Vitamin C: A Novel Regulator of Neutrophil Extracellular Trap Formation

Abstract
:1. Introduction
2. Experimental Section
2.1. Animals
2.1.1. Feces Induced Peritonitis
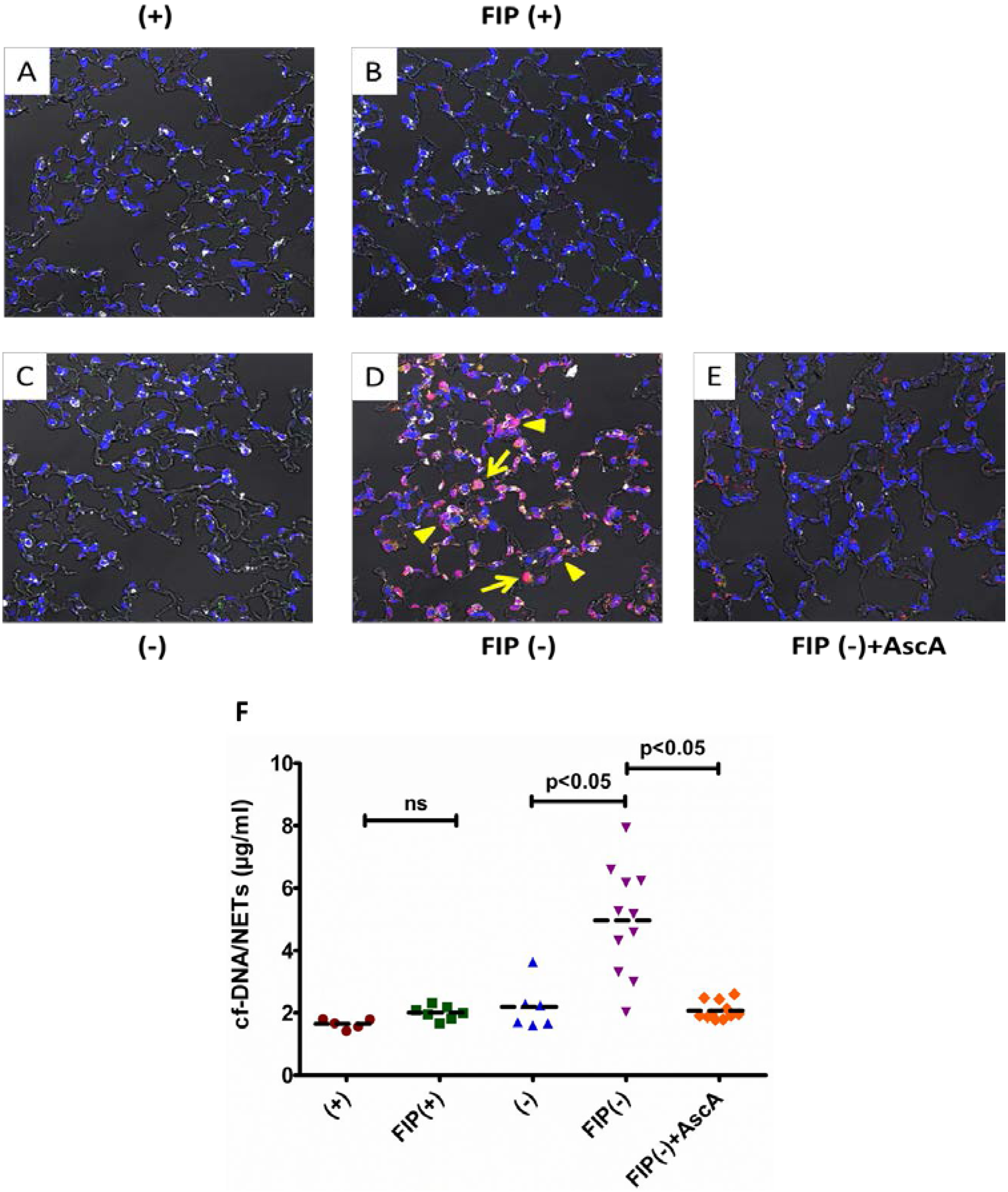
2.1.2. Gulo−/− Mice Were Divided into Five Groups
- (1)
- (+): vitamin C sufficient mice received saline alone (0.4 mL, i.p.)
- (2)
- FIP(+): vitamin C sufficient mice received fecal stem solution (0.4 mL, i.p.) followed 30 min later by saline (0.1 mL, i.p.)
- (3)
- (−): vitamin C deficient mice received saline alone (0.4 mL, i.p.)
- (4)
- FIP(−): vitamin C deficient mice received fecal stem solution (0.4 mL, i.p.) followed 30 min later by saline (0.1 mL, i.p.)
- (5)
- FIP(−) + AscA: vitamin C deficient mice received fecal stem solution (0.4 mL, i.p.) followed 30 min later by ascorbic acid (0.1 mL, i.p.)
2.2. Isolation of Mouse Peritoneal Neutrophils
2.3. Immunofluorescence and Differential Interference Contrast Imaging of Lung NETs
2.4. RNA Isolation and Real-Time Quantitative PCR (QPCR) Analysis

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Sequence 5′ to 3′ |
|---|---|
| ATF4 forward | CCTAGGTCTCTTAGATGACTATCTGGAGG |
| ATF4 reverse | CCAGGTCATCCATTCGAAACAGAGCATCG |
| BiP forward | GTGCAGCAGGACATCAAGTTCTTGCC |
| BiP reverse | TTCCCAAATACGCCTCAGCAGTCTCC |
| CHOP forward | CACCTATATCTCATCCCCAGGAAACG |
| CHOP reverse | TTCCTTGCTCTTCCTCCTCTTCCTCC |
| EDEM forward | GCCCTTTGGTGACATGACAATTGAGG |
| EDEM reverse | TCATTATTGCTGTCAGGAGGAACACC |
| XBP-1s forward | TGAGTCCGCAGCAGGTGC |
| XBP-1s reverse | CAACTTGTCCAGAATGCCCAAAAGG |
| XBP-1un forward | AAGAACACGCTTGGGAATGGACACGC |
| XBP-1un reverse | ACCTGCTGCAGAGGTGCACATAGTC |
| PAD4 forward | ACAGGTGAAAGCAGCCAGC |
| PAD4 reverse | AGTGATGTAGATCAGGGCTTGG |
| ATG3 forward | CACCACTGTCCAACATGGC |
| ATG3 reverse | GTTTACACCGCTTGTAGCATGG |
| ATG5 forward | ACAAGCAGCTCTGGATGGG |
| ATG5 reverse | GGAGGATATTCCATGAGTTTCCG |
| ATG6 forward | CACGAGCTTCAAGATCCTGG |
| ATG6 reverse | TCCTGAGTTAGCCTCTTCCTCC |
| ATG7 forward | ACGATGACGACACTGTTCTGG |
| ATG7 reverse | AGGTTACAGGGATCGTACACACC |
