Hyperspectral imagery (HSI) has found many applications in various fields, such as military, agriculture, and mineralogy [
1]. As one of the traditional hyperspectral analysis techniques, spectral matching classification is often used for signature discrimination, which concentrates on recognizing absorption band and shape of spectra [
2]. Many spectral matching classification techniques have been proposed. They often depend on a scalar metric to estimate how close two spectra are. In general, they fall into two categories [
3,
4,
5,
6]. In the first category, traditional algorithms estimate the similarity of two spectra by comparing signature distance. For example, minimum Euclidean distance (MED) classification method divides the classes of land covers by calculating the Euclidean distance of spectral vectors. The smaller the distance value is, the more similar the land covers are. MED provides a similarity scale to estimate spectral brightness with high efficiency. Binary coding (BC) is another method comparing spectral signature brightness [
7]. It calculates the mean value of spectral curve, and then the spectral curve is encoded to a sequence of zeroes and ones through mean-value-threshold. It may not provide a precise spectral matching result. Spectral distance based classification methods may lead the misclassification when image sunlight is insufficient or shadow exists [
8,
9]. In the second category, spectra are regarded as multidimensional vectors. The similarity of two spectra is estimated by comparing shapes. For example, spectral angle mapper (SAM) method calculates the generalized angle of two vectors. The smaller the angle is, the more similar the spectrums are [
10,
11]. Spectral shape based matching methods can avoid the partial noises, so a high accuracy of spectral matching result can be acquired. However, it is worth noting that some important details may be smoothed in these kinds of methods [
12]. In addition, there are some other encoding works with successful performance are proposed for image processing. Spatial pyramid matching (SPM) divides image into blocks in different scales, then the histograms of local features in each block represent the image [
13]. This approach achieved impressive performance of image classification. SCSPM incorporates sparse code (SC) into the framework of SPM. This variant of SPM achieves a higher recognition rate than traditional SPM [
