1. Introduction
Montelukast, a leukotriene receptor (cysLT1) antagonist, is used for the treatment of asthma. In mono-therapy it is used as an alternative to low-dose inhaled corticosteroids (ICS) or in addition to ICS for improving clinical management of the disease in various phenotypes of asthma patients [
1]. The heterogeneity of asthma is receiving increasing attention in order to better customize treatment according to different clinical and biological phenotypes. Montelukast, itself, is subject to high inter-individual variability in response which limits its effectiveness [
2]. In contrast, it is noteworthy that following 10 mg oral administration of montelukast to healthy volunteers, the reported oral bioavailability is in the region of 60% with low variability and low clearance of approximately 2% liver blood flow, again with low variability [
3,
4]. Despite this, Organic Anion Transporter Protein 2B1 (OATP2B1)-dependent human intestinal absorption (specifically the single nucleotide polymorphism (SNP) SLCO2B1*3) has been suggested as a possible cause for observed patient variability in effect [
5]. OATP2B1 is an uptake transporter located in the luminal membranes of human small intestinal enterocytes, in the sinusoidal membrane of hepatocytes, mammary glands, placenta, heart, and skeletal muscle [
6]. The claim relating variability in montelukast effect to OATP2B1 expression cited above has been disputed [
7]. Since it is important to understand if this is a point of differentiation for novel drug discovery projects which aim to provide more comprehensive allergic asthma therapy, we performed experiments in an attempt to provide further clarity.
2. Experimental Section
2.1. Materials
[3H]-Montelukast was supplied by The American Radiolabeled Chemical Company (St. Louis, MO, USA) and non-radiolabeled montelukast was supplied by Chemtronica (Stockholm, Sweden). Montelukast is light sensitive and was stored in darkness. [3H]-Estrone 3-sulfate (E13S) and [3H]-estradiol 17-beta-glucuronide (E217βG) were obtained from PerkinElmer (Waltham, MA, USA), whilst non-radiolabeled E13S, E217βG, rifampicin, dimethyl sulfoxide (DMSO), 2-(N-morpholino)ethanesulfonic acid (MES), and fetal calf serum (FCS) were obtained from Sigma–Aldrich (St. Louis, MO, USA). Erlotinib was obtained from Chemtronica (Stockholm, Sweden); all cell culture reagents, Leibovitz medium supplemented with l-glutamine, Hank’s balanced salt solution (HBSS), phosphate buffered saline (PBS), and HEPES from Invitrogen Life Technologies (Paisley, UK); sodium hydroxide (NaOH) and hydrochloric acid (HCl) from Merck KgaA (Darmstadt, Germany), and the scintillation cocktail Optiphase “Hisafe” 2 was provided by PerkinElmer (Waltham, MA, USA). Pierce® BCA Protein Assay kit was used for protein determination (Thermo Scientific, Waltham, MA, USA).
2.2. Cell Culture
Stable HEK293 cell lines, either expressing human OATP2B1/SLCO2B1 (HEK-OATP2B1) or transfected with empty vector (HEK-mock) were produced at the University of Uppsala, Sweden, as detailed previously [
8]. OATP1B1/SLCO1B1 (HEK-OATP1B1) and corresponding mock transfected HEK293 cells were produced in-house at AstraZeneca [
9]. The cells were thawed and cultured in 25 cm
2 flasks (Nunc, Thermo Scientific, Waltham, MA, USA) at 37 °C in an atmosphere of 95% air and 5% CO
2 using antibiotic free cell culture medium for the first passage: Dulbecco’s Modified Eagle Medium (DMEM) with Glutamax (500 mL) containing heat inactivated FCS (50 mL). In the continuous culture hygromycin B (75 µg/mL) was used for the HEK-OATP2B1 and 1% G418 for the HEK-OATP1B1 culture. In the OATP2B1 uptake studies, the cells were seeded in CellBIND 24-well plates (Corning Costar
® Corporation, Corning, NY, USA) at a density of 600,000 cells/well and experiments were conducted two days after seeding. HEK-OATP2B1 and HEK mock cells were used at passages 4 to 10 and passages 15 to 22, respectively. The OATP1B1 uptake studies were performed two days following cell seeding (density of 250,000 cells/well) in poly-lysine-
d-coated 24-well plates (BD Biosciences, San José, CA, USA) with the cells used at passages 33 to 40 (HEK-OATP1B1) and passages 13 to 20 (HEK-mock cells).
