Chronic Vitamin C Deficiency Promotes Redox Imbalance in the Brain but Does Not Alter Sodium-Dependent Vitamin C Transporter 2 Expression


Abstract
:1. Introduction
2. Experimental Section
2.1. Animal Experimentation
2.2. Euthanasia
2.3. Biochemistry Analyses
2.4. RNA Extraction and RT-PCR
2.5. Gene Expression Analysis

{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Sequence | Product size (bp) | NCBI Accession No. |
|---|---|---|---|
| Beta-actin | (F): GTAAGGACCTCTATGCCAACACA (R): ATGCCAATCTCATCTCGTTTTCT | 346 | [GenBank:AF508792] |
| S18 | (F): ATGTGGTGTTGAGGAAAGCAG (R): GCTTGTTGTCCAGACCGTTG | 195 | [GenBank:XM_003473925.1] |
| SVCT2 | (F): GTCCATCGGTGACTACTA (R): ATGCCATCAAGAACACAGGA | 114 | [GenBank:AF411585] |
2.6. Protein Extraction and Western Blot
2.7. Statistics
3. Results
3.1. Biochemical Analyses
| Biochemical results | CTRL | REPL | DEF | Effect |
|---|---|---|---|---|
| Plasma VitC (nmol/mL) | 51.8 ± 22.4 a | 57.3 ± 13.6 a | 4.0 ± 3.2 b | *** |
| Plasma DHA% (% of total VitC) | 10.2 ± 4.9 a | 7.7 ± 2.2 a | 19.1 ± 3.8 b | *** |
| Brain VitC (nmol/g tissue) | 1399 ± 143 a | 1498 ± 42 a | 495 ± 252 b | *** |
| Brain DHA% (% of total VitC) | 4.5 ± 2.8 a | 3.1 ± 2.2 a | 9.5 ± 4.6 b | *** |
| Brain MDA (nmol/g tissue) | 313 ± 124 a | 258 ± 58.9 a | 476 ±106 b | *** |
| Brain GSH (nmol/g tissue) | 1348 ± 112 | 1407 ± 44.6 | 1341 ± 101.0 | - |
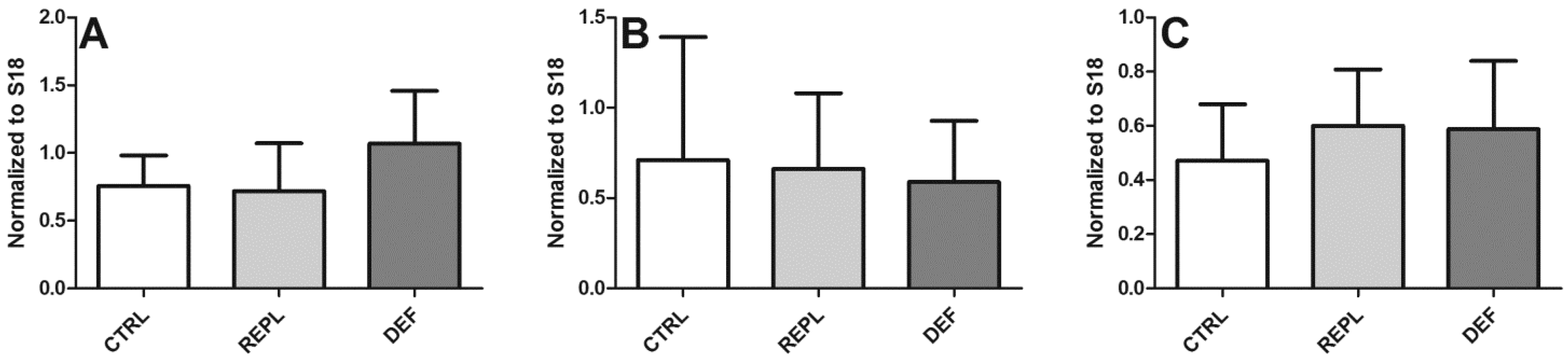
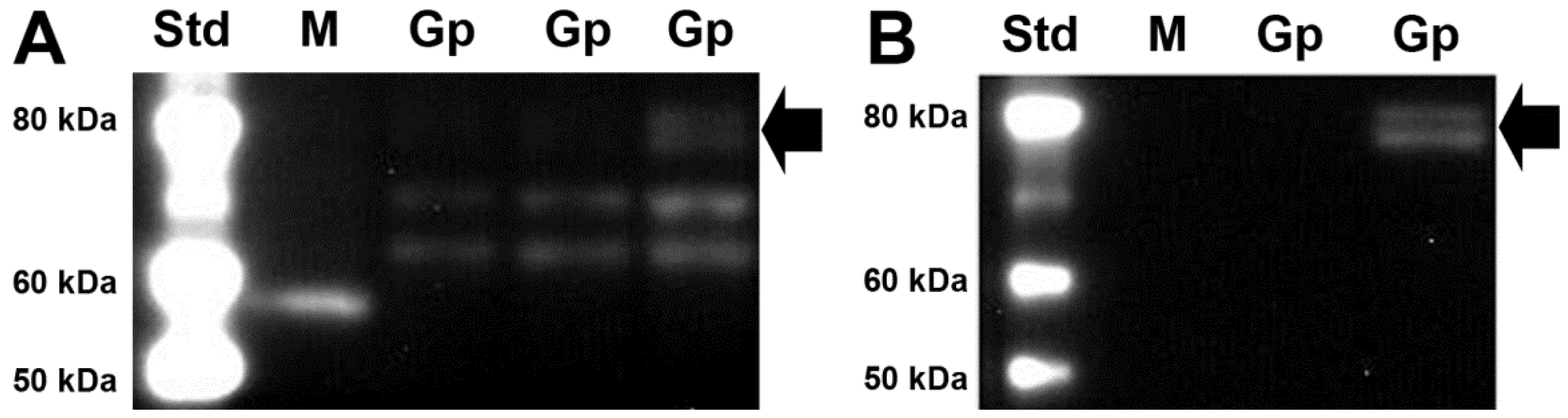
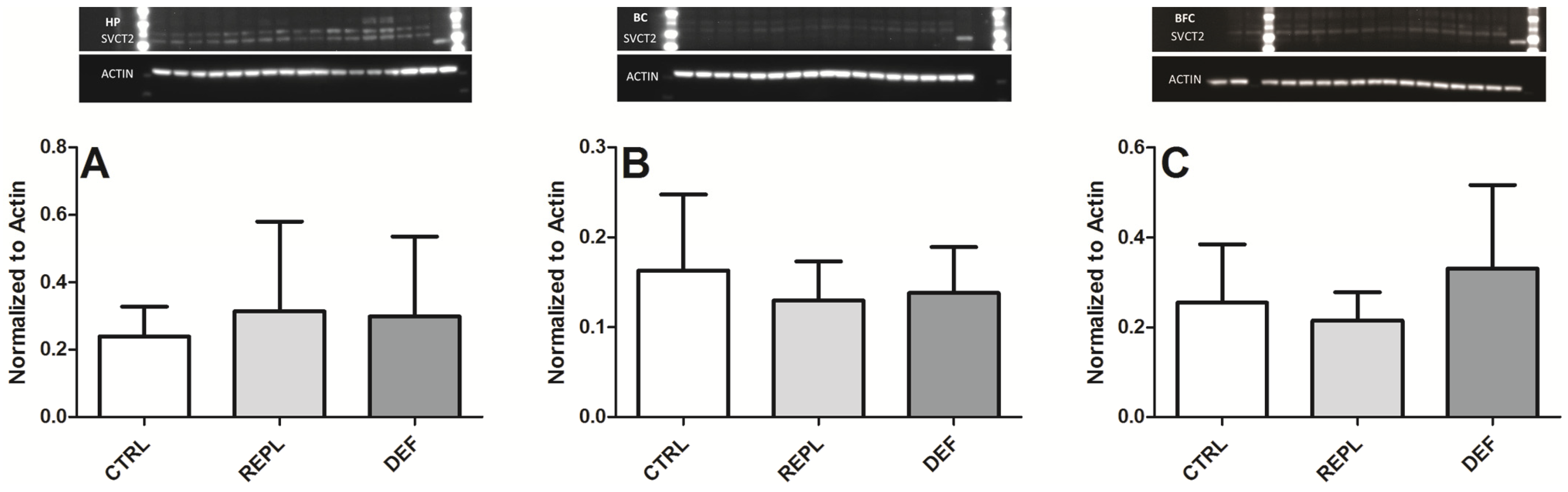
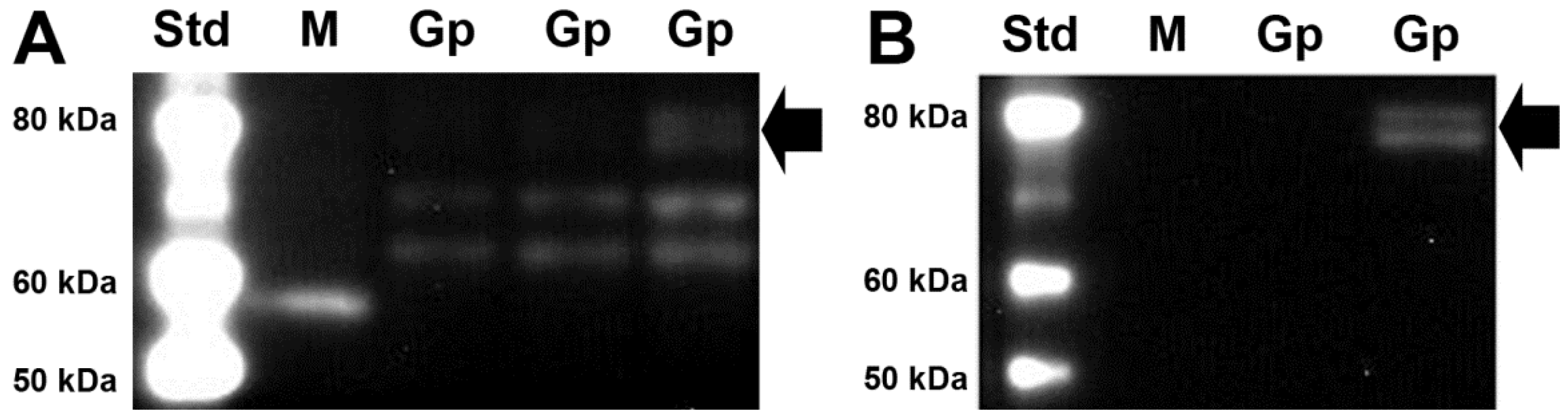
3.2. SVCT2 Expression
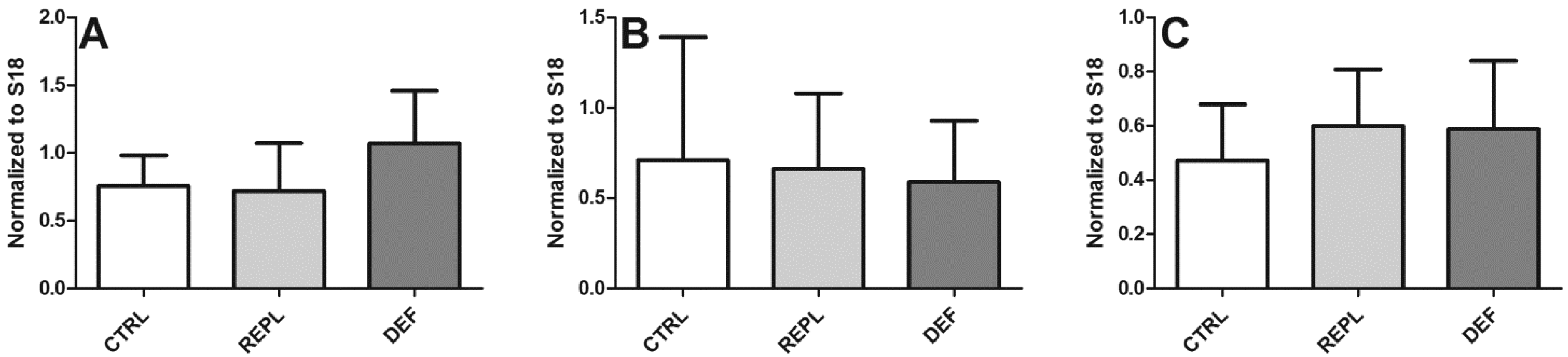
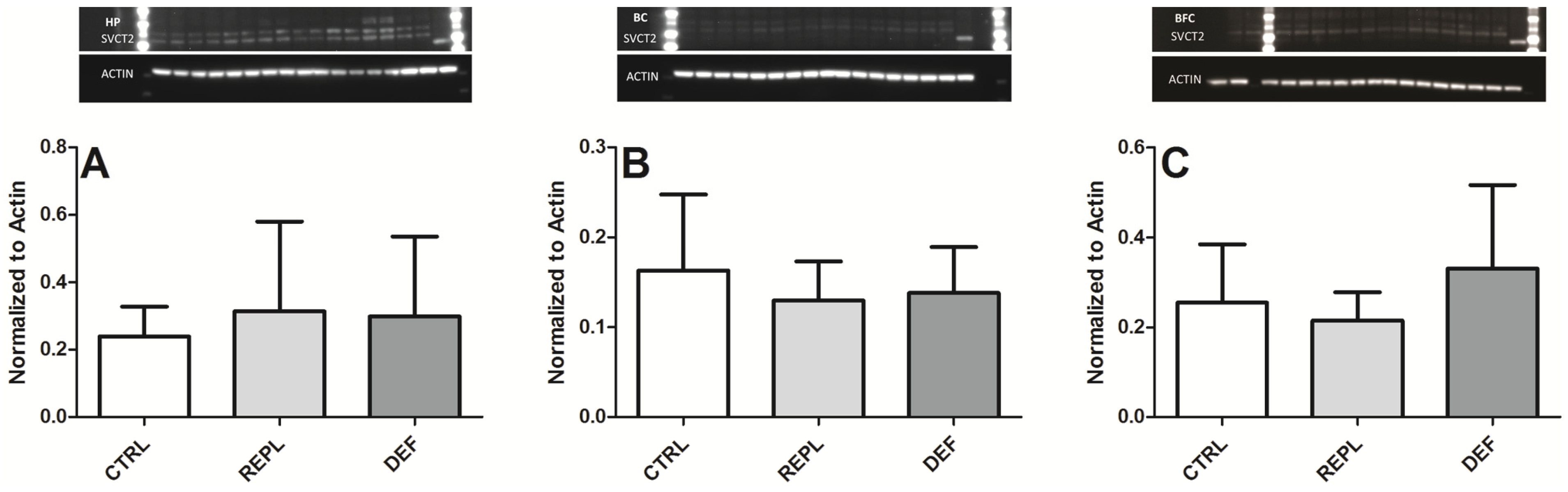
| Densitometry | HP | BC | BFG |
|---|---|---|---|
| CTRL | 0.2393 ± 0.0883 | 0.1632 ± 0.084 | 0.2549 ± 0.1294 |
| REPL | 0.3139 ± 0.2667 0.1299 | 0.1299 ± 0.043 | 0.2146 ± 0.063 |
| DEF | 0.2991 ± 0.2365 | 0.1380 ± 0.051 | 0.3305 ± 0.1858 |