14]. However, it consumes more time for encoding the local descriptors. Low rank representation (Lrr) based spatial pyramid matching (SPM) method encodes the descriptors under the framework of SPM and calculates the representation in the matrix space directly [
15,
16]. It improves the robustness of the variants of SPM. LrrSPM achieves a better efficiency than the variants of SPM.
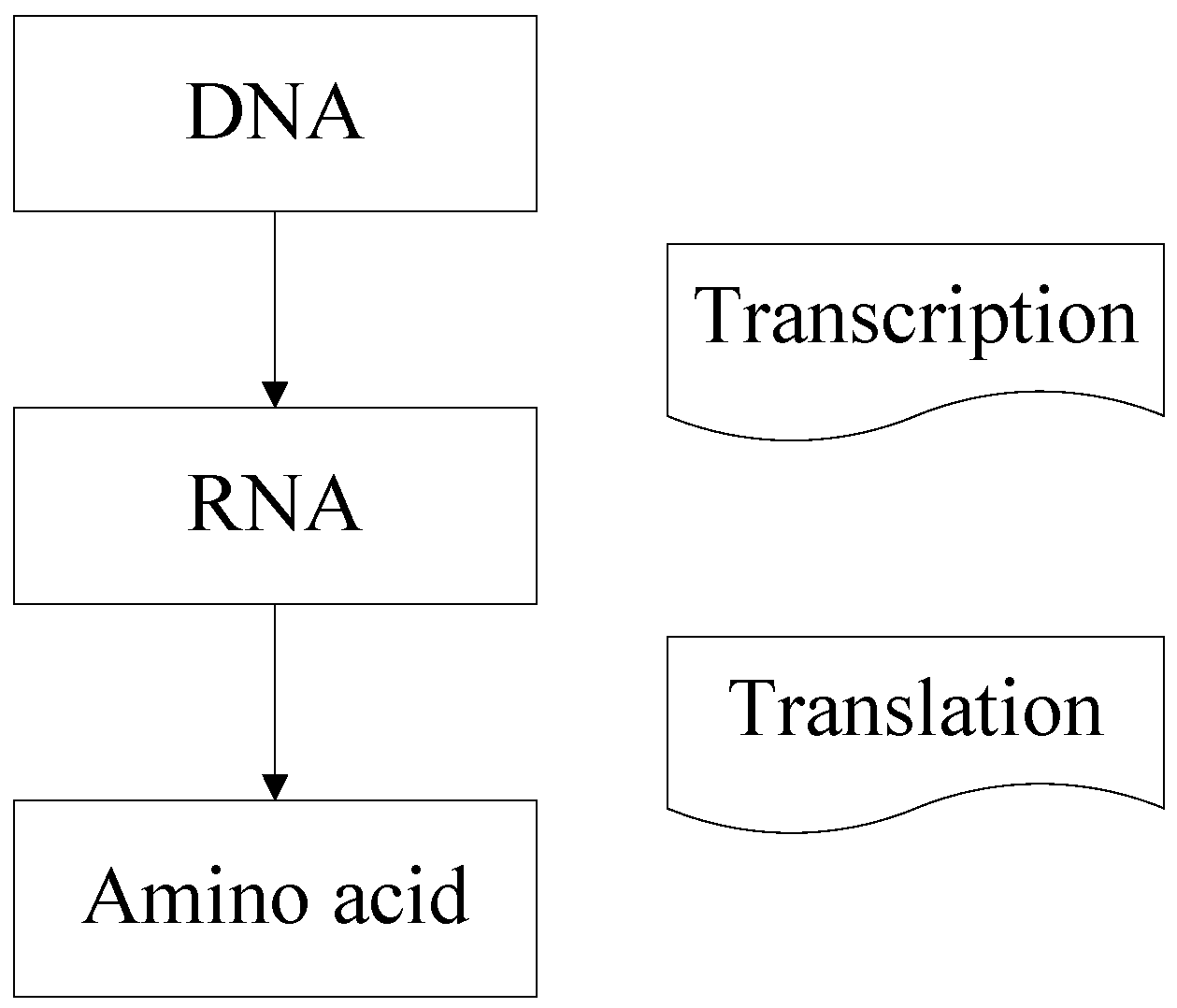
Recently, a novel classification strategy inspired by the artificial deoxyribonucleic acid (DNA) technology has been presented for hyperspectral remote sensing imagery [
17,
18,
19,
20]. As a branch of computational intelligence, artificial DNA encoding and matching (ADEM) method has the strong computing and matching capability to discriminate the tiny differences in DNA strands [
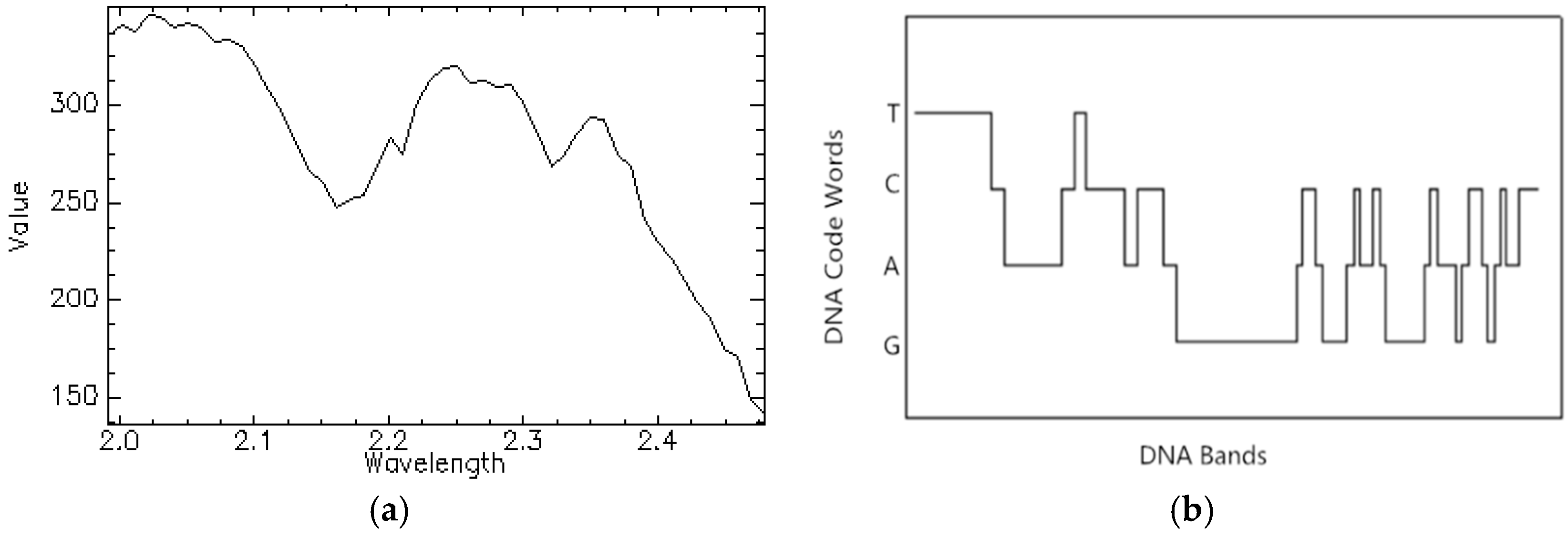
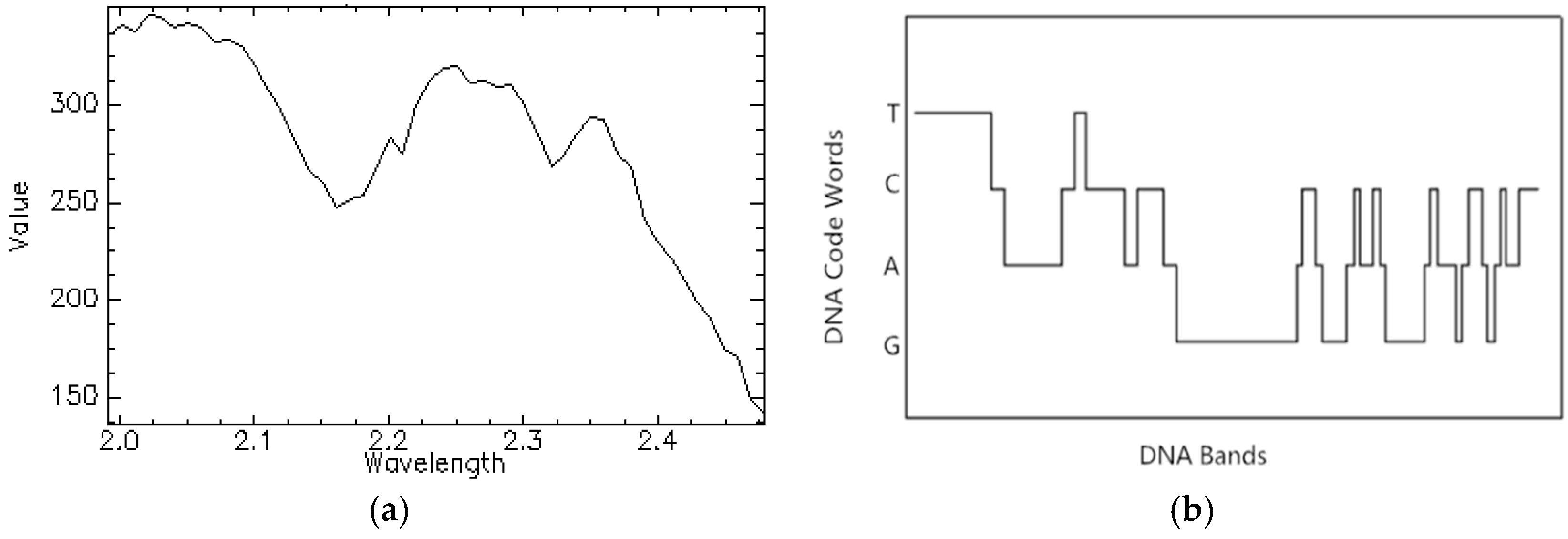
21]. It can discriminate the tiny differences in DNA strands by DNA encoding and matching in the molecule layer. DNA encoding method in this classifier plays an important role. It not only reduces the influence of the noise caused by sunlight and weather but also takes both brightness and shape information into consideration [
