Dietary Fructose Feeding Increases Adipose Methylglyoxal Accumulation in Rats in Association with Low Expression and Activity of Glyoxalase-2

Abstract
:1. Introduction

2. Experimental Section
2.1. Materials
2.2. Animal Studies and Design

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ingredient | Control | Fructose | Fructose + 0.5% GTE | Fructose + 1.0% GTE |
|---|---|---|---|---|
| Egg whites | 200 | 200 | 199 | 198 |
| Corn Starch | 530.7 | 30.7 | 30.5 | 30.4 |
| Fructose | 100 | 600 | 597 | 594 |
| Cellulose | 50.0 | 50.0 | 49.8 | 49.5 |
| Soybean Oil | 70.0 | 70.0 | 69.7 | 69.3 |
| t-Butylhydroquinone (tBHQ) | 0.014 | 0.014 | 0.014 | 0.014 |
| Mineral Mix (AIN93G-EGG-MX) | 35.0 | 35.0 | 34.8 | 34.7 |
| Vitamin Mix (AIN-93-VX) | 10.0 | 10.0 | 10.0 | 9.9 |
| Biotin premix (1 mg/g sucrose) | 1.8 | 1.8 | 1.8 | 1.8 |
| Choline bitartrate | 2.5 | 2.5 | 2.5 | 2.5 |
| Green tea extract (GTE) b | 0.0 | 0.0 | 5.0 | 10.0 |
2.3. Plasma Chemistries
2.4. Hepatic Lipids and Glycogen
2.5. Tissue and Plasma Methylglyoxal
2.6. Hepatic and Adipose Glyoxalase-1 and Glyoxalase-2 Activities
2.7. Quantitative Real-Time PCR
2.8. Statistical Analyses
3. Results
3.1. Body Weight and Composition, Liver Injury, and Clinical Chemistries
| Parameter | Control | Fructose | Fructose + 0.5% GTE | Fructose + 1.0% GTE | P |
|---|---|---|---|---|---|
| Food Intake, g/day | 18.8 ± 0.5 | 21.8 ± 2.1 | 25.2 ± 1.4 | 26.4 ± 3.3 | NS |
| Body Mass, g | 397.0 ± 8.6 | 388.0 ± 29.5 | 404.5 ± 7.1 | 394.9 ± 11.3 | NS |
| Adipose Mass, % body mass | 1.48 ± 0.07 a | 1.20 ± 0.58 b,c | 1.28 ± 0.05 b | 1.06 ± 0.04 c | 0.0001 |
| Liver Mass, % body mass | 3.63 ± 0.11 b | 4.10 ± 0.06 a | 4.25 ± 0.04 a | 4.15 ± 0.06 a | <0.001 |