| ATG8 forward | ACAAAGAGTGGAAGATGTCCG |
| ATG8 reverse | GGAACTTGGTCTTGTCCAGG |
| TNFα forward | GATGAGAAGTTCCCAAATGGC |
| TNFα reverse | TTGGTGGTTTGCTACGACG |
| IL-1β forward | CTGAACTCAACTGTGAAATGCC |
| IL-1β reverse | CAGGTCAAAGGTTTGGAAGC |
| 18S forward | GATAGCTCTTTCTCGATTCCG |
| 18S reverse | AGAGTCTCGTTCGTTATCGG |
2.5. Western Blot Analysis
2.6. Isolation of Human Neutrophils and NETs Release
2.7. Immunofluorescence Staining of Human NETs
2.8. Quantification of Cell Free DNA
2.9. Statistical Analysis

3. Results
3.1. Vitamin C Sufficient Mice Demonstrate Reduced Lung NETs and Lower cf-DNA Following Peritonitis-Induced Sepsis
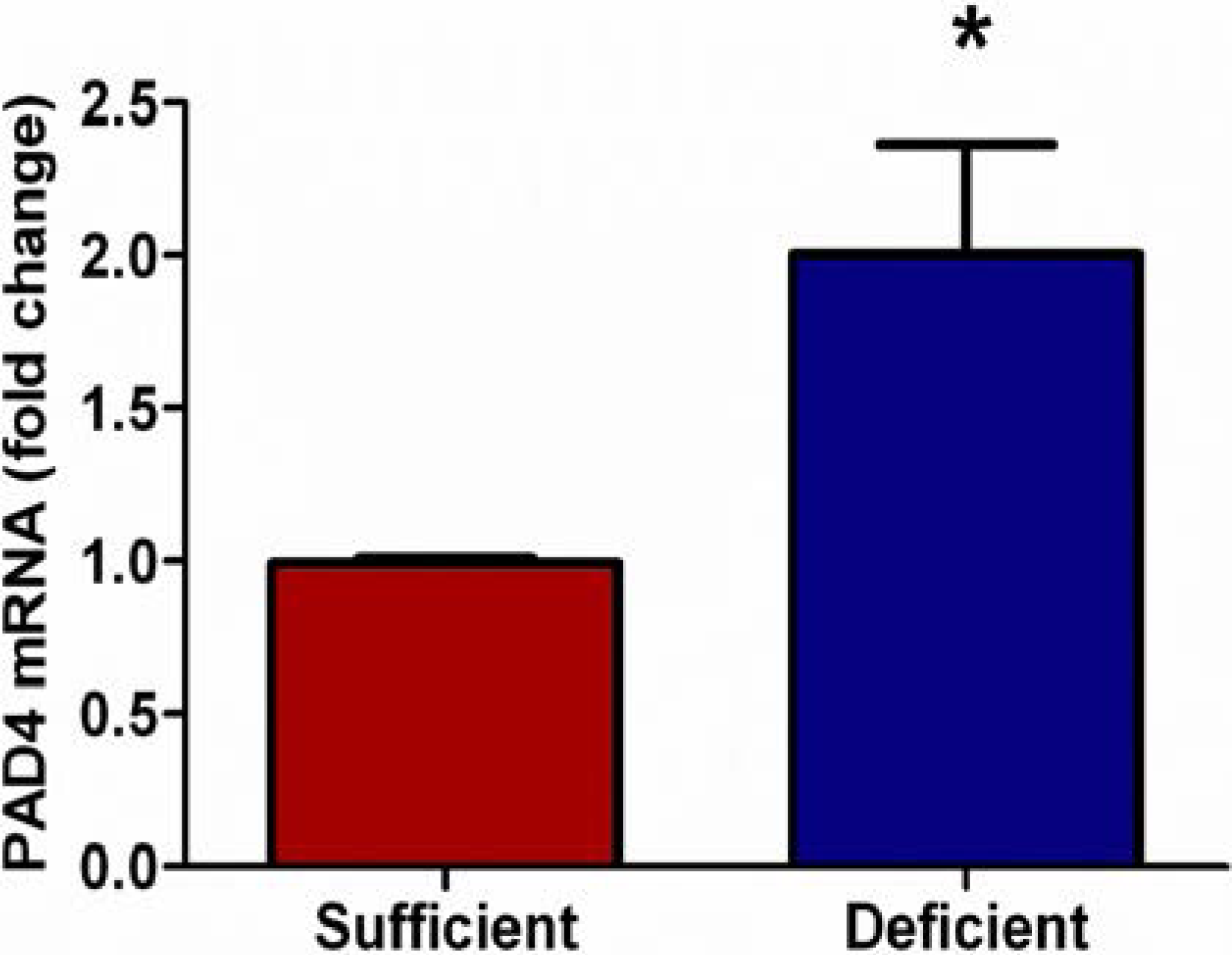
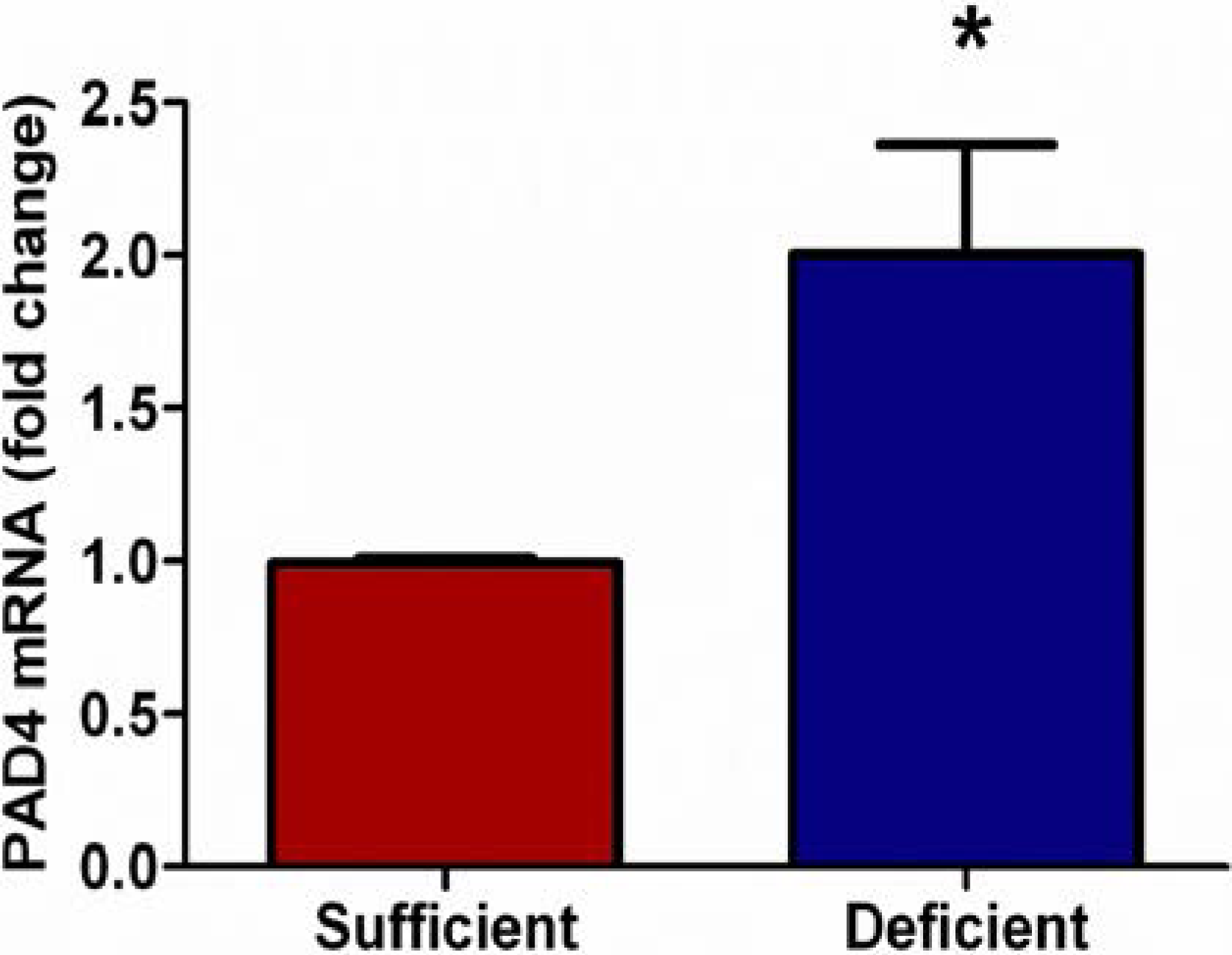
3.2. Vitamin C Deficient Neutrophils Show Increased PAD4 mRNA
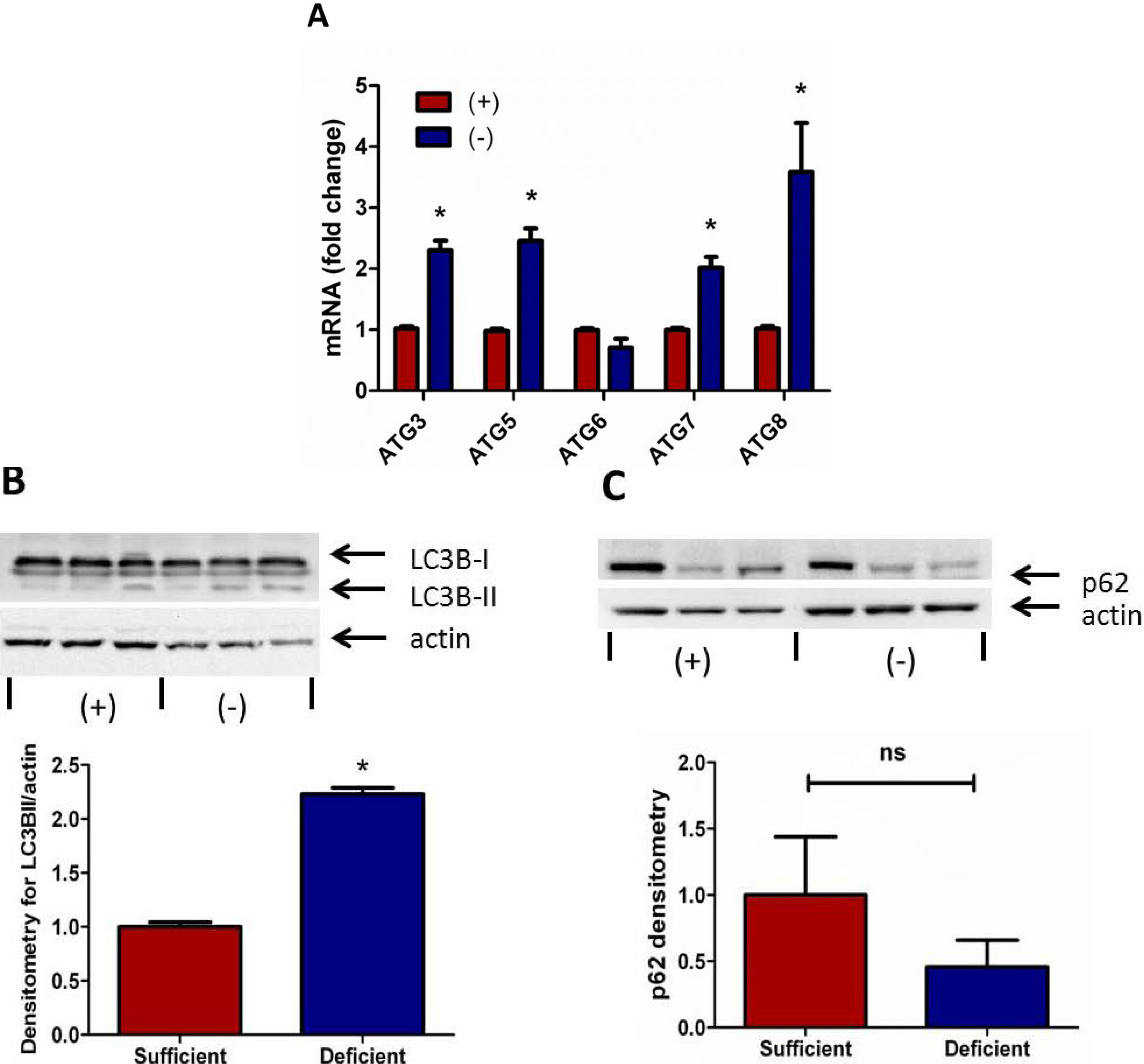
3.3. Autophagy Signaling Is Induced in Vitamin C Deficient Neutrophils


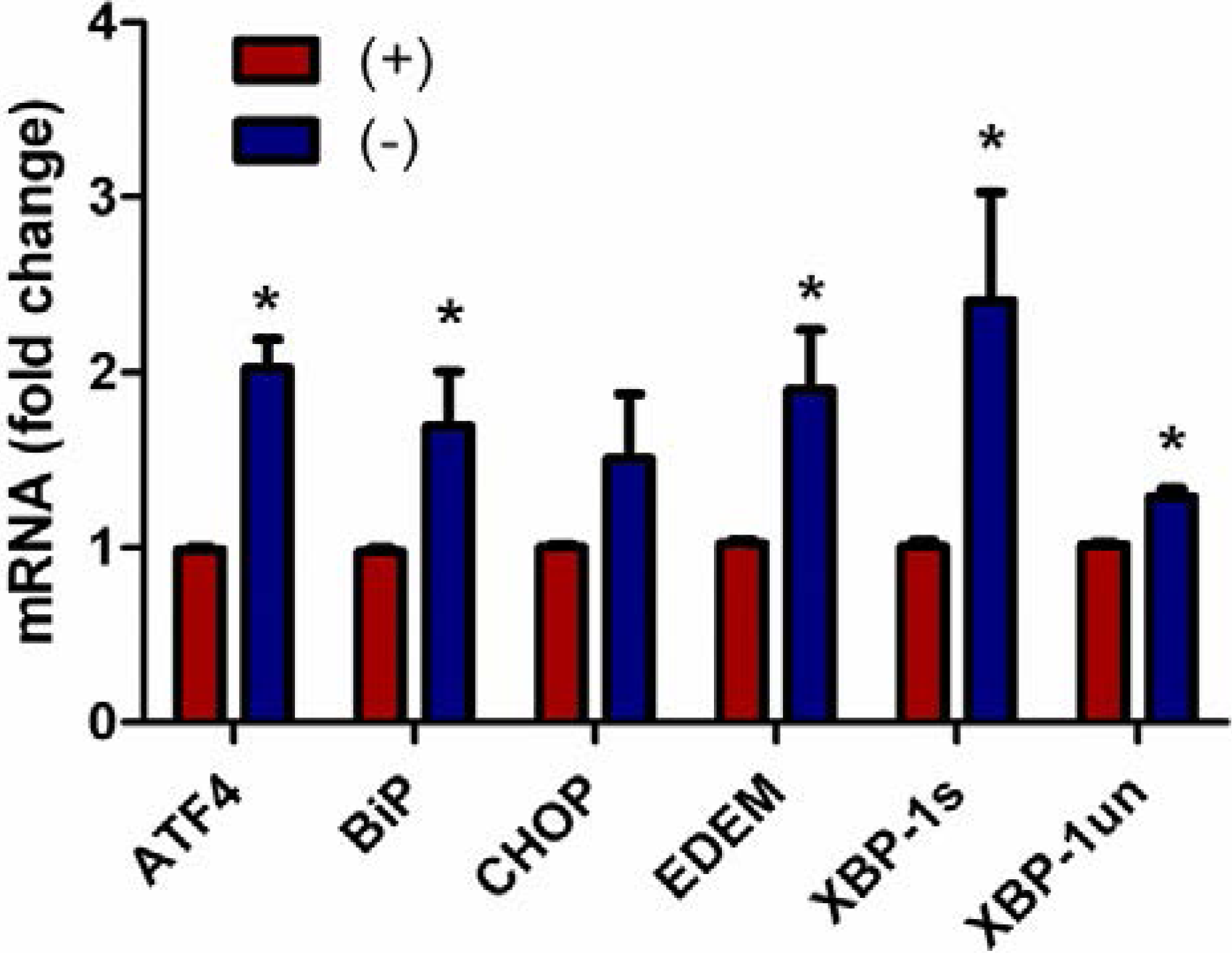
3.4. Endoplasmic Reticulum Stress Associated Gene Expression Is Up-Regulated in Vitamin C Deficient Neutrophils
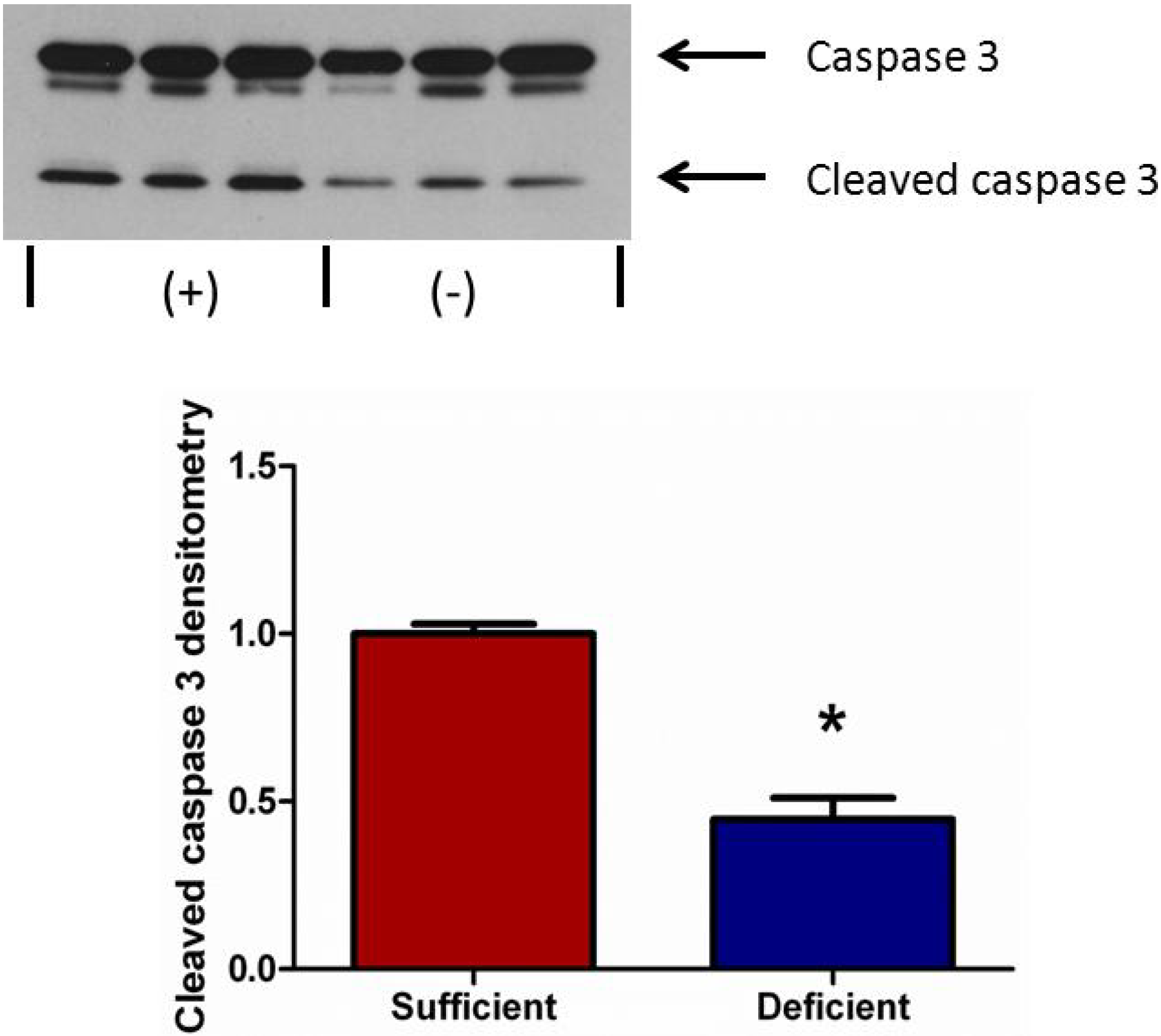
3.5. Vitamin C Deficient Neutrophils Undergo Attenuated Apoptosis

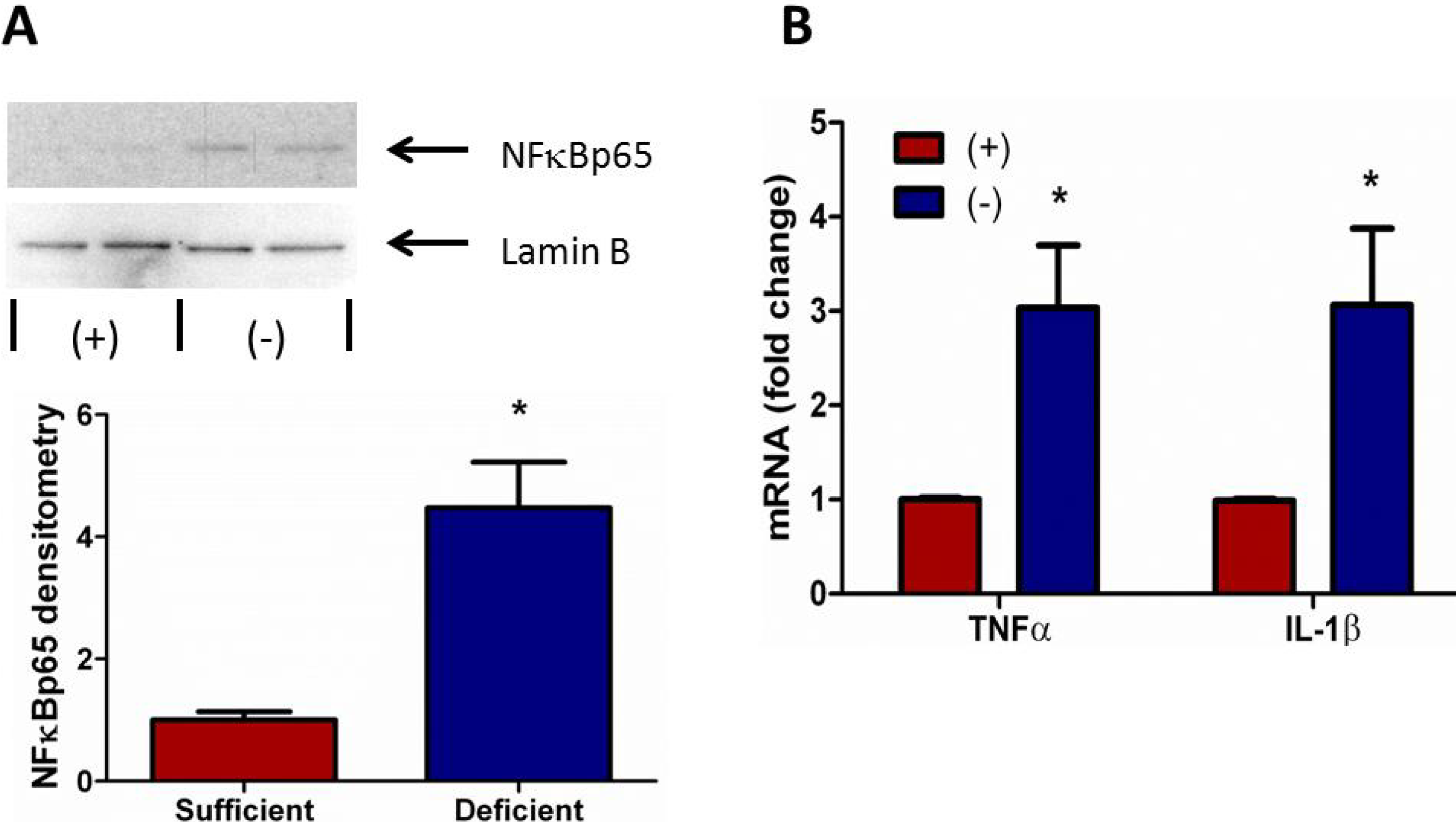
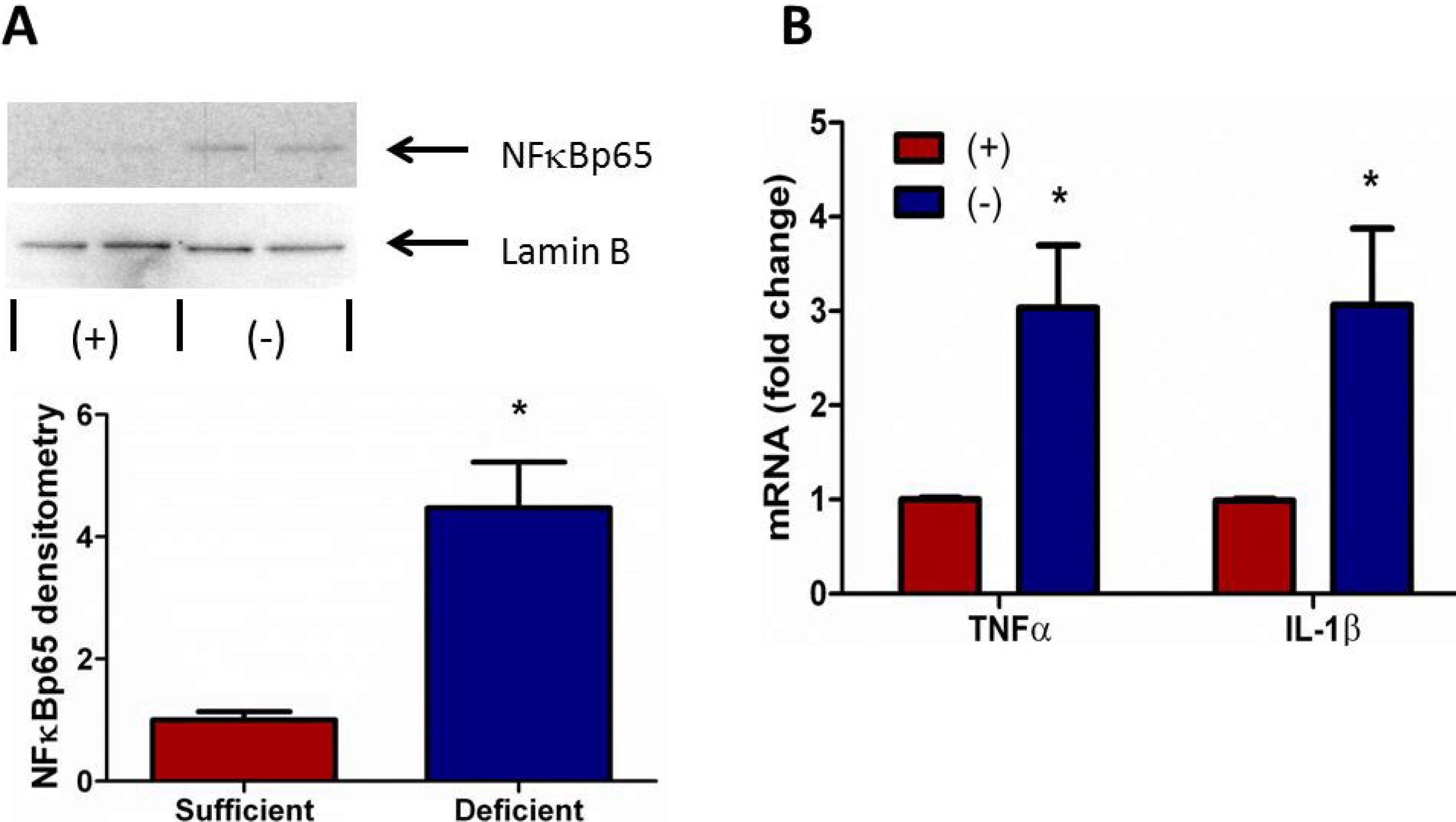
3.6. Vitamin C Deficient Neutrophils Exhibit Increased NFκB Activation
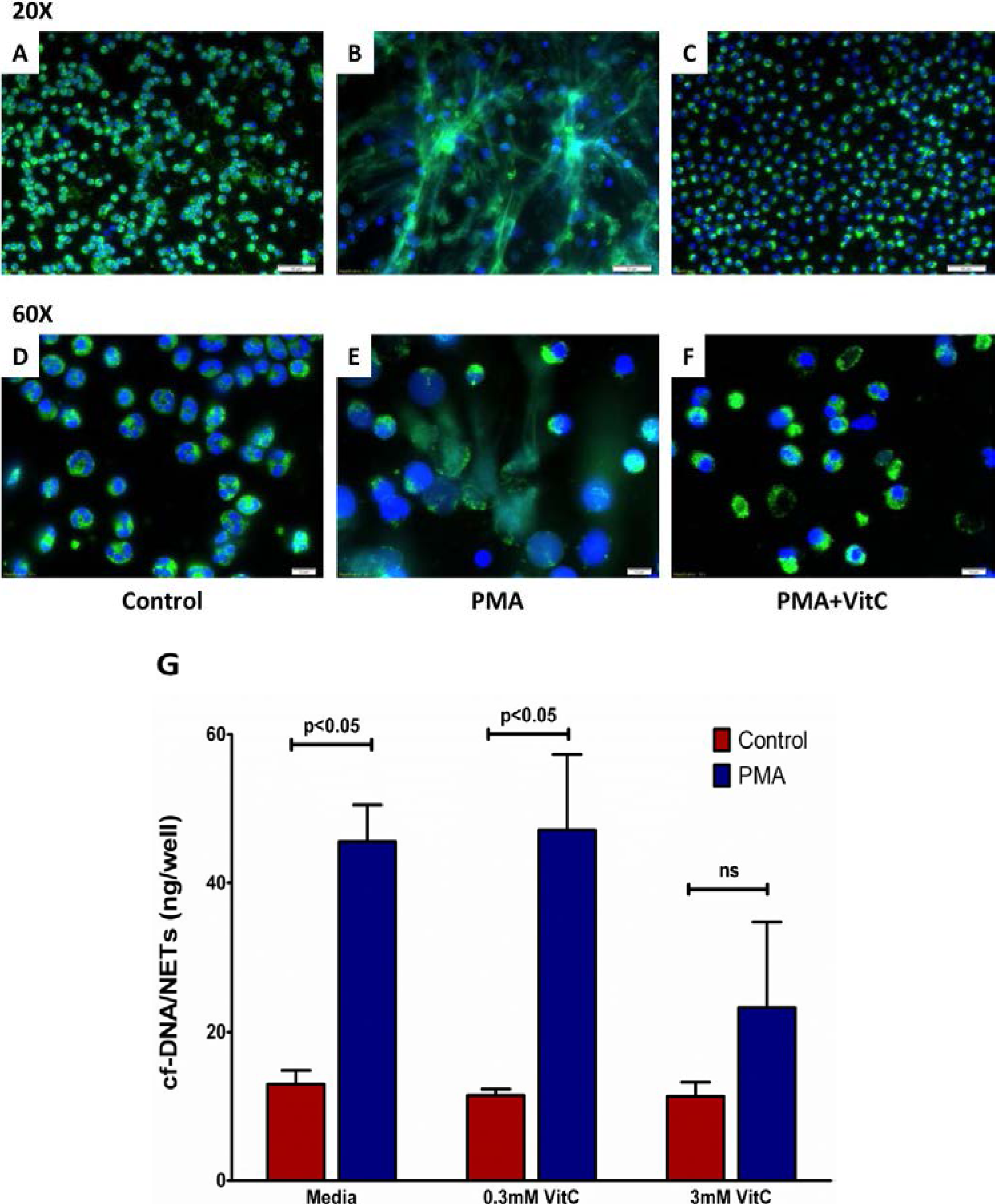
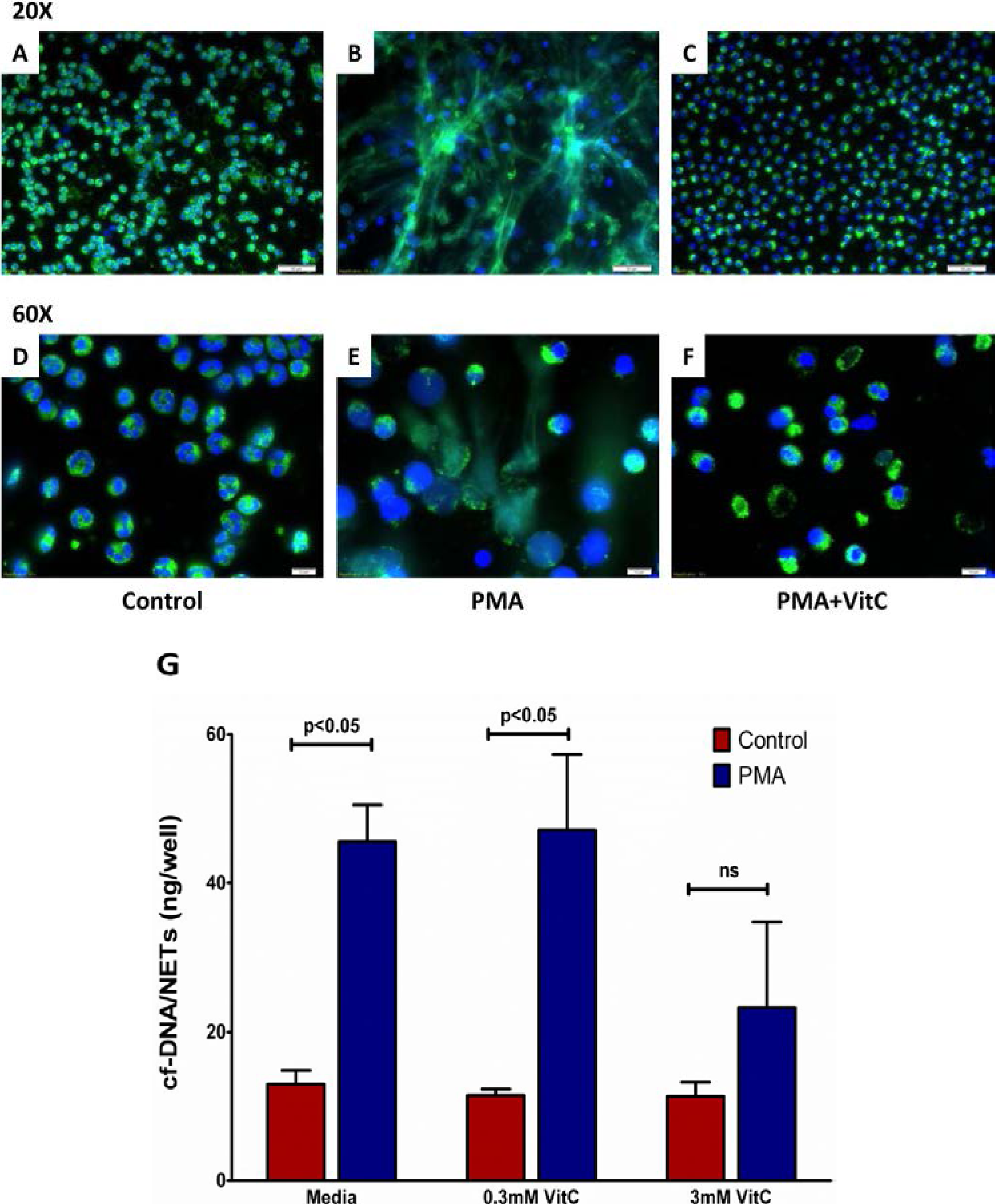
3.7. Vitamin C Attenuates NET Formation in Activated Human Neutrophils
4. Discussion

5. Conclusions
Acknowledgments
Conflict of Interest
References
- Winterbourn, C.C.; Kettle, A.J. Redox reactions and microbial killing in the neutrophil phagosome. Antioxid. Redox Signal. 2013, 18, 642–660. [Google Scholar] [CrossRef]
- Borregaard, N. Neutrophils, from marrow to microbes. Immunity 2010, 33, 657–670. [Google Scholar] [CrossRef]
- Takei, H.; Araki, A.; Watanabe, H.; Ichinose, A.; Sendo, F. Rapid killing of human neutrophils by the potent activator phorbol 12-myristate 13-acetate (PMA) accompanied by changes different from typical apoptosis or necrosis. J. Leukoc. Biol. 1996, 59, 229–240. [Google Scholar]
