The Emerging Role of Nutritional Vitamin D in Secondary Hyperparathyroidism in CKD

 , , ,
, , , {kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
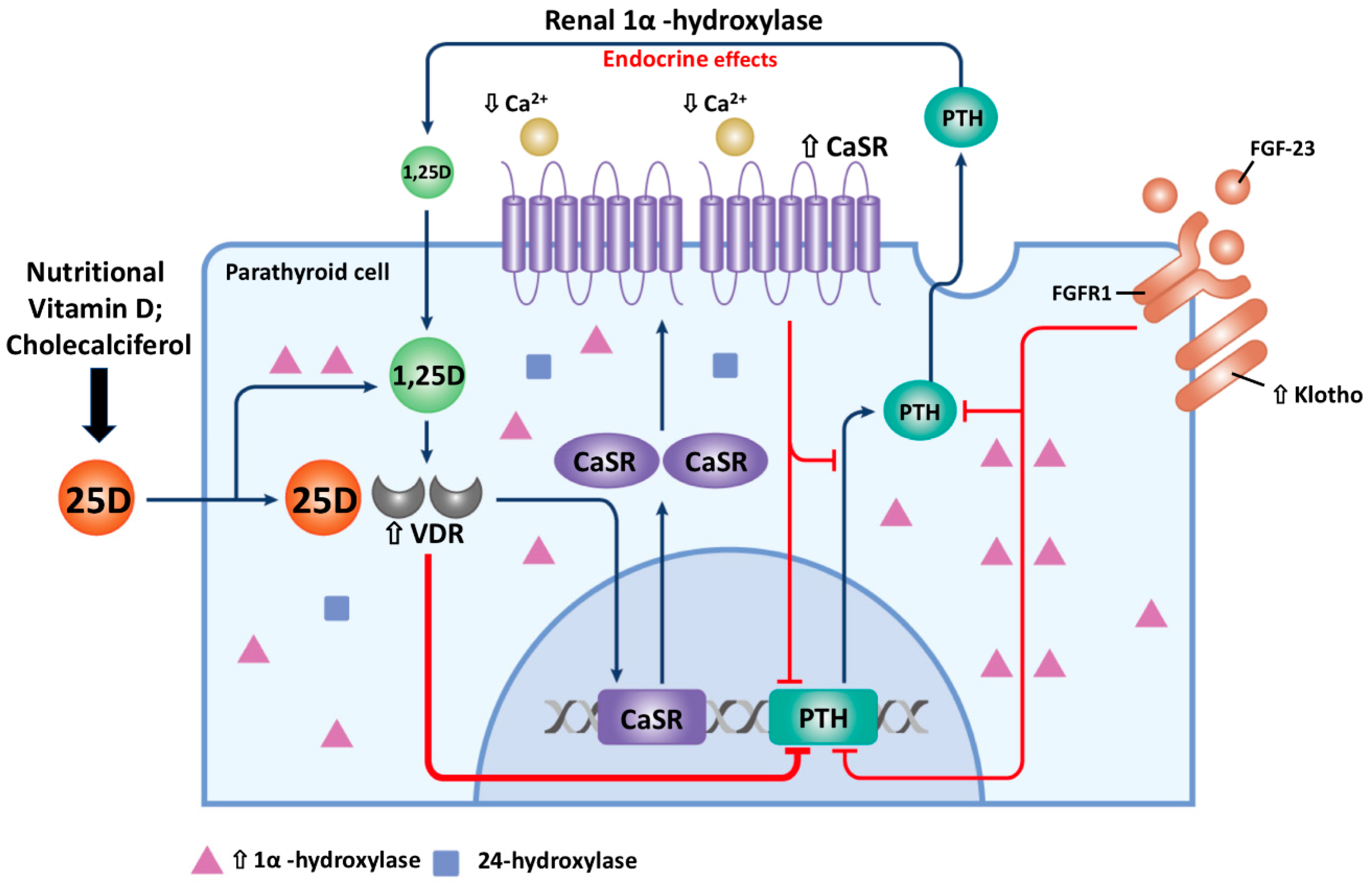
2. The Pathophysiology of Secondary Hyperparathyroidism
3. The Alteration of Vitamin D Metabolism in CKD
3.1. Decrease Vitamin D Synthesis and Increase Vitamin-D Catabolism in CKD
3.2. Nutritional Vitamin D Hunger in the PTG
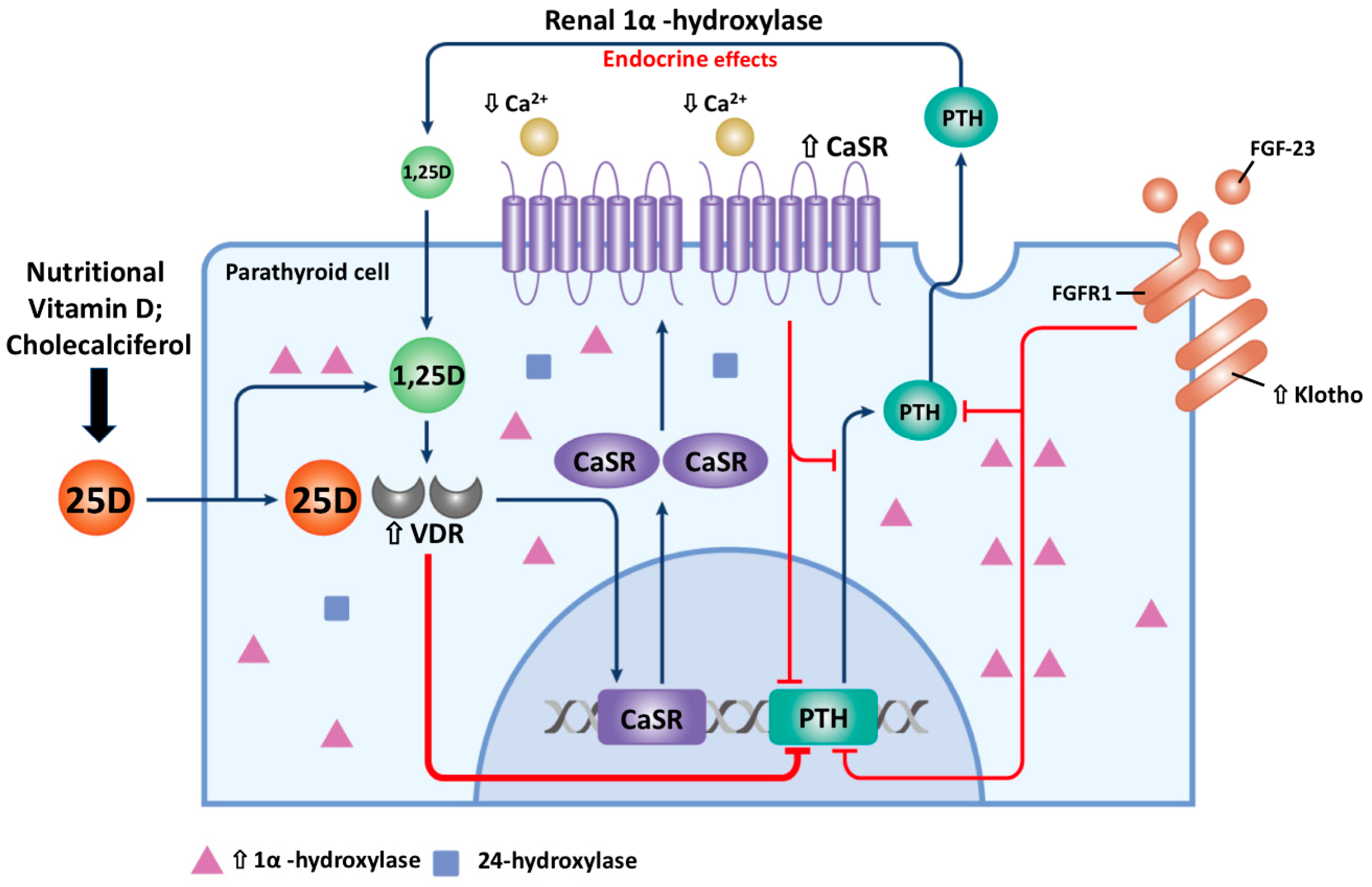
4. The Change of Parathyroid Gland Cell Membrane CaSR and Nuclear VDR Expression in SHPT
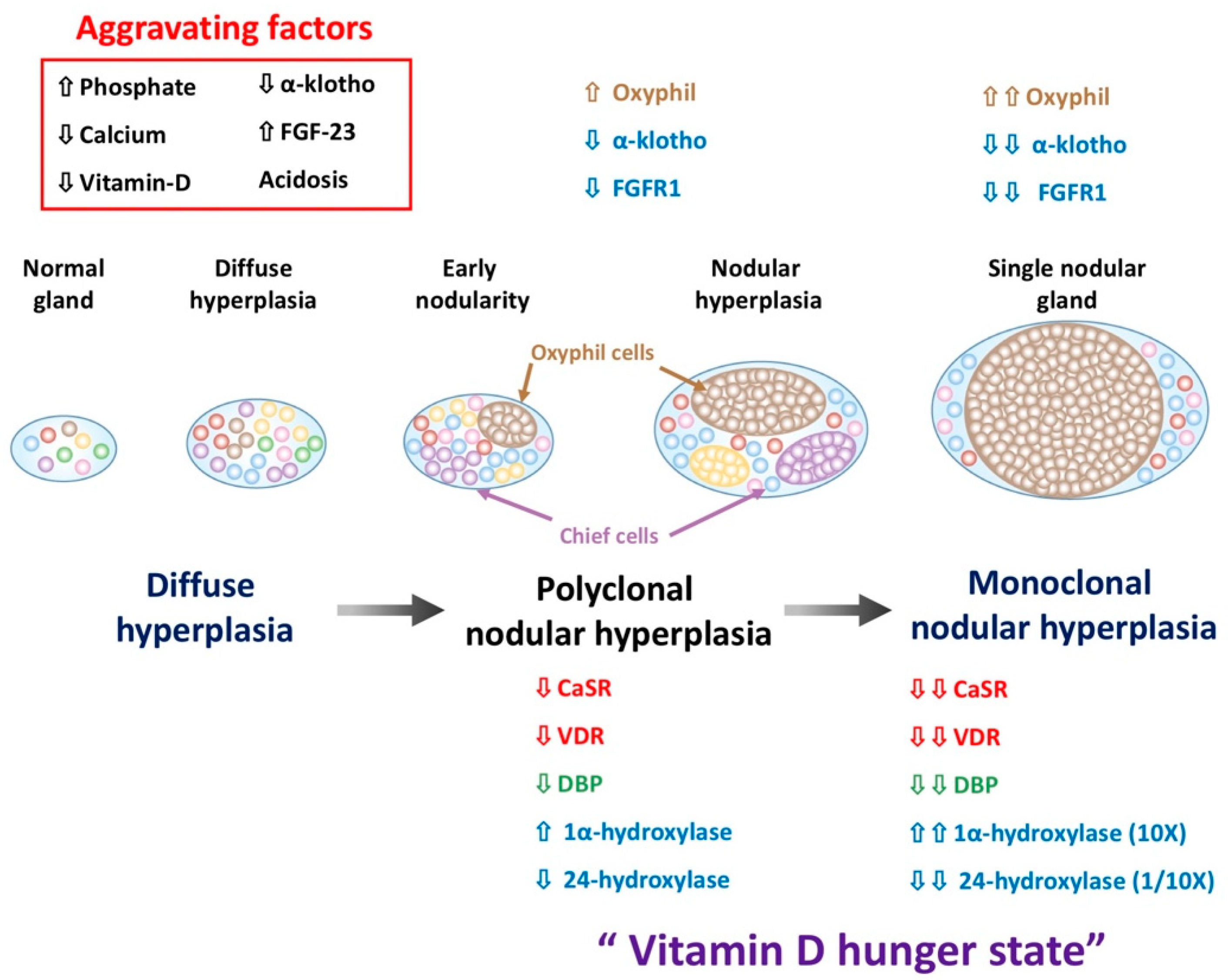
5. The Change of Parathyroid Cells Composition in SHPT
5.1. Increase Oxyphil Cells Proporation in PTG of SHPT
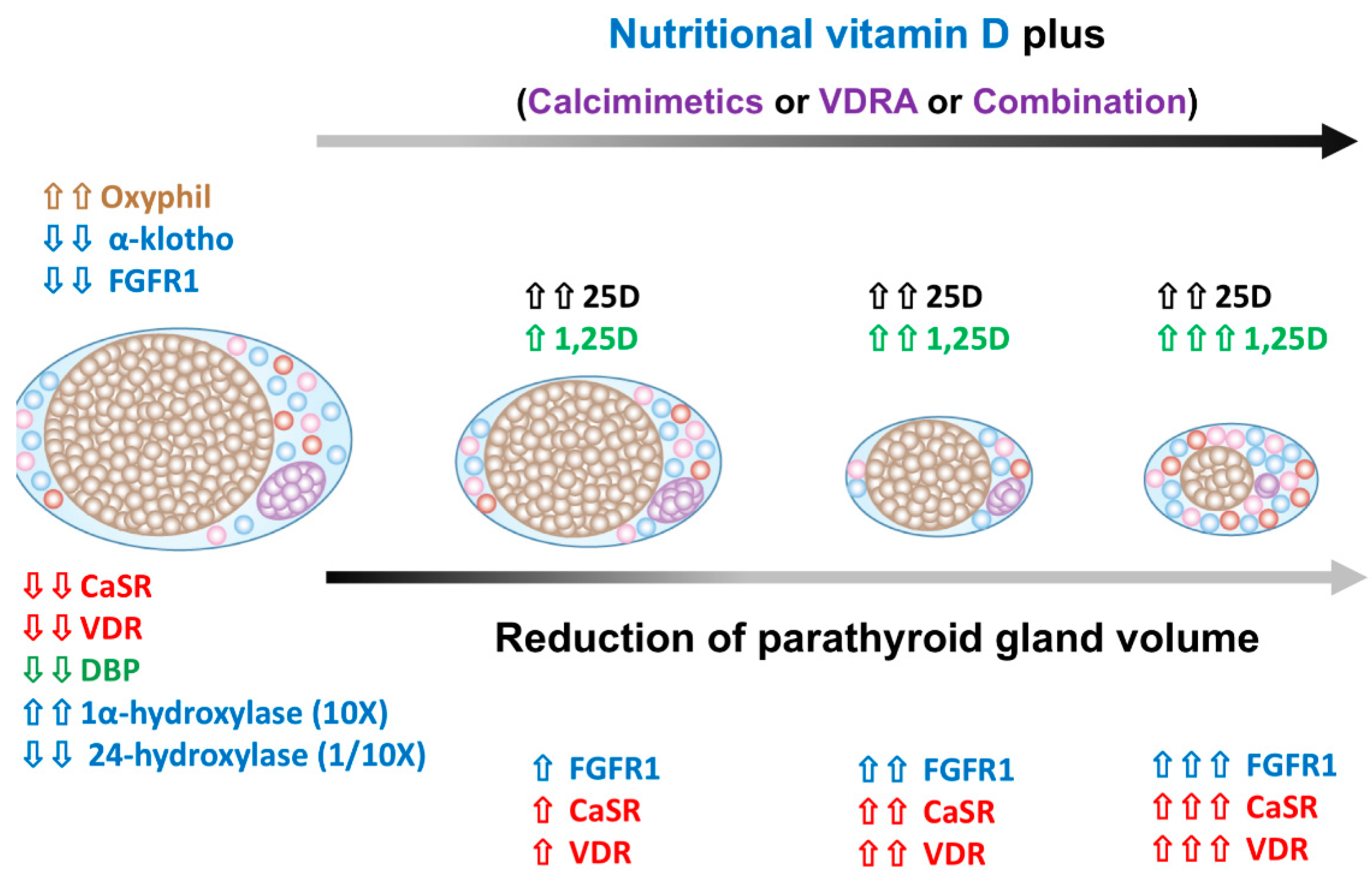
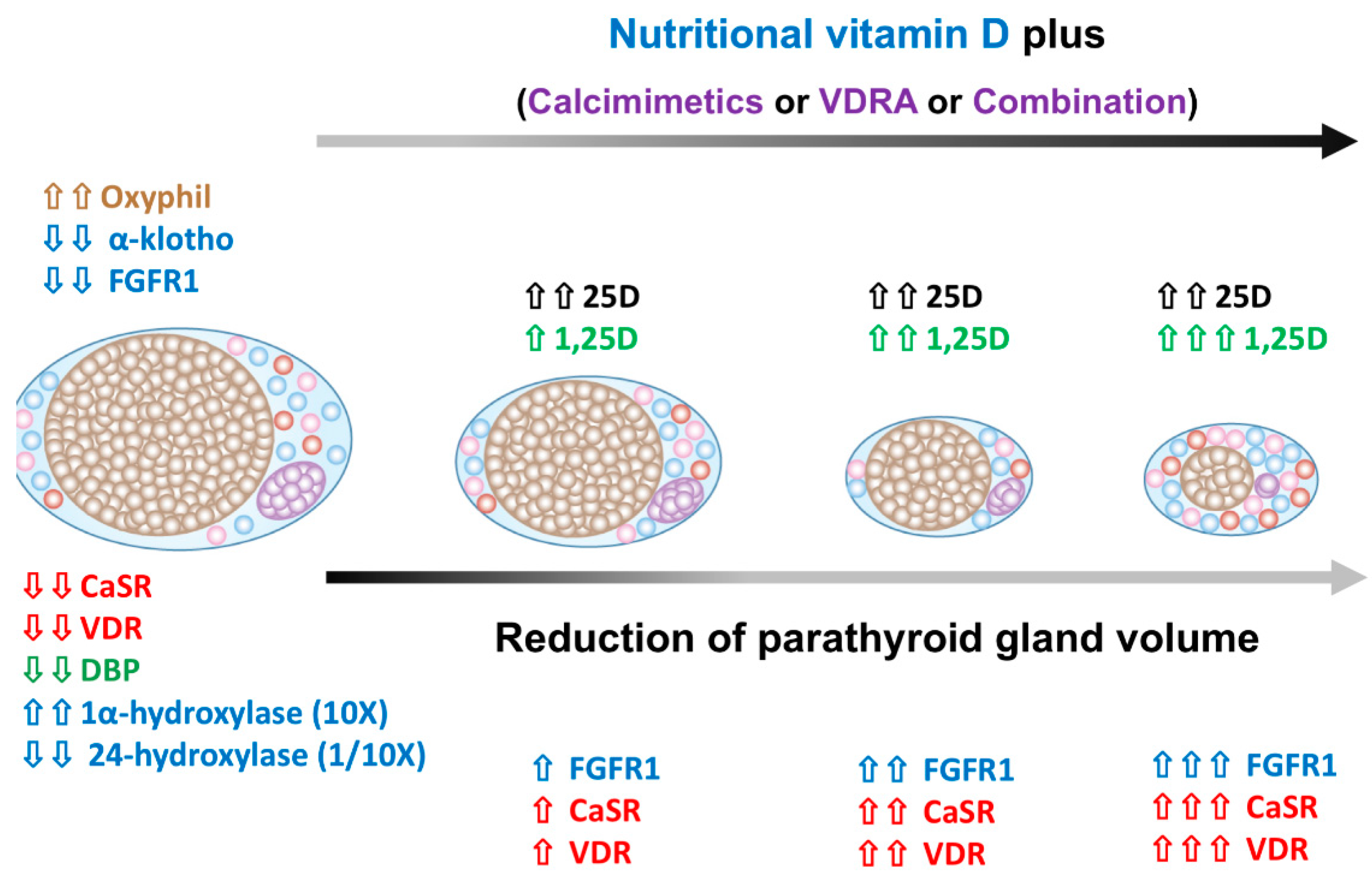
5.2. The Alteration of Parathyroid Cell Composition after SHPT Treatment
5.3. The Regression of Parathyroid Hyperplasia after SHPT Treatment
6. Vitamin D Binding Protein (DBP)
6.1. Role of DBP in Parathyroid 1,25D Synthesis
6.2. Do DBP Genotypes Influence Intrinsic 1,25D Production in the Parathyroid Gland?
7. The Negative Control of 1,25D on Vitamin D Metabolism
8. Synergistic Action of 25D and 1,25D on PTH Suppression
9. The Role of Vitamin-D Deficiency (VDD) on PTH Metabolism
10. Nutritional Vitamin D Prevents Developing SHPT in Early CKD
11. The Role of NVD in the PTH-Lowering Effect in Dialysis Patients Remains Controversial
12. NVD Has Adjuvant Benefits in Treating SHPT in Dialysis Patients
13. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviation
| 25D | 25-hydroxy vitamin D |
| 1,25D | 1,25-dihydroxy vitamin D |
| CKD | chronic kidney disease |
| CKD-MBD | chronic kidney disease-mineral bone disorders |
| CaSR | calcium sensing receptor |
| DBP | vitamin D binding protein |
| FGF-23 | fibroblast growth factor-23 |
| FGFR1 | fibroblast growth factor receptor 1 |
| HD | hemodialysis |
| PTH | parathyroid hormone |
| NVD | nutritional vitamin D |
| PTG | parathyroid gland |
| SHPT | secondary hyperparathyroidism |
| VDD | vitamin D deficiency |
| VDR | vitamin D receptor |
| VDRA | vitamin D receptor activator |
References
- Bureo, J.C.; Arevalo, J.C.; Anton, J.; Adrados, G.; Jimenez Morales, J.L.; Robles, N.R. Prevalence of secondary hyperparathyroidism in patients with stage 3 and 4 chronic kidney disease seen in internal medicine. Endocrinol. Nutr. 2015, 62, 300–305. [Google Scholar] [CrossRef] [PubMed]
- Hedgeman, E.; Lipworth, L.; Lowe, K.; Saran, R.; Do, T.; Fryzek, J. International burden of chronic kidney disease and secondary hyperparathyroidism: A systematic review of the literature and available data. Int. J. Nephrol. 2015, 2015, 184321. [Google Scholar] [CrossRef] [PubMed]
- Isakova, T.; Nickolas, T.L.; Denburg, M.; Yarlagadda, S.; Weiner, D.E.; Gutierrez, O.M.; Bansal, V.; Rosas, S.E.; Nigwekar, S.; Yee, J.; et al. KDOQI US Commentary on the 2017 KDIGO Clinical Practice Guideline Update for the Diagnosis, Evaluation, Prevention, and Treatment of Chronic Kidney Disease-Mineral and Bone Disorder (CKD-MBD). Am. J. Kidney Dis. 2017, 70, 737–751. [Google Scholar] [CrossRef] [PubMed]
- Negri, A.L.; Brandenburg, V.M. Calcitriol resistance in hemodialysis patients with secondary hyperparathyroidism. Int. Urol. Nephrol. 2014, 46, 1145–1151. [Google Scholar] [CrossRef]
- Llach, F.; Yudd, M. Paricalcitol in dialysis patients with calcitriol-resistant secondary hyperparathyroidism. Am. J. Kidney Dis. 2001, 38 (Suppl. 5), S45–S50. [Google Scholar] [CrossRef] [PubMed]
- Kuczera, P.; Adamczak, M.; Wiecek, A. Safety and efficiency of treatment with cinacalcet of haemodialysed patients with chronic kidney disease and secondary hyperparathyroidism. Endokrynol. Pol. 2013, 64, 176–181. [Google Scholar] [PubMed]
- Gois, P.H.F.; Ferreira, D.; Olenski, S.; Seguro, A.C. Vitamin D and Infectious Diseases: Simple Bystander or Contributing Factor? Nutrients 2017, 9, 651. [Google Scholar] [CrossRef] [PubMed]
- Mithal, A.; Wahl, D.A.; Bonjour, J.P.; Burckhardt, P.; Dawson-Hughes, B.; Eisman, J.A.; El-Hajj Fuleihan, G.; Josse, R.G.; Lips, P.; Morales-Torres, J. Global vitamin D status and determinants of hypovitaminosis D. Osteoporos. Int. 2009, 20, 1807–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holick, M.F. High prevalence of vitamin D inadequacy and implications for health. Mayo Clin. Proc. 2006, 81, 353–373. [Google Scholar] [CrossRef] [PubMed]
- Bischoff-Ferrari, H.A.; Giovannucci, E.; Willett, W.C.; Dietrich, T.; Dawson-Hughes, B. Estimation of optimal serum concentrations of 25-hydroxyvitamin D for multiple health outcomes. Am. J. Clin. Nutr. 2006, 84, 18–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Holick, M.F. Vitamin D for health and in chronic kidney disease. Semin. Dial. 2005, 18, 266–275. [Google Scholar] [CrossRef]
