Dietary Chrysin Suppresses Formation of Actin Cytoskeleton and Focal Adhesion in AGE-Exposed Mesangial Cells and Diabetic Kidney: Role of Autophagy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals
2.2. Culture of Human Renal Mesangial Cells (HRMC)
2.3. In Vivo Animal Experiments
2.4. Western Blot Analysis
2.5. Rhodamine-Phalloidin Staining of F-Actin
2.6. Mesangial Cell Motility
2.7. Immunocytochemical Staining
2.8. Data Analysis
3. Results
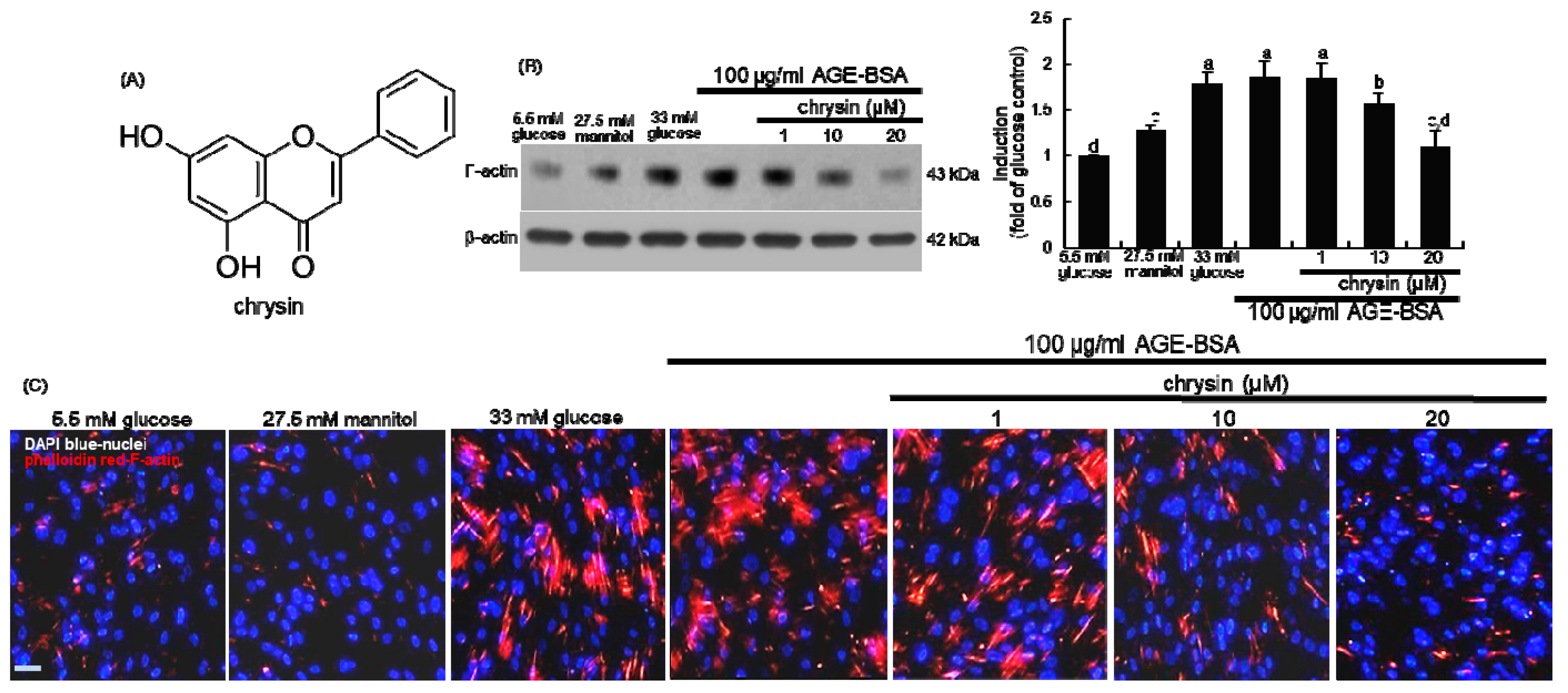
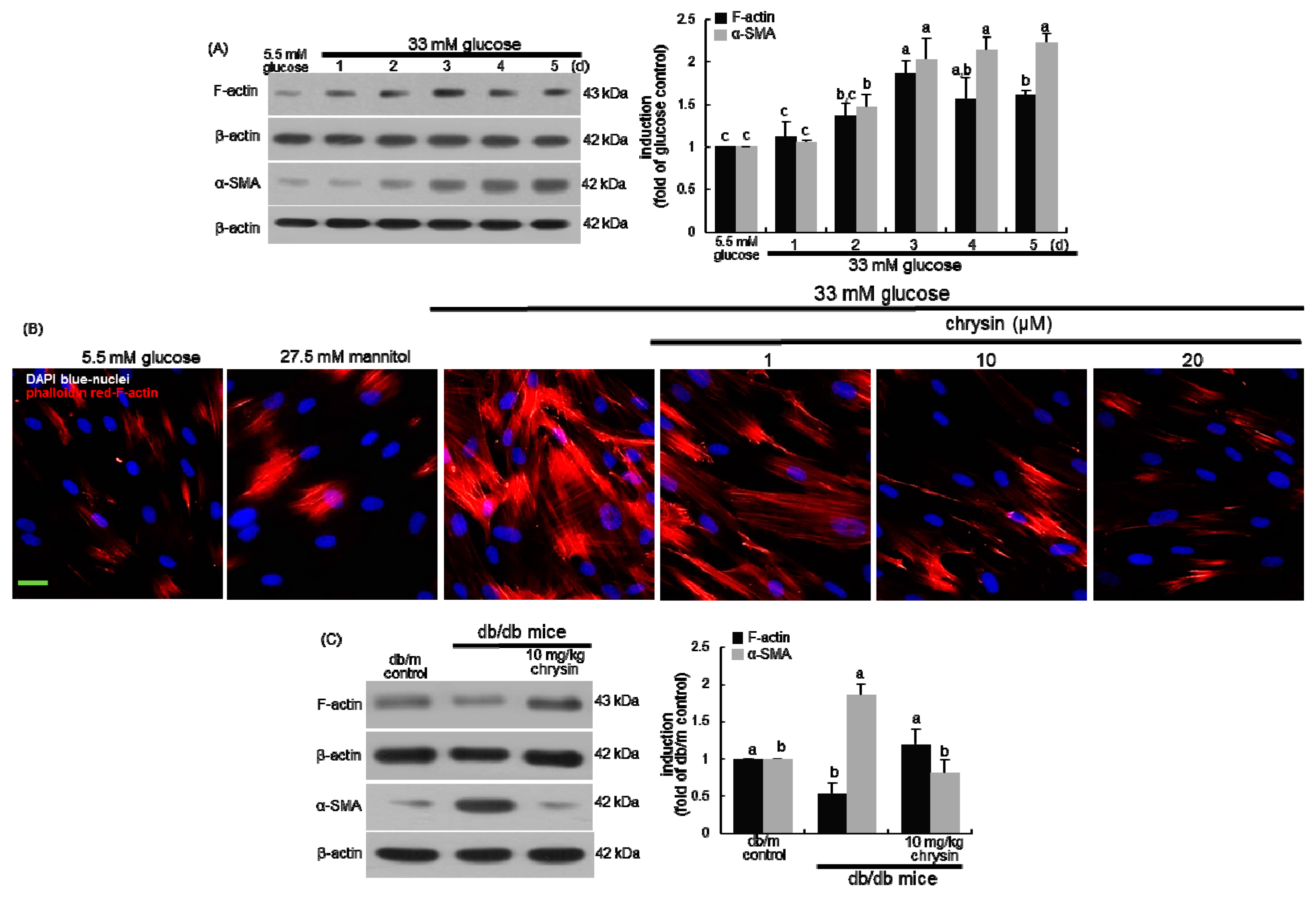
3.1. Effect of Chrysin on the Induction of Cytoskeletal Actin Proteins
3.2. Blockade of Focal Adhesion Formation by Chrysin
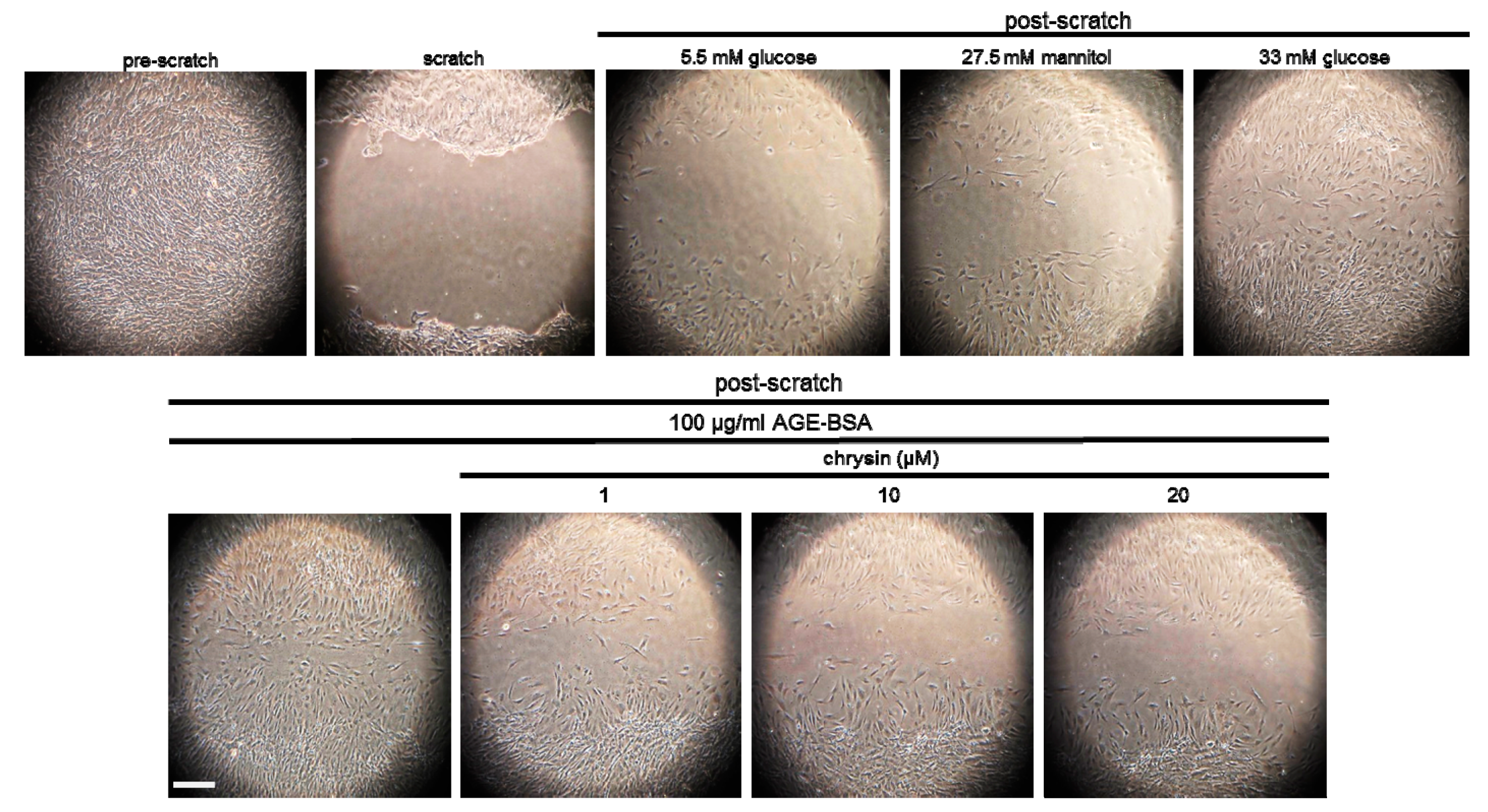
3.3. Inhibition of F-Actin Bundling and Mesangial Migration by Chrysin
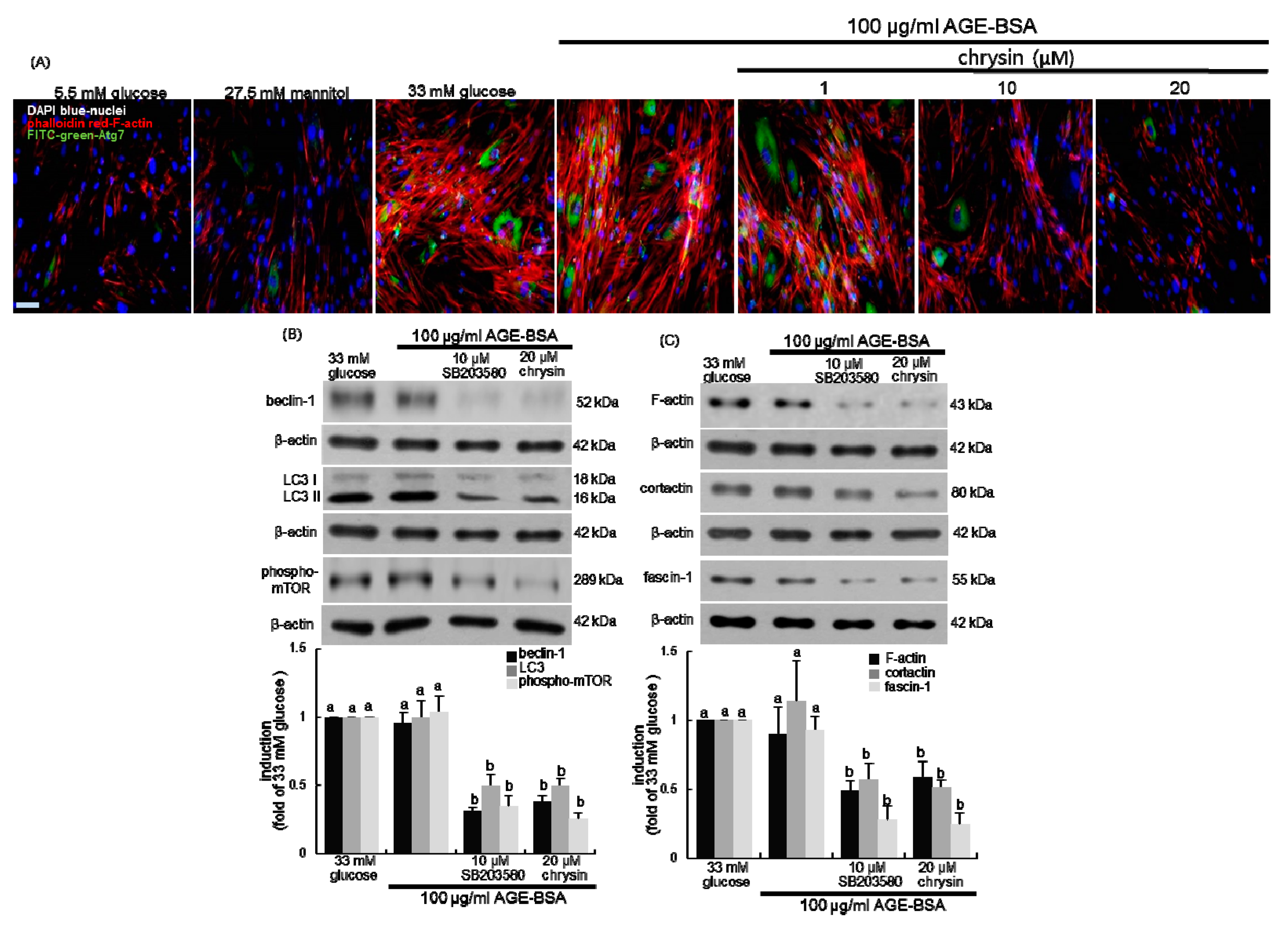
3.4. Inhibition of Autophagic Responses to AGE by Chrysin
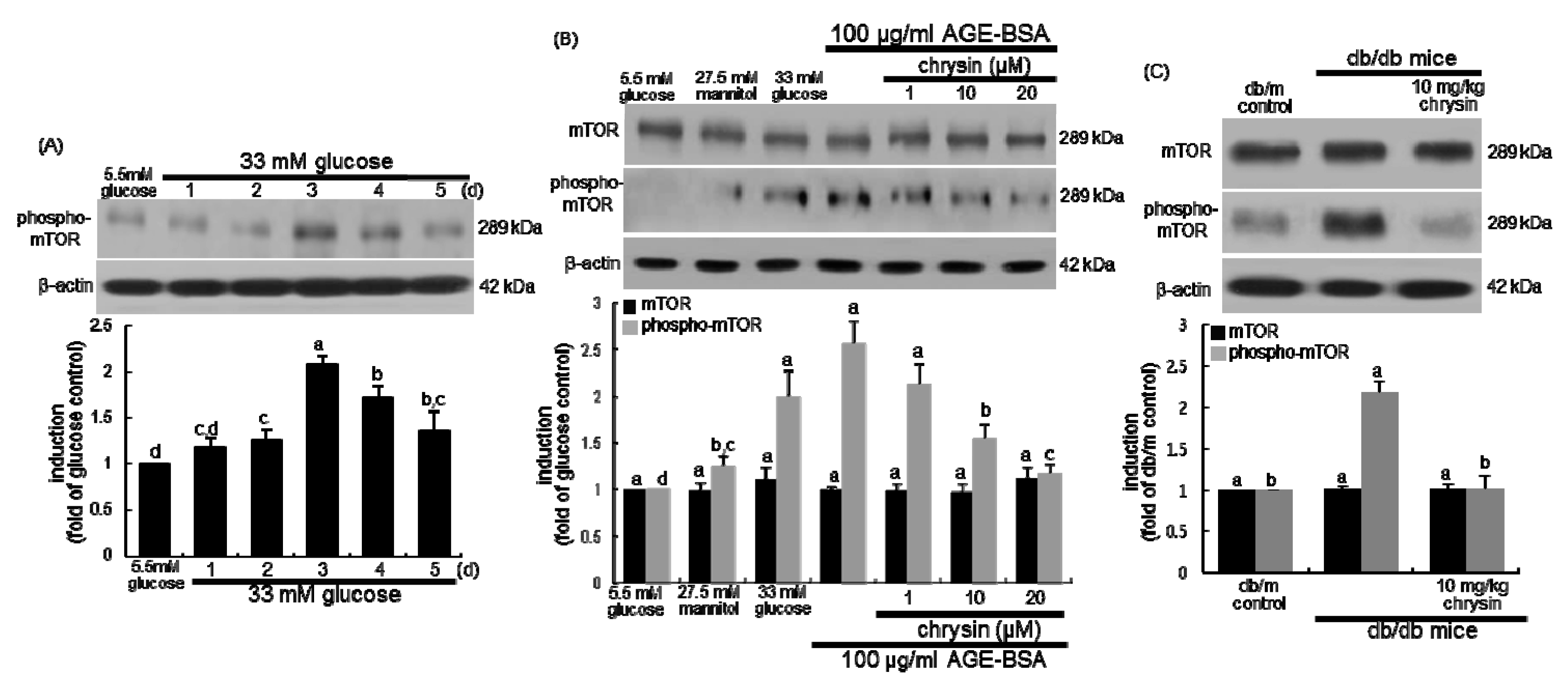
3.5. Suppression of AGE-Induced mTOR Activation by Chrysin
3.6. Association of Autophagy with Mesangial Motility
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Takano, K.; Kawasaki, Y.; Imaizumi, T.; Matsuura, H.; Nozawa, R.; Tannji, M.; Suyama, K.; Isome, M.; Suzuki, H.; Hosoya, M. Development of glomerular endothelial cells, podocytes and mesangial cells in the human fetus and infant. Tohoku J. Exp. Med. 2007, 212, 81–90. [Google Scholar] [CrossRef]
- Schlondorff, D.; Banas, B. The mesangial cell revisited: No cell is an island. J. Am. Soc. Nephrol. 2009, 20, 1179–1187. [Google Scholar] [CrossRef] [PubMed]
- Yahata, K.; Kikuchi, Y.; Koizumi, M.; Seta, K.; Wakui, H.; Komatsuda, A.; Shimizu, A. Membranoproliferative glomerulonephritis with unusual deposits of parallel arrangement striated structure: A new pathological entity? Clin. Nephrol. 2018, 89, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Tsurumi, H.; Kurihara, H.; Miura, K.; Tanego, A.; Ohta, Y.; Igarashi, T.; Oka, A.; Horita, S.; Hattori, M.; Harita, Y. Afadin is localized at cell-cell contact sites in mesangial cells and regulates migratory polarity. Lab. Investig. 2016, 96, 49–59. [Google Scholar] [CrossRef] [PubMed]