4. Discussion
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bayir, H.; Kagan, V.E.; Tyurina, Y.Y.; Tyurin, V.; Ruppel, R.A.; Adelson, P.D.; Graham, S.H.; Janesko, K.; Clark, R.S.B.; Kochanek, P.M. Assessment of antioxidant reserves and oxidative stress in cerebrospinal fluid after severe traumatic brain injury in infants and children. Pediatr. Res. 2002, 51, 571–578. [Google Scholar] [CrossRef]
- Miura, S.; Ishida-Nakajima, W.; Ishida, A.; Kawamura, M.; Ohmura, A.; Oguma, R.; Sato, Y.; Takahashi, T. Ascorbic acid protects the newborn rat brain from hypoxic-ischemia. Brain Dev. 2009, 31, 307–317. [Google Scholar] [CrossRef]
- Rice, M.E. Ascorbate regulation and its neuroprotective role in the brain. Trends Neurosci. 2000, 23, 209–216. [Google Scholar] [CrossRef]
- Rebec, G.V.; Pierce, R. A vitamin as neuromodulator: Ascorbate release into the extracellular fluid of the brain regulates dopaminergic and glutamatergic transmission. Prog. Neurobiol. 1994, 43, 537–565. [Google Scholar] [CrossRef]
- Hughes, R.; Hurley, R.; Jones, P. The retention of ascorbic acid by guinea-pig tissues. Br. J. Nutr. 1971, 26, 433–438. [Google Scholar] [CrossRef]
- Rice, M.; Russo-Menna, I. Differential compartmentalization of brain ascorbate and glutathione between neurons and glia. Neuroscience 1997, 82, 1213–1223. [Google Scholar] [CrossRef]
- Hilscherova, K.; Blankenship, A.; Nie, M.; Coady, K.; Upham, B.; Trosko, J.; Giesy, J. Oxidative stress in liver and brain of the hatchling chicken (Gallus domesticus) following in ovo injection with TCDD. Comp. Biochem. Physiol. C Pharmacol. Toxicol. Endocrinol. 2003, 136, 29–45. [Google Scholar] [CrossRef]
- Rice, M.; Forman, R.; Chen, B.; Avshalumov, M.; Cragg, S.; Drew, K. Brain antioxidant regulation in mammals and anoxia-tolerant reptiles: Balanced for neuroprotection and neuromodulation. Comp. Biochem. Physiol. C Pharmacol. Toxicol. Endocrinol. 2002, 133, 515–525. [Google Scholar] [CrossRef]