- Gonzalez, E.A.; Sachdeva, A.; Oliver, D.A.; Martin, K.J. Vitamin D insufficiency and deficiency in chronic kidney disease. A single center observational study. Am. J. Nephrol. 2004, 24, 503–510. [Google Scholar] [CrossRef]
- Fournier, A.; Fardellone, P.; Achard, J.M.; Ghazali, A.; Pruna, A.; El Esper, N.; Moriniere, P. Importance of vitamin D repletion in uraemia. Nephrol. Dial. Transplant. 1999, 14, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Eknoyan, G.; Levin, A.; Levin, N.W. Bone metabolism and disease in chronic kidney disease. Am. J. Kidney Dis. 2003, 42, 1–201. [Google Scholar] [CrossRef]
- Franca Gois, P.H.; Wolley, M.; Ranganathan, D.; Seguro, A.C. Vitamin D Deficiency in Chronic Kidney Disease: Recent Evidence and Controversies. Int. J. Environ. Res. Public Health 2018, 15, 1773. [Google Scholar] [CrossRef] [PubMed]
- Zheng, C.M.; Wu, C.C.; Hung, C.F.; Liao, M.T.; Shyu, J.F.; Hsu, Y.H.; Lu, C.L.; Wang, Y.H.; Zheng, J.Q.; Chang, T.J.; et al. Cholecalciferol Additively Reduces Serum Parathyroid Hormone and Increases Vitamin D and Cathelicidin Levels in Paricalcitol-Treated Secondary Hyperparathyroid Hemodialysis Patients. Nutrients 2016, 8, 708. [Google Scholar] [CrossRef]
- Zheng, C.M.; Wu, C.C.; Hung, C.F.; Liao, M.T.; Shyu, J.F.; Hsu, Y.H.; Lu, C.L.; Wang, Y.H.; Zheng, J.Q.; Chang, T.J.; et al. Cholecalciferol Additively Reduces Serum Parathyroid Hormone Levels in Severe Secondary Hyperparathyroidism Treated with Calcitriol and Cinacalcet among Hemodialysis Patients. Nutrients 2018, 10, 196. [Google Scholar] [CrossRef] [PubMed]
- Lang, C.L.; Wang, M.H.; Hung, K.Y.; Chiang, C.K.; Lu, K.C. Altered molecular repertoire of immune system by renal dysfunction in the elderly: Is prediction and targeted prevention in the horizon? EPMA J. 2013, 4, 17. [Google Scholar] [CrossRef]
- Madar, A.A.; Knutsen, K.V.; Stene, L.C.; Brekke, M.; Lagerlov, P.; Meyer, H.E.; Macdonald, H.M. Effect of vitamin D3-supplementation on bone markers (serum P1NP and CTX): A randomized, double blinded, placebo controlled trial among healthy immigrants living in Norway. Bone Rep. 2015, 2, 82–88. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.; Huang, Q.-R.; Gu, J.-M.; Hu, W.-W.; Liu, Y.-J.; Hu, Y.-Q.; Zhang, Z.-L. Comparison of the effects of cholecalciferol and calcitriol on calcium metabolism and bone turnover in Chinese postmenopausal women with vitamin D insufficiency. Acta Pharmacol. Sin. 2012, 33, 490–495. [Google Scholar] [CrossRef] [Green Version]
- Bang, U.C.; Kolte, L.; Hitz, M.; Schierbeck, L.L.; Nielsen, S.D.; Benfield, T.; Jensen, J.E. The effect of cholecalciferol and calcitriol on biochemical bone markers in HIV type 1-infected males: Results of a clinical trial. AIDS Res. Hum. Retrovir. 2013, 29, 658–664. [Google Scholar] [CrossRef]
- Rodriguez, M.; Nemeth, E.; Martin, D. The calcium-sensing receptor: A key factor in the pathogenesis of secondary hyperparathyroidism. Am. J. Physiol. Renal Physiol. 2005, 288, F253–F264. [Google Scholar] [CrossRef]
- K/DOQI clinical practice guidelines for bone metabolism and disease in chronic kidney disease. Am. J. Kidney Dis. 2003, 42 (Suppl. 3), S1–S201. [CrossRef]
- Aladren Regidor, M.J. Cinacalcet reduces vascular and soft tissue calcification in secondary hyperparathyroidism (SHPT) in hemodialysis patients. Clin. Nephrol. 2009, 71, 207–213. [Google Scholar] [CrossRef]
- Zheng, C.M.; Zheng, J.Q.; Wu, C.C.; Lu, C.L.; Shyu, J.F.; Yung-Ho, H.; Wu, M.Y.; Chiu, I.J.; Wang, Y.H.; Lin, Y.F.; et al. Bone loss in chronic kidney disease: Quantity or quality? Bone 2016, 87, 57–70. [Google Scholar] [CrossRef]
- Lu, K.C.; Wu, C.C.; Yen, J.F.; Liu, W.C. Vascular calcification and renal bone disorders. Sci. World J. 2014. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, H.; Nagano, N.; Urakawa, I.; Yamazaki, Y.; Iijima, K.; Fujita, T.; Yamashita, T.; Fukumoto, S.; Shimada, T. Direct evidence for a causative role of FGF23 in the abnormal renal phosphate handling and vitamin D metabolism in rats with early-stage chronic kidney disease. Kidney Int. 2010, 78, 975–980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perwad, F.; Portale, A.A. Vitamin D metabolism in the kidney: Regulation by phosphorus and fibroblast growth factor 23. Mol. Cell. Endocrinol. 2011, 347, 17–24. [Google Scholar] [CrossRef]