- Kohno, M.; Yasunari, K.; Minami, M.; Kano, H.; Maeda, K.; Mandal, A.K.; Inoki, K.; Haneda, M.; Yoshikawa, J. Regulation of rat mesangial cell migration by platelet-derived growth factor, angiotensin II, and adrenomedullin. J. Am. Soc. Nephrol. 1999, 10, 2495–2502. [Google Scholar] [PubMed]
- Tsurumi, H.; Harita, Y.; Kurihara, H.; Kosako, H.; Hayashi, K.; Matsunaga, A.; Kajiho, Y.; Kanda, S.; Miura, K.; Sekine, T.; et al. Epithelial protein lost in neoplasm modulates platelet-derived growth factor-mediated adhesion and motility of mesangial cells. Kidney Int. 2014, 86, 548–557. [Google Scholar] [CrossRef]
- Daniel, C.; Lüdke, A.; Wagner, A.; Todorov, V.T.; Hohenstein, B.; Hugo, C. Transgelin is a marker of repopulating mesangial cells after injury and promotes their proliferation and migration. Lab. Investig. 2012, 92, 812–826. [Google Scholar] [CrossRef] [Green Version]
- Bartlett, C.S.; Scott, R.P.; Carota, I.A.; Wnuk, M.L.; Kanwar, Y.S.; Miner, J.H.; Quaggin, S.E. Glomerular mesangial cell recruitment and function require the co-receptor neuropilin-1. Am. J. Physiol. Ren. Physiol. 2017, 313, F1232–F1242. [Google Scholar] [CrossRef]
- Chaki, S.P.; Barhoumi, R.; Berginski, M.E.; Sreenivasappa, H.; Trache, A.; Gomez, S.M.; Rivera, G.M. Nck enables directional cell migration through the coordination of polarized membrane protrusion with adhesion dynamics. J. Cell. Sci. 2013, 126, 1637–1649. [Google Scholar] [CrossRef]
- Sung, B.H.; Ketova, T.; Hoshino, D.; Zijlstra, A.; Weaver, A.M. Directional cell movement through tissues is controlled by exosome secretion. Nat. Commun. 2015, 6, 7164. [Google Scholar] [CrossRef]