- Brinkmann, V.; Reichard, U.; Goosmann, C.; Fauler, B.; Uhlemann, Y.; Weiss, D.S.; Weinrauch, Y.; Zychlinsky, A. Neutrophil extracellular traps kill bacteria. Science 2004, 303, 1532–1535. [Google Scholar] [CrossRef]
- Narasaraju, T.; Yang, E.; Samy, R.P.; Ng, H.H.; Poh, W.P.; Liew, A.A.; Phoon, M.C.; van Rooijen, N.; Chow, V.T. Excessive neutrophils and neutrophil extracellular traps contribute to acute lung injury of influenza pneumonitis. Am. J. Pathol. 2011, 179, 199–210. [Google Scholar] [CrossRef]
- Grommes, J.; Soehnlein, O. Contribution of neutrophils to acute lung injury. Mol. Med. 2011, 17, 293–307. [Google Scholar]
- Saffarzadeh, M.; Juenemann, C.; Queisser, M.A.; Lochnit, G.; Barreto, G.; Galuska, S.P.; Lohmeyer, J.; Preissner, K.T. Neutrophil extracellular traps directly induce epithelial and endothelial cell death: A predominant role of histones. PLoS One 2012, 7, e32366. [Google Scholar] [CrossRef]
- Douda, D.N.; Jackson, R.; Grasemann, H.; Palaniyar, N. Innate immune collectin surfactant protein D simultaneously binds both neutrophil extracellular traps and carbohydrate ligands and promotes bacterial trapping. J. Immunol. 2011, 187, 1856–1865. [Google Scholar] [CrossRef]
- Gupta, A.K.; Hasler, P.; Holzgreve, W.; Gebhardt, S.; Hahn, S. Induction of neutrophil extracellular DNA lattices by placental microparticles and IL-8 and their presence in preeclampsia. Hum. Immunol. 2005, 66, 1146–1154. [Google Scholar] [CrossRef]
- Papayannopoulos, V.; Zychlinsky, A. NETs: A new strategy for using old weapons. Trends Immunol. 2009, 30, 513–521. [Google Scholar] [CrossRef]
- Dwivedi, D.J.; Toltl, L.J.; Swystun, L.J.; Pogue, J.; Liaw, K.L.; Weitz, J.I.; Cook, D.J.; Fox-Robichaud, A.E.; Liaw, P.C.; Canadian Critical Care Translational Biology Group. Prognostic utility and characterization of cell-free DNA in patients with severe sepsis. Crit. Care 2012, 16, R151. [Google Scholar] [CrossRef]
- Fisher, B.J.; Seropian, I.M.; Kraskauskas, D.; Thakkar, J.N.; Voelkel, N.F.; Fowler, A.A.; Natarajan, R. Ascorbic acid attenuates lipopolysaccharide-induced acute lung injury. Crit. Care Med. 2011, 39, 1454–1460. [Google Scholar]