- Parfitt, A.M. The hyperparathyroidism of chronic renal failure: A disorder of growth. Kidney Int. 1997, 52, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Vulpio, C.; Bossola, M. Parathyroid Nodular Hyperplasia and Responsiveness to Drug Therapy in Renal Secondary Hyperparathyroidism: An Open Question. Ther. Apher. Dial. 2018, 22, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Jager, M.D.; Serttas, M.; Beneke, J.; Muller, J.A.; Schrem, H.; Kaltenborn, A.; Ramackers, W.; Ringe, B.P.; Gwiasda, J.; Trankenschuh, W.; et al. Risk-factors for nodular hyperplasia of parathyroid glands in sHPT patients. PLoS ONE 2017, 12, e0186093. [Google Scholar] [CrossRef] [PubMed]
- Ritter, C.S.; Finch, J.L.; Slatopolsky, E.A.; Brown, A.J. Parathyroid hyperplasia in uremic rats precedes down-regulation of the calcium receptor. Kidney Int. 2001, 60, 1737–1744. [Google Scholar] [CrossRef]
- Brown, A.J.; Ritter, C.S.; Finch, J.L.; Slatopolsky, E.A. Decreased calcium-sensing receptor expression in hyperplastic parathyroid glands of uremic rats: Role of dietary phosphate. Kidney Int. 1999, 55, 1284–1292. [Google Scholar] [CrossRef] [PubMed]
- Restrepo Valencia, C.A.; David, S.P.; Pinilla, C.; Elias, C.; Cardona, C.; Arnoby, J. Ultrasound Detection of Parathyroid Hyperplasia and Correlation with Clinical and Laboratory Findings in Patients with Chronic Kidney Disease. Rev. Colomb. Radiol. 2011, 22, 3341–3347. [Google Scholar]
- Tominaga, Y.; Tanaka, Y.; Sato, K.; Nagasaka, T.; Takagi, H. Histopathology, pathophysiology, and indications for surgical treatment of renal hyperparathyroidism. Semin. Surg. Oncol. 1997, 13, 78–86. [Google Scholar] [CrossRef]
- Tominaga, Y.; Matsuoka, S.; Sato, T.; Uno, N.; Goto, N.; Katayama, A.; Haba, T. Clinical features and hyperplastic patterns of parathyroid glands in hemodialysis patients with advanced secondary hyperparathyroidism refractory to maxacalcitol treatment and required parathyroidectomy. Ther. Apher. Dial. 2007, 11, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, S.; Tominaga, Y.; Sato, T.; Uno, N.; Hiramitu, T.; Goto, N.; Nagasaka, T.; Uchida, K. Relationship between the dimension of parathyroid glands estimated by ultrasonography and the hyperplastic pattern in patients with renal hyperparathyroidism. Ther. Apher. Dial. 2008, 12, 391–395. [Google Scholar] [CrossRef] [PubMed]
- Kaseda, R.; Hosojima, M.; Sato, H.; Saito, A. Role of megalin and cubilin in the metabolism of vitamin D(3). Ther. Apher. Dial. 2011, 15 (Suppl. 1), 14–17. [Google Scholar] [CrossRef]
- Axen, E. Purification from pig kidney of a microsomal cytochrome P450 catalyzing 1 alpha-hydroxylation of 25-hydroxyvitamin D3. FEBS Lett. 1995, 375, 277–279. [Google Scholar] [CrossRef]
- Takeyama, K.; Kitanaka, S.; Sato, T.; Kobori, M.; Yanagisawa, J.; Kato, S. 25-Hydroxyvitamin D3 1alpha-hydroxylase and vitamin D synthesis. Science 1997, 277, 1827–1830. [Google Scholar] [CrossRef]
- Ritter, C.S.; Armbrecht, H.J.; Slatopolsky, E.; Brown, A.J. 25-Hydroxyvitamin D(3) suppresses PTH synthesis and secretion by bovine parathyroid cells. Kidney Int. 2006, 70, 654–659. [Google Scholar] [CrossRef] [PubMed]
- Van Driel, M.; Koedam, M.; Buurman, C.J.; Hewison, M.; Chiba, H.; Uitterlinden, A.G.; Pols, H.A.; van Leeuwen, J.P. Evidence for auto/paracrine actions of vitamin D in bone: 1alpha-hydroxylase expression and activity in human bone cells. FASEB J. 2006, 20, 2417–2419. [Google Scholar] [CrossRef]
- Atkins, G.J.; Anderson, P.H.; Findlay, D.M.; Welldon, K.J.; Vincent, C.; Zannettino, A.C.; O’Loughlin, P.D.; Morris, H.A. Metabolism of vitamin D3 in human osteoblasts: Evidence for autocrine and paracrine activities of 1 alpha,25-dihydroxyvitamin D3. Bone 2007, 40, 1517–1528. [Google Scholar] [CrossRef] [PubMed]
- Zehnder, D.; Bland, R.; Chana, R.S.; Wheeler, D.C.; Howie, A.J.; Williams, M.C.; Stewart, P.M.; Hewison, M. Synthesis of 1,25-dihydroxyvitamin D(3) by human endothelial cells is regulated by inflammatory cytokines: A novel autocrine determinant of vascular cell adhesion. J. Am. Soc. Nephrol. 2002, 13, 621–629. [Google Scholar] [PubMed]
- Eyles, D.W.; Smith, S.; Kinobe, R.; Hewison, M.; McGrath, J.J. Distribution of the vitamin D receptor and 1 alpha-hydroxylase in human brain. J. Chem. Neuroanat. 2005, 29, 21–30. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.C.; Wu, C.C.; Hung, Y.M.; Liao, M.T.; Shyu, J.F.; Lin, Y.F.; Lu, K.C.; Yeh, K.C. Pleiotropic effects of vitamin D in chronic kidney disease. Clin. Chim. Acta 2016, 453, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Zehnder, D.; Evans, K.N.; Kilby, M.D.; Bulmer, J.N.; Innes, B.A.; Stewart, P.M.; Hewison, M. The ontogeny of 25-hydroxyvitamin D(3) 1alpha-hydroxylase expression in human placenta and decidua. Am. J. Pathol. 2002, 161, 105–114. [Google Scholar] [CrossRef]