| Hepatic Total Lipid, mg/g liver | 52.7 ± 3.2 | 52.6 ± 2.1 | 54.6 ± 2.4 | 48.7 ± 2.1 | NS |
| Hepatic Triglyceride, µmol/g liver | 26.6 ± 1.2 | 28.0 ± 1.5 | 29.1 ± 2.1 | 24.6 ± 1.4 | NS |
| Hepatic Glycogen, mg/g liver | 33.0 ± 1.5 | 34.2 ± 1.4 | 33.2 ± 1.4 | 37.0 ± 2.1 | NS |
| Plasma Glucose, mg/dL | 10.5 ± 0.9 | 11.6 ± 1.1 | 12.2 ± 0.6 | 13.1 ± 0.5 | NS |
| Plasma Cholesterol, mg/dL | 96.1 ± 6.0 | 102.6 ± 5.6 | 108.0 ± 4.9 | 104.0 ± 4.5 | NS |
| Plasma Triglycerides, mmol/L | 118.3 ± 12.1 b | 227.6 ± 16.5 a | 214.1 ± 22.6 a | 187.4 ± 12.3 a | <0.001 |
| Plasma β-hydroxybutyrate, mmol/L | 0.265 ± 0.018 b | 0.423 ± 0.044 a | 0.441 ± 0.036 a | 0.353 ± 0.015 a | <0.01 |
| Plasma Insulin, pmol/L | 375.5 ± 171.4 | 340.9 ± 153.4 | 260.8 ± 62.0 | 330.1 ± 74.5 | NS |
| Plasma ALT, U/L | 23.9 ± 4.5 | 41.4 ± 6.2 | 27.6 ± 3.8 | 29.5 ± 4.0 | 0.08 |
| Plasma AST, U/L | 46.7 ± 8.4 | 71.1 ± 11.5 | 41.1 ± 4.9 | 45.1 ± 5.6 | 0.06 |
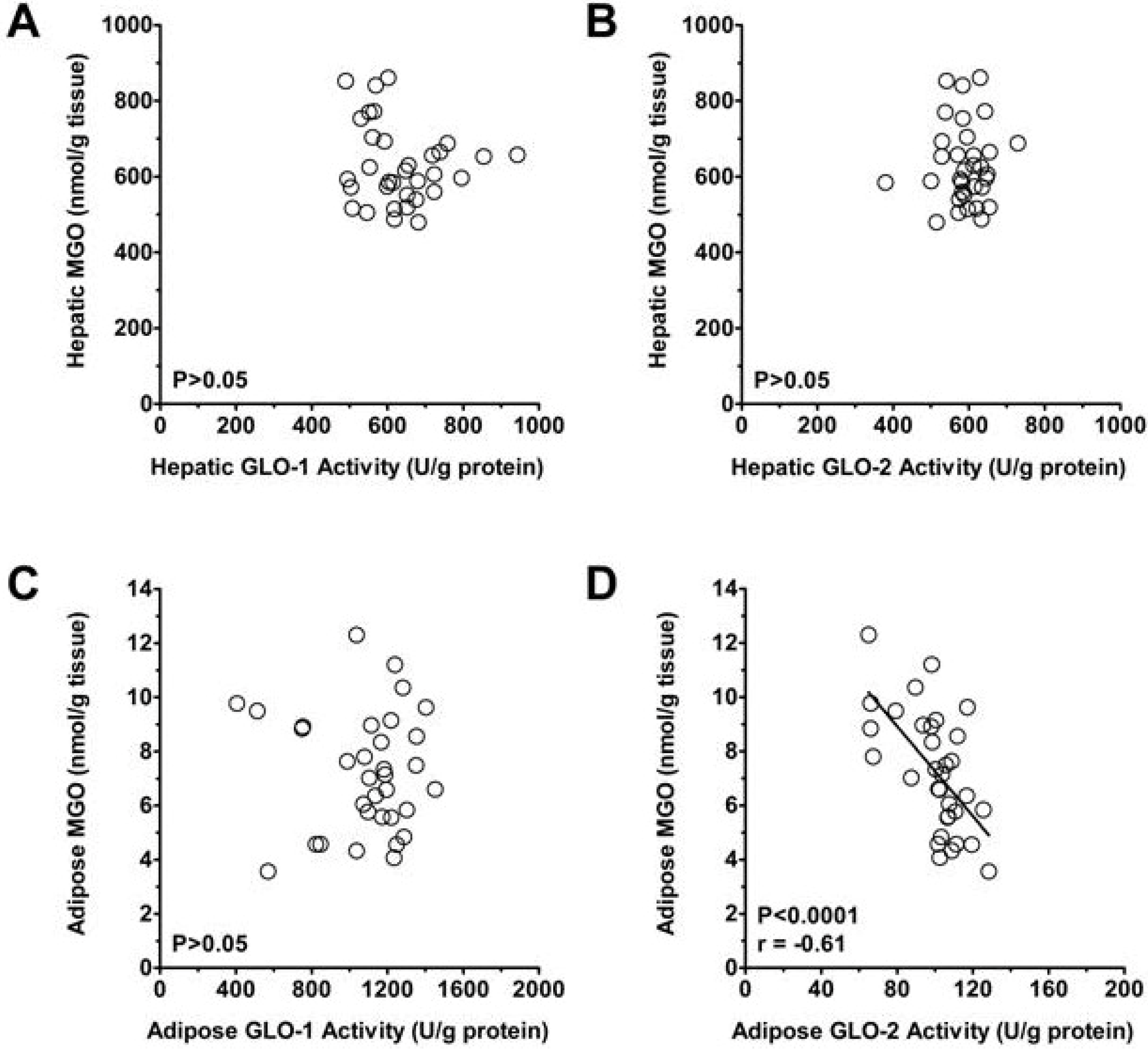
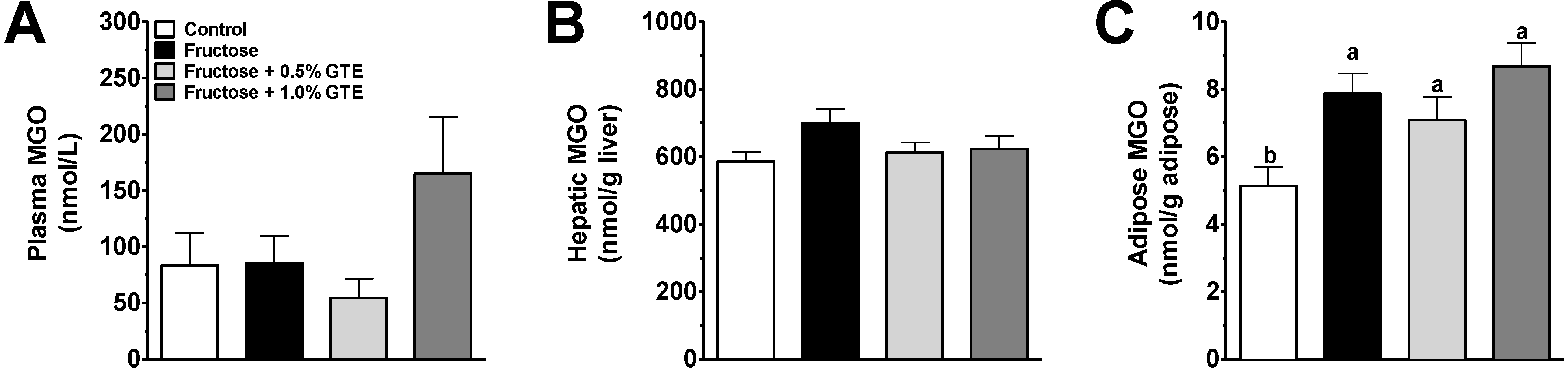
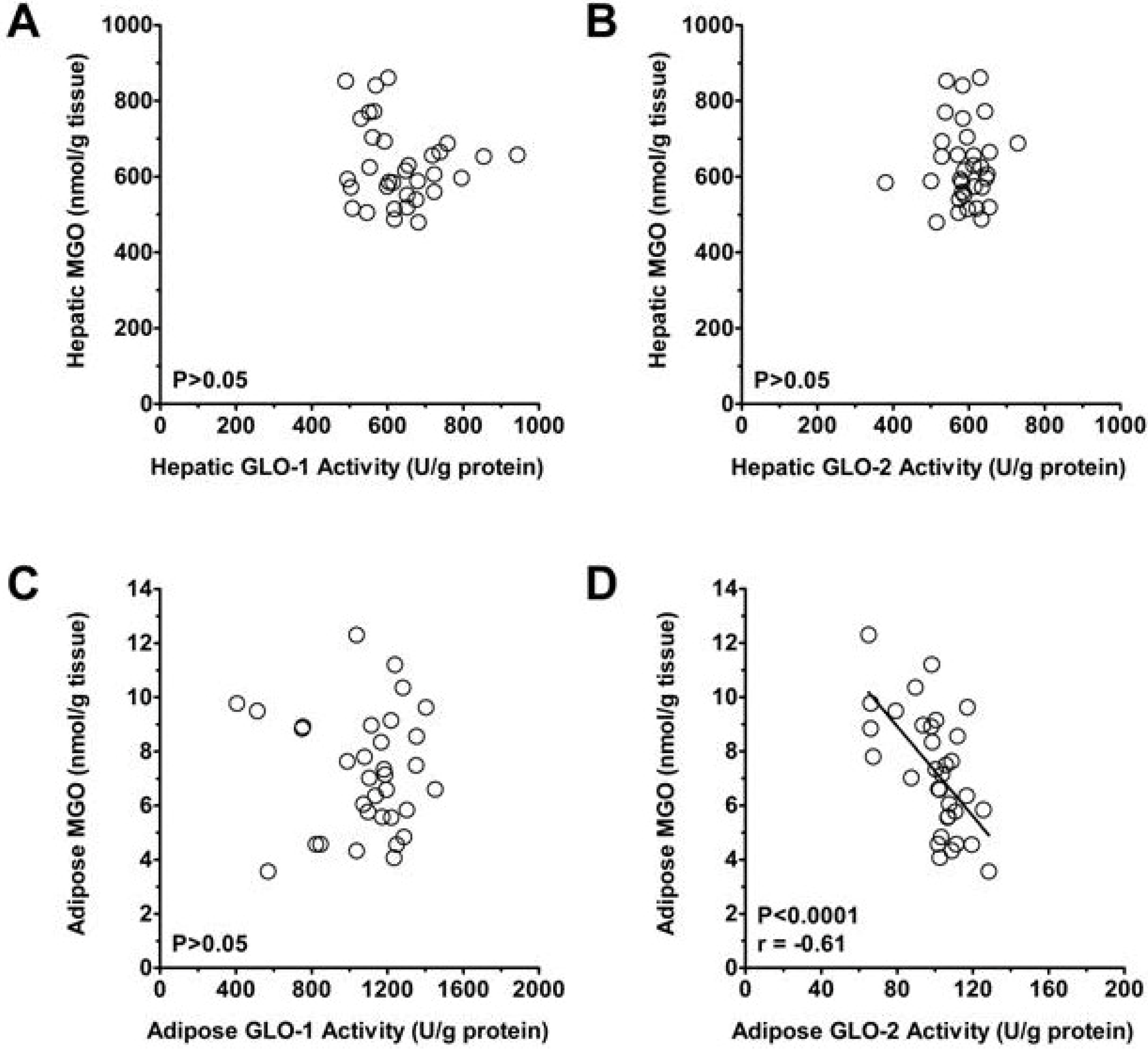
3.2. Plasma and Tissue MGO
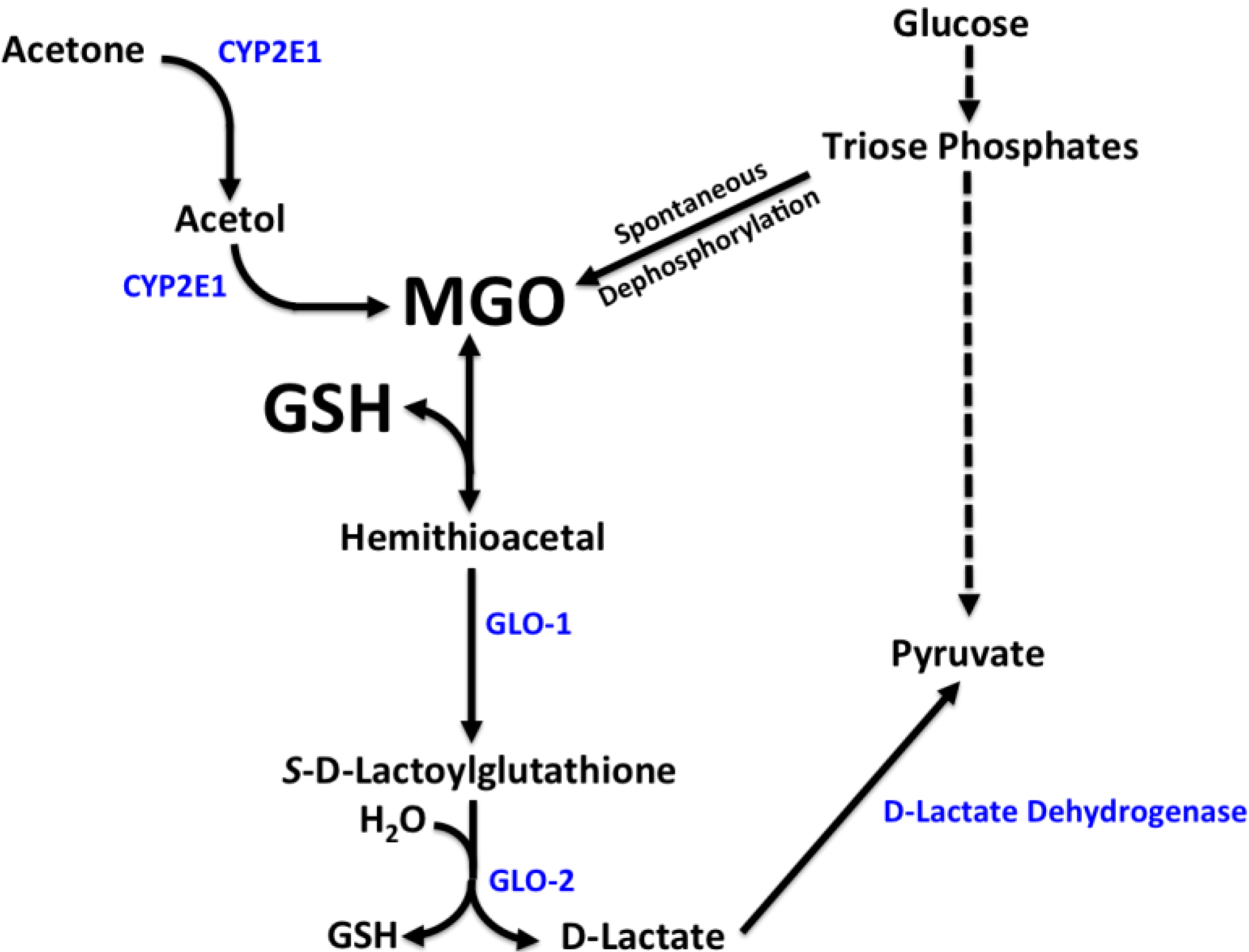
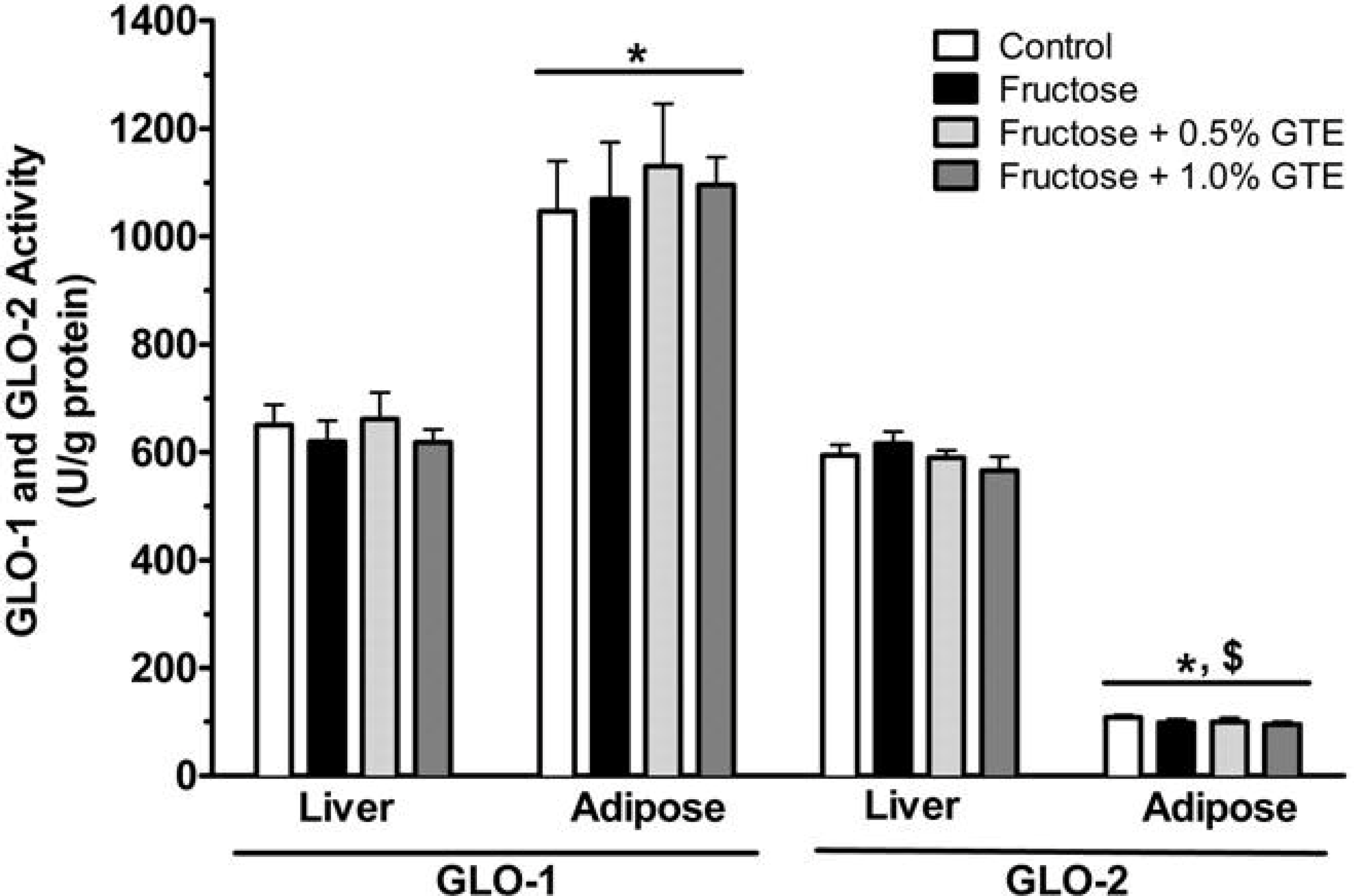
3.3. Methylglyoxal Metabolism

4. Discussion
5. Conclusions
Acknowledgments
Conflict of Interest
References
- Matsumura, Y.; Iwasawa, A.; Kobayashi, T.; Kamachi, T.; Ozawa, T.; Kohno, M. The reactivity of alpha-oxoaldehyde with reactive oxygen species in diabetes complications. J. Clin. Biochem. Nutr. 2013, 52, 128–132. [Google Scholar] [CrossRef]
- Thornalley, P.J. Glyoxalase I—Structure, function and a critical role in the enzymatic defence against glycation. Biochem. Soc. Trans. 2003, 31, 1343–1348. [Google Scholar] [CrossRef]