- Fisher, B.J.; Kraskauskas, D.; Martin, E.J.; Farkas, D.; Wegelin, J.A.; Brophy, D.; Ward, K.R.; Voelkel, N.F.; Fowler, A.A.; Natarajan, R. Mechanisms of attenuation of abdominal sepsis induced acute lung injury by ascorbic acid. Am. J. Physiol. Lung Cell. Mol. Physiol. 2012, 303, L20–L32. [Google Scholar] [CrossRef]
- Padayatty, S.J.; Katz, A.; Wang, Y.; Eck, P.; Kwon, O.; Lee, J.H.; Chen, S.; Corpe, C.; Dutta, A.; Dutta, S.K.; et al. Vitamin C as an antioxidant: evaluation of its role in disease prevention. J. Am. Coll. Nutr. 2003, 22, 18–35. [Google Scholar] [CrossRef]
- Vissers, M.C.; Wilkie, R.P. Ascorbate deficiency results in impaired neutrophil apoptosis and clearance and is associated with up-regulation of hypoxia-inducible factor 1α. J. Leukoc. Biol. 2007, 81, 1236–1244. [Google Scholar] [CrossRef]
- Nishikimi, M.; Fukuyama, R.; Minoshima, S.; Shimizu, N.; Yagi, K. Cloning and chromosomal mapping of the human nonfunctional gene for l-gulono-γ-lactone oxidase, the enzyme for l-ascorbic acid biosynthesis missing in man. J. Biol. Chem. 1994, 269, 13685–13688. [Google Scholar]
- Remijsen, Q.; Vanden Berghe, T.; Wirawan, E.; Asselbergh, B.; Parthoens, E.; de Rycke, R.; Noppen, S.; Delforge, M.; Willems, J.; Vandenabeele, P. Neutrophil extracellular trap cell death requires both autophagy and superoxide generation. Cell Res. 2011, 21, 290–304. [Google Scholar] [CrossRef]
- Wang, Y.; Li, M.; Stadler, S.; Correll, S.; Li, P.; Wang, D.; Hayama, R.; Leonelli, L.; Han, H.; Grigoryev, S.A.; et al. Histone hypercitrullination mediates chromatin decondensation and neutrophil extracellular trap formation. J. Cell Biol. 2009, 184, 205–213. [Google Scholar] [CrossRef]
- Fisher, B.J.; Kraskauskas, D.; Martin, E.J.; Farkas, D.; Puri, P.; Massey, H.D.; Idowu, M.O.; Brophy, D.; Voelkel, N.F.; Fowler, A.A.; et al. Attenuation of sepsis induced organ injury by vitamin C. J. Parenter. Enter. Nutr. 2013, in press. [Google Scholar]