- Sánchez-Moreno, C.; Paniagua, M.; Madrid, A.; Martín, A. Protective effect of vitamin C against the ethanol mediated toxic effects on human brain glial cells. J. Nutr. Biochem. 2003, 14, 606–613. [Google Scholar] [CrossRef]
- Hart, J.-M.; Chang, B.-J.; Li, T.-Z.; Choe, N.-H.; Quan, F.-S.; Jang, B.-J.; Cho, I.-H.; Hong, H.-N.; Lee, J.-H. Protective effects of ascorbic acid against lead-induced apoptotic neurodegeneration in the developing rat hippocampus in vivo. Brain Res. 2007, 1185, 68–74. [Google Scholar]
- Markesbery, W.; Lovell, M. Four-hydroxynonenal, a product of lipid peroxidation, is increased in the brain in Alzheimer’s disease. Neurobiol. Aging 1998, 19, 33–36. [Google Scholar] [CrossRef]
- Butterfield, D.A.; Reed, T.; Perluigi, M.; de Marco, C.; Coccia, R.; Cini, C.; Sultana, R. Elevated protein-bound levels of the lipid peroxidation product, 4-hydroxy-2-nonenal, in brain from persons with mild cognitive impairment. Neurosci. Lett. 2006, 397, 170–173. [Google Scholar] [CrossRef]
- Dhariwal, K.; Hartzell, W.; Levine, M. Ascorbic acid and dehydroascorbic acid measurements in human plasma and serum. Am. J. Clin. Nutr. 1991, 54, 712–716. [Google Scholar]
- Lindblad, M.; Tveden-Nyborg, P.; Lykkesfeldt, J. Regulation of Vitamin C Homeostasis during Deficiency. Nutrients 2013, 5, 2860–2879. [Google Scholar] [CrossRef]
- Harrison, F.; May, J. Vitamin C function in the brain: vital role of the ascorbate transporter SVCT2. Free Radic. Biol. Med. 2009, 46, 719–730. [Google Scholar] [CrossRef]
- Tsukaguchi, H.; Tokui, T.; Mackenzie, B.; Berger, U.V.; Chen, X.Z.; Wang, Y.; Brubaker, R.F.; Hediger, M.A. A family of mammalian Na+-dependent L-ascorbic acid transporters. Nature 1999, 399, 70–75. [Google Scholar] [CrossRef]