- McLellan, A.C.; Thornalley, P.J.; Benn, J.; Sonksen, P.H. Glyoxalase system in clinical diabetes mellitus and correlation with diabetic complications. Clin. Sci. (Lond.) 1994, 87, 21–29. [Google Scholar]
- Dhar, A.; Desai, K.M.; Wu, L. Alagebrium attenuates acute methylglyoxal-induced glucose intolerance in Sprague-Dawley rats. Br. J. Pharmacol. 2010, 159, 166–175. [Google Scholar] [CrossRef]
- Dhar, A.; Dhar, I.; Jiang, B.; Desai, K.M.; Wu, L. Chronic methylglyoxal infusion by minipump causes pancreatic beta-cell dysfunction and induces type 2 diabetes in Sprague-Dawley rats. Diabetes 2011, 60, 899–908. [Google Scholar]
- Brouwers, O.; Niessen, P.M.; Haenen, G.; Miyata, T.; Brownlee, M.; Stehouwer, C.D.; De Mey, J.G.; Schalkwijk, C.G. Hyperglycaemia-induced impairment of endothelium-dependent vasorelaxation in rat mesenteric arteries is mediated by intracellular methylglyoxal levels in a pathway dependent on oxidative stress. Diabetologia 2010, 53, 989–1000. [Google Scholar] [CrossRef]
- Giesecke, D.; Fabritius, A.; Wallenberg, P.V. A quantitative study on the metabolism of d(−) lactic acid in the rat and the rabbit. Comp. Biochem. Physiol. B 1981, 69, 85–89. [Google Scholar]
- Bray, G.A.; Nielsen, S.J.; Popkin, B.M. Consumption of high-fructose corn syrup in beverages may play a role in the epidemic of obesity. Am. J. Clin. Nutr. 2004, 79, 537–543. [Google Scholar]
- Ford, E.S.; Li, C.; Zhao, G.; Tsai, J. Trends in obesity and abdominal obesity among adults in the United States from 1999–2008. Int. J. Obes. (Lond.) 2011, 35, 736–743. [Google Scholar] [CrossRef]
