar] [CrossRef]
- Khaliq, A.; Li, X.F.; Shams, M.; Sisi, P.; Acevedo, C.A.; Whittle, M.J.; Weich, H.; Ahmed, A. Localisation of placenta growth factor (PIGF) in human term placenta. Growth Factors 1996, 13, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Torry, D.S.; Ahn, H.; Barnes, E.L.; Torry, R.J. Placenta growth factor: Potential role in pregnancy. Am. J. Reprod. Immunol. 1999, 41, 79–85. [Google Scholar] [CrossRef] [PubMed]
- Carmeliet, P.; Moons, L.; Luttun, A.; Vincenti, V.; Compernolle, V.; De Mol, M.; Wu, Y.; Bono, F.; Devy, L.; Beck, H.; et al. Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat. Med. 2001, 7, 575–583. [Google Scholar] [CrossRef] [PubMed]
- Luttun, A.; Tjwa, M.; Moons, L.; Wu, Y.; Angelillo-Scherrer, A.; Liao, F.; Nagy, J.A.; Hooper, A.; Priller, J.; De Klerck, B.; et al. Revascularization of ischemic tissues by PlGF treatment, and inhibition of tumor angiogenesis, arthritis and atherosclerosis by anti-Flt1. Nat. Med. 2002, 8, 831–840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edirimanne, V.E.; Woo, C.W.; Siow, Y.L.; Pierce, G.N.; Xie, J.Y.; O, K. Homocysteine stimulates NADPH oxidase-mediated superoxide production leading to endothelial dysfunction in rats. Can. J. Physiol. Pharmacol. 2007, 85, 1236–1247. [Google Scholar] [CrossRef] [PubMed]
- Outinen, P.A.; Sood, S.K.; Pfeifer, S.I.; Pamidi, S.; Podor, T.J.; Li, J.; Weitz, J.I.; Austin, R.C. Homocysteine-induced endoplasmic reticulum stress and growth arrest leads to specific changes in gene expression in human vascular endothelial cells. Blood 1999, 94, 959–967. [Google Scholar] [PubMed]
- Jacques, P.F.; Bostom, A.G.; Wilson, P.W.; Rich, S.; Rosenberg, I.H.; Selhub, J. Determinants of plasma total homocysteine concentration in the Framingham Offspring cohort. Am. J. Clin. Nutr. 2001, 73, 613–621. [Google Scholar] [PubMed]
- Miller, J.W.; Nadeau, M.R.; Smith, J.; Smith, D.; Selhub, J. Folate-deficiency-induced homocysteinaemia in rats: Disruption of S-adenosylmethionine’s co-ordinate regulation of homocysteine metabolism. Biochem. J. 1994, 298, 415–419. [Google Scholar] [PubMed]
- Lentz, S.R.; Sadler, J.E. Inhibition of thrombomodulin surface expression and protein C activation by the thrombogenic agent homocysteine. J. Clin. Invest. 1991, 88, 1906–1914. [Google Scholar] [CrossRef] [PubMed]
- Malhotra, J.D.; Kaufman, R.J. The endoplasmic reticulum and the unfolded protein response. Semin. Cell Dev. Biol. 2007, 18, 716–731. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Cai, Y.; Adachi, M.T.; Oshiro, S.; Aso, T.; Kaufman, R.J.; Kitajima, S. Homocysteine induces programmed cell death in human vascular endothelial cells through activation of the unfolded protein response. J. Biol. Chem. 2001, 276, 35867–35874. [Google Scholar] [CrossRef] [PubMed]
- Oosterbaan, A.M.; Steegers, E.A.; Ursem, N.T. The effects of homocysteine and folic acid on angiogenesis and VEGF expression during chicken vascular development. Microvasc. Res. 2012, 83, 98–104. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.T.; Li, Q.; Zhang, X.H.; Wu, W.K.; Sun, J.; Li, L.; Zhang, Q.; Tan, H.M. Homocysteine impaired endothelial function through compromised vascular endothelial growth factor/Akt/endothelial nitric oxide synthase signalling. Clin. Exp. Pharmacol. Physiol. 2010, 37, 1071–1077. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Dey, S.K. Roadmap to embryo implantation: Clues from mouse models. Nat. Rev. Genet. 2006, 7, 185–199. [Google Scholar] [CrossRef] [PubMed]
- Bagchi, I.C.; Li, Q.; Cheon, Y.P. Role of steroid hormone-regulated genes in implantation. Ann. N. Y. Acad. Sci. 2001, 943, 68–76. [Google Scholar] [CrossRef] [PubMed]