- Garabedian, M.; Holick, M.F.; Deluca, H.F.; Boyle, I.T. Control of 25-hydroxycholecalciferol metabolism by parathyroid glands. Proc. Natl. Acad. Sci. USA 1972, 69, 1673–1676. [Google Scholar] [CrossRef] [PubMed]
- Murayama, A.; Takeyama, K.; Kitanaka, S.; Kodera, Y.; Kawaguchi, Y.; Hosoya, T.; Kato, S. Positive and negative regulations of the renal 25-hydroxyvitamin D3 1alpha-hydroxylase gene by parathyroid hormone, calcitonin, and 1alpha,25(OH)2D3 in intact animals. Endocrinology 1999, 140, 2224–2231. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Deluca, H.F. The control of 25-hydroxyvitamin D metabolism by inorganic phosphorus. Arch. Biochem. Biophys. 1973, 154, 566–574. [Google Scholar] [CrossRef]
- Tanaka, Y.; DeLuca, H.F. Rat renal 25-hydroxyvitamin D3 1- and 24-hydroxylases: Their in vivo regulation. Am. J. Physiol. 1984, 246 Pt 1, E168–E173. [Google Scholar] [CrossRef]
- Horiuchi, N.; Suda, T.; Takahashi, H.; Shimazawa, E.; Ogata, E. In vivo evidence for the intermediary role of 3′,5′-cyclic AMP in parathyroid hormone-induced stimulation of 1alpha,25-dihydroxyvitamin D3 synthesis in rats. Endocrinology 1977, 101, 969–974. [Google Scholar] [CrossRef] [PubMed]
- Henry, H.L. Parathyroid hormone modulation of 25-hydroxyvitamin D3 metabolism by cultured chick kidney cells is mimicked and enhanced by forskolin. Endocrinology 1985, 116, 503–510. [Google Scholar] [CrossRef] [PubMed]
- Brenza, H.L.; DeLuca, H.F. Regulation of 25-hydroxyvitamin D3 1alpha-hydroxylase gene expression by parathyroid hormone and 1,25-dihydroxyvitamin D3. Arch. Biochem. Biophys. 2000, 381, 143–152. [Google Scholar] [CrossRef] [PubMed]
- Tyson, D.R.; Swarthout, J.T.; Jefcoat, S.C.; Partridge, N.C. PTH induction of transcriptional activity of the cAMP response element-binding protein requires the serine 129 site and glycogen synthase kinase-3 activity, but not casein kinase II sites. Endocrinology 2002, 143, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Helvig, C.F.; Cuerrier, D.; Hosfield, C.M.; Ireland, B.; Kharebov, A.Z.; Kim, J.W.; Ramjit, N.J.; Ryder, K.; Tabash, S.P.; Herzenberg, A.M.; et al. Dysregulation of renal vitamin D metabolism in the uremic rat. Kidney Int. 2010, 78, 463–472. [Google Scholar] [CrossRef] [PubMed]
- Inoue, Y.; Segawa, H.; Kaneko, I.; Yamanaka, S.; Kusano, K.; Kawakami, E.; Furutani, J.; Ito, M.; Kuwahata, M.; Saito, H.; et al. Role of the vitamin D receptor in FGF23 action on phosphate metabolism. Biochem. J. 2005, 390 Pt 1, 325–331. [Google Scholar] [CrossRef]
- Jones, G.; Strugnell, S.A.; DeLuca, H.F. Current understanding of the molecular actions of vitamin D. Physiol. Rev. 1998, 78, 1193–1231. [Google Scholar] [CrossRef] [PubMed]
- St-Arnaud, R. Chapter 4—CYP24A1: Structure, Function, and Physiological Role. In Vitamin D, 3rd ed.; Feldman, D., Pike, J.W., Adams, J.S., Eds.; Academic Press: San Diego, CA, USA, 2011; pp. 43–56. [Google Scholar]
- Bikle, D.D. Vitamin D metabolism, mechanism of action, and clinical applications. Chem. Boil. 2014, 21, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Zierold, C.; Mings, J.A.; DeLuca, H.F. Regulation of 25-hydroxyvitamin D3-24-hydroxylase mRNA by 1,25-dihydroxyvitamin D3 and parathyroid hormone. J. Cell. Biochem. 2003, 88, 234–237. [Google Scholar] [CrossRef] [PubMed]
- Jones, G.; Prosser, D.E.; Kaufmann, M. 25-Hydroxyvitamin D-24-hydroxylase (CYP24A1): Its important role in the degradation of vitamin D. Arch. Biochem. Biophys. 2012, 523, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Shimada, T.; Hasegawa, H.; Yamazaki, Y.; Muto, T.; Hino, R.; Takeuchi, Y.; Fujita, T.; Nakahara, K.; Fukumoto, S.; Yamashita, T. FGF-23 is a potent regulator of vitamin D metabolism and phosphate homeostasis. J. Bone Miner. Res. 2004, 19, 429–435. [Google Scholar] [CrossRef] [PubMed]
- Levin, A.; Bakris, G.L.; Molitch, M.; Smulders, M.; Tian, J.; Williams, L.A.; Andress, D.L. Prevalence of abnormal serum vitamin D, PTH, calcium, and phosphorus in patients with chronic kidney disease: Results of the study to evaluate early kidney disease. Kidney Int. 2007, 71, 31–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, S.W.; Russell, J.; Avioli, L.V. 25-hydroxycholecalciferol to 1,25-dihydroxycholecalciferol: Conversion impaired by systemic metabolic acidosis. Science 1977, 195, 994–996. [Google Scholar] [CrossRef] [PubMed]
- Hsu, C.H.; Patel, S.R.; Young, E.W.; Vanholder, R. Effects of purine derivatives on calcitriol metabolism in rats. Am. J. Physiol. 1991, 260 Pt 2, F596–F601. [Google Scholar] [CrossRef]
- Vanholder, R.; Patel, S.; Hsu, C.H. Effect of uric acid on plasma levels of 1,25(OH)2D in renal failure. J. Am. Soc. Nephrol. 1993, 4, 1035–1038. [Google Scholar] [PubMed]