- Cowie, C.C.; Rust, K.F.; Ford, E.S.; Eberhardt, M.S.; Byrd-Holt, D.D.; Li, C.; Williams, D.E.; Gregg, E.W.; Bainbridge, K.E.; Saydah, S.H.; et al. Full accounting of diabetes and pre-diabetes in the U.S. population in 1988–1994 and 2005–2006. Diabetes Care 2009, 32, 287–294. [Google Scholar]
- Malik, V.S.; Hu, F.B. Sweeteners and risk of obesity and type 2 diabetes: The role of sugar-sweetened beverages. Curr. Diabetes Rep. 2012. [Google Scholar] [CrossRef]
- Jia, X.; Wu, L. Accumulation of endogenous methylglyoxal impaired insulin signaling in adipose tissue of fructose-fed rats. Mol. Cell. Biochem. 2007, 306, 133–139. [Google Scholar] [CrossRef]
- Ackerman, Z.; Oron-Herman, M.; Grozovski, M.; Rosenthal, T.; Pappo, O.; Link, G.; Sela, B.A. Fructose-induced fatty liver disease: Hepatic effects of blood pressure and plasma triglyceride reduction. Hypertension 2005, 45, 1012–1018. [Google Scholar] [CrossRef]
- Wang, X.; Jia, X.; Chang, T.; Desai, K.; Wu, L. Attenuation of hypertension development by scavenging methylglyoxal in fructose-treated rats. J. Hypertens. 2008, 26, 765–772. [Google Scholar] [CrossRef]
- Lo, C.Y.; Li, S.; Tan, D.; Pan, M.H.; Sang, S.; Ho, C.T. Trapping reactions of reactive carbonyl species with tea polyphenols in simulated physiological conditions. Mol. Nutr. Food Res. 2006, 50, 1118–1128. [Google Scholar] [CrossRef]
- Sang, S.; Shao, X.; Bai, N.; Lo, C.Y.; Yang, C.S.; Ho, C.T. Tea polyphenol (−)-epigallocatechin-3-gallate: A new trapping agent of reactive dicarbonyl species. Chem. Res. Toxicol. 2007, 20, 1862–1870. [Google Scholar] [CrossRef]