- Daroux, M.; Prévost, G.; Maillard-Lefebvre, H.; Gaxatte, C.; D’Agati, V.D.; Schmidt, A.M.; Boulanger, E. Advanced glycation end-products: Implications for diabetic and non-diabetic nephropathies. Diabetes Metab. 2010, 36, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Scindia, Y.; Deshmukh, U.; Bagavant, H. Mesangial pathology in glomerular disease: Targets for therapeutic intervention. Adv. Drug Deliv. Rev. 2010, 62, 1337–1343. [Google Scholar] [CrossRef] [PubMed]
- Kanwar, Y.; Wada, J.; Sun, L.; Xie, P.; Wallner, E.; Chen, S.; Chugh, S.; Danesh, F. Diabetic nephropathy: Mechanisms of renal disease progression. Exp. Biol. Med. 2008, 33, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Lan, L.; Han, Y.; Ren, W.; Jiang, J.; Wang, P.; Hu, Z. Advanced glycation end-products affect the cytoskeletal structure of rat glomerular endothelial cells via the Ras-related C3 botulinum toxin substrate 1 signaling pathway. Mol. Med. Rep. 2015, 11, 4321–4326. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kwon, J. Actin cytoskeletal rearrangement and dysfunction due to activation of the receptor for advanced glycation end products is inhibited by thymosin beta 4. J. Physiol. 2015, 593, 1873–1886. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, C.; Chang, Y.; Li, Y.; Chen, S.; Chen, Y.; Ye, N.; Dai, D.; Sun, Y. Advanced glycation end products promote the proliferation and migration of primary rat vascular smooth muscle cells via the upregulation of BAG3. Int. J. Mol. Med. 2017, 39, 1242–1254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.L.; Lu, S.; Szeto, K.W.; Sun, J.; Wang, Y.; Lasheras, J.C.; Chien, S. FAK and paxillin dynamics at focal adhesions in the protrusions of migrating cells. Sci. Rep. 2014, 4, 6024. [Google Scholar] [CrossRef] [PubMed]