- Takemoto, F.; Shinki, T.; Yokoyama, K.; Inokami, T.; Hara, S.; Yamada, A.; Kurokawa, K.; Uchida, S. Gene expression of vitamin D hydroxylase and megalin in the remnant kidney of nephrectomized rats. Kidney Int. 2003, 64, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Ben-Dov, I.Z.; Galitzer, H.; Lavi-Moshayoff, V.; Goetz, R.; Kuro-o, M.; Mohammadi, M.; Sirkis, R.; Naveh-Many, T.; Silver, J. The parathyroid is a target organ for FGF23 in rats. J. Clin. Investig. 2007, 117, 4003–4008. [Google Scholar] [CrossRef] [Green Version]
- Canalejo, R.; Canalejo, A.; Martinez-Moreno, J.M.; Rodriguez-Ortiz, M.E.; Estepa, J.C.; Mendoza, F.J.; Munoz-Castaneda, J.R.; Shalhoub, V.; Almaden, Y.; Rodriguez, M. FGF23 fails to inhibit uremic parathyroid glands. J. Am. Soc. Nephrol. 2010, 21, 1125–1135. [Google Scholar] [CrossRef]
- Lafage-Proust, M.H. Does the downregulation of the FGF23 signaling pathway in hyperplastic parathyroid glands contribute to refractory secondary hyperparathyroidism in CKD patients? Kidney Int. 2010, 77, 390–392. [Google Scholar] [CrossRef]
- Yan, J.; Jingbo, C.; Wang, D.; Xie, S.; Yuan, L.; Zhong, X.; Hao, L. A correlation between decreased parathyroid alpha-Klotho and fibroblast growth factor receptor 1 expression with pathological category and parathyroid gland volume in dialysis patients. Int. Urol. Nephrol. 2015, 47, 701–706. [Google Scholar] [CrossRef] [PubMed]
- Segersten, U.; Correa, P.; Hewison, M.; Hellman, P.; Dralle, H.; Carling, T.; Akerstrom, G.; Westin, G. 25-hydroxyvitamin D(3)-1alpha-hydroxylase expression in normal and pathological parathyroid glands. J. Clin. Endocrinol. Metab. 2002, 87, 2967–2972. [Google Scholar] [PubMed]
- Correa, P.; Segersten, U.; Hellman, P.; Akerstrom, G.; Westin, G. Increased 25-hydroxyvitamin D3 1alpha-hydroxylase and reduced 25-hydroxyvitamin D3 24-hydroxylase expression in parathyroid tumors--new prospects for treatment of hyperparathyroidism with vitamin d. J. Clin. Endocrinol. Metab. 2002, 87, 5826–5829. [Google Scholar] [CrossRef] [PubMed]
- Ritter, C.S.; Haughey, B.H.; Armbrecht, H.J.; Brown, A.J. Distribution and regulation of the 25-hydroxyvitamin D3 1alpha-hydroxylase in human parathyroid glands. J. Steroid Biochem. Mol. Biol. 2012, 130, 73–80. [Google Scholar] [CrossRef] [PubMed]
- Yano, S.; Sugimoto, T.; Tsukamoto, T.; Chihara, K.; Kobayashi, A.; Kitazawa, S.; Maeda, S.; Kitazawa, R. Association of decreased calcium-sensing receptor expression with proliferation of parathyroid cells in secondary hyperparathyroidism. Kidney Int. 2000, 58, 1980–1986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Muller-Hocker, J.; Schafer, S.; Krebs, S.; Blum, H.; Zsurka, G.; Kunz, W.S.; Prokisch, H.; Seibel, P.; Jung, A. Oxyphil cell metaplasia in the parathyroids is characterized by somatic mitochondrial DNA mutations in NADH dehydrogenase genes and cytochrome c oxidase activity-impairing genes. Am. J. Pathol. 2014, 184, 2922–2935. [Google Scholar] [CrossRef] [PubMed]
- Lomonte, C.; Vernaglione, L.; Chimienti, D.; Bruno, A.; Cocola, S.; Teutonico, A.; Cazzato, F.; Basile, C. Does vitamin D receptor and calcium receptor activation therapy play a role in the histopathologic alterations of parathyroid glands in refractory uremic hyperparathyroidism? Clin. J. Am. Soc. Nephrol. 2008, 3, 794–799. [Google Scholar] [CrossRef]
- Sumida, K.; Nakamura, M.; Ubara, Y.; Marui, Y.; Tanaka, K.; Takaichi, K.; Tomikawa, S.; Inoshita, N.; Ohashi, K. Histopathological alterations of the parathyroid glands in haemodialysis patients with secondary hyperparathyroidism refractory to cinacalcet hydrochloride. J. Clin. Pathol. 2011, 64, 756–760. [Google Scholar] [CrossRef]
- Tanaka, Y.; Funahashi, H.; Imai, T.; Seo, H.; Tominaga, Y.; Takagi, H. Oxyphil cell function in secondary parathyroid hyperplasia. Nephron 1996, 73, 580–586. [Google Scholar]
- Matsushita, H.; Hara, M.; Endo, Y.; Shishiba, Y.; Hara, S.; Ubara, Y.; Nakazawa, H.; Suzuki, N.; Kawaminami, K.; Kido, T.; et al. Proliferation of parathyroid cells negatively correlates with expression of parathyroid hormone-related protein in secondary parathyroid hyperplasia. Kidney Int. 1999, 55, 130–138. [Google Scholar] [CrossRef]
- Lewin, E.; Almaden, Y.; Rodriguez, M.; Olgaard, K. PTHrP enhances the secretory response of PTH to a hypocalcemic stimulus in rat parathyroid glands. Kidney Int. 2000, 58, 71–81. [Google Scholar] [CrossRef] [PubMed]
- Lewin, E.; Garfia, B.; Almaden, Y.; Rodriguez, M.; Olgaard, K. Autoregulation in the parathyroid glands by PTH/PTHrP receptor ligands in normal and uremic rats. Kidney Int. 2003, 64, 63–70. [Google Scholar] [CrossRef] [PubMed]