- Ramathal, C.Y.; Bagchi, I.C.; Taylor, R.N.; Bagchi, M.K. Endometrial decidualization of mice and men. Semin. Reprod. Med. 2010, 28, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Critchley, H.O.; Saunders, P.T. Hormone receptor dynamics in a receptive human endometrium. Reprod. Sci. 2009, 16, 191–199. [Google Scholar] [CrossRef] [PubMed]
- Shweiki, D.; Itin, A.; Neufeld, G.; Gitay-Goren, H.; Keshet, E. Patterns of expression of vascular endothelial growth factor (VEGF) and VEGF receptors in mice suggest a role in hormonally regulated angiogenesis. J. Clin. Invest. 1993, 91, 2235–2243. [Google Scholar] [CrossRef] [PubMed]
- Wetendorf, M.; DeMayo, F.J. The progesterone receptor regulates implantation, decidualization, and glandular development via a complex paracrine signaling network. Mol. Cell. Endocrinol. 2012, 357, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Park, S.; Bagchi, M.K.; Taylor, R.N.; Katzenellenbogen, B.S. The coregulator, repressor of estrogen receptor activity (REA), is a crucial regulator of the timing and magnitude of uterine decidualization. Endocrinology 2013, 154, 1349–1360. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Chen, X.; Liu, X.; Ding, Y.; Gao, R.; Qiu, Y.; Wang, Y.; He, J. Exposure of mice to benzo(a)pyrene impairs endometrial receptivity and reduces the number of implantation sites during early pregnancy. Food Chem. Toxicol. 2014, 69, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Kumar, T.R.; Wang, Y.; Lu, N.; Matzuk, M.M. Follicle stimulating hormone is required for ovarian follicle maturation but not male fertility. Nat. Genet. 1997, 15, 201–204. [Google Scholar] [CrossRef] [PubMed]
- Niswender, G.D.; Juengel, J.L.; Silva, P.J.; Rollyson, M.K.; McIntush, E.W. Mechanisms controlling the function and life span of the corpus luteum. Physiol. Rev. 2000, 80, 1–29. [Google Scholar] [PubMed]
- Petroff, M.G.; Petroff, B.K.; Pate, J.L. Mechanisms of cytokine-induced death of cultured bovine luteal cells. Reproduction 2001, 121, 753–760. [Google Scholar] [CrossRef] [PubMed]
- Mahesh, V.B.; Brann, D.W.; Hendry, L.B. Diverse modes of action of progesterone and its metabolites. J. Steroid Biochem. Mol. Biol. 1996, 56, 209–219. [Google Scholar] [CrossRef]
- Golub, M.S.; Kaufman, F.L.; Campbell, M.A.; Li, L.H.; Donald, J.M. “Natural” progesterone: Information on fetal effects. Birth Defects Res. B Dev. Reprod. Toxicol. 2006, 77, 455–470. [Google Scholar] [CrossRef] [PubMed]
- Da Silva, R.P.; Kelly, K.B.; Al Rajabi, A.; Jacobs, R.L. Novel insights on interactions between folate and lipid metabolism. Biofactors 2014, 40, 277–283. [Google Scholar] [CrossRef] [PubMed]
- Obeid, R.; Herrmann, W. Homocysteine and lipids: S-adenosyl methionine as a key intermediate. FEBS Lett. 2009, 583, 1215–1225. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Gao, R.; Liu, X.; Chen, X.; Liao, X.; Geng, Y.; Ding, Y.; Wang, Y.; He, J. Folate Deficiency Could Restrain Decidual Angiogenesis in Pregnant Mice. Nutrients 2015, 7, 6425-6445. https://doi.org/10.3390/nu7085284
Li Y, Gao R, Liu X, Chen X, Liao X, Geng Y, Ding Y, Wang Y, He J. Folate Deficiency Could Restrain Decidual Angiogenesis in Pregnant Mice. Nutrients. 2015; 7(8):6425-6445. https://doi.org/10.3390/nu7085284
Chicago/Turabian StyleLi, Yanli, Rufei Gao, Xueqing Liu, Xuemei Chen, Xinggui Liao, Yanqing Geng, Yubin Ding, Yingxiong Wang, and Junlin He. 2015. "Folate Deficiency Could Restrain Decidual Angiogenesis in Pregnant Mice" Nutrients 7, no. 8: 6425-6445. https://doi.org/10.3390/nu7085284