- Kim, H.; Bae, S.; Yu, Y.; Kim, Y.; Kim, H.R.; Hwang, Y.I.; Kang, J.S.; Lee, W.J. The analysis of vitamin C concentration in organs of gulo(−/−) mice upon vitamin C withdrawal. Immune Netw. 2012, 12, 18–26. [Google Scholar] [CrossRef]
- Vissers, M.C.; Bozonet, S.M.; Pearson, J.F.; Braithwaite, L.J. Dietary ascorbate intake affects steady state tissue concentrations in vitamin C-deficient mice: Tissue deficiency after suboptimal intake and superior bioavailability from a food source (kiwifruit). Am. J. Clin. Nutr. 2011, 93, 292–301. [Google Scholar] [CrossRef]
- Tsurubuchi, T.; Aratani, Y.; Maeda, N.; Koyama, H. Retardation of early-onset PMA-induced apoptosis in mouse neutrophils deficient in myeloperoxidase. J. Leukoc. Biol. 2001, 70, 52–58. [Google Scholar]
- Fowler, A.A.; Fisher, B.J.; Centor, R.M.; Carchman, R.A. Development of the adult respiratory distress syndrome: Progressive alteration of neutrophil chemotactic and secretory processes. Am. J. Pathol. 1984, 116, 427–435. [Google Scholar]
- Meng, W.; Paunel-Görgülü, A.; Flohé, S.; Witte, I.; Schädel-Höpfner, M.; Windolf, J.; Lögters, T.T. Deoxyribonuclease is a potential counter regulator of aberrant neutrophil extracellular traps formation after major trauma. Mediat. Inflamm. 2012, 2012. [Google Scholar] [CrossRef]
- Fuchs, T.A.; Abed, U.; Goosmann, C.; Hurwitz, R.; Schulze, I.; Wahn, V.; Weinrauch, Y.; Brinkmann, V.; Zychlinsky, A. Novel cell death program leads to neutrophil extracellular traps. J. Cell Biol. 2007, 176, 231–241. [Google Scholar]
- Asaga, H.; Nakashima, K.; Senshu, T.; Ishigami, A.; Yamada, M. Immunocytochemical localization of peptidylargininedeiminase in human eosinophils and neutrophils. J. Leukoc. Biol. 2001, 70, 46–51. [Google Scholar]
- Mastronardi, F.G.; Wood, D.D.; Mei, J.; Raijmakers, R.; Tseveleki, V.; Dosch, H.M.; Probert, L.; Casaccia-Bonnefil, P.; Moscarello, M.A. Increased citrullination of histone H3 in multiple sclerosis brain and animal models of demyelination: A role for tumor necrosis factor-induced peptidylargininedeiminase 4 translocation. J. Neurosci. 2006, 26, 11387–11396. [Google Scholar]