- Sotiriou, S.; Gispert, S.; Cheng, J.; Wang, Y.; Chen, A.; Hoogstraten-Miller, S.; Miller, G.; Kwon, O.; Levine, M.; Guttentag, S. Ascorbic-acid transporter Slc23a1 is essential for vitamin C transport into the brain and for perinatal survival. Nat. Med. 2002, 8, 514–517. [Google Scholar] [CrossRef]
- Harrison, F.; Dawes, S.; Meredith, M.; Babaev, V.; Li, L.; May, J. Low vitamin C increased oxidative stress cell death in mice that lack the sodium-dependent vitamin C transporter SVCT2. Free Radic. Biol. Med. 2010, 49, 821–829. [Google Scholar] [CrossRef]
- Tveden-Nyborg, P.; Hasselholt, S.; Miyashita, N.; Moos, T.; Poulsen, H.E.; Lykkesfeldt, J. Chronic Vitamin C Deficiency does not Accelerate Oxidative Stress in Ageing Brains of Guinea Pigs. Basic Clin. Pharmacol. Toxicol. 2012, 110, 524–529. [Google Scholar] [CrossRef]
- Harrison, F.; Green, R.; Dawes, S.; May, J. Vitamin C distribution and retention in the mouse brain. Brain Res. 2010, 1348, 181–186. [Google Scholar]
- Mardones, L.; Zúñiga, F.A.; Villagrán, M.; Sotomayor, K.; Mendoza, P.; Escobar, D.; González, M.; Ormazabal, V.; Maldonado, M.; Oñate, G. Essential role of intracellular glutathione in controlling ascorbic acid transporter expression and function in rat hepatocytes and hepatoma cells. Free Radic. Biol. Med. 2012, 52, 1874–1887. [Google Scholar] [CrossRef]
- Meredith, M.E.; Harrison, F.; May, J. Differential regulation of the ascorbic acid transporter SVCT2 during development and in response to ascorbic acid depletion. Biochem. Biophys. Res. Commun. 2011, 414, 737–742. [Google Scholar] [CrossRef]
- Amano, A.; Aigaki, T.; Maruyama, N.; Ishigami, A. Ascorbic acid depletion enhances expression of the sodium-dependent vitamin C transporters, SVCT1 and SVCT2, and uptake of ascorbic acid in livers of SMP30/GNL knockout mice. Arch. Biochem. Biophys. 2010, 496, 38–44. [Google Scholar] [CrossRef]