- Totlani, V.M.; Peterson, D.G. Epicatechin carbonyl-trapping reactions in aqueous maillard systems: Identification and structural elucidation. J. Agric. Food Chem. 2006, 54, 7311–7318. [Google Scholar] [CrossRef]
- Tan, D.; Wang, Y.; Lo, C.Y.; Sang, S.; Ho, C.T. Methylglyoxal: Its presence in beverages and potential scavengers. Ann. N. Y. Acad. Sci. 2008, 1126, 72–75. [Google Scholar]
- Noda, Y.; Peterson, D.G. Structure-reactivity relationships of flavan-3-ols on product generation in aqueous glucose/glycine model systems. J. Agric. Food Chem. 2007, 55, 3686–3691. [Google Scholar] [CrossRef]
- Park, H.J.; DiNatale, D.A.; Chung, M.Y.; Park, Y.K.; Lee, J.Y.; Koo, S.I.; O’Connor, M.; Manautou, J.E.; Bruno, R.S. Green tea extract attenuates hepatic steatosis by decreasing adipose lipogenesis and enhancing hepatic antioxidant defenses in ob/ob mice. J. Nutr. Biochem. 2011, 22, 393–400. [Google Scholar]
- Park, H.J.; Lee, J.Y.; Chung, M.Y.; Park, Y.K.; Bower, A.M.; Koo, S.I.; Giardina, C.; Bruno, R.S. Green tea extract suppresses NFkappaB activation and inflammatory responses in diet-induced obese rats with nonalcoholic steatohepatitis. J. Nutr. 2012, 142, 57–63. [Google Scholar] [CrossRef]
- Masterjohn, C.; Bruno, R.S. Therapeutic potential of green tea in nonalcoholic fatty liver disease. Nutr. Rev. 2012, 70, 41–56. [Google Scholar] [CrossRef]
- Lindeberg, S.; Berntorp, E.; Nilsson-Ehle, P.; Terent, A.; Vessby, B. Age relations of cardiovascular risk factors in a traditional Melanesian society: The Kitava Study. Am. J. Clin. Nutr. 1997, 66, 845–852. [Google Scholar]
- Casazza, J.P.; Felver, M.E.; Veech, R.L. The metabolism of acetone in rat. J. Biol. Chem. 1984, 259, 231–236. [Google Scholar]