- Cheng, O.Z.; Palaniyar, N. NET balancing: A problem in inflammatory lung diseases. Front. Immunol. 2013, 4, 1–13. [Google Scholar]
- Klionsky, D.J.; Abeliovich, H.; Agostinis, P.; Agrawal, D.K.; Aliev, G.; Askew, D.S.; Baba, M.; Baehrecke, E.H.; Bahr, B.A.; Ballabio, A.; et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy 2008, 4, 151–175. [Google Scholar]
- Bjorkoy, G.; Lamark, T.; Pankiv, S.; Overvatn, A.; Brech, A.; Johansen, T. Monitoring autophagic degradation of p62/SQSTM1. Methods Enzymol. 2009, 452, 181–197. [Google Scholar] [CrossRef]
- Deegan, S.; Saveljeva, S.; Gorman, A.M.; Samali, A. Stress-induced self-cannibalism: On the regulation of autophagy by endoplasmic reticulum stress. Cell. Mol. Life Sci. 2013, 70, 2425–2441. [Google Scholar] [CrossRef]
- Yang, K.Y.; Arcaroli, J.J.; Abraham, E. Early alterations in neutrophil activation are associated with outcome in acute lung injury. Am. J. Respir. Crit. Care Med. 2003, 167, 1567–1574. [Google Scholar] [CrossRef]
- Clarke, R.; Cook, K.L.; Hu, R.; Facey, C.O.; Tavassoly, I.; Schwartz, J.L.; Baumann, W.T.; Tyson, J.J.; Xuan, J.; Wang, Y.; et al. Endoplasmic reticulum stress, the unfolded protein response, autophagy, and the integrated regulation of breast cancer cell fate. Cancer Res. 2012, 72, 1321–1331. [Google Scholar]
- Ying, S.; Kojima, T.; Kawada, A.; Nachat, R.; Serre, G.; Simon, M.; Takahara, H. An intronic enhancer driven by NF-κB contributes to transcriptional regulation of peptidylargininedeiminase type I gene in human keratinocytes. J. Investig. Dermatol. 2010, 130, 2543–2552. [Google Scholar] [CrossRef]
- Cárcamo, J.M.; Pedraza, A.; Bórquez-Ojeda, O.; Golde, D.W. Vitamin C suppresses TNFα-induced NFκB activation by inhibiting IκBα phosphorylation. Biochemistry 2002, 41, 12995–13002. [Google Scholar] [CrossRef]
- Klionsky, D.J.; Emr, S.D. Autophagy as a regulated pathway of cellular degradation. Science 2000, 290, 1717–1721. [Google Scholar] [CrossRef]