- Nishikimi, M.; Kawai, T.; Yagi, K. Guinea pigs possess a highly mutated gene for l-gulono-gamma-lactone oxidase, the key enzyme for l-ascorbic acid biosynthesis missing in this species. J. Biol. Chem. 1992, 267, 21967–21972. [Google Scholar]
- Lykkesfeldt, J.; Perez Trueba, G.; Poulsen, H.; Christen, S. Vitamin C deficiency in weanling guinea pigs: Differential expression of oxidative stress and DNA repair in liver and brain. Br. J. Nutr. 2007, 98, 1116–1119. [Google Scholar]
- Tveden-Nyborg, P.; Johansen, L.K.; Raida, Z.; Villumsen, C.K.; Larsen, J.O.; Lykkesfeldt, J. Vitamin C deficiency in early postnatal life impairs spatial memory and reduces the number of hippocampal neurons in guinea pigs. Am. J. Clin. Nutr. 2009, 90, 540–546. [Google Scholar] [CrossRef]
- Tveden-Nyborg, P.; Vogt, L.; Schjoldager, J.G.; Jeannet, N.; Hasselholt, S.; Paidi, M.D.; Christen, S.; Lykkesfeldt, J. Maternal Vitamin C Deficiency during Pregnancy Persistently Impairs Hippocampal Neurogenesis in Offspring of Guinea Pigs. PLoS One 2012, 7, e48488. [Google Scholar] [CrossRef] [Green Version]
- Lykkesfeldt, J. Determination of Ascorbic Acid and Dehydroascorbic Acid in Biological Samples by High-Performance Liquid Chromatography Using Subtraction Methods: Reliable Reduction with Tris[2-carboxyethyl] phosphine Hydrochloride. Anals Biochem. 2000, 282, 89–93. [Google Scholar] [CrossRef]
- Lykkesfeldt, J. Determination of malondialdehyde as dithiobarbituric acid adduct in biological samples by HPLC with fluorescence detection: Comparison with ultraviolet-visible spectrophotometry. Clin. Chem. 2001, 47, 1725–1727. [Google Scholar]
- Hissin, P.J.; Hilf, R. A fluorometric method for determination of oxidized and reduced glutathione in tissues. An. Biochem. 1976, 74, 214–226. [Google Scholar] [CrossRef]
- Clark, A.G.; Rohrbaugh, A.L.; Otterness, I.; Kraus, V.B. The effects of ascorbic acid on cartilage metabolism in guinea pig articular cartilage explants. Matrix Biol. 2002, 21, 175–184. [Google Scholar] [CrossRef]