7]. In a word, it has been proven that ADEM can comprehensively compare the similarity of two spectra. However, there are some limitations in practice so that the classification result is affected. Because when all of the bands participate in the encoding procedure, data volume is expanded which may amplify redundancy. Moreover, the full-band comparison degrades the importance of key spectral bands carrying critical information in class separation [
22].
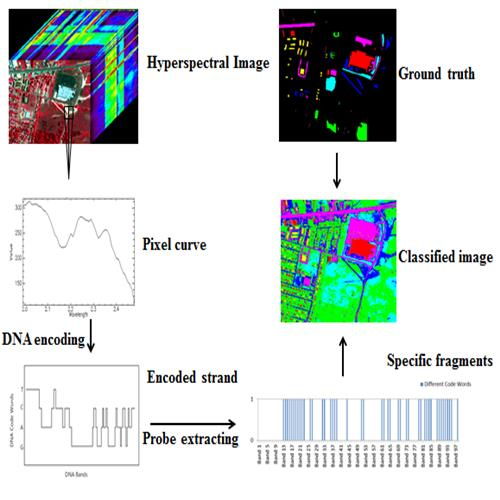
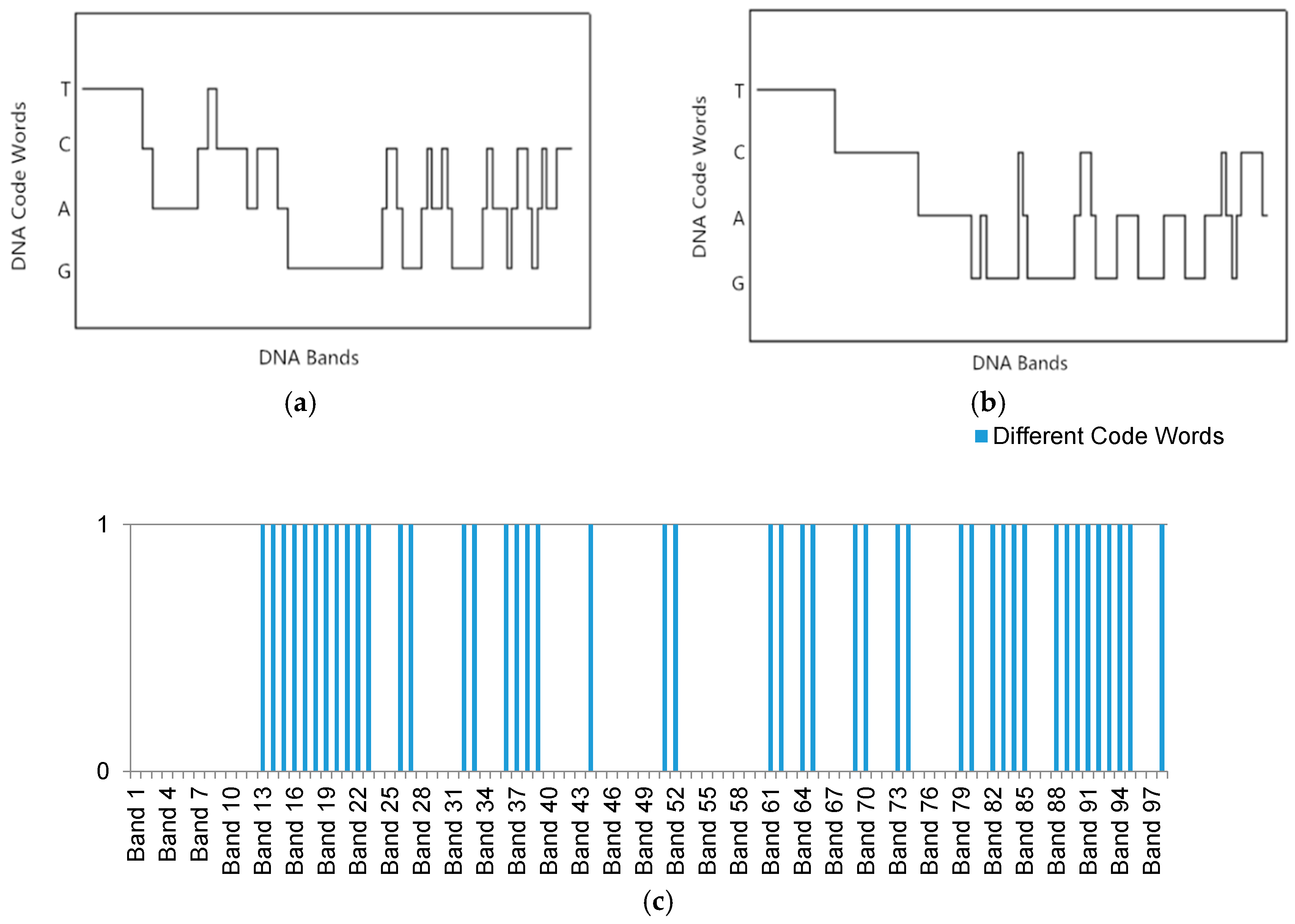
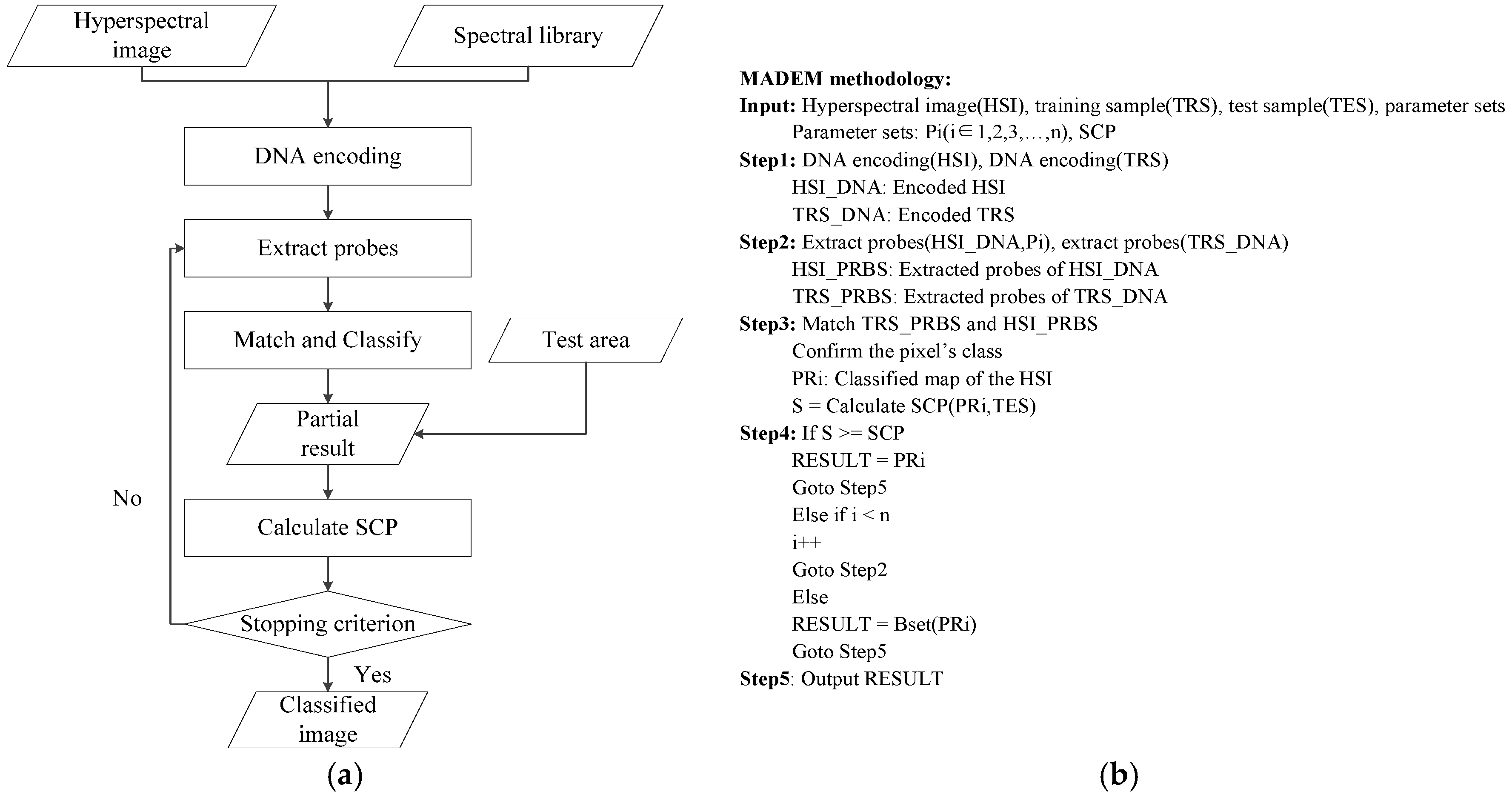
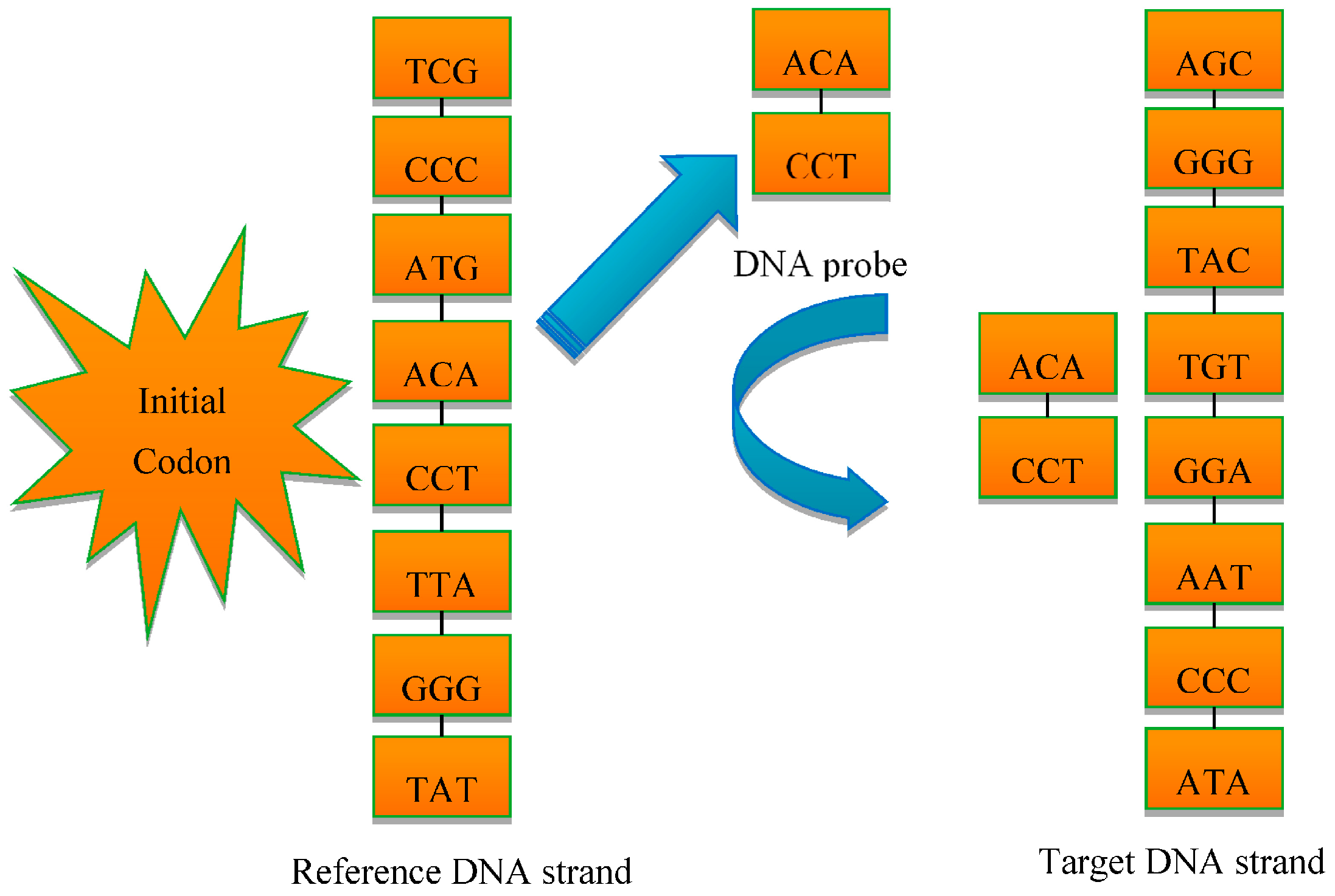
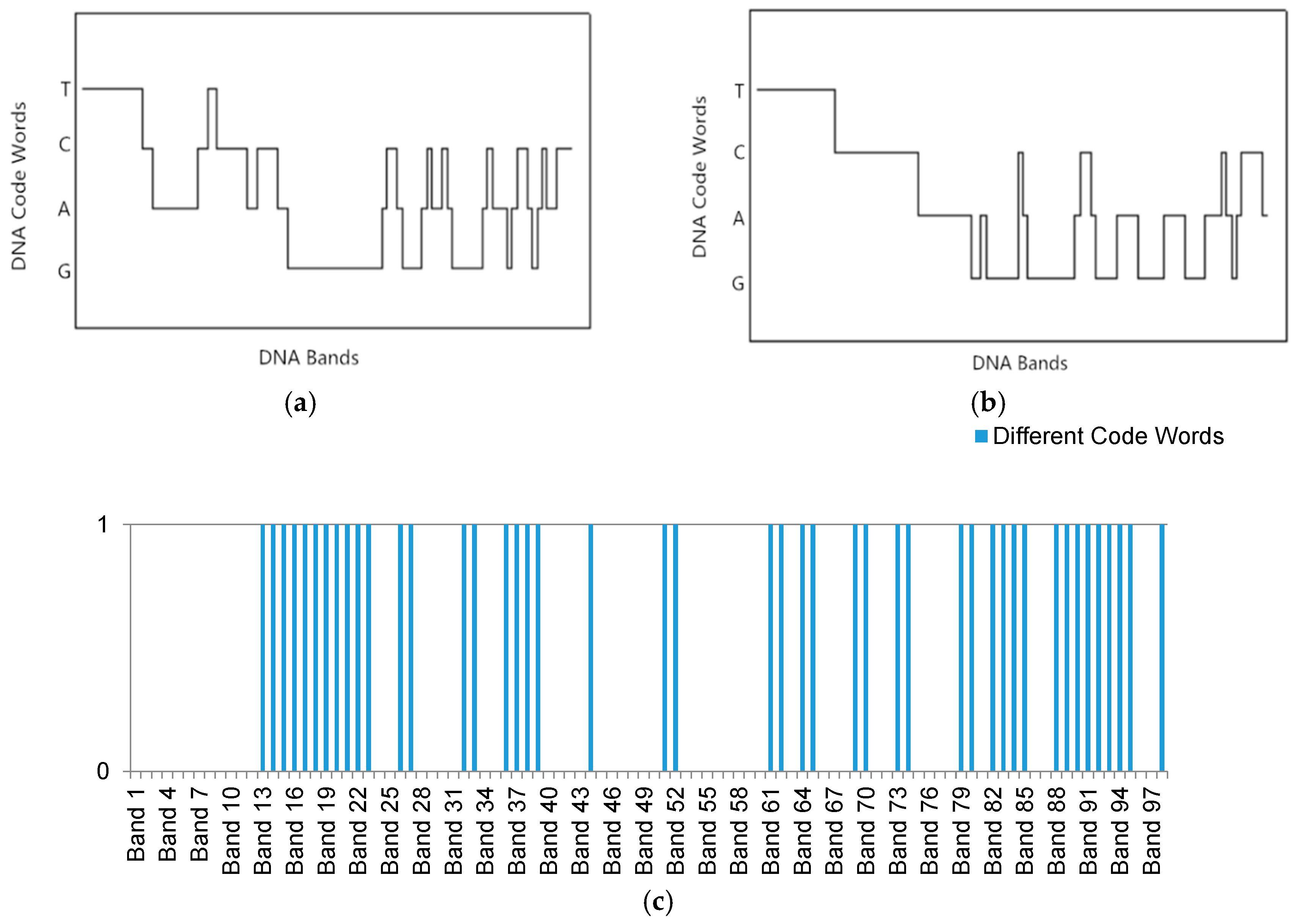
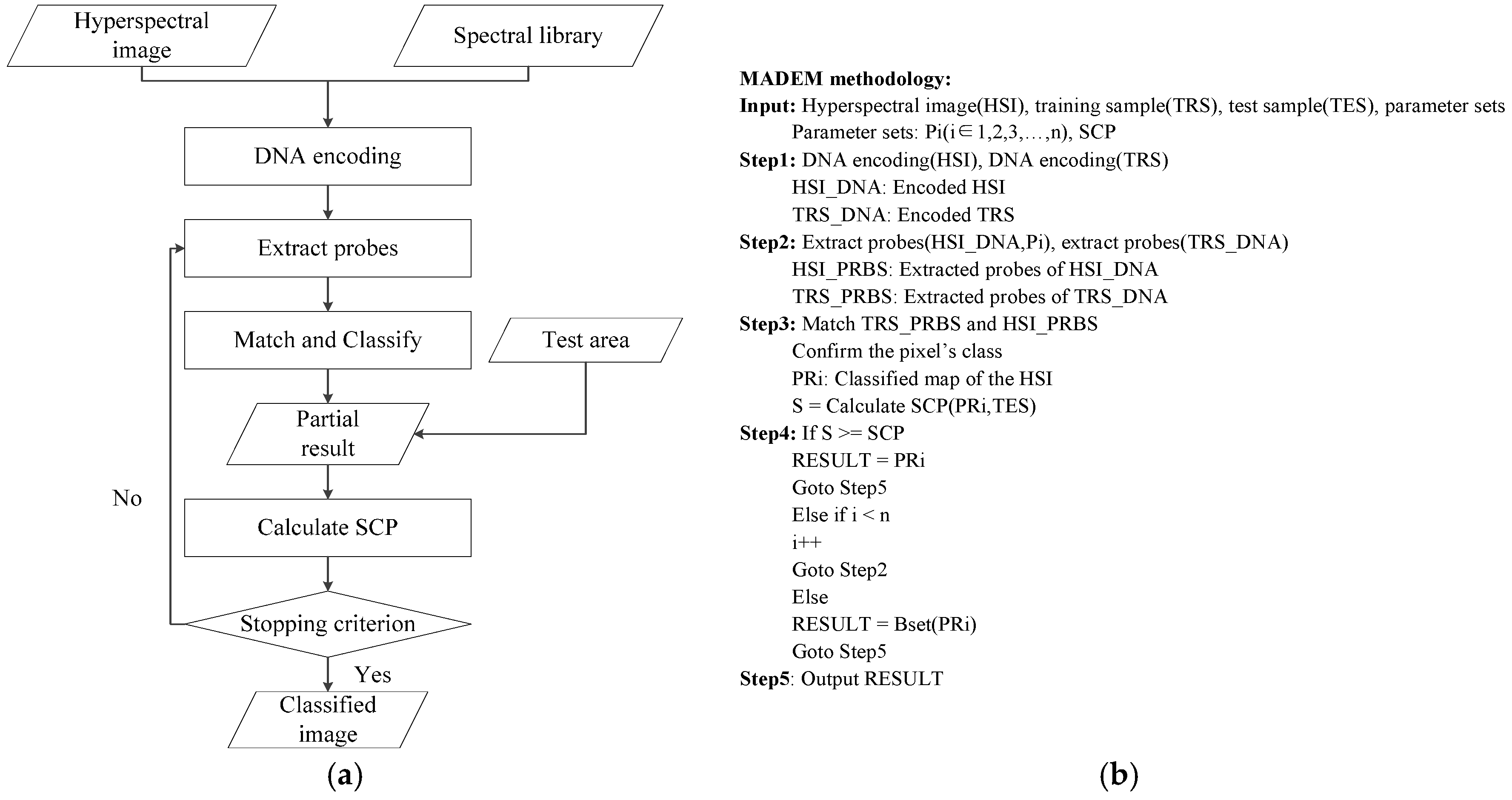
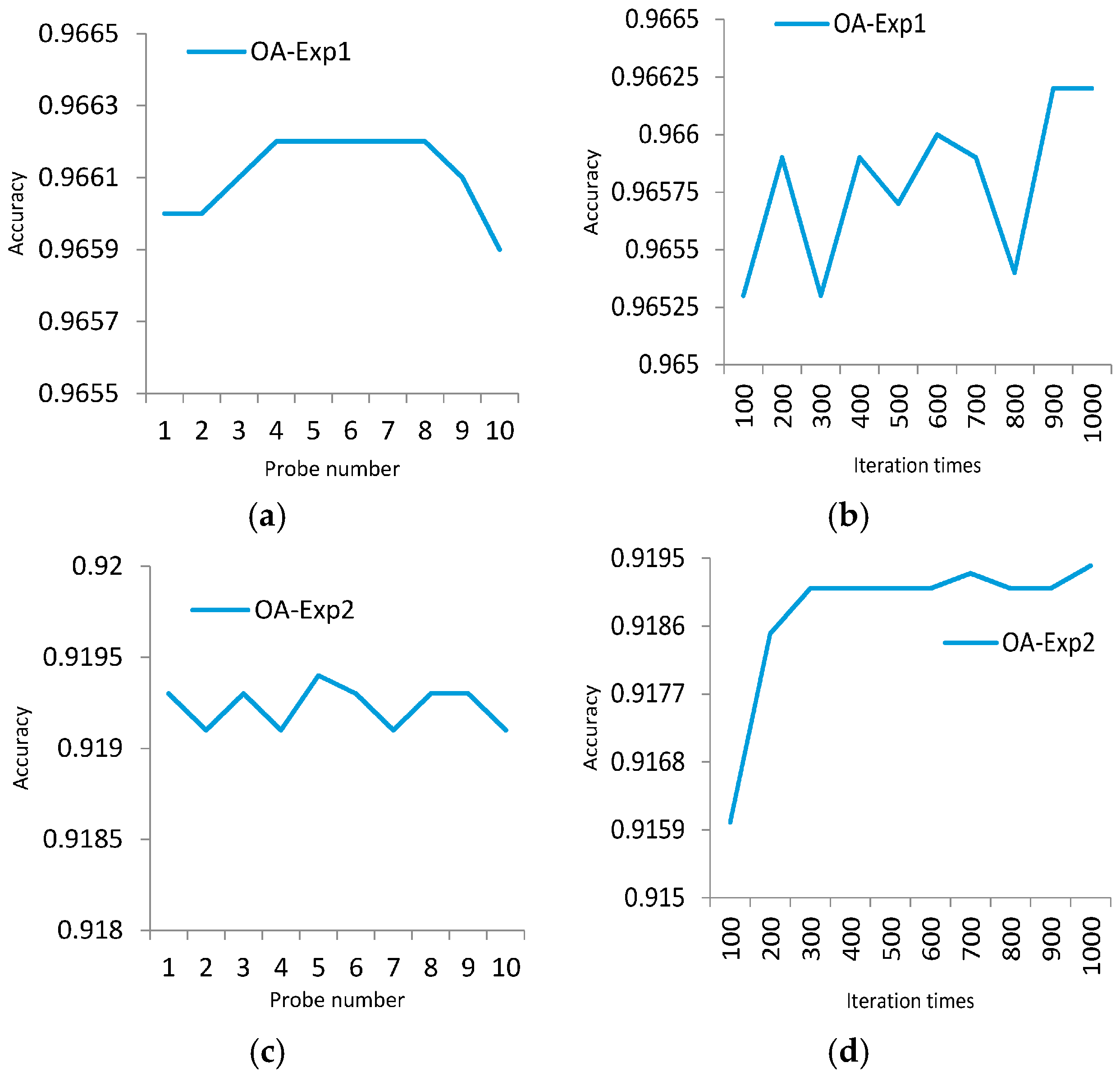
In order to overcome these shortcomings, a new multi-probe based artificial DNA encoding and matching (MADEM) method is proposed in this paper. The biggest difference between ADEM and MADEM is that the latter looks for and extracts the specific information/fragments of the encoded DNA strands, which can replace the full-encoded DNA strands for the matching and classification process in hyperspectral remote sensing imagery. In other words, MADEM aims at seeking out the temporal information of the encoded DNA strands. In this method, multiple probes, defined as the extracted specific fragments of the DNA strand, is put forward for HSI. They are the temporal information of the DNA strands and are used to copy genes and find the most characteristic element of a spectrum. It is well known that only genes can transfer the genetic information of creatures in biological sciences [
23]. Differences in genes can lead to various lives. Thus, the probes, which represent unique fragments of encoded strand, can be extracted to distinguish different spectra. This new method can help to improve the district division by discriminating spectra of different land covers. Meanwhile, the impact of redundant bands can be reduced. Generally, the number of probes is varied due to numerous genes on DNA strands. The probes are extracted randomly and do not share cross regions during the process. In order to generate the most effective probes, the algorithm can be iterated many times to achieve the best solution. The extracted probes provide a more precise discrimination of spectrums, which is favorable for matching and classification process.
The rest of this paper is organized as follows:
Section 2 introduces the background of biological DNA and DNA encoding method for hyperspectral remote sensing data.
Section 3 provides the background of DNA probe technology and describes the multi-probe selection strategy in detail.