- Thievessen, I.; Fakhri, N.; Steinwachs, J.; Kraus, V.; McIsaac, R.S.; Gao, L.; Chen, B.C.; Baird, M.A.; Davidson, M.W.; Betzig, E.; et al. Vinculin is required for cell polarization, migration, and extracellular matrix remodeling in 3D collagen. FASEB J. 2015, 29, 4555–4567. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, Y.S.; Kim, J.; Kim, K.M.; Jung, D.H.; Choi, S.; Kim, C.S.; Kim, J.S. Myricetin inhibits advanced glycation end product (AGE)-induced migration of retinal pericytes through phosphorylation of ERK1/2, FAK-1, and paxillin in vitro and in vivo. Biochem. Pharmacol. 2015, 93, 496–505. [Google Scholar] [CrossRef]
- Crean, J.K.; Furlong, F.; Finlay, D.; Mitchell, D.; Murphy, M.; Conway, B.; Brady, H.R.; Godson, C.; Martin, F. Connective tissue growth factor/CCN2 stimulates mesangial cell migration through integrated dissolution of focal adhesion complexes and activation of cell polarization. FASEB J. 2004, 18, 1541–1543. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, A.; Takabatake, Y.; Kimura, T.; Maejima, I.; Namba, T.; Yamamoto, T.; Matsuda, J.; Minami, S.; Kaimori, J.Y.; Matsui, I.; et al. Autophagy inhibits the accumulation of advanced glycation end products by promoting lysosomal biogenesis and function in the kidney proximal tubules. Diabetes 2017, 66, 1359–1372. [Google Scholar] [CrossRef] [PubMed]
- Xu, L.; Fan, Q.; Wang, X.; Zhao, X.; Wang, L. Inhibition of autophagy increased AGE/ROS-mediated apoptosis in mesangial cells. Cell Death Dis. 2016, 7, e2445. [Google Scholar] [CrossRef]
- Vargas, F.; Romecín, P.; García-Guillén, A.I.; Wangesteen, R.; Vargas-Tendero, P.; Paredes, M.D.; Atucha, N.M.; García-Estañ, J. Flavonoids in kidney health and disease. Front. Physiol. 2018, 9, 394. [Google Scholar] [CrossRef] [PubMed]
- Trujillo, J.; Chirino, Y.; Molina-Jijón, E.; Andérica-Romero, A.C.; Tapia, E.; Pedraza-Chaverrí, J. Renoprotective effect of the antioxidant curcumin: Recent findings. Redox Biol. 2013, 1, 448–456. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ojha, S.; Venkataraman, B.; Kurdi, A.; Mahgoub, E.; Sadek, B.; Rajesh, M. Plant-derived agents for counteracting cisplatin-induced nephrotoxicity. Oxid. Med. Cell. Longev. 2016, 2016, 4320374. [Google Scholar] [CrossRef] [PubMed]