- Basile, C.; Lomonte, C. The function of the parathyroid oxyphil cells in uremia: Still a mystery? Kidney Int. 2017, 92, 1046–1048. [Google Scholar] [CrossRef] [PubMed]
- Ritter, C.; Miller, B.; Coyne, D.W.; Gupta, D.; Zheng, S.; Brown, A.J.; Slatopolsky, E. Paricalcitol and cinacalcet have disparate actions on parathyroid oxyphil cell content in patients with chronic kidney disease. Kidney Int. 2017, 92, 1217–1222. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Mao, J.; Wang, M.; Zhang, M.; Ni, L.; Tao, Y.; Huang, B.; Chen, J. Comparative proteomic analysis of chief and oxyphil cell nodules in refractory uremic hyperparathyroidism by iTRAQ coupled LC-MS/MS. J. Proteom. 2018, 179, 42–52. [Google Scholar] [CrossRef] [PubMed]
- Fukagawa, M.; Okazaki, R.; Takano, K.; Kaname, S.; Ogata, E.; Kitaoka, M.; Harada, S.; Sekine, N.; Matsumoto, T.; Kurokawa, K. Regression of parathyroid hyperplasia by calcitriol-pulse therapy in patients on long-term dialysis. N. Engl. J. Med. 1990, 323, 421–422. [Google Scholar] [PubMed]
- Fukagawa, M.; Kitaoka, M. Yi, H.; Fukuda, N.; Matsumoto, T.; Ogata, E.; Kurokawa, K. Serial evaluation of parathyroid size by ultrasonography is another useful marker for the long-term prognosis of calcitriol pulse therapy in chronic dialysis patients. Nephron 1994, 68, 221–228. [Google Scholar] [CrossRef]
- Fukagawa, M.; Yumita, S.; Akizawa, T.; Uchida, E.; Tsukamoto, Y.; Iwasaki, M.; Koshikawa, S. Cinacalcet (KRN1493) effectively decreases the serum intact PTH level with favourable control of the serum phosphorus and calcium levels in Japanese dialysis patients. Nephrol. Dial. Transplant. 2008, 23, 328–335. [Google Scholar] [CrossRef]
- Colloton, M.; Shatzen, E.; Miller, G.; Stehman-Breen, C.; Wada, M.; Lacey, D.; Martin, D. Cinacalcet HCl attenuates parathyroid hyperplasia in a rat model of secondary hyperparathyroidism. Kidney Int. 2005, 67, 467–476. [Google Scholar] [CrossRef]
- Chin, J.; Miller, S.C.; Wada, M.; Nagano, N.; Nemeth, E.F.; Fox, J. Activation of the calcium receptor by a calcimimetic compound halts the progression of secondary hyperparathyroidism in uremic rats. J. Am. Soc. Nephrol. 2000, 11, 903–911. [Google Scholar]
- Mizobuchi, M.; Ogata, H.; Hatamura, I.; Saji, F.; Koiwa, F.; Kinugasa, E.; Koshikawa, S.; Akizawa, T. Activation of calcium-sensing receptor accelerates apoptosis in hyperplastic parathyroid cells. Biochem. Biophys. Res. Commun. 2007, 362, 11–16. [Google Scholar] [CrossRef] [PubMed]
- Mendoza, F.J.; Lopez, I.; Canalejo, R.; Almaden, Y.; Martin, D.; Aguilera-Tejero, E.; Rodriguez, M. Direct upregulation of parathyroid calcium-sensing receptor and vitamin D receptor by calcimimetics in uremic rats. Am. J. Physiol. Renal Physiol. 2009, 296, F605–F613. [Google Scholar] [CrossRef] [PubMed]
- Riccardi, D.; Martin, D. The role of the calcium-sensing receptor in the pathophysiology of secondary hyperparathyroidism. NDT Plus 2008, 1 (Suppl. 1), i7–i11. [Google Scholar] [CrossRef] [PubMed]
- Bikle, D.D.; Siiteri, P.K.; Ryzen, E.; Haddad, J.G. Serum protein binding of 1,25-dihydroxyvitamin D: A reevaluation by direct measurement of free metabolite levels. J. Clin. Endocrinol. Metab. 1985, 61, 969–975. [Google Scholar] [CrossRef] [PubMed]
- Mason, D.; Donabella, P.J.; Nnani, D.; Musteata, F.M. Normalized vitamin D metabolite concentrations are better correlated to pharmacological effects than measured concentrations. Future Sci. OA 2015, 1, Fso83. [Google Scholar] [CrossRef] [PubMed]
- Bhan, I.; Powe, C.E.; Berg, A.H.; Ankers, E.; Wenger, J.B.; Karumanchi, S.A.; Thadhani, R.I. Bioavailable vitamin D is more tightly linked to mineral metabolism than total vitamin D in incident hemodialysis patients. Kidney Int. 2012, 82, 84–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powe, C.E.; Ricciardi, C.; Berg, A.H.; Erdenesanaa, D.; Collerone, G.; Ankers, E.; Wenger, J.; Karumanchi, S.A.; Thadhani, R.; Bhan, I. Vitamin D-binding protein modifies the vitamin D-bone mineral density relationship. J. Bone Miner. Res. 2011, 26, 1609–1616. [Google Scholar] [CrossRef] [Green Version]
- Tsuprykov, O.; Chen, X.; Hocher, C.F.; Skoblo, R.; Lianghong, Y.; Hocher, B. Why should we measure free 25(OH) vitamin D? J. Steroid Biochem. Mol. Biol. 2018, 180, 87–104. [Google Scholar] [CrossRef]
- Bikle, D.D.; Malmstroem, S.; Schwartz, J. Current Controversies: Are Free Vitamin Metabolite Levels a More Accurate Assessment of Vitamin D Status than Total Levels? Endocrinol. Metab. Clin. N. Am. 2017, 46, 901–918. [Google Scholar] [CrossRef]