- May, J.M.; Qu, Z.; Qiao, H.; Koury, M.J. Maturational loss of the vitamin C transporter in erythrocytes. Biochem. Biophys. Res. Commun. 2007, 360, 295–298. [Google Scholar] [CrossRef]
- Savini, I.; Rossi, A.; Catani, M.V.; Ceci, R.; Avigliano, L. Redox regulation of vitamin C transporter SVCT2 in C2C12 myotubes. Biochem. Biophys. Res. Commun. 2007, 361, 385–390. [Google Scholar] [CrossRef]
- Schjoldager, J.G.; Tveden-Nyborg, P.; Lykkesfeldt, J. Prolonged maternal vitamin C deficiency overrides preferential fetal ascorbate transport but does not influence perinatal survival in guinea pigs. Br. J. Nutr. 2013, 110, 1573–1579. [Google Scholar] [CrossRef]
- Harrison, F.; Meredith, M.; Dawes, S.; Saskowski, J.; May, J. Low ascorbic acid and increased oxidative stress in Gulo−/− mice during development. Brain Res. 2010, 1349, 143–152. [Google Scholar]
- Johnston, C.S.; Meyer, C.; Srilakshmi, J. Vitamin C elevates red blood cell glutathione in healthy adults. Am. J. Clin. Nutr. 1993, 58, 103–105. [Google Scholar]
- Lykkesfeldt, J. Increased oxidative damage in vitamin C deficiency is accompanied by induction of ascorbic acid recycling capacity in young but not mature guinea pigs. Free Radic. Res. 2002, 36, 567–574. [Google Scholar] [CrossRef]
- Castro, M.; Caprile, T.; Astuya, A.; Millán, C.; Reinicke, K.; Vera, J.C.; Vásquez, O.; Aguayo, L.G.; Nualart, F. High-affinity sodium–vitamin C co-transporters (SVCT) expression in embryonic mouse neurons. J. Neurochem. 2001, 78, 815–823. [Google Scholar] [CrossRef]
- García, M.D.L.A.; Salazar, K.; Millán, C.; Rodríguez, F.; Montecinos, H.; Caprile, T.; Silva, C.; Cortes, C.; Reinicke, K.; Vera, J.C. Sodium vitamin C cotransporter SVCT2 is expressed in hypothalamic glial cells. Glia 2005, 50, 32–47. [Google Scholar] [CrossRef]