- Kang, M.K.; Park, S.H.; Choi, Y.J.; Shin, D.; Kang, Y.H. Chrysin inhibits diabetic renal tubulointerstitial fibrosis through blocking epithelial to mesenchymal transition. J. Mol. Med. (Berl.) 2015, 93, 759–772. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.Y.; Kang, M.K.; Lee, E.J.; Kim, Y.H.; Oh, H.; Kang, Y.H. Eucalyptol inhibits advanced glycation end products-induced disruption of podocyte slit junctions by suppressing Rage-Erk-C-Myc signaling pathway. Mol. Nutr. Food Res. 2018, 62, e1800302. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Kang, M.K.; Kim, J.K.; Kim, J.L.; Kang, S.W.; Lim, S.S.; Kang, Y.H. Purple corn anthocyanins retard diabetes-associated glomerulosclerosis in mesangial cells and db/db mice. Eur. J. Nutr. 2012, 51, 961–973. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Qin, H.; Shi, Q.; Zhang, Y.; Zhou, F.; Wu, H.; Ding, S.; Niu, Z.; Lu, Y.; Shen, P. Chrysin attenuates inflammation by regulating M1/M2 status via activating PPARγ. Biochem. Pharmacol. 2014, 89, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Anandhi, R.; Thomas, P.A.; Geraldine, P. Evaluation of the anti-atherogenic potential of chrysin in Wistar rats. Mol. Cell. Biochem. 2014, 385, 103–113. [Google Scholar] [CrossRef]
- Nabavi, S.F.; Braidy, N.; Habtemariam, S.; Orhan, I.E.; Daglia, M.; Manayi, A.; Gortzi, O.; Nabavi, S.M. Neuroprotective effects of chrysin: From chemistry to medicine. Neurochem. Int. 2015, 90, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Ali, B.H.; Adham, S.A.; Za’abi, M.A.; Waly, M.I.; Yasin, J.; Nemmar, A.; Schupp, N. Ameliorative effect of chrysin on adenine-induced chronic kidney disease in rats. PLoS ONE 2015, 10, e0125285. [Google Scholar] [CrossRef] [PubMed]
