Vitamin A Deficiency and Alterations in the Extracellular Matrix

Abstract
:1. Introduction
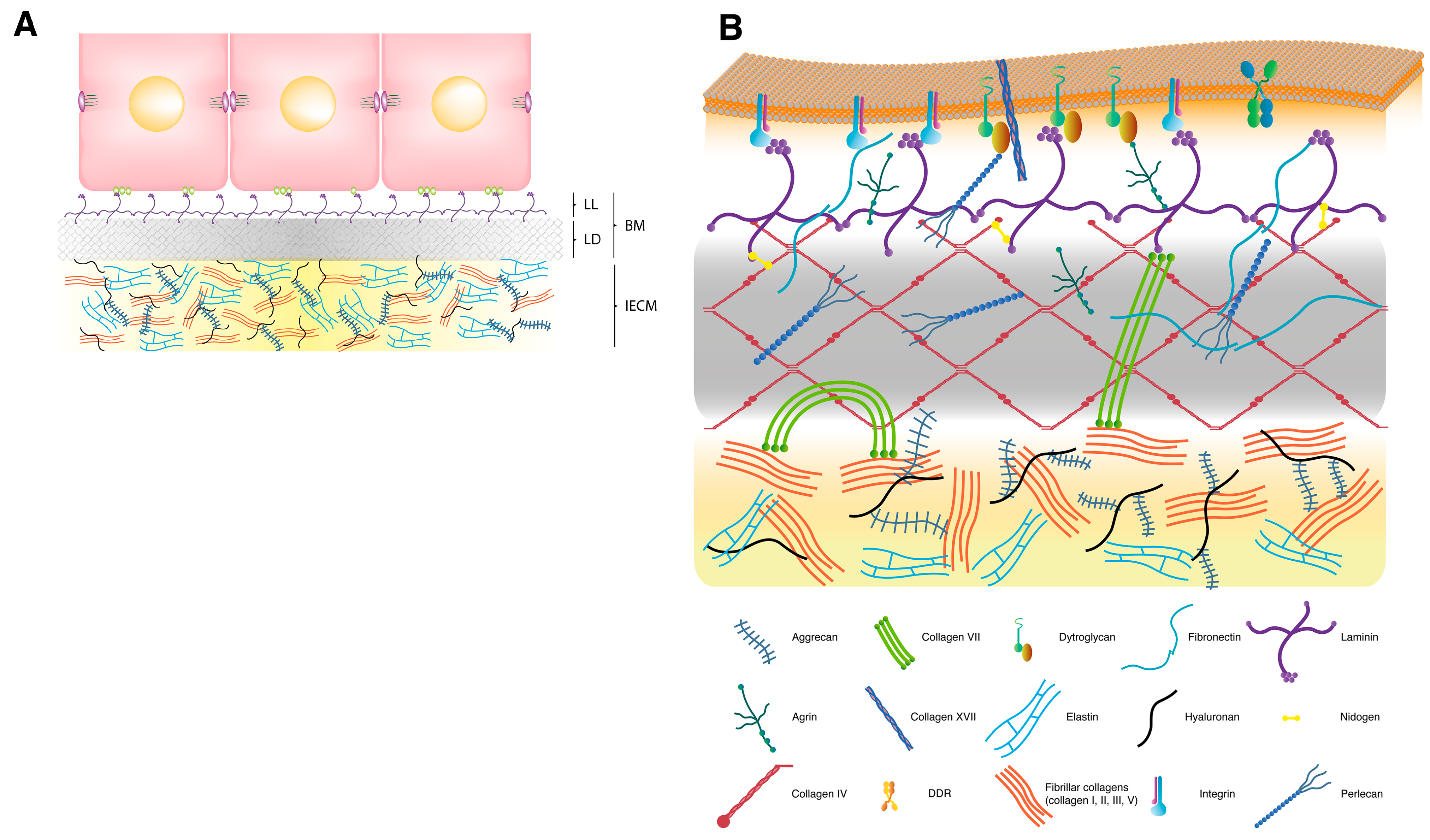
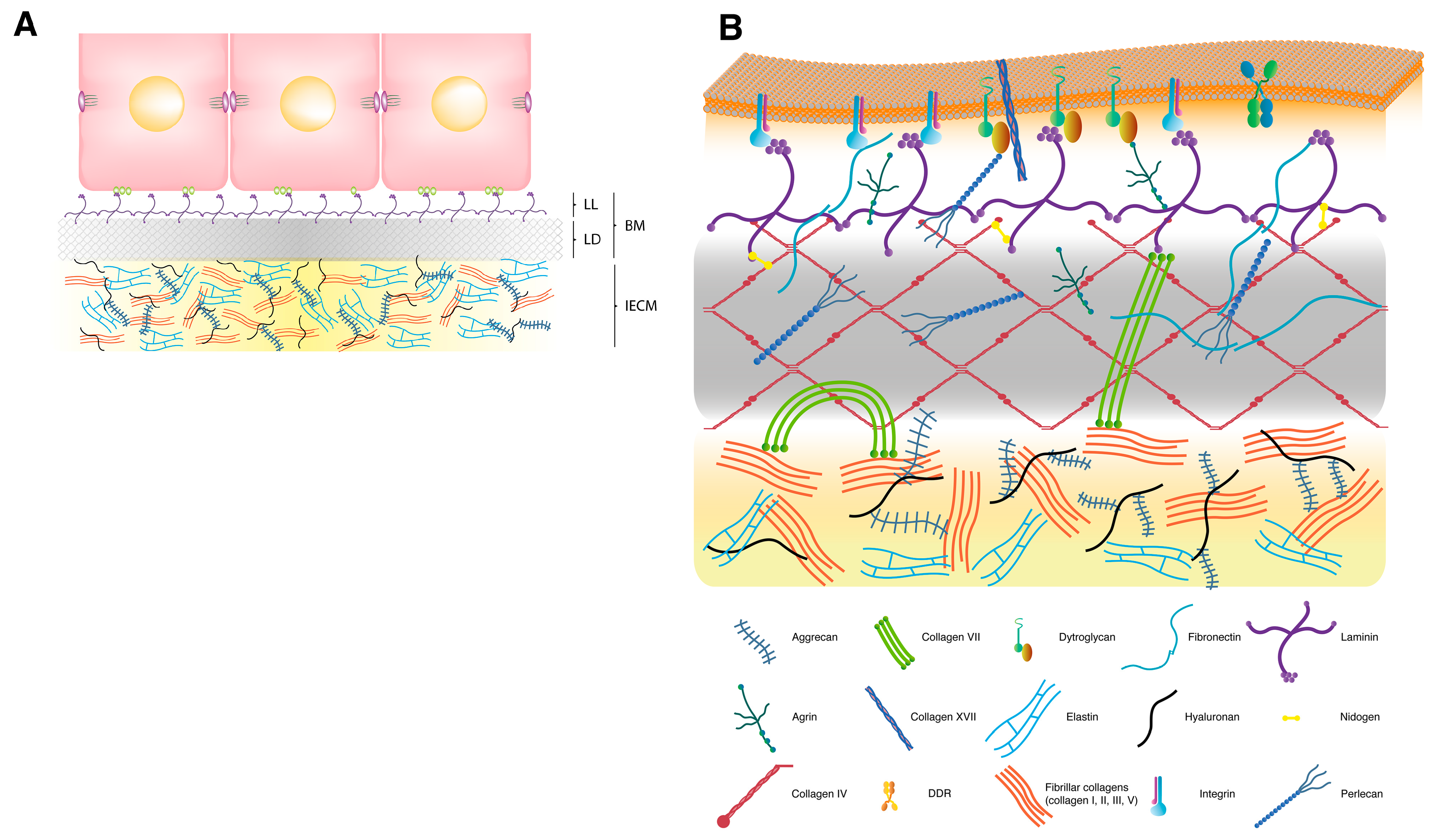
2. Extracellular Matrix
2.1. Interstitial Extracellular Matrix
2.2. Basement Membrane
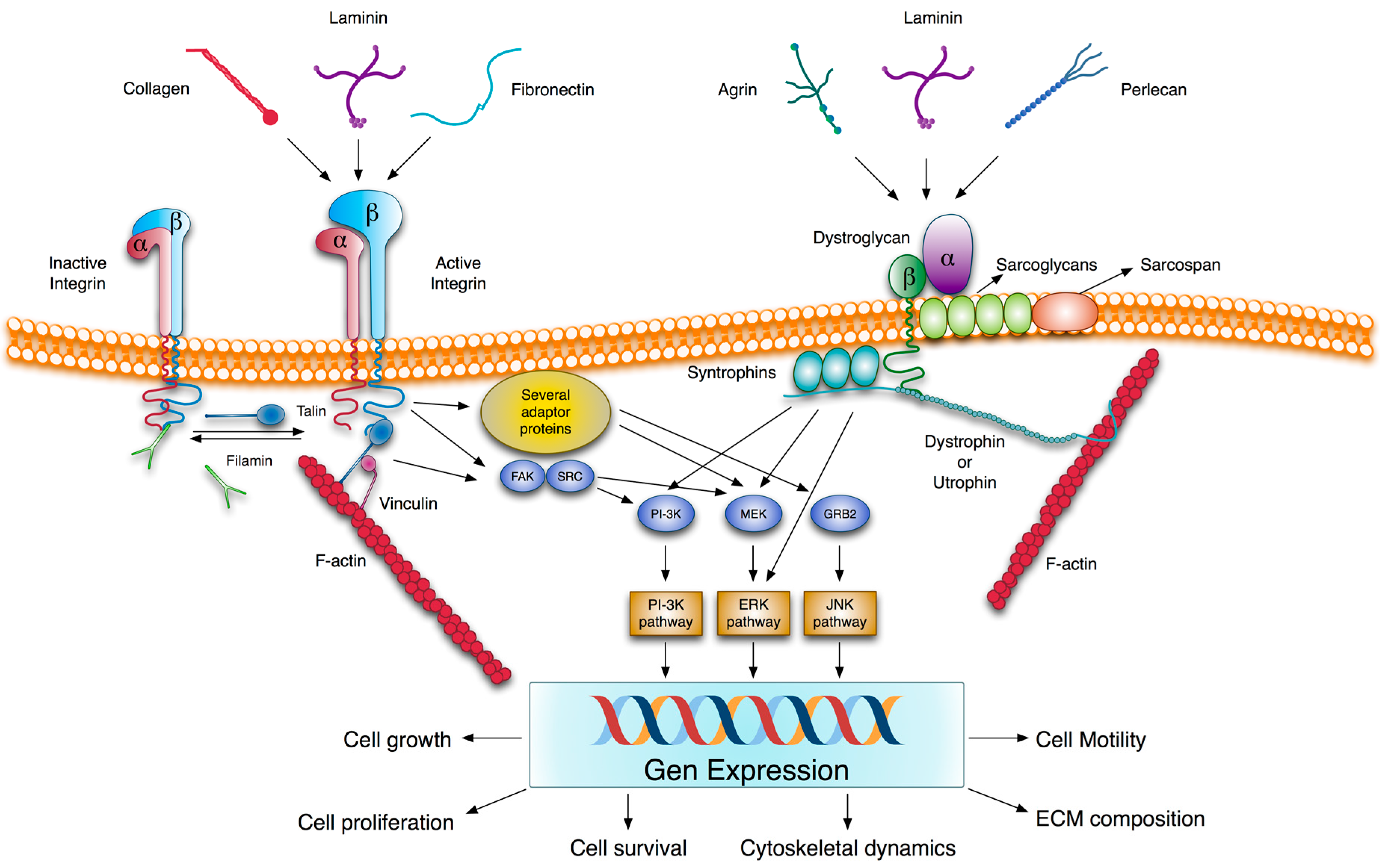
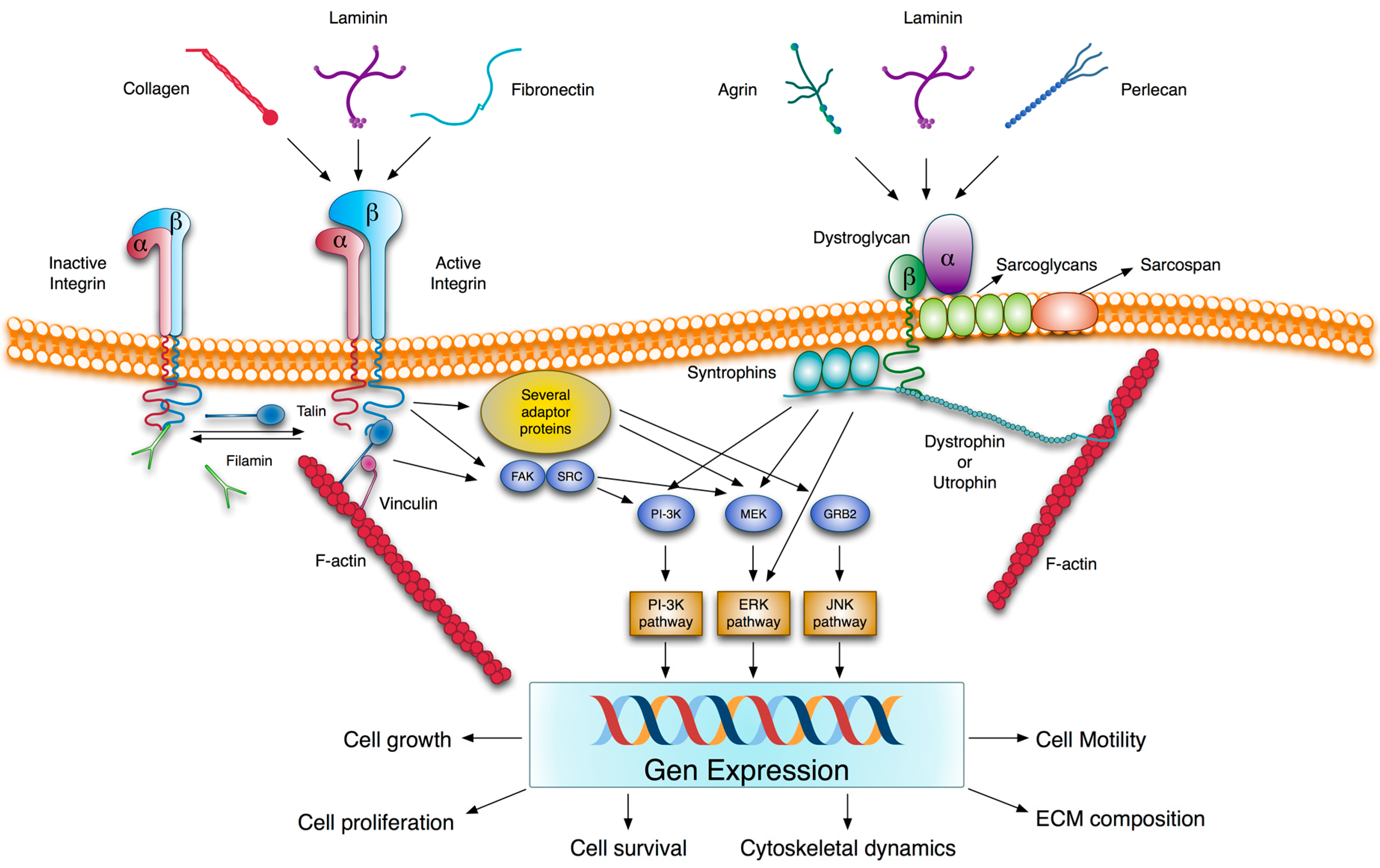
2.3. Transmembrane Extracellular Matrix Receptors
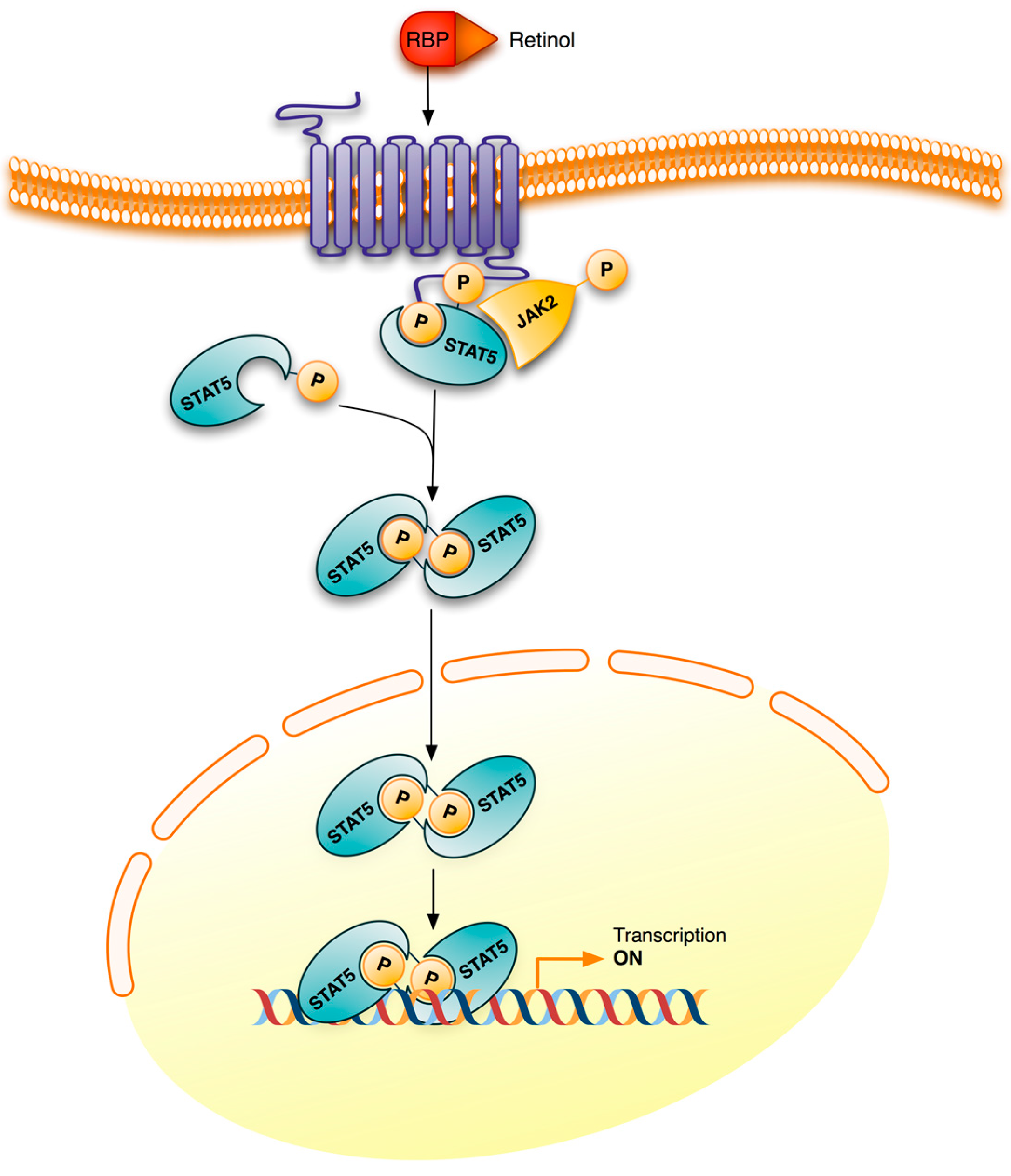
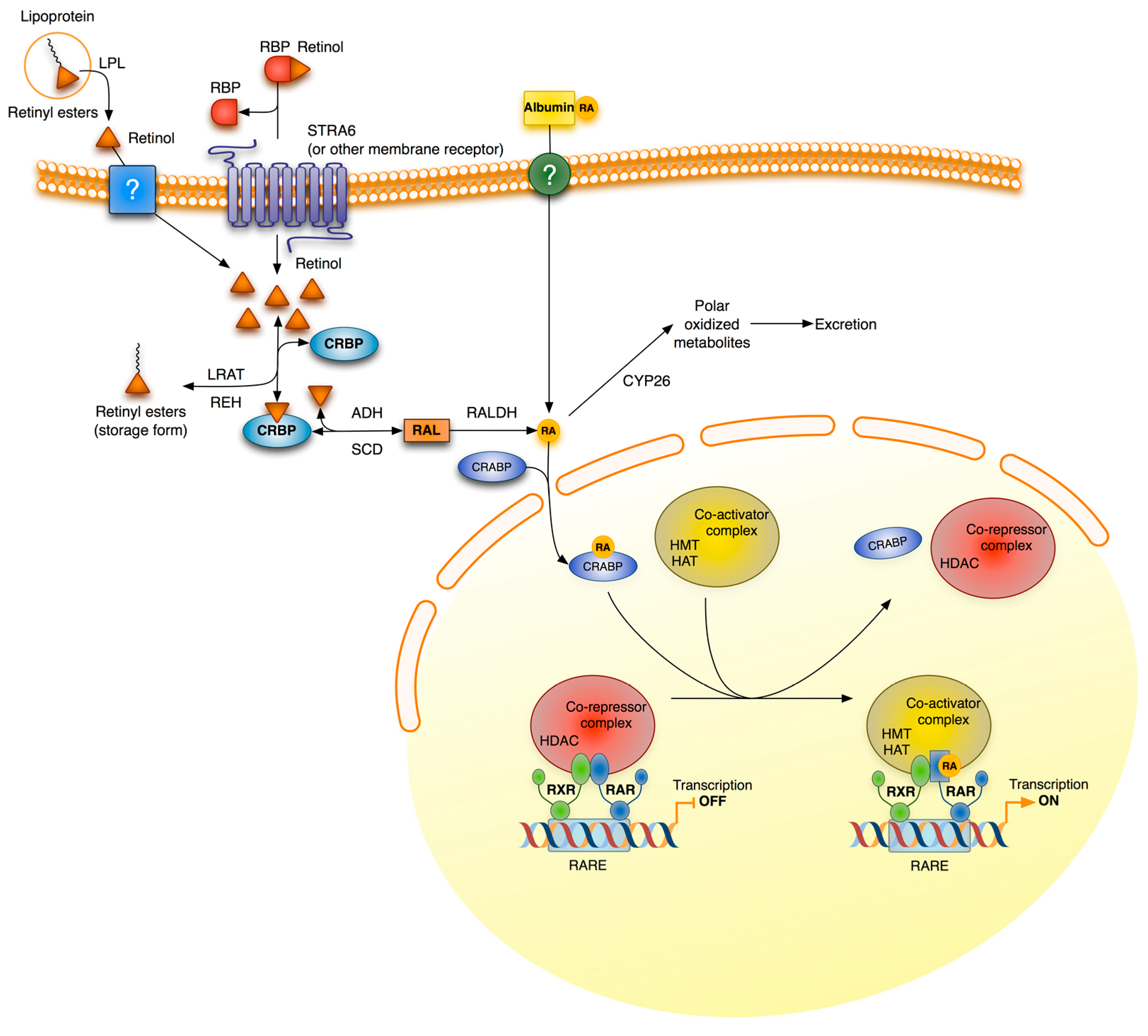
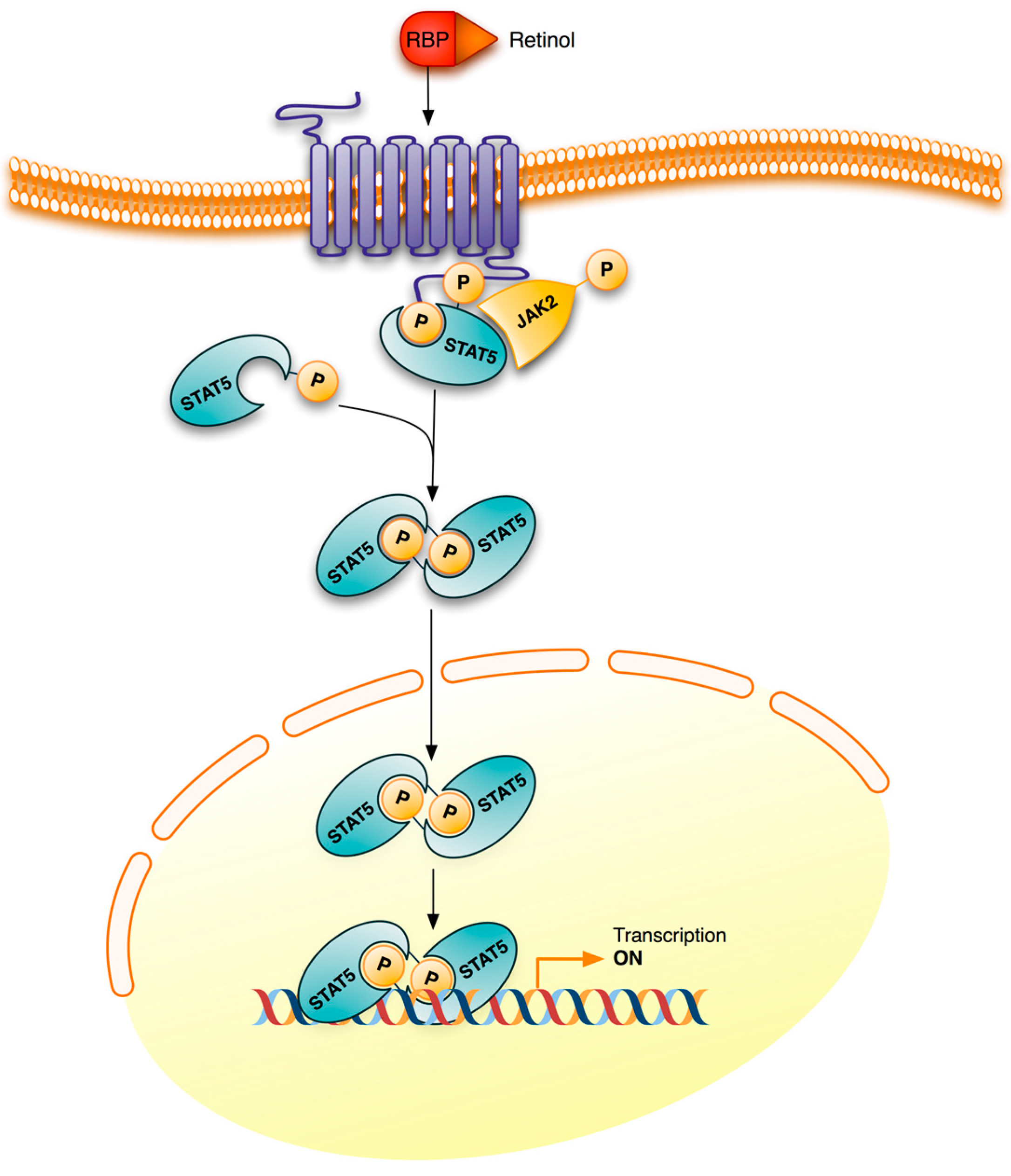
3. Retinoid Signaling

4. Vitamin A Deficiency
4.1. Overview and Clinical Manifestations
4.2. Epidemiology and Incidence
5. Vitamin A Deficiency and Alterations in Extracellular Matrix
5.1. Kidney

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ECM Component | Modification |
|---|---|
| Collagen IV (protein) | Increased |
| α1(IV) chain (protein and mRNA) | Increased |
| α2(IV) chain (protein and mRNA) | Lowered |
| α3(IV) chain (protein and mRNA) | Lowered |
| α4(IV) chain (protein and mRNA) | Increased |
| α5(IV) chain (protein and mRNA) | Increased |
| α6(IV) chain (protein and mRNA) | Increased |
| Matrix Metalloproteinases and Inhibitors | |
| MMP 2 (protein) | Lowered |
| MMP 9 (protein) | Lowered |
| TIMP 1 (protein) | Unaffected |
| TIMP 2 (protein) | Unaffected |
| Glomerular BM | Enlarged |
| Tubular BM | Enlarged and presence of collagen I fibrils |
5.2. Lung
| ECM Component | Modification | Refs. |
|---|---|---|
| Collagen (protein) | Increased | [135] |
| Lowered | [140] | |
| Collagen I (protein) | Increased | [138] |
| α1(I) chain (mRNA) | Increased | [138] |
| α2(I) chain (mRNA) | Increased | [138] |
| Elastin (protein) | Lowered | [135,139] |
| Collagen IV (protein) | Increased | [138] |
| α1(IV) chain (protein and mRNA) | Increased | [141] |
| α2(IV) chain (protein and mRNA) | Unaffected | [141] |
| α3(IV) chain (protein and mRNA) | Increased | [141] |
| α4(IV) chain (protein and mRNA) | Increased | [141] |
| α5(IV) chain (protein and mRNA) | Unaffected | [141] |
| Laminin | ||
| Laminin α2 chain (mRNA) | Lowered | [141] |
| Laminin α4 chain (mRNA) | Lowered | [141] |
| Laminin α5 chain (protein and mRNA) | Lowered | [141] |
| Laminin β1 chain (protein and mRNA) | Lowered | [141] |
| Laminin γ1 chain (protein and mRNA) | Lowered | [141] |
| Matrix metalloproteinases and inhibitors | ||
| MMP 2 (activity) | Lowered | [141] |
| MMP 9 (activity) | Lowered | [141] |
| TIMP 1 (protein) | Unaffected | [141] |
| TIMP 2 (protein) | Unaffected | [141] |
| Alveolar BM | Enlarged and presence of collagen I fibrils | [138] |
5.3. Liver
| ECM Component | Modification |
|---|---|
| Collagen (protein) | Increased |
| Collagen IV (protein) | Increased |
| Laminin 1 (protein) | Increased |
| Fibronectin (protein) | Increased |
| Perisinusoidal space | Enhanced extracellular material |
6. Mechanism of Extracellular Matrix Modifications in Vitamin A Deficiency
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- McLaren, D.S.; Kraemer, K. Vitamin A in nature. World Rev. Nutr. Diet 2012, 103, 7–17. [Google Scholar] [PubMed]
- De Luca, L.M. Retinoids and their receptors in differentiation, embryogenesis and neoplasia. FASEB J. 1991, 5, 2924–2933. [Google Scholar]
- Duester, G. Retinoic acid synthesis and signaling during early organogenesis. Cell 2008, 134, 921–931. [Google Scholar] [CrossRef] [PubMed]
- Mark, M.; Ghyselinck, N.B.; Chambon, P. Function of retinoic acid receptors during embryonic development. Nucl. Recept. Signal. 2009, 7, e002. [Google Scholar] [PubMed]
- Ross, A.C. Vitamin A and retinoic acid in T cell-related immunity. Am. J. Clin. Nutr. 2012, 96, 1166S–1172S. [Google Scholar] [CrossRef] [PubMed]
- Sommer, A.; Vyas, K.S. A global clinical view on vitamin A and carotenoids. Am. J. Clin. Nutr. 2012, 96, 1204S–1206S. [Google Scholar] [CrossRef] [PubMed]
- Rhinn, M.; Dolle, P. Retinoic acid signaling during development. Development 2012, 139, 843–858. [Google Scholar] [CrossRef] [PubMed]