2.3. HEK-OATP Uptake Experiments
Montelukast, E13S, and E217βG dose solutions were prepared as a mixture of [3H] compound (1 µCi/mL) and non-radiolabeled compound. Uptake experiments were performed in a thermostated shaker, set to 37 °C and 250 rpm (Thermostat; BMG Lab Vision, Rotenberg, Germany). At the start of the experiment, the cell culture medium was removed and the cells were washed twice with pre-heated transport media (700 µL per well). Thereafter, the cells were incubated in pre-warmed transport media (200 µL) for 10 min and experiments initiated by the addition of an equal volume of pre-warmed dose solution (200 μL) to each well. All uptake experiments were carried out in transport medium consisting of HBSS buffered with 25 mM MES adjusted to pH 6.5 with NaOH or buffered with 25 mM HEPES adjusted to pH 7.4 with NaOH. All experiments were performed in triplicate and the total concentration of DMSO and ethanol never exceeded 1% and 0.2% (v/v), respectively, in the incubations.
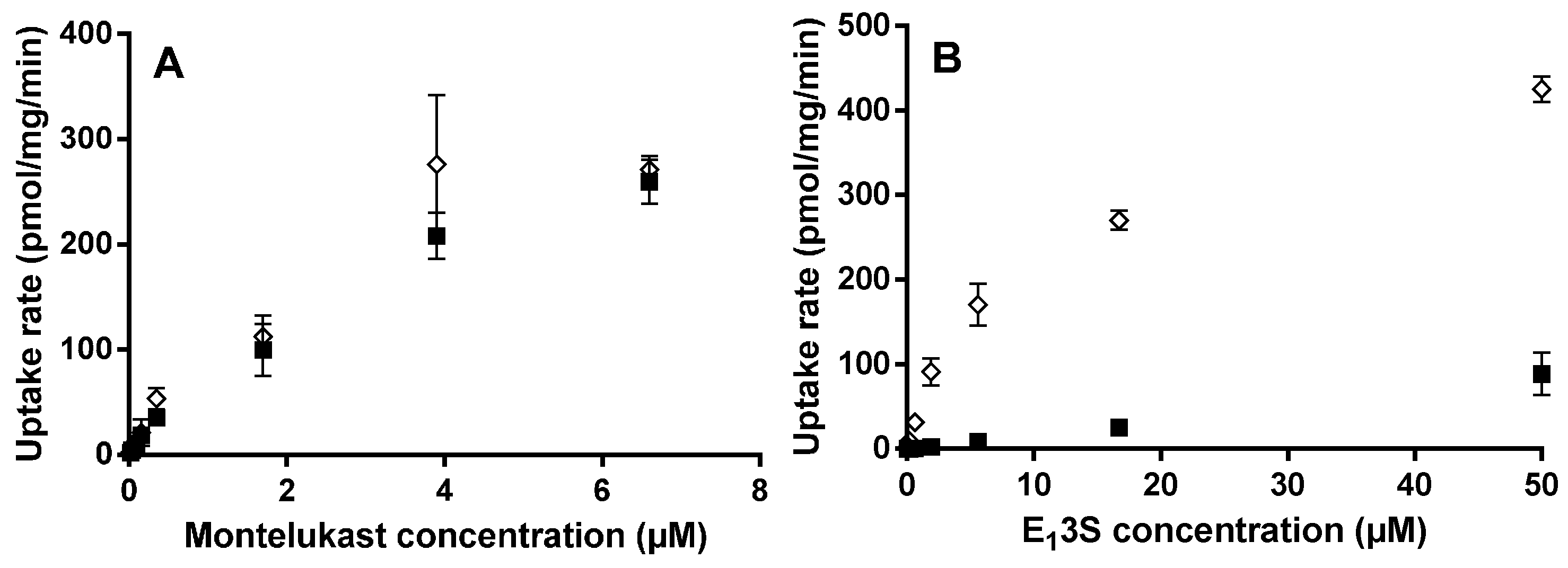
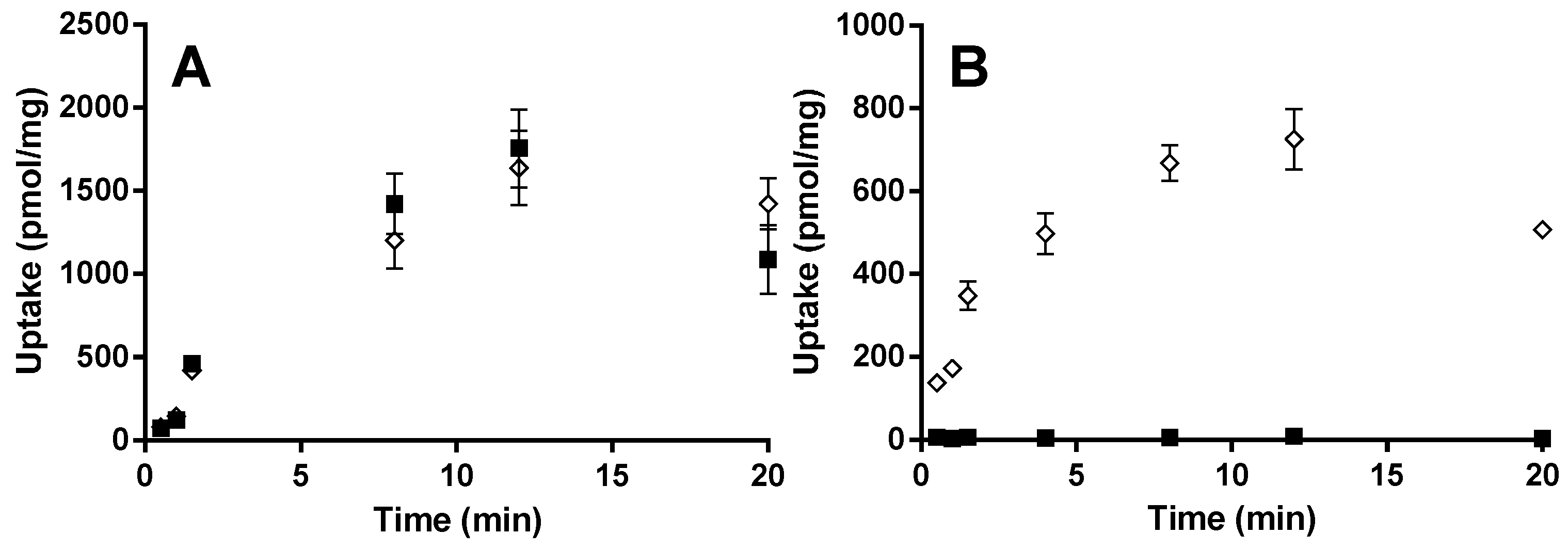
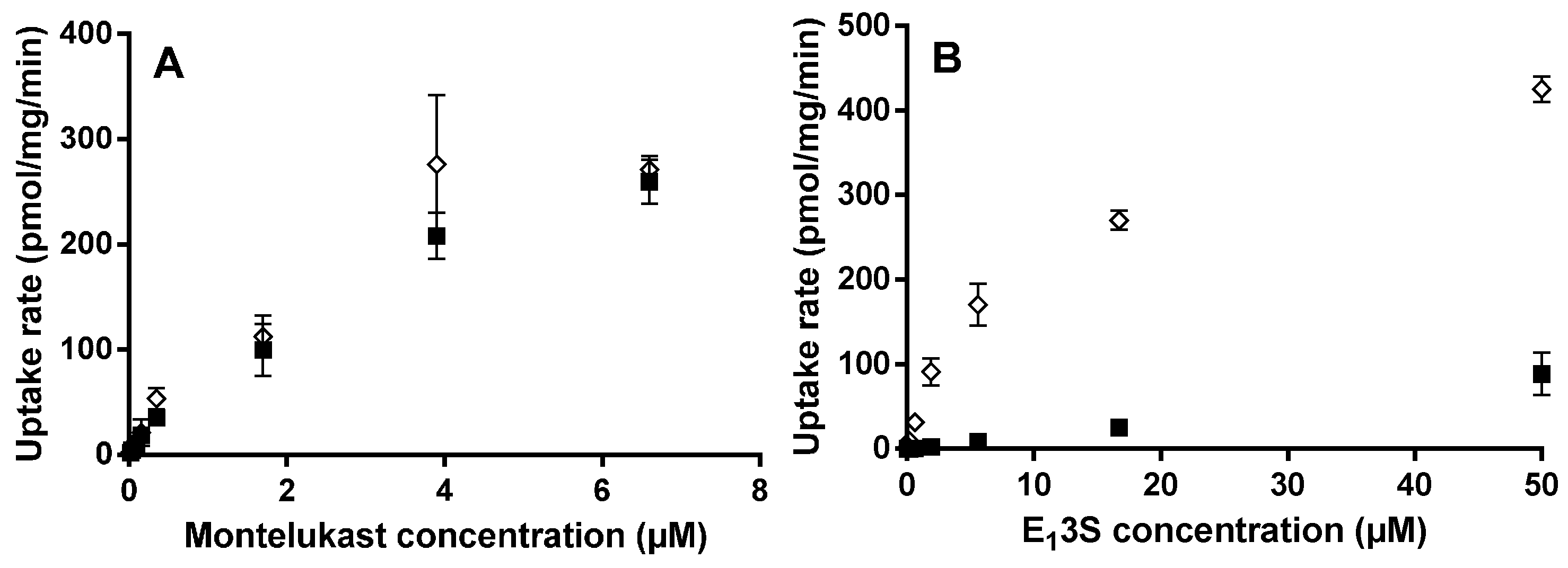
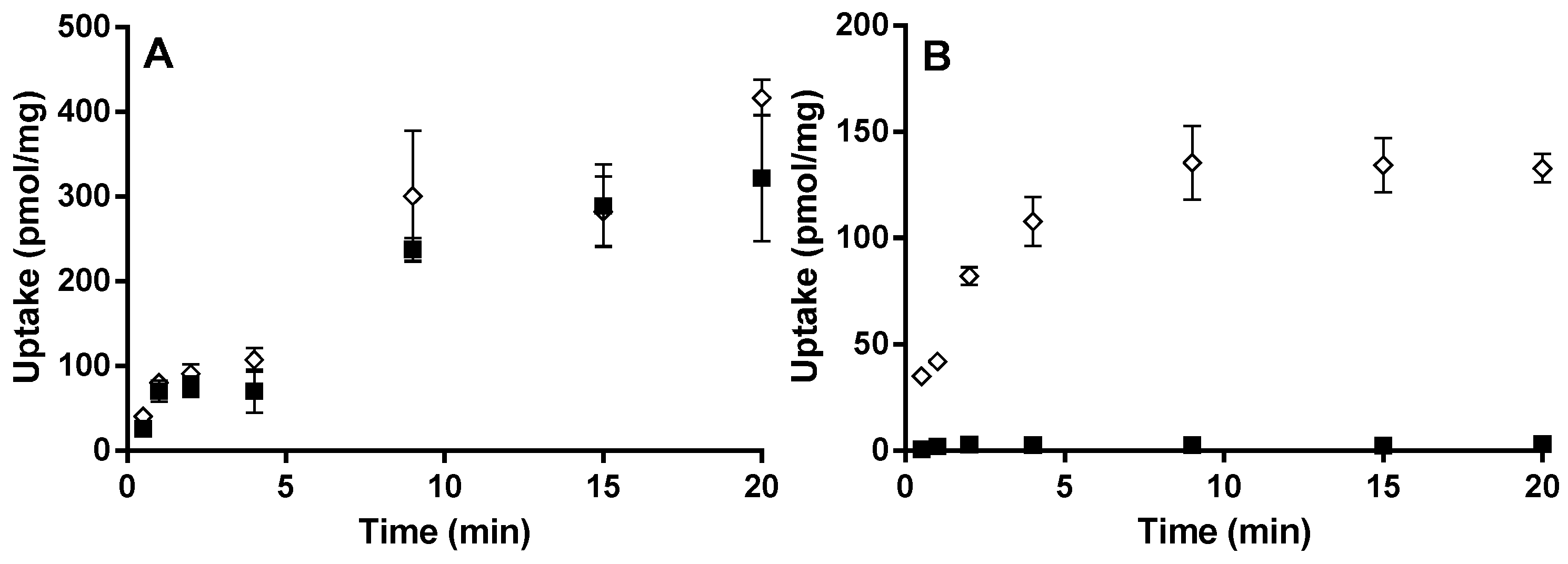
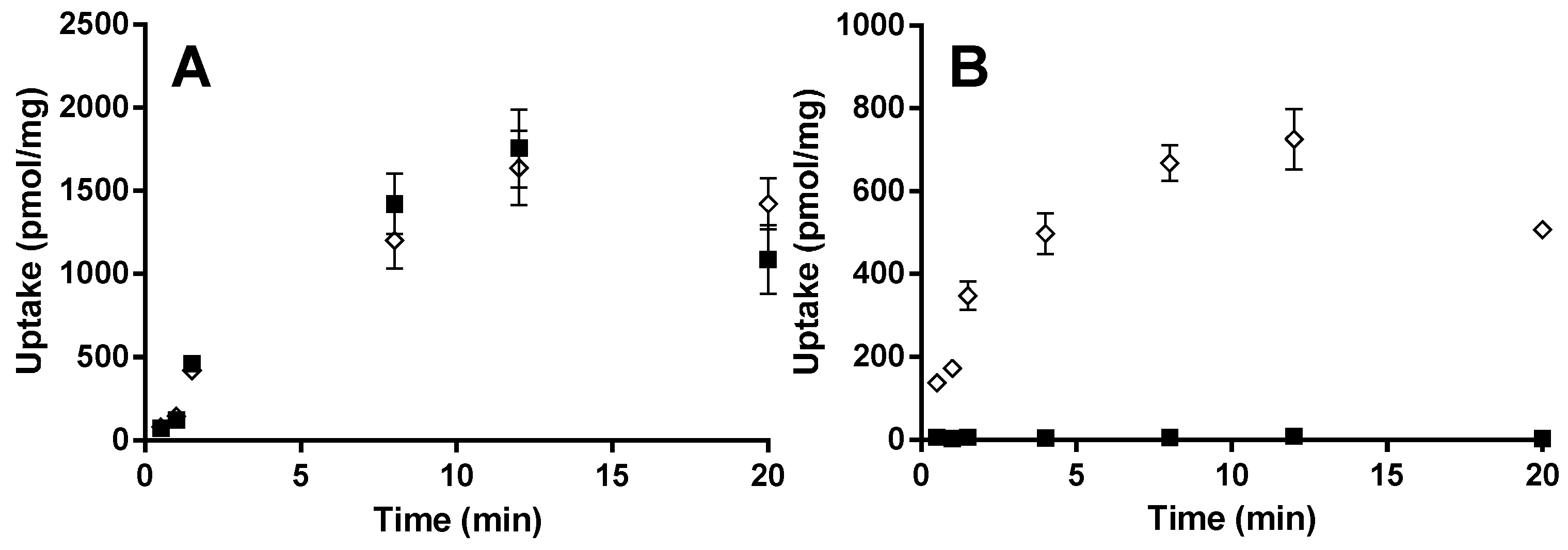