- Beertsen, W.; Willenborg, M.; Everts, V.; Zirogianni, A.; Podschun, R.; Schröder, B.; Eskelinen, E.L.; Saftig, P. Impaired phagosomal maturation in neutrophils leads to periodontitis in lysosomal-associated membrane protein-2 knockout mice. J. Immunol. 2008, 180, 475–482. [Google Scholar]
- Huang, J.; Canadien, V.; Lam, G.Y.; Steinberg, B.E.; Dinauer, M.C.; Magalhaes, M.A.; Glogauer, M.; Grinstein, S.; Brumell, J.H. Activation of antibacterial autophagy by NADPH oxidases. Proc. Natl. Acad. Sci. USA 2009, 106, 6226–6231. [Google Scholar] [CrossRef]
- Mitroulis, I.; Kourtzelis, I.; Kambas, K.; Rafail, S.; Chrysanthopoulou, A.; Speletas, M.; Ritis, K. Regulation of the autophagic machinery in human neutrophils. Eur. J. Immunol. 2010, 40, 1461–1472. [Google Scholar] [CrossRef]
- Pankiv, S.; Clausen, T.H.; Lamark, T.; Brech, A.; Bruun, J.A.; Outzen, H.; Overvatn, A.; Bjorkoy, G.; Johansen, T. P62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 2007, 282, 24131–24145. [Google Scholar] [CrossRef]
- Patel, A.S.; Morse, D.; Choi, A.M. Regulation and functional significance of autophagy in respiratory cell biology and disease. Am. J. Respir. Cell Mol. Biol. 2013, 48, 1–9. [Google Scholar] [CrossRef]
- Rouschop, K.M.; van den Beucken, T.; Dubois, L.; Niessen, H.; Bussink, J.; Savelkouls, K.; Keulers, T.; Mujcic, H.; Landuyt, W.; Voncken, J.W.; et al. The unfolded protein response protects human tumor cells during hypoxia through regulation of the autophagy genes MAP1LC3B and ATG5. J. Clin. Investig. 2012, 120, 127–141. [Google Scholar]
- Moine, P.; McIntyre, R.; Schwartz, M.D.; Kaneko, D.; Shenkar, R.; Le Tulzo, Y.; Moore, E.E.; Abraham, E. NF-κB regulatory mechanisms in alveolar macrophages from patients with acute respiratory distress syndrome. Shock 2000, 13, 85–91. [Google Scholar]
- Burstein, E.; Duckett, C.S. Dying for NF-κB? Control of cell death by transcriptional regulation of the apoptotic machinery. Curr. Opin. Cell Biol. 2003, 15, 732–737. [Google Scholar] [CrossRef]
- Caudrillier, A.; Kessenbrock, K.; Gilliss, B.M.; Nguyen, J.X.; Marques, M.B.; Monestier, M.; Toy, P.; Werb, Z.; Looney, M.R. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J. Clin. Investig. 2012, 122, 2661–2671. [Google Scholar] [CrossRef]