- Gudas, L.J. Retinoids induce stem cell differentiation via epigenetic changes. Semin. Cell Dev. Biol. 2013, 24, 701–705. [Google Scholar] [CrossRef] [PubMed]
- Chambon, P. A decade of molecular biology of retinoic acid receptors. FASEB J. 1996, 10, 940–954. [Google Scholar] [PubMed]
- Perissi, V.; Jepsen, K.; Glass, C.K.; Rosenfeld, M.G. Deconstructing repression: Evolving models of corepressor action. Nat. Rev. Genet. 2010, 11, 109–123. [Google Scholar] [CrossRef] [PubMed]
- Ziouzenkova, O.; Orasanu, G.; Sharlach, M.; Akiyama, T.E.; Berger, J.P.; Viereck, J.; Hamilton, J.A.; Tang, G.; Dolnokowski, G.G.; Vogel, S.; et al. Retnaldehyde represses adipogenesis and diet-induced obesity. Nat. Med. 2007, 13, 695–702. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.C.; Noy, N. Signaling by vitamin A and retinol-binding protein in regulation of insulin responses and lipid homeostasis. Biochim. Biophys. Acta 2012, 1821, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Wilson, J.G.; Roth, C.B.; Warkany, J. An analysis of the syndrome of malformations induced by maternal vitamin A deficiency. Effects of restoration of vitamin A at various times during gestation. Am. J. Anat. 1953, 92, 189–217. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, Y.I.; Smith, J.E.; Winick, M.; Goodman, D.S. Vitamin A deficiency and fetal growth and development in the rat. J. Nutr. 1975, 105, 1299–1305. [Google Scholar] [PubMed]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O.; Naba, A. Overview of the matrisome-an inventory of extracellular matrix constituents and functions. Cold Spring Harb. Perspect. Biol. 2012, 4, a004903. [Google Scholar] [CrossRef] [PubMed]
- Alam, N.; Goel, H.L.; Zarif, M.J.; Butterfield, J.E.; Perkins, H.M.; Sansoucy, B.G.; Sawyer, T.K.; Languino, L.R. The integrin-growth factor receptor duet. J. Cell. Physiol. 2007, 213, 649–653. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Turnbull, J.; Guimond, S. Extracellular matrix and cell signaling: The dynamic cooperation of integrin, proteoglycan and growth factor receptor. J. Endocrinol. 2011, 209, 139–151. [Google Scholar] [CrossRef] [PubMed]
- Van Dijk, M.; Göransson, S.A.; Strömblad, S. Cell to extracellular matrix interactions and their reciprocal nature in cancer. Exp. Cell Res. 2013, 319, 1663–1670. [Google Scholar] [CrossRef] [PubMed]
- Järveläinen, H.; Sainio, A.; Koulu, M.; Wight, T.N.; Penttinen, R. Extracellular matrix molecules: Potential targets in pharmacotherapy. Pharmacol. Rev. 2009, 61, 198–223. [Google Scholar] [CrossRef] [PubMed]
- Timpl, R. Structure and biological activity of basement membrane proteins. Eur. J. Biochem. 1989, 180, 487–502. [Google Scholar] [CrossRef] [PubMed]
- Ricard-Blum, S. The collagen family. Cold Spring Harb. Perspect. Biol. 2011, 3, a004978. [Google Scholar] [CrossRef] [PubMed]
- Baldwin, A.K.; Simpson, A.; Steer, R.; Cain, S.A.; Kielty, C.M. Elastic fibers in health and disease. Exp. Rev. Mol. Med. 2013, 15, e8. [Google Scholar] [CrossRef]
- Tsang, K.Y.; Cheung, M.C.H.; Chan, D.; Cheah, K.S.E. The developmental roles of the extracellular matrix: Beyond structure to regulation. Cell Tissue Res. 2010, 339, 93–110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pankov, R.; Yamada, K.M. Fibronectin at a glance. J. Cell Sci. 2002, 115, 3861–3863. [Google Scholar] [CrossRef] [PubMed]
- Ffrench-Constant, C. Alternative splicing of fibronectin-many different proteins but few different functions. Exp. Cell Res. 1995, 221, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Schwarzbauer, J.E. Fibronectin fibrillogenesis, a cell-mediated matrix assembly process. Matrix Biol. 2005, 24, 389–399. [Google Scholar] [CrossRef] [PubMed]
- Singh, P.; Carraher, C.; Schwarzbauer, J.E. Assembly of fibronectin extracellular matrix. Annu. Rev. Cell Dev. Biol. 2010, 26, 397–419. [Google Scholar] [CrossRef] [PubMed]