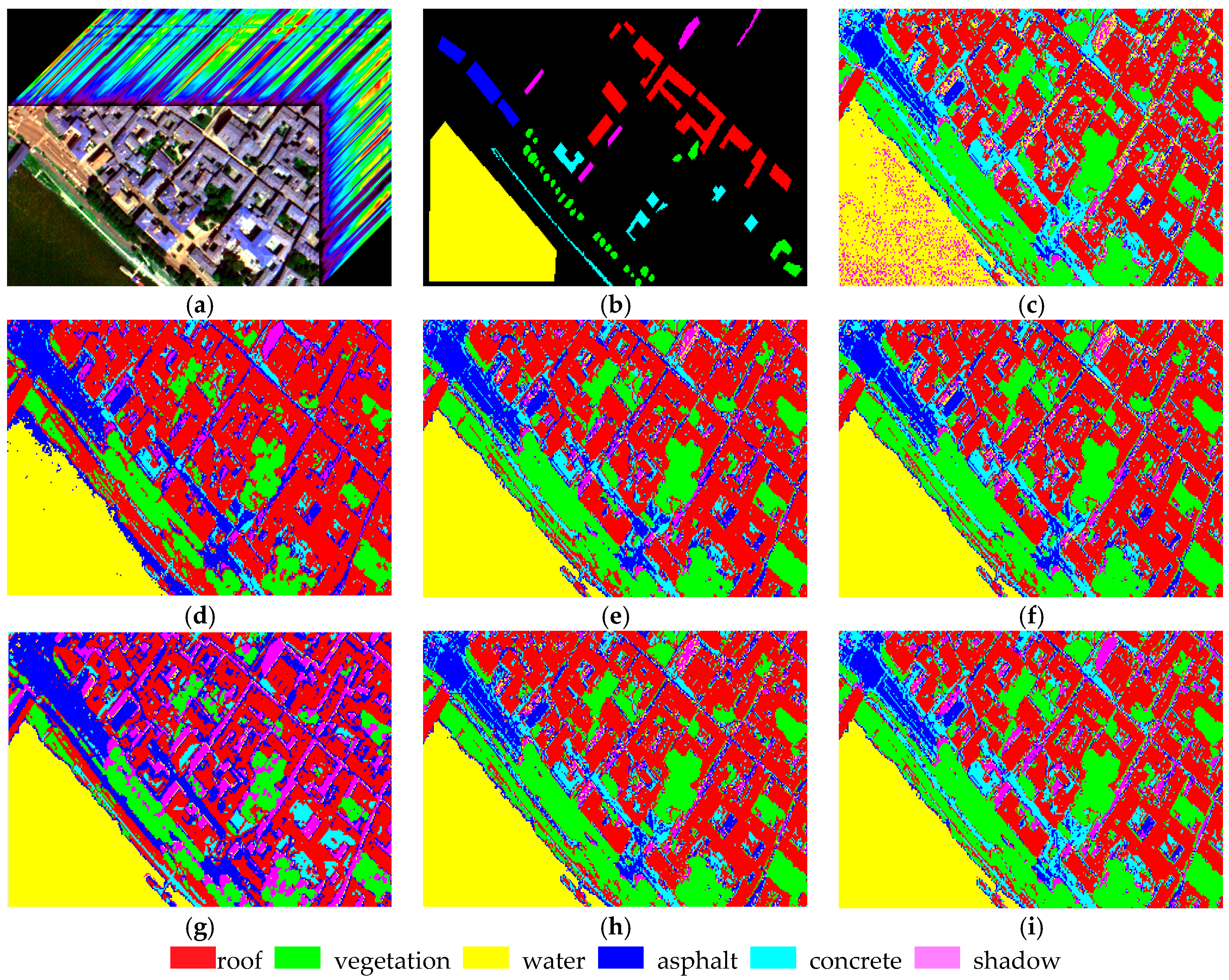
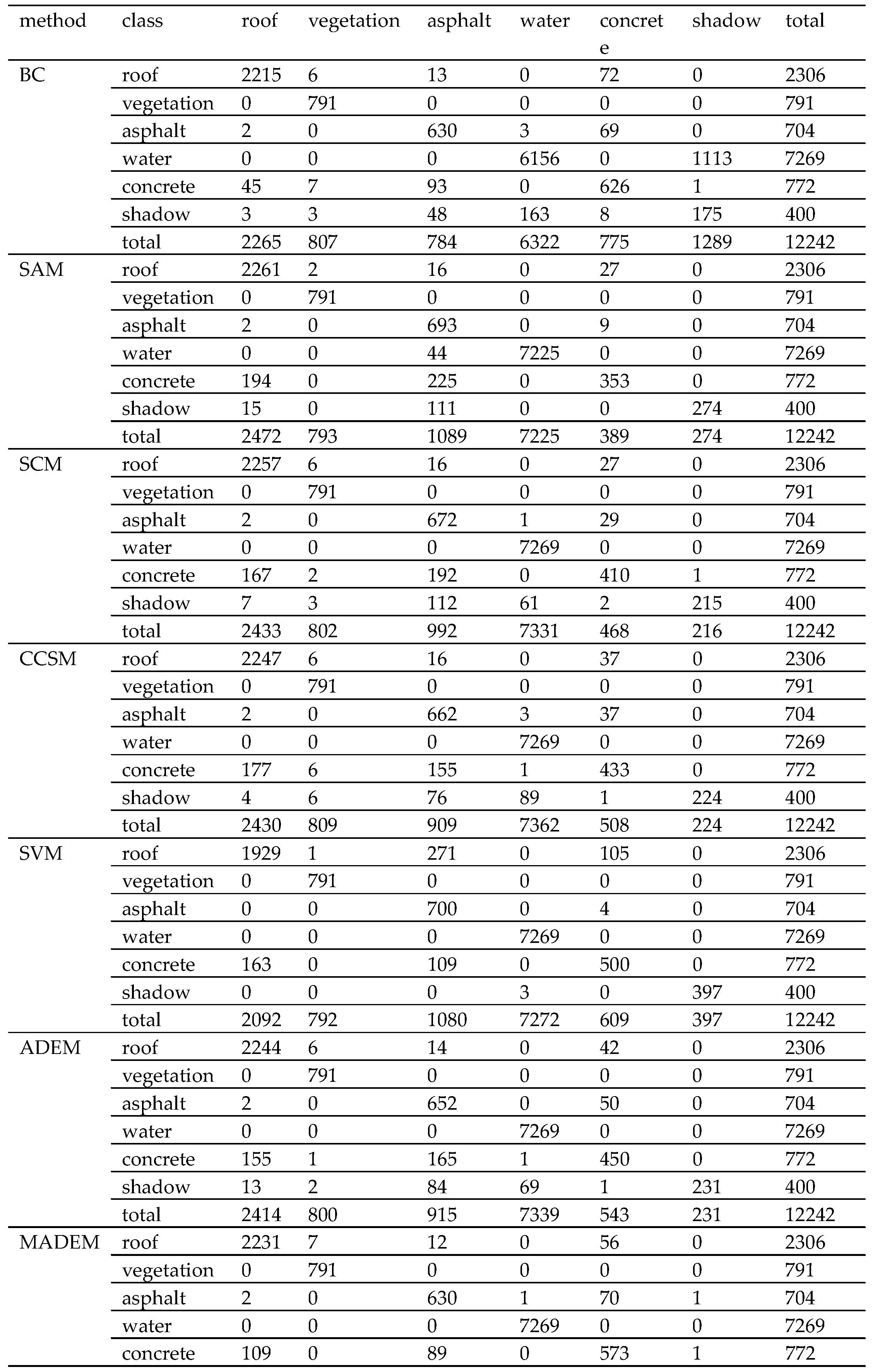
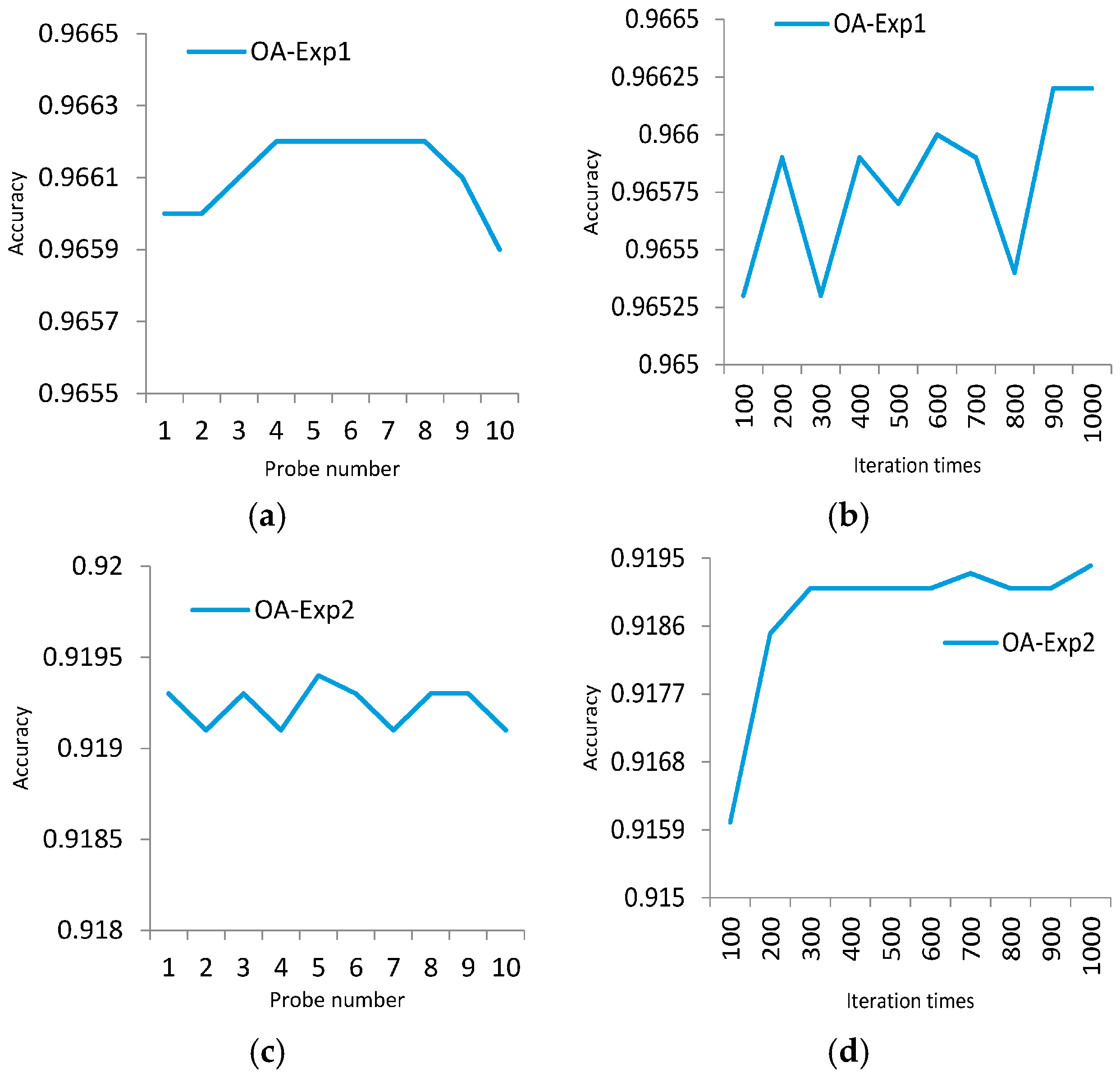
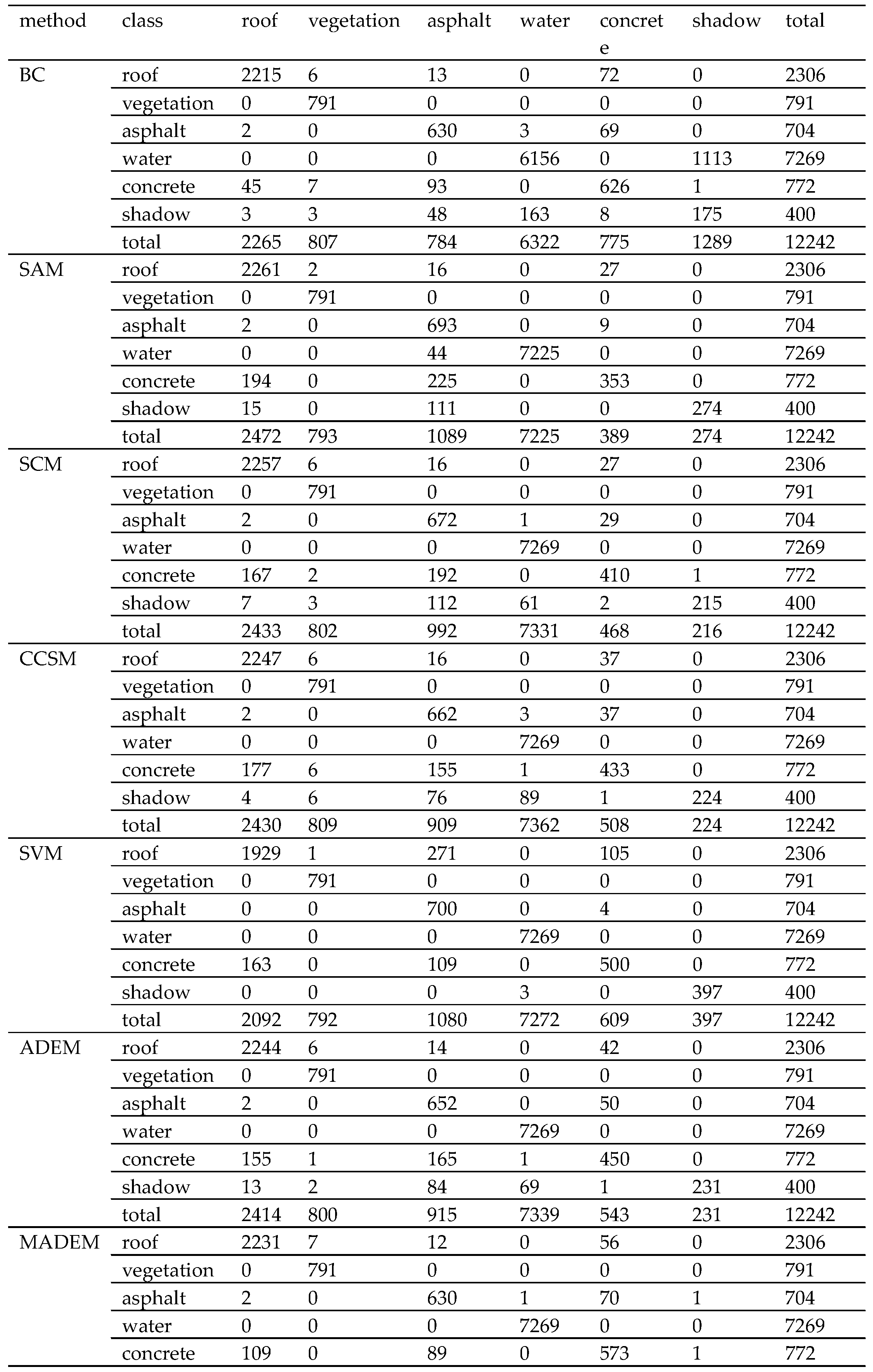