- Shrestha, S.; Ehlers, S.J.; Lee, J.Y.; Fernandez, M.L.; Koo, S.I. Dietary green tea extract lowers plasma and hepatic triglycerides and decreases the expression of sterol regulatory element-binding protein-1c mRNA and its responsive genes in fructose-fed, ovariectomized rats. J. Nutr. 2009, 139, 640–645. [Google Scholar]
- Klevay, L.M. The biotin requirement of rats fed 20% egg white. J. Nutr. 1976, 106, 1643–1646. [Google Scholar]
- Bruno, R.S.; Dugan, C.E.; Smyth, J.A.; DiNatale, D.A.; Koo, S.I. Green tea extract protects leptin-deficient, spontaneously obese mice from hepatic steatosis and injury. J. Nutr. 2008, 138, 323–331. [Google Scholar]
- Matsui, T.; Soya, S.; Okamoto, M.; Ichitani, Y.; Kawanaka, K.; Soya, H. Brain glycogen decreases during prolonged exercise. J. Physiol. 2011, 589, 3383–3393. [Google Scholar] [Green Version]
- Masterjohn, C.; Mah, E.; Guo, Y.; Koo, S.I.; Bruno, R.S. gamma-Tocopherol abolishes postprandial increases in plasma methylglyoxal following an oral dose of glucose in healthy, college-aged men. J. Nutr. Biochem. 2012, 23, 292–298. [Google Scholar] [CrossRef]
- Thornalley, P.J. Modification of the glyoxalase system in human red blood cells by glucose in vitro. Biochem. J. 1988, 254, 751–755. [Google Scholar]
- Yang, Y.; Seo, J.M.; Nguyen, A.; Pham, T.X.; Park, H.J.; Park, Y.; Kim, B.; Bruno, R.S.; Lee, J. Astaxanthin-rich extract from the green alga Haematococcus pluvialis lowers plasma lipid concentrations and enhances antioxidant defense in apolipoprotein E knockout mice. J. Nutr. 2011, 141, 1611–1617. [Google Scholar] [CrossRef]
- National Center for Biotechnology Information. Available online: http://www.ncbi.nlm.nih.gov (accessed on 07 November 2011).