- LeBleu, V.S.; MacDonald, B.; Kalluri, R. Structure and function of basement membranes. Exp. Biol. Med. 2007, 232, 1121–1129. [Google Scholar] [CrossRef]
- Hudson, B.G.; Tryggvason, K.; Sundaramoorthy, M.; Neilson, E.G. Alport’s syndrome, Goodpasture’s syndrome, and type IV collagen. N. Engl. J. Med. 2003, 348, 2543–2556. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Ding, M.; Zhao, Z.; Reeders, S.T. Complete primary structure of the sixth chain of human basement membrane collagen, α6(IV). Isolation of the cDNAs for α6(IV) and comparison with five other type IV collagen chains. J. Biol. Chem. 1994, 269, 13193–13199. [Google Scholar] [PubMed]
- Khoshnoodi, J.; Pedchenko, V.; Hudson, B.G. Mammalian collagen IV. Microsc. Res. Tech. 2008, 71, 357–370. [Google Scholar] [CrossRef] [PubMed]
- Timoneda, J.; Gunwar, S.; Monfort, G.; Saus, J.; Noelken, M.; Hudson, B.G. Unusual dissociative behaviour of the noncollagenous domain (hexamer) of basement membrane collagen during electrophoresis and chromatofocusing. Connect. Tissue Res. 1990, 24, 169–186. [Google Scholar] [CrossRef] [PubMed]
- Sundaramoorthy, M.; Meiyappan, M.; Todd, P.; Hudson, B.G. Crystal structure of NC1 domains: Structural basis for type IV collagen assembly in basement membranes. J. Biol. Chem. 2002, 277, 31142–31153. [Google Scholar] [CrossRef] [PubMed]
- Khoshnoodi, J.; Cartailler, J.P.; Alvares, K.; Veis, A.; Hudson, B.G. Molecular recognition in the assembly of collagens: Terminal non-collagenous domains are key recognition modules in the formation of triple-helical protomers. J. Biol. Chem. 2006, 281, 38117–38121. [Google Scholar] [CrossRef] [PubMed]
- Khoshnoodi, J.; Sigmundsson, K.; Cartailler, J.P.; Bondar, O.; Sundaramoorthy, M.; Hudson, B.G. Mechanism of chain selection in the assembly of collagen IV: A prominent role for the α2 chain. J. Biol. Chem. 2006, 281, 6058–6069. [Google Scholar] [CrossRef] [PubMed]
- Yurchenco, P.D. Basement membranes: Cell scaffoldings and signaling platforms. Cold Spring Harb. Perspect. Biol. 2011, 3, a004911. [Google Scholar] [CrossRef] [PubMed]
- Hohenester, E.; Yurchenco, P.D. Laminins in basement membrane assembly. Cell Adh. Migr. 2013, 7, 56–63. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.S.; Champliaud, M.F.; Burgeson, R.E.; Marinkovich, M.P.; Yurchenco, P.D. Self-assembly of laminin isoforms. J. Biol. Chem. 1997, 272, 31525–31532. [Google Scholar] [CrossRef] [PubMed]
- Iozzo, R.V.; Zoeller, J.J.; Nyström, A. Basement membrane proteoglycans: Modulators par excellence of cancer growth and angiogenesis. Mol. Cells 2009, 27, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Whitelock, J.M.; Melrose, J.; Iozzo, R.V. Diverse cell signaling events modulated by perlecan. Biochemistry 2008, 47, 11174–11183. [Google Scholar] [CrossRef] [PubMed]
- Gill, S.; Wight, T.N.; Frevert, C.W. Proteoglycans: Key regulators of pulmonary inflammation and the innate immune response to lung infection. Anat. Rec. 2010, 293, 968–981. [Google Scholar] [CrossRef]
- Ho, M.S.P.; Böse, K.; Mokkapati, S.; Nischt, R.; Smyth, N. Nidogens-Extracellular matrix linker molecules. Microsc. Res. Tech. 2008, 71, 387–395. [Google Scholar]
- McKee, K.K.; Harrison, D.; Capizzi, S.; Yurchenco, P.D. Role of laminin terminal globular domains in basement assembly. J. Biol. Chem. 2007, 282, 21437–21447. [Google Scholar] [CrossRef] [PubMed]
- Behrens, D.T.; Villone, D.; Koch, M.; Brunner, G.; Sorokin, L.; Robenek, H.; Bruckner-Tuderman, L.; Bruckner, P.; Hansen, U. The epidermal basement membrane is a composite of separate laminin- or collagen IV-containing networks connected by aggregated perlecan, but not by nidogens. J. Biol. Chem. 2012, 287, 18700–18709. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.D.; Humphries, M.J. Integrin structure, activation, and interactions. Cold Spring Harb. Perspect. Biol. 2011, 3, a004994. [Google Scholar] [CrossRef] [PubMed]
- Humpries, J.D.; Byron, A.; Humphries, J. Integrin ligands. J. Cell Sci. 2006, 119, 3901–3903. [Google Scholar] [CrossRef] [PubMed]