- Kandemir, F.M.; Kucukler, S.; Eldutar, E.; Caglayan, C.; Gülçin, I. Chrysin protects rat kidney from paracetamol-induced oxidative stress, inflammation, apoptosis, and autophagy: A multi-biomarker approach. Sci. Pharm. 2017, 85, 4. [Google Scholar] [CrossRef] [PubMed]
- Lee, E.J.; Kang, M.K.; Kim, D.Y.; Kim, Y.H.; Oh, H.; Kang, Y.H. Chrysin inhibits advanced glycation end products-induced kidney fibrosis in renal mesangial cells and diabetic kidneys. Nutrients 2018, 10, 882. [Google Scholar] [CrossRef] [PubMed]
- Uruno, T.; Liu, J.; Zhang, P.; Fan, Y.X.; Egile, C.; Li, R.; Mueller, S.C.; Zhan, X. Activation of Arp2/3 complex-mediated actin polymerization by cortactin. Nat. Cell Biol. 2001, 3, 259–266. [Google Scholar] [CrossRef] [PubMed]
- Jansen, S.; Collins, A.; Yang, C.; Rebowski, G.; Svitkina, T.; Dominguez, R. Mechanism of actin filament bundling by fascin. J. Biol. Chem. 2011, 286, 30087–30096. [Google Scholar] [CrossRef] [PubMed]
- Kajiwara, M.; Masuda, S. Role of mTOR inhibitors in kidney disease. Int. J. Mol. Sci. 2016, 17, 975. [Google Scholar] [CrossRef]
- Fantus, D.; Rogers, N.M.; Grahammer, F.; Huber, T.B.; Thomson, A.W. Roles of mTOR complexes in the kidney: Implications for renal disease and transplantation. Nat. Rev. Nephrol. 2016, 12, 587–609. [Google Scholar] [CrossRef]
- Kast, D.J.; Dominguez, R. The cytoskeleton–autophagy connection. Curr. Biol. 2017, 27, R318–R326. [Google Scholar] [CrossRef]
- Zhou, J.; Fan, Y.; Zhong, J.; Huang, Z.; Huang, T.; Lin, S.; Chen, H. TAK1 mediates excessive autophagy via p38 and ERK in cisplatin-induced acute kidney injury. J. Cell. Mol. Med. 2018, 22, 2908–2921. [Google Scholar] [CrossRef] [Green Version]
- Grossi, V.; Lucarelli, G.; Forte, G.; Peserico, A.; Matrone, A.; Germani, A.; Rutigliano, M.; Stella, A.; Bagnulo, R.; Loconte, D.; et al. Loss of STK11 expression is an early event in prostate carcinogenesis and predicts therapeutic response to targeted therapy against MAPK/p38. Autophagy 2015, 11, 2102–2113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kustermans, G.; Piette, J.; Legrand-Poels, S. Actin-targeting natural compounds as tools to study the role of actin cytoskeleton in signal transduction. Biochem. Pharmacol. 