- Juan, C.C.; Au, L.C.; Fang, V.S.; Kang, S.F.; Ko, Y.H.; Kuo, S.F.; Hsu, Y.P.; Kwok, C.F.; Ho, L.T. Suppressed gene expression of adipocyte resistin in an insulin-resistant rat model probably by elevated free fatty acids. Biochem. Biophys. Res. Commun. 2001, 289, 1328–1333. [Google Scholar]
- Hara, T.; Cameron-Smith, D.; Cooney, G.J.; Kusunoki, M.; Tsutsumi, K.; Storlien, L.H. The actions of a novel lipoprotein lipase activator, NO-1886, in hypertriglyceridemic fructose-fed rats. Metabolism 1998, 47, 149–153. [Google Scholar] [CrossRef]
- Blakely, S.R.; Akintilo, A.O.; Pointer, R.H. Effects of fructose, levamisole and vanadate on insulin action in rat adipose tissue. J. Nutr. 1987, 117, 559–566. [Google Scholar]
- Shapiro, A.; Mu, W.; Roncal, C.; Cheng, K.Y.; Johnson, R.J.; Scarpace, P.J. Fructose-induced leptin resistance exacerbates weight gain in response to subsequent high-fat feeding. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2008, 295, R1370–R1375. [Google Scholar] [CrossRef]
- Riby, J.E.; Fujisawa, T.; Kretchmer, N. Fructose absorption. Am. J. Clin. Nutr. 1993, 58, 748S–753S. [Google Scholar]
- Blaak, E.E.; Saris, W.H. Postprandial thermogenesis and substrate utilization after ingestion of different dietary carbohydrates. Metabolism 1996, 45, 1235–1242. [Google Scholar]
- Thorburn, A.W.; Storlien, L.H.; Jenkins, A.B.; Khouri, S.; Kraegen, E.W. Fructose-induced in vivo insulin resistance and elevated plasma triglyceride levels in rats. Am. J. Clin. Nutr. 1989, 49, 1155–1163. [Google Scholar]
- Jerzykowski, T.; Winter, R.; Matuszewski, W.; Piskorska, D. A re-evaluation of studies on the distribution of glyoxalases in animal and tumour tissues. Int. J. Biochem. 1978, 9, 853–860. [Google Scholar] [CrossRef]
- Kawase, M.; Kondoh, C.; Matsumoto, S.; Teshigawara, M.; Chisaka, Y.; Higashiura, M.; Nakata, K.; Ohmori, S. Contents of d-lactate and its related metabolites as well as enzyme activities in the liver, muscle and blood plasma of aging rats. Mech. Ageing Dev. 1995, 84, 55–63. [Google Scholar] [CrossRef]
- Larsen, K.; Aronsson, A.C.; Marmstal, E.; Mannervik, B. Immunological comparison of glyoxalase I from yeast and mammals and quantitative determination of the enzyme in human tissues by radioimmunoassay. Comp. Biochem. Physiol. B 1985, 82, 625–638. [Google Scholar]
- Xue, M.; Rabbani, N.; Thornalley, P.J. Glyoxalase in ageing. Semin. Cell. Dev. Biol. 2011, 22, 293–301. [Google Scholar]
- Xue, M.; Rabbani, N.; Momiji, H.; Imbasi, P.; Anwar, M.M.; Kitteringham, N.; Park, B.K.; Souma, T.; Moriguchi, T.; Yamamoto, M.; et al. Transcriptional control of glyoxalase 1 by Nrf2 provides a stress-responsive defence against dicarbonyl glycation. Biochem. J. 2012, 443, 213–222. [Google Scholar] [CrossRef]
- Castro, M.C.; Massa, M.L.; Schinella, G.; Gagliardino, J.J.; Francini, F. Lipoic acid prevents liver metabolic changes induced by administration of a fructose-rich diet. Biochim. Biophys. Acta 2013, 1830, 2226–2232. [Google Scholar] [CrossRef]
- Ranganathan, S.; Ciaccio, P.J.; Walsh, E.S.; Tew, K.D. Genomic sequence of human glyoxalase-I: Analysis of promoter activity and its regulation. Gene 1999, 240, 149–155. [Google Scholar] [CrossRef]
- Phillips, S.A.; Thornalley, P.J. The formation of methylglyoxal from triose phosphates. Investigation using a specific assay for methylglyoxal. Eur. J. Biochem. 1993, 212, 101–105. [Google Scholar] [CrossRef]