- Giancotti, F.G.; Ruoslahti, E. Integrin signaling. Science 1999, 285, 1028–1032. [Google Scholar] [CrossRef] [PubMed]
- Lock, J.G.; Wehrle-Haller, B.; Strömblad, S. Cell-matrix adhesion complexes: Master control machinery of cell migration. Semin. Cancer Biol. 2008, 18, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Legate, K.R.; Wickström, S.A.; Fässler, R. Genetic and cell biological analysis of integrin outside-in signaling. Genes Dev. 2009, 23, 397–418. [Google Scholar] [CrossRef] [PubMed]
- Spence, H.J.; Dhillon, A.S.; James, M.; Winder, S.J. Dystroglycan, a scaffold for the ERK-MAP kinase cascade. EMBO Rep. 2004, 5, 484–489. [Google Scholar] [CrossRef] [PubMed]
- Moore, C.J.; Winder, S.J. The inside and out of dystroglycan post-translational modification. Neuromuscul. Disord. 2012, 22, 959–965. [Google Scholar] [CrossRef] [PubMed]
- Vogel, W.F.; Abdulhussein, R.; Ford, C.E. Sensing extracellular matrix: An update on discoidin domain receptor function. Cell Signal. 2006, 18, 1108–1116. [Google Scholar] [CrossRef] [PubMed]
- Leitinger, B. Transmembrane collagen receptors. Annu. Rev. Cell Dev. Biol. 2011, 27, 265–290. [Google Scholar] [CrossRef] [PubMed]
- Vogel, W.; Gish, G.D.; Alves, F.; Pawson, T. The discoidin domain receptor tyrosine kinases are activated by collagen. Mol. Cell 1997, 1, 13–23. [Google Scholar] [CrossRef] [PubMed]
- Shrivastava, A.; Radziejewski, C.; Campbell, E.; Kovac, L.; McGlynn, M.; Ryan, T.E.; Davis, S.; Goldfarb, M.P.; Glass, D.J.; Lemke, G.; et al. An orphan receptor tyrosine kinase family whose members serve as nonintegrin collagen receptors. Mol. Cell 1997, 1, 25–34. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Z.; Su, H.W.; Hsu, Y.C.; Shen, M.R.; Tang, M.J. A discoidin domain receptor 1/SHP-2 signaling complex inhibits α2β1-integrim-mediated signal transducers and activators of transcription 1/3 activation and cell migration. Mol. Biol. Cell 2006, 17, 2839–2852. [Google Scholar] [CrossRef] [PubMed]
- Xu, H.; Bihan, D.; Chang, F.; Huang, P.H.; Farndale, R.W.; Leitinger, B. Discoidin domain receptors promote α1β1- and α2β1-integrin mediated cell adhesion to collagen by enhancing integrin activation. PLoS One 2012, 7, e52209. [Google Scholar] [CrossRef] [PubMed]
- Dejmek, J.; Dib, K.; Jönsson, M.; Andersson, T. Wnt-5a and G-protein signaling are required for collagen-induced DDR1 receptor activation and normal mammary cell adhesion. Int. J. Cancer 2003, 103, 344–351. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.G.; Hwang, S.Y.; Aaronson, S.A.; Mandinova, A.; Lee, S.W. DDR1 receptor tyrosine kinase promotes prosurvival pathway through Notch1 activation. J. Biol. Chem. 2011, 286, 17672–17681. [Google Scholar] [CrossRef] [PubMed]
- Iwai, L.K.; Chang, F.; Huang, P.H. Phosphoproteomic analysis identifies insulin enhancement of discoidin domain receptor 2 phosphorylation. Cell Adh. Migr. 2013, 7, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Fu, H.L.; Valiathan, R.R.; Arkwright, R.; Sohail, A.; Mihai, C.; Kumarasi, M.; Mahasenan, K.V.; Mobashery, S.; Huang, P.; Agarwal, G.; et al. Discoidin domain receptors: Unique receptor tyrosine kinases in collagen-mediated signaling. J. Biol. Chem. 2013, 288, 7430–7437. [Google Scholar] [CrossRef] [PubMed]
- Jandrot-Perrus, M.; Busfield, S.; Lagrue, A.H.; Xiong, X.; Debili, N.; Chickering, T.; Le Couedic, J.P.; Goodearl, A.; Dussault, B.; Fraser, C.; et al. Cloning, characterization, and functional studies of human and mouse glycoprotein VI: A platelet-specific collagen receptor from the immunoglobulin superfamily. Blood 2000, 96, 1798–1807. [Google Scholar] [PubMed]
- Lebbink, R.J.; de Ruiter, T.; Adelmeijer, J.; Brenkman, A.B.; van Helvoort, J.M.; Koch, M.; Farndale, R.W.; Lisman, T.; Sonnenberg, A.; Lenting, P.J.; et al. Collagens are functional, high affinity ligands for the inhibitory immune receptor LAIR-1. J. Exp. Med. 2006, 203, 1419–1425. [Google Scholar] [CrossRef] [PubMed]