- Mun, G.; Kim, M.; Lee, J.; Kim, H.; Chung, Y.; Chung, Y.; Kang, J.; Hwang, Y.; Oh, S.; Kim, J. Immunohistochemical study of the distribution of sodium-dependent vitamin C transporters in adult rat brain. J. Neurosci. Res. 2006, 83, 919–928. [Google Scholar]
- May, J.M. Vitamin C Transport and Its Role in the Central Nervous System. In Water Soluble Vitamins; Olaf, S., Ed.; Springer: Dordrech, The Netherlands, 2012; Volume 56, pp. 85–103. [Google Scholar]
- Savini, I.; Catani, M.V.; Arnone, R.; Rossi, A.; Frega, G.; Del Principe, D.; Avigliano, L. Translational control of the ascorbic acid transporter SVCT2 in human platelets. Free Radic. Biol. Med. 2007, 42, 608–616. [Google Scholar] [CrossRef]
- Caprile, T.; Salazar, K.; Astuya, A.; Cisternas, P.; Silva‐Alvarez, C.; Montecinos, H.; Millán, C.; García, M.A.; Nualart, F. The Na+-dependent l-ascorbic acid transporter SVCT2 expressed in brainstem cells, neurons, and neuroblastoma cells is inhibited by flavonoids. J. Neurochem. 2009, 108, 563–577. [Google Scholar] [CrossRef]
- Nualart, F.; Castro, T.; Low, M.; Henríquez, J.P.; Oyarce, K.; Cisternas, P.; García, A.; Yáñez, A.J.; Bertinat, R.; Montecinos, V.P. Dynamic expression of the sodium-vitamin C co-transporters, SVCT1 and SVCT2, during perinatal kidney development. Histochem. Cell Biol. 2013, 139, 233–247. [Google Scholar] [CrossRef]
- Harrison, F.; Yu, S.; van den Bossche, K.; Li, L.; May, J.; McDonald, M. Elevated oxidative stress and sensorimotor deficits but normal cognition in mice that cannot synthesize ascorbic acid. J. Neurochem. 2008, 106, 1198–1208. [Google Scholar] [CrossRef]
- Paidi, M.D.; Schjoldager, J.G.; Lykkesfeldt, J.; Tveden-Nyborg, P. Prenatal vitamin C deficiency results in differential levels of oxidative stress during late gestation in foetal guinea pig brains. Redox Biol. 2014, 2, 361–367. [Google Scholar] [CrossRef]