- Yoshinari, K.; Sato, T.; Okino, N.; Sugatani, J.; Miwa, M. Expression and induction of cytochromes p450 in rat white adipose tissue. J. Pharmacol. Exp. Ther. 2004, 311, 147–154. [Google Scholar]
- Song, B.J.; Veech, R.L.; Park, S.S.; Gelboin, H.V.; Gonzalez, F.J. Induction of rat hepatic N-nitrosodimethylamine demethylase by acetone is due to protein stabilization. J. Biol. Chem. 1989, 264, 3568–3572. [Google Scholar]
- Dhar, A.; Desai, K.; Kazachmov, M.; Yu, P.; Wu, L. Methylglyoxal production in vascular smooth muscle cells from different metabolic precursors. Metabolism 2008, 57, 1211–1220. [Google Scholar]
- Beisswenger, B.G.; Delucia, E.M.; Lapoint, N.; Sanford, R.J.; Beisswenger, P.J. Ketosis leads to increased methylglyoxal production on the Atkins diet. Ann. N. Y. Acad. Sci. 2005, 1043, 201–210. [Google Scholar] [CrossRef]
- Nagai, R.; Nagai, M.; Shimasaki, S.; Baynes, J.W.; Fujiwara, Y. Citric acid inhibits development of cataracts, proteinuria and ketosis in streptozotocin (type 1) diabetic rats. Biochem. Biophys. Res. Commun. 2010, 393, 118–122. [Google Scholar]
- Abdel-Sayed, A.; Binnert, C.; Le, K.A.; Bortolotti, M.; Schneiter, P.; Tappy, L. A high-fructose diet impairs basal and stress-mediated lipid metabolism in healthy male subjects. Br. J. Nutr. 2008, 100, 393–399. [Google Scholar]
- Wolfe, B.E.; Jimerson, D.C.; Orlova, C.; Mantzoros, C.S. Effect of dieting on plasma leptin, soluble leptin receptor, adiponectin and resistin levels in healthy volunteers. Clin. Endocrinol. (Oxf.) 2004, 61, 332–338. [Google Scholar] [CrossRef]
- Reichard, G.A., Jr.; Haff, A.C.; Skutches, C.L.; Paul, P.; Holroyde, C.P.; Owen, O.E. Plasma acetone metabolism in the fasting human. J. Clin. Investig. 1979, 63, 619–626. [Google Scholar] [CrossRef]
- Musa-Veloso, K.; Likhodii, S.S.; Rarama, E.; Benoit, S.; Liu, Y.M.; Chartrand, D.; Curtis, R.; Carmant, L.; Lortie, A.; Comeau, F.J.; et al. Breath acetone predicts plasma ketone bodies in children with epilepsy on a ketogenic diet. Nutrition 2006, 22, 1–8. [Google Scholar] [CrossRef]
- Kizhner, T.; Werman, M.J. Long-term fructose intake: Biochemical consequences and altered renal histology in the male rat. Metabolism 2002, 51, 1538–1547. [Google Scholar] [CrossRef]
- Creighton, D.J.; Migliorini, M.; Pourmotabbed, T.; Guha, M.K. Optimization of efficiency in the glyoxalase pathway. Biochemistry 1988, 27, 7376–7384. [Google Scholar] [CrossRef]
- Madian, A.G.; Myracle, A.D.; Diaz-Maldonado, N.; Rochelle, N.S.; Janle, E.M.; Regnier, F.E. Determining the effects of antioxidants on oxidative stress induced carbonylation of proteins. Anal. Chem. 2011, 83, 9328–9336. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Masterjohn, C.; Park, Y.; Lee, J.; Noh, S.K.; Koo, S.I.; Bruno, R.S. Dietary Fructose Feeding Increases Adipose Methylglyoxal Accumulation in Rats in Association with Low Expression and Activity of Glyoxalase-2. Nutrients 2013, 5, 3311-3328. https://doi.org/10.3390/nu5083311
Masterjohn C, Park Y, Lee J, Noh SK, Koo SI, Bruno RS. Dietary Fructose Feeding Increases Adipose Methylglyoxal Accumulation in Rats in Association with Low Expression and Activity of Glyoxalase-2. Nutrients. 2013; 5(8):3311-3328. https://doi.org/10.3390/nu5083311
Chicago/Turabian StyleMasterjohn, Christopher, Youngki Park, Jiyoung Lee, Sang K. Noh, Sung I. Koo, and Richard S. Bruno. 2013. "Dietary Fructose Feeding Increases Adipose Methylglyoxal Accumulation in Rats in Association with Low Expression and Activity of Glyoxalase-2" Nutrients 5, no. 8: 3311-3328. https://doi.org/10.3390/nu5083311