For the OATP2B1 time-dependent experiments, E13S (0.5 µM) and montelukast (1 µM) were incubated at pH 6.5 and 7.4 from 0.5 to 20 min. Concentration-dependent and transport inhibition experiments were performed for 3 min at pH 6.5, with E13S (0.02 to 50 μM) and montelukast (0.02 to 6.6 μM). E217βG (1 µM) and montelukast (1 µM) were incubated at pH 7.4 from 0.5 to 20 min with HEK-OATP1B1 and corresponding mock transfected cells. Concentration-dependent experiments were performed for 1 min with E217βG (0.02 to 50 µM).
The uptake experiments were terminated by removing the incubation media and washing the cells twice with PBS. Ice-cold NaOH (0.2 M, 500 μL) was added to each well and the plates were placed in a refrigerator for at least one hour to lyse the cells. The cell homogenate was neutralized with an equal volume of HCl (0.2 M) prior to sampling for protein determination and scintillation counting for radioactivity analysis. The absorbance was read at 562 nm using a SpectraMax 190 plate reader (Molecular Devices, Sunnyvale CA, USA). The samples were assayed for radioactivity using a Wallac WinSpectral™ 1414 liquid scintillation counter and a Tri-Carb 3100TR Liquid Scintillation analyzer, both supplied by PerkinElmer (Waltham, MA, USA). Scintillation cocktail Optiphase “Hisafe” 2 was added to each sample before analysis.