- Chun, R.F.; Lauridsen, A.L.; Suon, L.; Zella, L.A.; Pike, J.W.; Modlin, R.L.; Martineau, A.R.; Wilkinson, R.J.; Adams, J.; Hewison, M. Vitamin D Binding Protein Is Not Involved in Vitamin D Deficiency in Patients with Chronic Kidney Disease. Biomed. Res. Int. 2015, 2015, 492365. [Google Scholar]
- Chun, R.F.; Lauridsen, A.L.; Suon, L.; Zella, L.A.; Pike, J.W.; Modlin, R.L.; Martineau, A.R.; Wilkinson, R.J.; Adams, J.; Hewison, M. Vitamin D-binding protein directs monocyte responses to 25-hydroxy- and 1,25-dihydroxyvitamin D. J. Clin. Endocrinol. Metab. 2010, 95, 3368–3376. [Google Scholar] [CrossRef] [PubMed]
- Bikle, D.D.; Gee, E. Free, and not total, 1,25-dihydroxyvitamin D regulates 25-hydroxyvitamin D metabolism by keratinocytes. Endocrinology 1989, 124, 649–654. [Google Scholar] [CrossRef] [PubMed]
- Hewison, M. An update on vitamin D and human immunity. Clin. Endocrinol. 2012, 76, 315–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauridsen, A.L.; Vestergaard, P.; Hermann, A.P.; Brot, C.; Heickendorff, L.; Mosekilde, L.; Nexo, E. Plasma concentrations of 25-hydroxy-vitamin D and 1,25-dihydroxy-vitamin D are related to the phenotype of Gc (vitamin D-binding protein): A cross-sectional study on 595 early postmenopausal women. Calcif. Tissue Int. 2005, 77, 15–22. [Google Scholar] [CrossRef]
- Wang, T.J.; Zhang, F.; Richards, J.B.; Kestenbaum, B.; van Meurs, J.B.; Berry, D.; Kiel, D.P.; Streeten, E.A.; Ohlsson, C.; Koller, D.L.; et al. Common genetic determinants of vitamin D insufficiency: A genome-wide association study. Lancet 2010, 376, 180–188. [Google Scholar] [CrossRef]
- Kagi, L.; Bettoni, C.; Pastor-Arroyo, E.M.; Schnitzbauer, U.; Hernando, N.; Wagner, C.A. Regulation of vitamin D metabolizing enzymes in murine renal and extrarenal tissues by dietary phosphate, FGF23, and 1,25(OH)2D3. PLoS ONE 2018, 13, e0195427. [Google Scholar] [CrossRef]
- Kim, M.S.; Fujiki, R.; Kitagawa, H.; Kato, S. 1alpha,25(OH)2D3-induced DNA methylation suppresses the human CYP27B1 gene. Mol. Cell. Endocrinol. 2007, 265–266, 168–173. [Google Scholar] [CrossRef]
- St-Arnaud, R.; Arabian, A.; Travers, R.; Barletta, F.; Raval-Pandya, M.; Chapin, K.; Depovere, J.; Mathieu, C.; Christakos, S.; Demay, M.B.; et al. Deficient mineralization of intramembranous bone in vitamin D-24-hydroxylase-ablated mice is due to elevated 1,25-dihydroxyvitamin D and not to the absence of 24,25-dihydroxyvitamin D. Endocrinology 2000, 141, 2658–2666. [Google Scholar] [CrossRef]
- Kawahara, M.; Iwasaki, Y.; Sakaguchi, K.; Taguchi, T.; Nishiyama, M.; Nigawara, T.; Tsugita, M.; Kambayashi, M.; Suda, T.; Hashimoto, K. Predominant role of 25OHD in the negative regulation of PTH expression: Clinical relevance for hypovitaminosis D. Life Sci. 2008, 82, 677–683. [Google Scholar] [CrossRef]
- Arcidiacono, M.V.; Yang, J.; Fernandez, E.; Dusso, A. The induction of C/EBPβ contributes to vitamin D inhibition of ADAM17 expression and parathyroid hyperplasia in kidney disease. Nephrol. Dial. Transplant. 2015, 30, 423–433. [Google Scholar] [CrossRef]
- Evenepoel, P.; Bover, J.; Urena Torres, P. Parathyroid hormone metabolism and signaling in health and chronic kidney disease. Kidney Int. 2016, 90, 1184–1190. [Google Scholar] [CrossRef] [PubMed]
- Disthabanchong, S.; Hassan, H.; McConkey, C.L.; Martin, K.J.; Gonzalez, E.A. Regulation of PTH1 receptor expression by uremic ultrafiltrate in UMR 106-01 osteoblast-like cells. Kidney Int. 2004, 65, 897–903. [Google Scholar] [CrossRef] [PubMed]
- Sage, A.P.; Lu, J.; Atti, E.; Tetradis, S.; Ascenzi, M.G.; Adams, D.J.; Demer, L.L.; Tintut, Y. Hyperlipidemia induces resistance to PTH bone anabolism in mice via oxidized lipids. J. Bone Miner. Res. 2011, 26, 1197–1206. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nii-Kono, T.; Iwasaki, Y.; Uchida, M.; Fujieda, A.; Hosokawa, A.; Motojima, M.; Yamato, H.; Kurokawa, K.; Fukagawa, M. Indoxyl sulfate induces skeletal resistance to parathyroid hormone in cultured osteoblastic cells. Kidney Int. 2007, 71, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.L.; Shyu, J.F.; Wu, C.C.; Hung, C.F.; Liao, M.T.; Liu, W.C.; Zheng, C.M.; Hou, Y.C.; Lin, Y.F.; Lu, K.C. Association of Anabolic Effect of Calcitriol with Osteoclast-Derived Wnt 10b Secretion. Nutrients 2018, 10, 1164. [Google Scholar] [CrossRef] [PubMed]
- Wolf, M.; Shah, A.; Gutierrez, O.; Ankers, E.; Monroy, M.; Tamez, H.; Steele, D.; Chang, Y.; Camargo, C.A., Jr.; Tonelli, M.; et al. Vitamin D levels and early mortality among incident hemodialysis patients. Kidney Int. 2007, 72, 1004–1013. [Google Scholar] [CrossRef]