- Berger, U.V.; Lu, X.C.M.; Liu, W.; Tang, Z.; Slusher, B.S.; Hediger, M.A. Effect of middle cerebral artery occlusion on mRNA expression for the sodium-coupled vitamin C transporter SVCT2 in rat brain. J. Neurochem. 2003, 86, 896–906. [Google Scholar] [CrossRef]
- Gess, B.; Sevimli, S.; Strecker, J.K.; Young, P.; Schäbitz, W.R. Sodium-dependent vitamin C transporter 2 (SVCT2) expression and activity in brain capillary endothelial cells after transient ischemia in mice. PLoS One 2011, 6, e17139. [Google Scholar]
- Subramanian, V.S.; Marchant, J.S.; Reidling, J.C.; Said, H.M. N-Glycosylation is required for Na+-dependent vitamin C transporter functionality. Biochem. Biophys. Res. Commun. 2008, 374, 123–127. [Google Scholar] [CrossRef]
- Chi, X.; May, J.M. Oxidized lipoprotein induces the macrophage ascorbate transporter (SVCT2): Protection by intracellular ascorbate against oxidant stress and apoptosis. Arch. Biochem. Biophys. 2009, 485, 174–182. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Paidi, M.D.; Schjoldager, J.G.; Lykkesfeldt, J.; Tveden-Nyborg, P. Chronic Vitamin C Deficiency Promotes Redox Imbalance in the Brain but Does Not Alter Sodium-Dependent Vitamin C Transporter 2 Expression. Nutrients 2014, 6, 1809-1822. https://doi.org/10.3390/nu6051809
Paidi MD, Schjoldager JG, Lykkesfeldt J, Tveden-Nyborg P. Chronic Vitamin C Deficiency Promotes Redox Imbalance in the Brain but Does Not Alter Sodium-Dependent Vitamin C Transporter 2 Expression. Nutrients. 2014; 6(5):1809-1822. https://doi.org/10.3390/nu6051809
Chicago/Turabian StylePaidi, Maya D., Janne G. Schjoldager, Jens Lykkesfeldt, and Pernille Tveden-Nyborg. 2014. "Chronic Vitamin C Deficiency Promotes Redox Imbalance in the Brain but Does Not Alter Sodium-Dependent Vitamin C Transporter 2 Expression" Nutrients 6, no. 5: 1809-1822. https://doi.org/10.3390/nu6051809