2008, 76, 1310–1322. [Google Scholar] [CrossRef] [PubMed]
- Allingham, J.S.; Klenchin, V.A.; Rayment, I. Actin-targeting natural products: Structures, properties and mechanisms of action. Cell. Mol. Life Sci. 2006, 63, 2119–2134. [Google Scholar] [CrossRef] [PubMed]
- Hou, B.; Qiang, G.; Zhao, Y.; Yang, X.; Chen, X.; Yan, Y.; Wang, X.; Liu, C.; Zhang, L.; Du, G. Salvianolic acid A protects against diabetic nephropathy through ameliorating glomerular endothelial dysfunction via inhibiting AGE-RAGE signaling. Cell. Physiol. Biochem. 2017, 44, 2378–2394. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.J.; Liu, H.T.; Lai, Y.H.; Jan, T.R.; Nomura, N.; Chang, H.W.; Chou, C.C.; Lee, Y.J.; Tsai, P.J. Honokiol, a polyphenol natural compound, attenuates cisplatin-induced acute cytotoxicity in renal epithelial cells through cellular oxidative stress and cytoskeleton modulations. Front. Pharmacol. 2018, 9, 357. [Google Scholar] [CrossRef] [PubMed]
- Krause, M.; Dent, E.W.; Bear, J.E.; Loureiro, J.J.; Gertler, F.B. Ena/VASP proteins: Regulators of the actin cytoskeleton and cell migration. Annu. Rev. Cell Dev. Biol. 2003, 19, 541–564. [Google Scholar] [CrossRef]
- Ding, Y.; Choi, M.E. Autophagy in diabetic nephropathy. J. Endocrinol. 2015, 224, R15–R30. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, M.N.; Mowers, E.E.; Drake, L.E.; Collier, C.; Chen, H.; Zamora, M.; Mui, S.; Macleod, K.F. Autophagy promotes focal adhesion disassembly and cell motility of metastatic tumor cells through the direct interaction of paxillin with LC3. Cell Rep. 2016, 15, 1660–1672. [Google Scholar] [CrossRef] [PubMed]