- Thomas, G.M.; Carbo, C.; Curtis, B.R.; Martinod, K.; Mazo, I.B.; Schatzberg, D.; Cifuni, S.M.; Fuchs, T.A.; von Andrian, U.H.; Hartwig, J.H.; et al. Extracellular DNA traps are associated with the pathogenesis of TRALI in humans and mice. Blood 2012, 119, 6335–6343. [Google Scholar] [CrossRef]
- Drifte, G.; Dunn-Siegrist, I.; Tissières, P.; Pugin, J. Innate immune functions of immature neutrophils in patients with sepsis and severe systemic inflammatory response syndrome. Crit. Care Med. 2013, 41, 820–832. [Google Scholar] [CrossRef]
- Rhodes, A.; Wort, S.J.; Thomas, H.; Collinson, P.; Bennett, E.D. Plasma DNA concentration as a predictor of mortality and sepsis in critically ill patients. Crit. Care 2006, 10, R60. [Google Scholar] [CrossRef]
- Saukkonen, K.; Lakkisto, P.; Pettila, V.; Varpula, M.; Karlsson, S.; Ruokonen, E.; Pulkki, K. Cell-free plasma DNA as a predictor of outcome in severe sepsis and septic shock. Clin. Chem. 2008, 54, 1000–1007. [Google Scholar] [CrossRef]
- Guimarães-Costa, A.B.; Nascimento, M.T.; Wardini, A.B.; Pinto-da-Silva, L.H.; Saraiva, E.M. ETosis: A Microbicidal Mechanism beyond Cell Death. J. Parasitol. Res. 2012, 2012, 929743. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mohammed, B.M.; Fisher, B.J.; Kraskauskas, D.; Farkas, D.; Brophy, D.F.; Fowler, A.A.; Natarajan, R. Vitamin C: A Novel Regulator of Neutrophil Extracellular Trap Formation. Nutrients 2013, 5, 3131-3150. https://doi.org/10.3390/nu5083131
Mohammed BM, Fisher BJ, Kraskauskas D, Farkas D, Brophy DF, Fowler AA, Natarajan R. Vitamin C: A Novel Regulator of Neutrophil Extracellular Trap Formation. Nutrients. 2013; 5(8):3131-3150. https://doi.org/10.3390/nu5083131
Chicago/Turabian StyleMohammed, Bassem M., Bernard J. Fisher, Donatas Kraskauskas, Daniela Farkas, Donald F. Brophy, Alpha A. Fowler, and Ramesh Natarajan. 2013. "Vitamin C: A Novel Regulator of Neutrophil Extracellular Trap Formation" Nutrients 5, no. 8: 3131-3150. https://doi.org/10.3390/nu5083131