- Meyaard, L. LAIR and collagens in immune regulation. Immunol. Lett. 2010, 128, 26–28. [Google Scholar] [CrossRef] [PubMed]
- Watson, S.P.; Herbert, J.M.J.; Pollitt, A.Y. GPVI and CLEC-2 in hemostasis and vascular integrity. J. Thromb. Haemost. 2010, 8, 1457–1467. [Google Scholar] [CrossRef]
- Berry, D.C.; Noy, N. All-trans-retinoic acid represses obesity and insulin resistance by activating both peroxisome proliferation-activated receptor β/δ and retinoic acid receptor. Mol. Cell. Biol. 2009, 29, 3286–3296. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.Y.; Clifford, J.; Zusi, C.; Starrett, J.; Tortolani, D.; Ostrowski, J.; Reczek, P.R.; Chambon, P.; Gronemeyer, H. Two distinct actions of retinoid-receptor ligands. Nature 1996, 382, 819–822. [Google Scholar] [CrossRef] [PubMed]
- Bruck, N.; Vitoux, D.; Ferry, C.; Duong, V.; Bauer, A.; de The, H.; Rochette-Egly, C. A coordinated phosphorylation cascade initiated by p38MAPK/MSK1 directs RARα to target promoters. EMBO J. 2009, 28, 34–47. [Google Scholar] [CrossRef] [PubMed]
- Al Tanoury, Z.; Piskunov, A.; Rochette-Egly, C. Vitamin A and retinoid signaling: Genomic and non-genomic effects. J. Lipid Res. 2013, 54, 1744–1760. [Google Scholar] [CrossRef] [PubMed]
- Cañón, E.; Cosgaya, J.M.; Scsucova, S.; Aranda, A. Rapid effects of retinoic acid on CREB and ERK phosphorylation in neuronal cells. Mol. Biol. Cell 2004, 15, 5583–5592. [Google Scholar] [CrossRef] [PubMed]
- Masiá, S.; Alvarez, S.; de Lera, A.R.; Barettino, D. Rapid, non-genomic actions of retinoic acid on phosphatidylinositol-3-kinase signaling pathway mediated by the retinoic acid receptor. Mol. Endocrinol. 2007, 21, 2391–2402. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Tao, J.; Zhu, Q.; Jia, P.-M.; Dou, A.-X.; Li, X.; Cheng, F.; Waxman, S.; Chen, G.-Q.; Chen, S.-J.; et al. Rapid induction of cAMP/PKA pathway during retinoic acid-induced acute promyelocytic leukemia cell differentiation. Leukemia 2004, 18, 285–292. [Google Scholar] [CrossRef] [PubMed]
- Kambhampati, S.; Li, Y.; Verma, A.; Sassano, A.; Majchrzak, B.; Deb, D.K.; Parmar, S.; Giafis, N.; Kalvakolanu, D.V.; Rahman, A.; et al. Activation of protein kinase Cδ by all-trans-retinoic acid. J. Biol. Chem. 2003, 278, 32544–32551. [Google Scholar] [CrossRef] [PubMed]
- Pendaries, V.; Verrecchia, F.; Michel, S.; Mauviel, A. Retinoic acid receptors interfere with the TGF-β/Smad signaling Pathway in a ligand-specific manner. Oncogene 2003, 22, 8212–8220. [Google Scholar] [CrossRef] [PubMed]
- Cao, Z.; Flanders, K.C.; Bertolette, D.; Lyakh, L.A.; Wurthner, J.U.; Parks, T.W.; Letterio, J.J.; Ruscetti, F.W.; Roberts, A.B. Levels of phosphor-Smad2/3 are sensors of the interplay between effects of TGF-β and retinoic acid on monocytic and granulocytic differentiation of HL-60 cells. Blood 2003, 101, 498–507. [Google Scholar] [CrossRef] [PubMed]
- Berry, D.C.; Jin, H.; Majumdar, A.; Noy, N. Signaling by vitamin A and retinol-binding protein regulates gene expression to inhibit insulin responses. Proc. Natl. Acad. Sci. USA 2011, 108, 4340–4345. [Google Scholar] [CrossRef] [PubMed]
- Petrosino, J.M.; Disilvestro, D.; Ziouzenkova, O. Aldehyde dehydrogenase 1A1: Friend or foe to female metabolism? Nutrients 2014, 6, 950–973. [Google Scholar] [CrossRef]
- National Academy of Sciences (NAS). The Essential Guide to Nutrient Requirements. Food and Nutrition Board; National Academic Press: Washington, DC, USA, 2006. Available online: http://www.nap.edu/catalog.php?record_id=11537 (accessed on 20 January 2013).
- World Health Organization (WHO). Global prevalence of vitamin A deficiency in populations at risk 1995–2005. In WHO Global Database on Vitamin A Deficiency; World Health Organization: Geneva, Switzerland, 2009. [Google Scholar]
- World Health Organization (WHO). Micronutrients Deficiencies; World Health Organization: Geneva, Switzerland, 2014. Available online: http://www.who.int/nutrition/topics/vad/en/ (accessed on 10 January 2014).