- Schwartz, J.B.; Kane, L.; Bikle, D. Response of Vitamin D Concentration to Vitamin D3 Administration in Older Adults without Sun Exposure: A Randomized Double-Blind Trial. J. Am. Geriatr. Soc. 2016, 64, 65–72. [Google Scholar] [CrossRef]
- Aloia, J.; Dhaliwal, R.; Mikhail, M.; Shieh, A.; Stolberg, A.; Ragolia, L.; Fazzari, M.; Abrams, S.A. Free 25(OH)D and Calcium Absorption, PTH, and Markers of Bone Turnover. J. Clin. Endocrinol. Metab. 2015, 100, 4140–4145. [Google Scholar] [CrossRef]
- Jemielita, T.O.; Leonard, M.B.; Baker, J.; Sayed, S.; Zemel, B.S.; Shults, J.; Herskovitz, R.; Denburg, M.R. Association of 25-hydroxyvitamin D with areal and volumetric measures of bone mineral density and parathyroid hormone: Impact of vitamin D-binding protein and its assays. Osteoporos. Int. 2016, 27, 617–626. [Google Scholar] [CrossRef]
- Shieh, A.; Ma, C.; Chun, R.F.; Wittwer-Schegg, J.; Swinkels, L.; Huijs, T.; Wang, J.; Donangelo, I.; Hewison, M.; Adams, J.S. Associations Between Change in Total and Free 25-Hydroxyvitamin D with 24,25-Dihydroxyvitamin D and Parathyroid Hormone. J. Clin. Endocrinol. Metab. 2018, 103, 3368–3375. [Google Scholar] [CrossRef]
- Chandra, P.; Binongo, J.N.; Ziegler, T.R.; Schlanger, L.E.; Wang, W.; Someren, J.T.; Tangpricha, V. Cholecalciferol (vitamin D3) therapy and vitamin D insufficiency in patients with chronic kidney disease: A randomized controlled pilot study. Endocr. Pract. 2008, 14, 10–17. [Google Scholar] [CrossRef] [PubMed]
- Westerberg, P.A.; Sterner, G.; Ljunggren, O.; Isaksson, E.; Elvarson, F.; Dezfoolian, H.; Linde, T. High doses of cholecalciferol alleviate the progression of hyperparathyroidism in patients with CKD Stages 3–4: Results of a 12-week double-blind, randomized, controlled study. Nephrol. Dial. Transplant. 2018, 33, 466–471. [Google Scholar] [CrossRef] [PubMed]
- Mangoo-Karim, R.; Da Silva Abreu, J.; Yanev, G.P.; Perez, N.N.; Stubbs, J.R.; Wetmore, J.B. Ergocalciferol versus Cholecalciferol for Nutritional Vitamin D Replacement in CKD. Nephron 2015, 130, 99–104. [Google Scholar] [CrossRef] [PubMed]
- Blair, D.; Byham-Gray, L.; Lewis, E.; McCaffrey, S. Prevalence of vitamin D [25(OH)D] deficiency and effects of supplementation with ergocalciferol (vitamin D2) in stage 5 chronic kidney disease patients. J. Ren. Nutr. 2008, 18, 375–382. [Google Scholar] [CrossRef] [PubMed]
- Bouchard, J.; Ouimet, D.; Vallee, M.; Leblanc, M.; Pichette, V. Effect of vitamin D supplementation on calcidiol and parathyroid hormone levels. Perit. Dial. Int. 2008, 28, 565. [Google Scholar] [PubMed]
- Bucharles, S.; Barberato, S.H.; Stinghen, A.E.; Gruber, B.; Piekala, L.; Dambiski, A.C.; Custodio, M.R.; Pecoits-Filho, R. Impact of cholecalciferol treatment on biomarkers of inflammation and myocardial structure in hemodialysis patients without hyperparathyroidism. J. Ren. Nutr. 2012, 22, 284–291. [Google Scholar] [CrossRef] [PubMed]
- Kandula, P.; Dobre, M.; Schold, J.D.; Schreiber, M.J., Jr.; Mehrotra, R.; Navaneethan, S.D. Vitamin D supplementation in chronic kidney disease: A systematic review and meta-analysis of observational studies and randomized controlled trials. Clin. J. Am. Soc. Nephrol. 2011, 6, 50–62. [Google Scholar] [CrossRef] [PubMed]
- Jean, G.; Terrat, J.C.; Vanel, T.; Hurot, J.M.; Lorriaux, C.; Mayor, B.; Chazot, C. Evidence for persistent vitamin D 1-alpha-hydroxylation in hemodialysis patients: Evolution of serum 1,25-dihydroxycholecalciferol after 6 months of 25-hydroxycholecalciferol treatment. Nephron Clin. Pract. 2008, 110, c58–c65. [Google Scholar] [CrossRef] [PubMed]
- Bhan, I.; Dobens, D.; Tamez, H.; Deferio, J.J.; Li, Y.C.; Warren, H.S.; Ankers, E.; Wenger, J.; Tucker, J.K.; Trottier, C.; et al. Nutritional vitamin D supplementation in dialysis: A randomized trial. Clin. J. Am. Soc. Nephrol. 2015, 10, 611–619. [Google Scholar] [CrossRef] [PubMed]

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lu, C.-L.; Yeih, D.-F.; Hou, Y.-C.; Jow, G.-M.; Li, Z.-Y.; Liu, W.-C.; Zheng, C.-M.; Lin, Y.-F.; Shyu, J.-F.; Chen, R.; et al. The Emerging Role of Nutritional Vitamin D in Secondary Hyperparathyroidism in CKD. Nutrients 2018, 10, 1890. https://doi.org/10.3390/nu10121890
Lu C-L, Yeih D-F, Hou Y-C, Jow G-M, Li Z-Y, Liu W-C, Zheng C-M, Lin Y-F, Shyu J-F, Chen R, et al. The Emerging Role of Nutritional Vitamin D in Secondary Hyperparathyroidism in CKD. Nutrients. 2018; 10(12):1890. https://doi.org/10.3390/nu10121890
Chicago/Turabian StyleLu, Chien-Lin, Dong-Feng Yeih, Yi-Chou Hou, Guey-Mei Jow, Zong-Yu Li, Wen-Chih Liu, Cai-Mei Zheng, Yuh-Feng Lin, Jia-Fwu Shyu, Remy Chen, and et al. 2018. "The Emerging Role of Nutritional Vitamin D in Secondary Hyperparathyroidism in CKD" Nutrients 10, no. 12: 1890. https://doi.org/10.3390/nu10121890