- Zhuo, C.; Ji, Y.; Chen, Z.; Kitazato, K.; Xiang, Y.; Zhong, M.; Wang, Q.; Pei, Y.; Ju, H.; Wang, Y. Proteomics analysis of autophagy-deficient Atg7−/− MEFs reveals a close relationship between F-actin and autophagyBiochem. Biophys. Res. Commun. 2013, 437, 482–488. [Google Scholar] [CrossRef]
- Chen, J.; Zhao, D.; Zhu, M.; Zhang, M.; Hou, X.; Ding, W.; Sun, S.; Bu, W.; Feng, L.; Ma, S.; et al. Paeoniflorin ameliorates AGEs-induced mesangial cell injury through inhibiting RAGE/mTOR/autophagy pathway. Biomed. Pharmacother. 2017, 89, 1362–1369. [Google Scholar] [CrossRef] [PubMed]
- Inoki, K. mTOR signaling in autophagy regulation in the kidney. Semin. Nephrol. 2014, 34, 2–8. [Google Scholar] [CrossRef] [PubMed]
- Sato, T.; Ishii, J.; Ota, Y.; Sasaki, E.; Shibagaki, Y.; Hattori, S. Mammalian target of rapamycin (mTOR) complex 2 regulates filamin A-dependent focal adhesion dynamics and cell migration. Genes Cells 2016, 21, 579–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]







© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lee, E.-J.; Kang, M.-K.; Kim, Y.-H.; Kim, D.Y.; Oh, H.; Kim, S.-I.; Oh, S.Y.; Kang, Y.-H. Dietary Chrysin Suppresses Formation of Actin Cytoskeleton and Focal Adhesion in AGE-Exposed Mesangial Cells and Diabetic Kidney: Role of Autophagy. Nutrients 2019, 11, 127. https://doi.org/10.3390/nu11010127
Lee E-J, Kang M-K, Kim Y-H, Kim DY, Oh H, Kim S-I, Oh SY, Kang Y-H. Dietary Chrysin Suppresses Formation of Actin Cytoskeleton and Focal Adhesion in AGE-Exposed Mesangial Cells and Diabetic Kidney: Role of Autophagy. Nutrients. 2019; 11(1):127. https://doi.org/10.3390/nu11010127
Chicago/Turabian StyleLee, Eun-Jung, Min-Kyung Kang, Yun-Ho Kim, Dong Yeon Kim, Hyeongjoo Oh, Soo-Il Kim, Su Yeon Oh, and Young-Hee Kang. 2019. "Dietary Chrysin Suppresses Formation of Actin Cytoskeleton and Focal Adhesion in AGE-Exposed Mesangial Cells and Diabetic Kidney: Role of Autophagy" Nutrients 11, no. 1: 127. https://doi.org/10.3390/nu11010127