- Wolbach, S.B.; Howe, P.R. Tissue changes following deprivation of fat soluble A vitamin. J. Exp. Med. 1925, 42, 753–777. [Google Scholar] [CrossRef] [PubMed]
- Underwood, B.A. The Retinoids; Sporn, M.B., Roberts, A., Goodman, D.S., Eds.; Academic Press: New York, NY, USA, 1984; Volume 1, p. 281. [Google Scholar]
- Bendich, A.; Langseth, L. Safety of vitamin A. Am. J. Clin. Nutr. 1989, 49, 358–371. [Google Scholar] [PubMed]
- Lieber, C.S. Alcohol: Its metabolism and interaction with nutrients. Annu. Rev. Nutr. 2000, 20, 395–430. [Google Scholar] [CrossRef] [PubMed]
- Matsui, T. Differential activation of the murine laminin B1 gene promoter by RARα, RORα, and AP-1. Biochem. Biophys. Res. Commun. 1996, 220, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Axel, D.I.; Frigge, A.; Dittmann, J.; Runge, H.; Spyridopoulps, I.; Riessen, R.; Viebahn, R.; Karsch, K.R. All-trans retinoic acid regulates proliferation, migration, differentiation, and extracellular matrix turnover of human arterial smooth muscle cells. Cardiovasc. Res. 2001, 49, 851–862. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.S.; Jeong, W.I. Retinoic acids and hepatic stellate cells in liver disease. J. Gastroenterol. Hepatol. 2012, 27, 75–79. [Google Scholar]
- Aguilar, R.P.; Genta, S.; Oliveros, L.; Anzulovich, A.; Giménez, M.S.; Sánchez, S.S. Vitamin A deficiency injures liver parenchyma and alters the expression of hepatic extracellular matrix. J. Appl. Toxicol. 2009, 29, 214–222. [Google Scholar] [CrossRef] [PubMed]
- Barber, T.; Esteban-Pretel, G.; Marín, M.P.; Timoneda, J. Vitamin A and Carotenoids: Chemistry, Analysis, Function and Effects; Preedy, V.R., Ed.; The Royal Society of Chemistry: London, UK, 2012; p. 396. [Google Scholar]
- Xu, Q.; Lucio-Cazana, J.; Kitamura, M.; Ruan, X.; Fine, L.G.; Norman, J.T. Retinoids in nephrology: Promises and pitfalls. Kidney Int. 2004, 66, 2119–2131. [Google Scholar] [CrossRef] [PubMed]
- Lelièvre-Pégorier, M.; Vilar, J.; Ferrier, M.L.; Moreau, E.; Freund, N.; Gilbert, T.; Merlet-Bénichou, C. Mild vitamin A deficiency leads to inborn nephron deficit in the rat. Kidney Int. 1998, 54, 1455–1462. [Google Scholar] [CrossRef] [PubMed]
- Moreau, E.; Vilar, J.; Lelièvre-Pégorier, M.; Merlet-Benichou, C.; Gilbert, T. Regulation of c-ret expression by retinoic acid in rat metanephros: Implication in nephron mass control. Am. J. Physiol. 1998, 275, F938–F945. [Google Scholar] [PubMed]
- Mendelsohn, C.; Batourina, E.; Fung, S.; Gilbert, T.; Dodd, J. Stromal cells mediated retinoid-dependent functions essential for renal development. Development 1999, 126, 1139–1148. [Google Scholar] [PubMed]
- Batourina, E.; Gim, S.; Bello, N.; Shy, M.; Clagett-Dame, M.; Srinivas, S.; Costantini, F.; Mendelsohn, C. Vitamin A controls epithelial/mesenchymal interactions through Ret expression. Nat. Genet. 2001, 27, 74–78. [Google Scholar] [PubMed]
- Vilar, J.; Gilbert, T.; Moreau, E.; Merlet-Bénichou, C. Metanephros organogenesis is highly stimulated by vitamin A derivatives in organ culture. Kidney Int. 1996, 49, 1478–1487. [Google Scholar] [CrossRef] [PubMed]
- Merlet-Bénichou, C.; Vilar, J.; Lelièvre-Pégorier, M.; Gilbert, T. Role of retinoids in renal development: Pathophysiological implication. Curr. Opin. Nephrol. Hypertens. 1999, 8, 39–43. [Google Scholar] [CrossRef] [PubMed]
- Nelson, R.G. Intrauterine determinants of diabetic kidney disease in disadvantaged populations. Kidney Int. 2003, 83, S13–S16. [Google Scholar] [CrossRef]
- Atadzhanov, U.Zh.; Utegeov, N.U. Structural-functional damage to cellular membranes in deficiency of vitamins A, E, B2, PP in children with calculous pyelonephritis. Urologiia 2003, 1, 35–41. [Google Scholar] [PubMed]
- Sakly, R.; Fekih, M.; Ben Amor, A.; Najjar, M.F.; Mbazaa, M. Possible role of vitamin A and E deficiency in human idiopathic lithiasis. Ann. Urol. 2003, 37, 217–219. [Google Scholar] [CrossRef]
- Yang, H.; Chen, K.; Zhang, X.; Wang, L.; Li, C.; Tao, H.; Wang, L.; Li, Q. Vitamin A deficiency results in dysregulation of lipid efflux pathway in rat kidney. Pediatr. Nephrol. 2010, 25, 1435–1444. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J. Potential role of retinoids in the therapy of renal disease. Nephrol. Dial. Transplant. 2001, 16, 441–444. [Google Scholar] [CrossRef] [PubMed]
- Müller, U.; Brändli, A.W. Cell adhesion molecules and extracellular matrix constituents in kidney development and disease. J. Cell Sci. 1999, 112, 3855–3867. [Google Scholar] [PubMed]
- Gustafsson, E.; Fassler, R. Insights into extracellular matrix functions from mutant mouse models. Exp. Cell. Res. 2000, 261, 52–68. [Google Scholar] [CrossRef] [PubMed]
- Van Vliet, A.; Van Alderwegen, I.E.; Baelde, H.J.; de Heer, E.; Killen, P.D.; Kalluri, R.K.; Bruijn, J.A.; Bergijk, E.C. Differential expression of collagen type IV alpha-chains in the tubulointerstitial compartment in experimental chronic serum sickness nephritis. J. Pathol. 1999, 189, 279–287. [Google Scholar] [CrossRef] [PubMed]
- Hudson, B.G. The molecular basis of goodpasture and alport syndromes: Beacons for the Discovery of the collagen IV family. J. Am. Soc. Nephrol. 2004, 15, 2514–2527. [Google Scholar] [CrossRef] [PubMed]
- Miner, J.H.; Sanes, J.R. Molecular and functional defects in kidneys of mice lacking collagen 3 (IV): Implications for Alport syndrome. J. Cell Biol. 1996, 135, 1403–1413. [Google Scholar] [CrossRef] [PubMed]
- Mooney, A.; Jackson, K.; Bacon, R.; Strenli, C.; Edwards, G.; Bassuk, J.; Savill, J. Type IV collagen and laminin regulate glomerular mesangial cell susceptibility to apoptosis via 1 integrin-mediated survival signals. Am. J. Pathol. 1999, 155, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Marín, M.P.; Esteban-Pretel, G.; Alonso, R.; Sado, Y.; Barber, T.; Renau-Piqueras, J.; Timoneda, J. Vitamin A deficiency alters the structure and collagen IV composition of rat renal basement membranes. J. Nutr. 2005, 135, 695–701. [Google Scholar] [PubMed]
- Yang, C.W.; Hattori, M.; Vlassara, H.; He, C.J.; Carome, M.A.; Yamato, E.; Elliot, S.; Striker, G.E.; Striker, L.J. Overexpression of transforming growth factor-beta 1 mRNA is associated with upregulation of glomerular tenascin and laminin gene expression in nonobese dibetic mice. J. Am. Soc. Nephrol. 1995, 5, 1610–1617. [Google Scholar] [PubMed]
- Sayers, R.; Kalluri, R.; Rodgers, K.D.; Shield, C.F.; Meehan, D.T.; Cosgrove, D. Role of transforming growth factor-beta1 in Alport renal disease progression. Kidney Int. 1999, 56, 1662–1673. [Google Scholar] [CrossRef] [PubMed]
- Noel, L.H. Renal pathology and ultrastructural findings in Alport’s syndrome. Ren. Fail. 2000, 22, 751–758. [Google Scholar] [CrossRef] [PubMed]
- Tsilibary, E.C. Microvascular basement membranes in diabetes mellitus. J. Pathol. 2003, 200, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Striker, L.J.; Killen, P.D.; Chi, E.; Striker, G.E. The composition of glomerulosclerosis I. Studies in focal sclerosis, cresenteric glomerulonephritis and membranoproliferative glomerulonephritis. Lab. Investig. 1984, 5, 181–192. [Google Scholar]
- Ebihara, I.; Suzuk, S.; Nakamura, T.; Fukui, M.; Yaguchi, Y.; Tomino, Y.; Koide, H. Extracellular matrix component mRNA expression in glomeruli in experimental focal glomerulosclerosis. J. Am. Soc. Nephrol. 1993, 3, 1387–1397. [Google Scholar] [PubMed]
- Bergijk, E.C.; van Alderwegen, I.E.; Baelde, H.J.; de Heer, E.; Funabiki, K.; Miyai, H.; Killen, P.D.; Kalluri, R.K.; Bruijn, J.A. Differential expression of collagen IV isoforms in experimental glomerulosclerosis. J. Pathol. 1998, 184, 307–315. [Google Scholar] [CrossRef] [PubMed]
- Yagame, M.; Kim, Y.; Zhu, D.; Suzuki, D.; Eguchi, K.; Nomoto, Y.; Sakai, H.; Groppoli, T.; Steffes, M.W.; Mauer, S.M. Differential distribution of type IV collagen chains in patients with diabetic nephropathy in non-insulin dependent diabetes mellitus. Nephron 1995, 70, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Barsotti, P.; Muda, A.O.; Mazzucco, G.; Massella, L.; Basolo, B.; de Marchi, M.; Rizzoni, G.; Monga, G.; Faraggiana, T. Distribution of a-chains of type I V collagen in glomerular basement membranes with ultrastructural alterations suggestive of Alport syndrome. Nephrol. Dial. Transplant. 2001, 16, 945–952. [Google Scholar] [CrossRef] [PubMed]
- McLennan, S.V.; Martel, S.K.; Yue, D.K. Effects of mesangium glycation on matrix metalloproteinase activities: Possible role in diabetic nephropathy. Diabetes 2002, 51, 2612–2618. [Google Scholar] [CrossRef] [PubMed]
- Akool, El-S.; Kleinert, H.; Hamada, F.M.; Abdelwahab, M.H.; Forstermann, U.; Pfeilschifter, J.; Eberhardt, W. Nitric oxide increases the decay of matrix metalloproteinase 9 mRNA by inhibiting the expression of mRNA-stabilizing factor HuR. Mol. Cell. Biol. 2003, 23, 4901–4916. [Google Scholar] [CrossRef] [PubMed]
- Cheng, S.; Lovett, D.H. Gelatinase A (MMP-2) is necessary and sufficient for renal tubular cell epithelial-mesenchymal transformation. Am. J. Pathol. 2003, 162, 1937–1949. [Google Scholar] [CrossRef] [PubMed]
- Camp, T.M.; Smiley, L.M.; Hayden, M.R.; Tyagi, S.C. Mechanism of matrix accumulation and glomerulosclerosis in spontaneously hypertensive rats. J. Hypertens. 2003, 21, 1719–1727. [Google Scholar] [CrossRef] [PubMed]