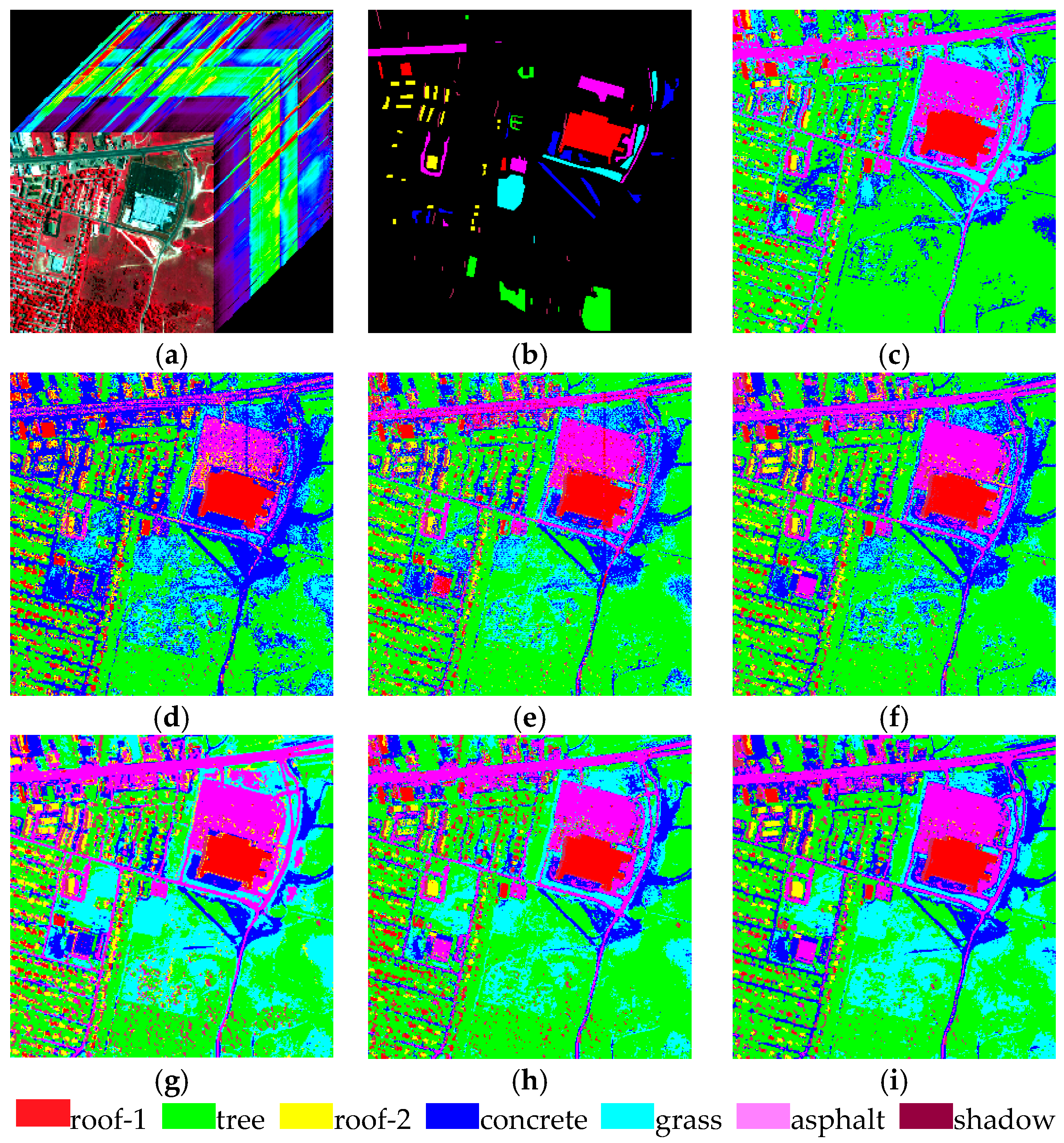
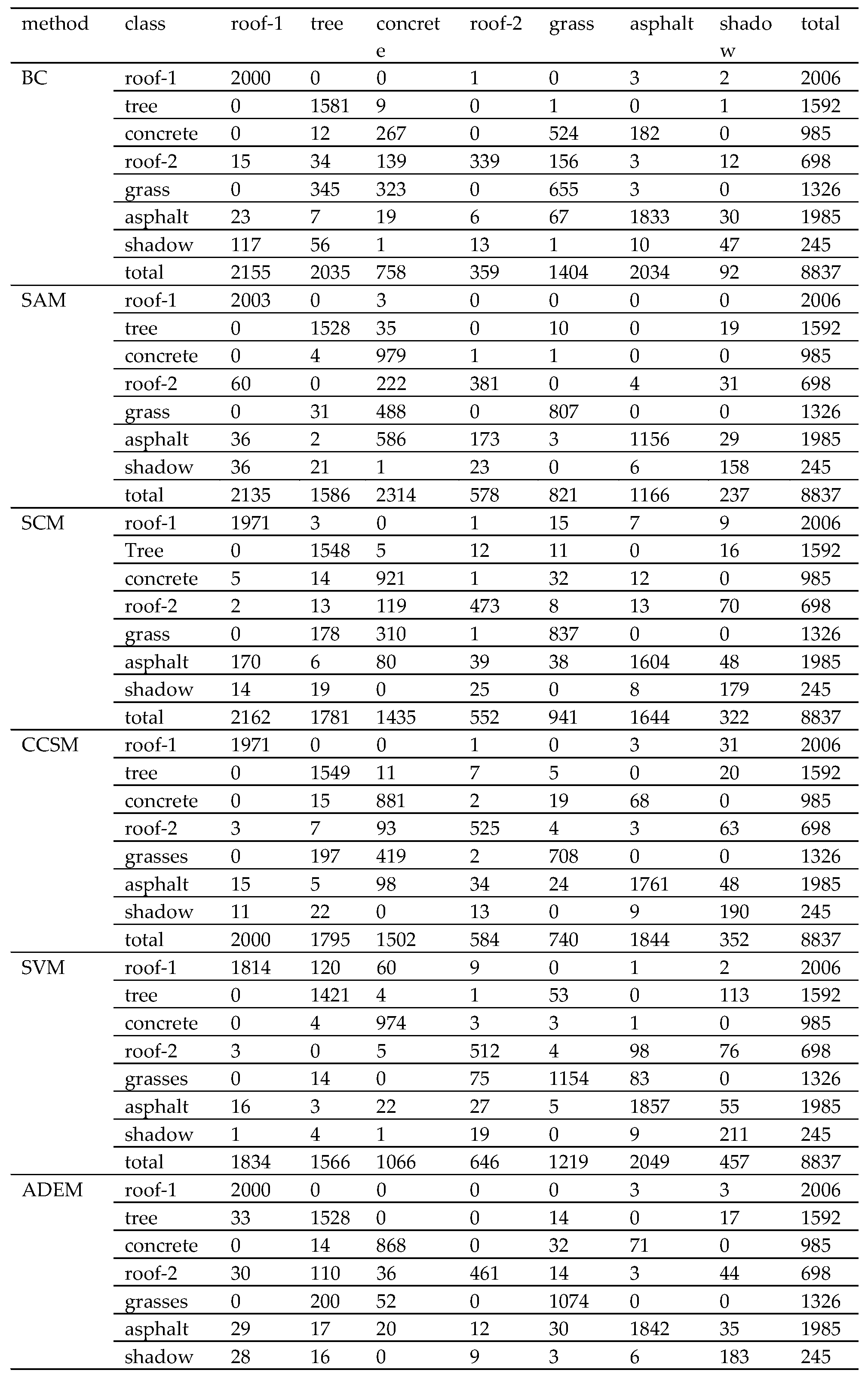
Section 4 shows two experiments using several traditional algorithms compared with MADEM. The computational complexity of these methods is provided in
Section 5. Finally, conclusion is drawn in
Section 6.








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}