- Rao, V.H.; Lee, G.E.; Kashtan, C.E.; Nemori, R.; Singh, R.K.; Meehan, D.T.; Rodgers, K.; Berridge, B.R.; Bhattacharya, G.; Cosgrove, D. Increased expression of MMP-2, MMP-9 (type IV collagenases/gelatinases), and MT1-MMP in canine X-linked Alport syndrome (XLAS). Kidney Int. 2003, 63, 1736–1748. [Google Scholar] [CrossRef] [PubMed]
- Chytil, F. Retinoids in lung development. FASEB J. 1996, 10, 986–992. [Google Scholar] [PubMed]
- Massaro, D.; Massaro, G.D. Pulmonary alveoli: Formation, “the call for oxygen”, and other regulators. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L345–L358. [Google Scholar] [PubMed]
- Maden, M.; Hind, M. Retinoic acid in alveolar development, maintenance and regeneration. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2004, 359, 799–808. [Google Scholar] [CrossRef] [PubMed]
- Zeltner, T.B.; Caduff, J.H.; Gehr, P.; Pfenninger, J.; Burri, P.H. The postnatal development and growth of the human lung. I. Morphometry. Respir. Physiol. 1987, 67, 247–267. [Google Scholar] [CrossRef] [PubMed]
- Shenai, J.P.; Chytil, F.; Stahlman, M.T. Vitamin A status of neonates with bronchopulmonary dysplasia. Pediatr. Res. 1985, 19, 185–188. [Google Scholar] [CrossRef] [PubMed]
- Herzog, E.L.; Brody, A.R.; Colby, T.V.; Mason, R.; Williams, M.C. Knowns and unknowns of the alveolus. Proc. Am. Thorac. Soc. 2008, 5, 778–782. [Google Scholar] [CrossRef] [PubMed]
- Pelosi, P.; Rocco, P.R.; Negrini, D.; Passi, A. The extra- cellular matrix of the lung and its role in edema formation. An. Acad. Bras. Cienc. 2007, 79, 285–297. [Google Scholar] [CrossRef] [PubMed]
- Salbert, G.; Fanjul, A.; Piedrafita, F.J.; Lu, X.P.; Kim, S.J.; Tran, P.; Pfahl, M. Retinoic acid receptors and retinoid X receptor-α down-regulate the transforming growth factor-β1 promoter by antagonizing AP-1 activity. Mol. Endocrinol. 1993, 7, 1347–1356. [Google Scholar] [PubMed]
- Antipatis, C.; Ashworth, C.J.; Grant, G.; Lea, R.G.; Hay, S.M.; Rees, W.D. Effects of maternal vitamin A status on fetal heart and lung: Changes in expression of key developmental genes. Am. J. Physiol. 1998, 275, L1184–L1191. [Google Scholar]
- McGowan, S.E.; Smith, J.; Holmes, A.J.; Smith, L.A.; Businga, T.R.; Madsen, M.T.; Kopp, U.C.; Kline, J.N. Vitamin A deficiency promotes bronchial hyperreactivity in rats by altering muscarinic M2 receptor function. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 282, L1031–L1039. [Google Scholar] [CrossRef] [PubMed]
- Baybutt, R.C.; Molteni, A. Vitamin A and emphysema. Vitam. Horm. 2007, 75, 385–401. [Google Scholar] [PubMed]
- Baybutt, R.C.; Hu, L.; Molteni, A. Vitamin A deficiency injures lung and liver parenchyma and impairs function of rat type II pneumocytes. J. Nutr. 2000, 130, 1159–1165. [Google Scholar] [PubMed]
- Biesalski, H.K.; Nohr, D. Importance of vitamin-A for lung function and development. Mol. Aspects Med. 2003, 24, 431–440. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Huang, H.M.; Li, T.Y.; Qu, P.; Liu, Y.X.; Chen, J. Marginal vitamin A deficiency affects lung maturation in rats from prenatal to adult stage. J. Nutr. Sci. Vitaminol. Tokyo 2009, 55, 208–214. [Google Scholar] [CrossRef] [PubMed]
- Esteban-Pretel, G.; Marín, M.P.; Renau-Piqueras, J.; Barber, T.; Timoneda, J. Vitamin A deficiency alters rat lung alveolar basement membrane: Reversibility by retinoic acid. J. Nutr. Biochem. 2010, 21, 227–236. [Google Scholar] [CrossRef] [PubMed]
- McGowan, S.E.; Takle, E.J.; Holmes, A.J. Vitamin A deficiency alters the pulmonary parenchymal elastic modulus and elastic fiber concentration in rats. Respir. Res. 2005, 6, 77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGowan, S.E.; Holmes, A.J. Vitamin A deficiency alters pulmonary parenchymal collagen and tissue mechanics. Respir. Physiol. Neurobiol. 2007, 156, 312–319. [Google Scholar] [CrossRef]
- Esteban-Pretel, G.; Marín, M.P.; Renau-Piqueras, J.; Sado, Y.; Barber, T.; Timoneda, J. Vitamin A deficiency disturbs collagen IV and laminin composition and decreases matrix metalloproteinase concentrations in rat lung. Partial reversibility by retinoic acid. J. Nutr. Biochem. 2013, 24, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.; Zolfaghari, R. Regulation of hepatic retinol metabolism: Perspectives from studies on vitamin A status. J. Nutr. 2004, 134, 269S–275S. [Google Scholar] [PubMed]
- Olson, J.A. Recommended dietary intakes (RDI) of vitamin A in Humans. Am. J. Clin. Nutr. 1987, 45, 704–7l6. [Google Scholar] [PubMed]
- Clugston, R.D.; Blaner, W.S. The adverse effects of alcohol on vitamin A metabolism. Nutrients 2012, 4, 356–371. [Google Scholar] [CrossRef] [PubMed]
- Iredale, J.P.; Thompson, A.; Henderson, N.C. Extracellular matrix degradation in liver fibrosis: Biochemistry and regulation. Biochim. Biophys. Acta 2013, 1832, 876–883. [Google Scholar] [CrossRef] [PubMed]
- Clement, B.; Grimaud, J.A.; Campion, J.P.; Deugnier, Y.; Guillouzo, A. Cell types involved in collagen and fibronectin production in normal and fibrotic human liver. Hepatology 1986, 6, 225–234. [Google Scholar] [PubMed]
- Friedman, S.L. Molecular regulation of hepatic fibrosis, an integrated cellular response to tissue injury. J. Biol. Chem. 2000, 275, 2247–2250. [Google Scholar] [CrossRef] [PubMed]
- Geerts, A. History, heterogeneity, developmental biology, and functions of quiescent hepatic stellate cells. Semin. Liver Dis. 2001, 21, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Mallat, A.; Lotersztajn, S. Cellular mechanisms of tissue fibrosis. 5. Novel insights into liver fibrosis. Am. J. Physiol. Cell. Physiol. 2013, 305, C789–C799. [Google Scholar] [CrossRef] [PubMed]
- Yin, C.H.; Evason, K.J.; Asahina, K.; Stainier, D. Hepatic stellate cells in liver development, regeneration, and cancer. J. Clin. Investig. 2013, 123, 1902–1910. [Google Scholar] [CrossRef] [PubMed]
- Forbes, S.J.; Parola, N. Liver fibrogenic cells. Best Pract. Res. Clin. Gastroenterol. 2011, 25, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Taura, K.; Miura, K.; Iwaisako, K.; Osterreicher, C.H.; Kodama, Y.; Penz-Osterreicher, M.; Brenner, D.A. Hepatocytes do not undergo epithelial-mesenchymal transition in liver fibrosis in mice. Hepatology 2010, 51, 1027–1036. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Liu, H.; Meyer, C.; Li, J.; Nadalin, S.; Königsrainer, A.; Weng, H.; Dooley, S.; Dijke, P. TGF-β mediated connective tissue growth factor (CTGF) expression in hepatic stellate cells requires Stat3 activation. J. Biol. Chem. 2013, 288, 30708–30719. [Google Scholar] [CrossRef] [PubMed]
- Yanagitani, A.; Yamada, S.; Yasui, S.; Shimomura, T.; Murai, R.; Murawaki, Y.; Hashiguchi, K.; Kanbe, T.; Saeki, T.; Ichiba, M.; et al. Retinoic acid receptor a dominant negative form causes steatohepatitis and liver tumors in transgenic mice. Hepatology 2004, 40, 366–375. [Google Scholar] [CrossRef] [PubMed]
- Shirakami, Y.; Lee, S.A.; Clugston, R.D.; Blaner, W.S. Hepatic metabolism of retinoids and disease associations. Biochim. Biophys. Acta 2012, 1821, 124–136. [Google Scholar] [CrossRef] [PubMed]
- Esteban-Pretel, G.; Marín, M.P.; Cabezuelo, F.; Moreno, V.; Renau-Piqueras, J.; Timoneda, J.; Barber, T. Vitamin A deficiency increases protein catabolism and induces urea cycle enzymes in rats. J. Nutr. 2010, 140, 792–779. [Google Scholar] [CrossRef] [PubMed]
- Tsukamoto, H.; She, H.; Hazra, S.; Cheng, J.; Wang, J. Fat paradox of steatohepatitis. J. Gastroenterol. Hepatol. 2008, 23, S104–S107. [Google Scholar] [PubMed]
- Kim, H.Y.; Wolf, G. Vitamin A deficiency alters genomic expression for fibronectin in liver and hepatocytes. J. Biol. Chem. 1987, 262, 365–371. [Google Scholar] [PubMed]
- Seifert, W.F.; Bosma, A.; Brouwer, A.; Hendriks, H.F.; Roholl, P.J.; van Leeuwen, R.E.; van Thiel-de Ruiter, G.C.; Seifert-Bock, I.; Knook, D.L. Vitamin A deficiency potentiates carbon tetrachloride-induced liver fibrosis in rats. Hepatology 1994, 19, 193–201. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Tankersley, L.R.; Tang, M.; Potter, J.J.; Mezey, E. Regulation of the murine α2(I) collagen promoter by retinoic acid and retinoid X receptors. Arch. Biochem. Biophys. 2002, 401, 262–270. [Google Scholar] [CrossRef] [PubMed]
- Meisler, N.T.; Parrelli, J.; Gendimenico, G.J.; Mezick, J.A.; Cutroneo, K.R. All-trans retinoic acid inhibition of pro-α1(I) collagen gene expression in fetal rat skin fibroblasts: Identification of a retinoic acid response element in the pro- α1(I) collagen gene. J. Investig. Dermatol. 1997, 108, 476–481. [Google Scholar] [CrossRef] [PubMed]
- Cohen, A.J.; Lassová, L.; Golden, E.B.; Niu, Z.; Adams, S.L. Retinoids directly activate the collagen X promoter in prehypertrophic chondrocytes through a distal retinoic acid response element. J. Cell. Biochem. 2006, 99, 269–278. [Google Scholar] [CrossRef] [PubMed]
- Pan, L.; Eckhoff, C.; Binckerhoff, C.E. Suppression of collagenase gene expression by all-trans and 9-cis retinoic acid is ligand dependent and requires both RARs and RXRs. J. Cell. Biochem. 1995, 57, 575–589. [Google Scholar] [CrossRef] [PubMed]
- Zaragozá, R.; Gimeno, A.; Miralles, V.J.; García-Trevijano, E.R.; Carmena, R.; García, C.; Mata, M.; Puertes, I.R.; Torres, L.; Viña, J.R. Retinoids induce MMP-9 expression through RARα during mammary gland remodeling. Am. J. Physiol. Endocrinol. Metab. 2007, 292, E1140–E1148. [Google Scholar] [CrossRef] [PubMed]
- Lackey, D.E.; Hoag, K.A. Vitamin A upregulates matrix metalloproteinase-9 activity by murine myeloid dendritic cells through a nonclassical transcriptional mechanism. J. Nutr. 2010, 140, 1502–1508. [Google Scholar] [CrossRef] [PubMed]
- Glick, A.B.; McCune, B.K.; Abdulkarem, N.; Flanders, K.C.; Lumadue, J.A.; Smith, J.M.; Sporn, M.B. Complex regulation of TGFβ expression by retinoic acid in the vitamin A-deficient rat. Development 1991, 111, 1081–1086. [Google Scholar] [PubMed]
- Mercier, T.; Gaillard-Sanchez, I.; Martel, P.; Heberden, C. TGF-β receptors are diminished after retinoid exposure in rat liver epithelial cells. J. Cell. Biochem. 1996, 61, 230–237. [Google Scholar] [CrossRef] [PubMed]
- Morath, C.V.; Dechow, C.; Lehrke, I.; Haxsen, V.; Waldherr, R.; Floege, J.; Ritz, E.; Wagner, J. Effetcs of retinoids on the TGF-β system and extracellular matrix in experimental glomerulonephritis. J. Am. Soc. Nephrol. 2001, 12, 2300–2309. [Google Scholar] [PubMed]
- Verrecchia, F.; Chu, M-L.; Mauviel, A. Identification of novel TGF-β/Smad gene targets in dermal fibroblast using a combined cDNA microarray/promoter transactivation approach. J. Biol. Chem. 2001, 276, 17058–17062. [Google Scholar] [CrossRef] [PubMed]
- Zeisberg, L.; Kalluri, R. The role of epithelial-to-mesenchymal transition in renal fibrosis. J. Mol. Med. 2004, 82, 175–181. [Google Scholar] [CrossRef] [PubMed]
- Willis, B.C.; duBois, R.M.; Borok, Z. Epithelial origin of myofibroblasts during fibrosis in the lung. Proc. Am. Thorac. Soc. 2006, 3, 377–382. [Google Scholar] [CrossRef] [PubMed]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef] [PubMed]
- Gatica, L.V.; Oliveros, L.B.; Pérez Díaz, M.F.; Domínguez, N.S.; Fornes, M.W.; Gimenez, M.S. Implication of vitamin A deficiency on vascular injury related to inflammation and oxidative stress. Effects on the ultrastructure of rat aorta. Eur. J. Nutr. 2012, 51, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Shimada, J.; Suzuki, Y.; Kim, S.J.; Wang, P.C.; Matsumura, M.; Kojima, S. Transactivation via 632 RAR/RXR-Sp1 interaction: Characterization of binding between Sp1 and GC box 633 motif. Mol. Endocrinol. 2001, 15, 1677–1692. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, C.; Fischer, G.; Kadner, H.; Genersch, E.; Kühn, K.; Pöschl, E. Differential effects of DNA-binding proteins on bidirectional transcription from the common promoter region of human collagen type IV genes COL4A1 and COL4A2. Biochim. Biophys. Acta 1993, 1174, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Tsang, K.J.; Crowe, D.L. Retinoic acid and extracellular matrix inhibition of matrix metalloproteinase 9 expression is mediated by mitogen activated protein kinase pathway. Int. J. Oncol. 2001, 18, 369–374. [Google Scholar] [PubMed]
- Barber, T.; Borrás, E.; Torres, L.; García, C.; Cabezuelo, F.; Lloret, A.; Pallardó, F.V.; Viña, J.R. Vitamin A deficiency causes oxidative damage to liver mitochondria in rats. Free Radic. Biol. Med. 2000, 29, 1–7. [Google Scholar] [PubMed]
- Iglesias de la Cruz, M.C.; Ruiz-Torres, P.; Alcammí, J.; Díez-Marqués, L.; Ortega-Velazquez, R.; Chen, S.; Rodríguez-Puyol, M.; Ziyadeh, F.N.; Rodríguez-Puyol, D. Hydrogen peroxide increases extracelular matrix mRNA through TGF-β in human mesangial cells. Kidney Int. 2001, 59, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.M.; Gaston Pravia, K.A. Oxidative stress and glutathione in TGF-β-mediated fibrogenesis. Free Radic. Biol. Med. 2010, 48, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Rhyu, D.Y.; Park, J.; Sharma, B.R.; Ha, H. Role of reactive oxygen species in transforming groth factor-beta-1-induced extracellular matrix accumulation in renal tubular epithelial cells. Transplant. Proc. 2012, 44, 625–628. [Google Scholar] [CrossRef] [PubMed]
- Samarakoon, R.; Overstreet, J.M.; Higgins, P.J. TGF-β signaling in tissue fibrosis: Redox controls, target genes and therapeutic opportunities. Cell. Signal. 2013, 25, 264–268. [Google Scholar] [CrossRef] [PubMed]
- Aoujehane, L.; Pissaia, A.; Scatton, O.; Podevin, P.; Massault, P.-P.; Chouzenoux, S.; Soubrane, O.; Calmus, Y.; Conti, F. Interleukine-4 induces the activation and collagen production of cultured human intrahepatic fibroblast via the STAT-6 pathway. Lab. Investig. 2008, 88, 973–985. [Google Scholar] [CrossRef] [PubMed]
- Mir, S.A.; Chatterjee, A.; Mitra, A.; Pathak, K.; Mahata, S.; Sarkar, S. Inhibition of signal transducer and activator of transcription 3 (STAT3) attenuates interleukin-6 (IL-6)-induced collagen synthesis and resultant hypertrophy in rat heart. J. Biol. Chem. 2012, 287, 2666–2677. [Google Scholar] [CrossRef] [PubMed]
- Matsui, F.; Rhee, A.; Hile, K.L.; Zhang, H.; Meldrum, K.K. IL-18 induces profibrotic renal tubular cell injury via STAT3 activation. Am. J. Physiol. Renal Physiol. 2013, 305, F1014–F1021. [Google Scholar] [CrossRef] [PubMed]
- Dai, B.; Cui, M.; Zhu, M.; Su, W.L.; Qiu, M.C.; Zhang, H. STAT 1/3 and ERK1/2 synergistically regulate cardiac fibrosis induced by high glucose. Cell. Physiol. Biochem. 2013, 32, 960–971. [Google Scholar] [CrossRef] [PubMed]
- Robledo, T.; Arriaga-Pizano, L.; Lopez-Pérez, M.; Pérez Salazar, E. Type IV collagen induces STAT5 activation in MCF7 human breast cancer cells. Matrix Biol. 2005, 24, 469–477. [Google Scholar] [CrossRef] [PubMed]
- Cortes-Reynosa, P.; Robledo, T.; Macias-Silva, M.; Vincent Wu, S.; Perez Salazar, E. Src kinase regulates metalloproteinase-9 secretion induced by type IV collagen in MCF-7 human breast cancer cells. Matrix Biol. 2008, 27, 220–231. [Google Scholar] [CrossRef] [PubMed]
- Shintani, Y.; Maeda, M.; Chaika, N.; Johnson, K.R.; Wheelock, M.J. Collagen I promotes epithelial-to-mesenchimal transition in lung cancer cells via transforming growth factor-β signaling. Am. J. Respir. Cell. Mol. Biol. 2008, 38, 95–104. [Google Scholar] [CrossRef] [PubMed]
- Zhou, T.B.; Drummen, G.P.; Qin, Y.H. The controversial role of retinoic acid in fibrotic diseases: Analysis of involved signaling pathways. Int. J. Mol. Sci. 2012, 14, 226–243. [Google Scholar] [CrossRef] [PubMed]
- Massaro, D.; Massaro, G.D. Retinoic acid treatment abrogates elastase-induced pulmonary emphysema in rats. Nat. Med. 1997, 3, 675–677. [Google Scholar] [CrossRef] [PubMed]
- Wagner, J.; Dechow, C.; Morath, C.; Lehrke, I.; Amann, K.; Waldherr, R.; Floege, J.; Ritz, E. Retinoic acid reduces glomerular injury in a rat model of glomerular damage. J. Am. Soc. Nephrol. 2000, 11, 1479–1487. [Google Scholar] [PubMed]
- Veness-Meehan, K.A.; Pierce, R.A.; Moats-Staats, B.M.; Stiles, A.D. Retinoic acid attenuates O2-induced inhibition of lung septation. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002, 283, L971–L980. [Google Scholar] [CrossRef] [PubMed]
- Massaro, D.; Massaro, G.D. Toward therapeutic pulmonary alveolar regeneration in humans. Proc. Am. Thorac. Soc. 2006, 3, 709–712. [Google Scholar] [CrossRef] [PubMed]
- James, M.L.; Ross, A.C.; Nicola, T.; Steele, C.; Ambalavanan, N. VARA attenuates hyperoxia-induced impaired alveolar development and lung function in newborn mice. Am. J. Physiol. Lung Cell. Mol. Physiol. 2013, 304, L803–L812. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, N.; Takasu, S. A close relationship between type 1 diabetes and vitamin A-deficiency and matrix metalloproteinase and hyaluronidase activities in skin tissues. Exp. Dermatol. 2011, 20, 899–904. [Google Scholar] [CrossRef] [PubMed]
- Oseto, S.; Moriyama, T.; Kawda, N.; Nagatoya, K.; Takeji, M.; Ando, A.; Yamamoto, T.; Imai, E.; Hori, M. Therapeutics effects of all-trans retinoic acid on rats with anti-GBM antibody glomerulonephritis. Kidney Int. 2003, 64, 1241–1252. [Google Scholar] [CrossRef] [PubMed]
- Davis, B.H.; Kramer, R.T.; Davidson, N.O. Retinoic acid modulates rat Ito cell proliferation, collagen, and transforming growth factor beta production. J. Clin. Investig. 1990, 86, 2062–2070. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Dan, Z. All-trans retinoic acid diminishes collagen production in a hepatic stellate cell line via suppression of active protein-1 and c-Jun N-terminal kinase signal. J. Huazhong Univ. Sci. Technol. Med. Sci. 2010, 30, 726–733. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barber, T.; Esteban-Pretel, G.; Marín, M.P.; Timoneda, J. Vitamin A Deficiency and Alterations in the Extracellular Matrix. Nutrients 2014, 6, 4984-5017. https://doi.org/10.3390/nu6114984
Barber T, Esteban-Pretel G, Marín MP, Timoneda J. Vitamin A Deficiency and Alterations in the Extracellular Matrix. Nutrients. 2014; 6(11):4984-5017. https://doi.org/10.3390/nu6114984
Chicago/Turabian StyleBarber, Teresa, Guillermo Esteban-Pretel, María Pilar Marín, and Joaquín Timoneda. 2014. "Vitamin A Deficiency and Alterations in the Extracellular Matrix" Nutrients 6, no. 11: 4984-5017. https://doi.org/10.3390/nu6114984