Km and Vmax for OATP2B1 and OATP1B1 substrates were obtained by fitting to a Michaelis–Menten equation (GraphPad Prism, Version 6.01, GraphPad Software Inc., San Diego, CA, USA). OATP-mediated uptake, calculated by subtracting the uptake in HEK-mock from that in HEK-OATP cells was used for the Michaelis–Menten fit.
The enterocyte blood flow method [
10] was used to calculate the intestinal drug concentration experienced by the drug transporter after a 10 mg dose (drug concentration = fraction absorbed × absorption rate constant × dose / enterocyte blood flow of 18L/h).
2.4. Human Hepatocyte Uptake Experiments
Pooled cryopreserved primary human hepatocytes were obtained from Bioreclamation IVT (10-donor batch IRK, Brussels, Belgium). Hepatocyte uptake was assessed using an “oil-spin” procedure, as previously described [
11]. Incubations with montelukast or E
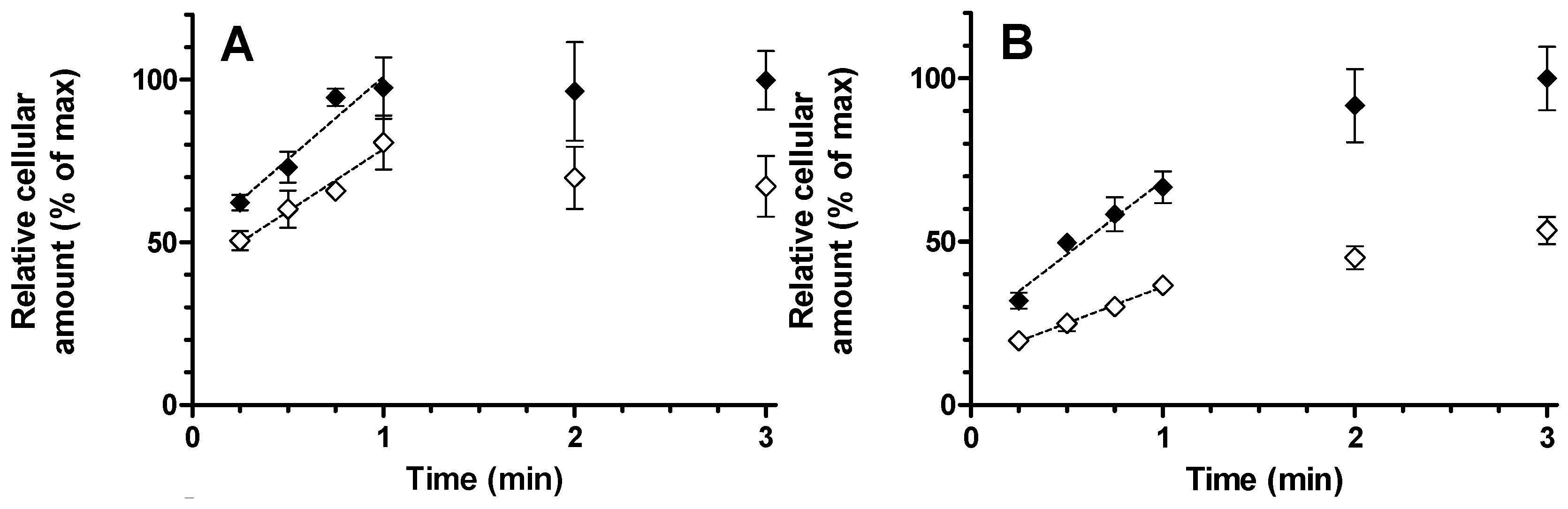
13S (1 µM) were performed in serum free Leibovitz L-15 medium in absence and presence of OATP transport inhibitors rifampicin (10 µM) or erlotinib (4 µM) at a final hepatocyte concentration of 1,000,000 cells/mL. Briefly, cells were thawed and re-suspended in Leibovitz medium according to the manufacturer’s protocol, counted using a CASY cell counter (Innovatis AG, Bielefeld, Germany) and further diluted to reach a concentration three times that of the final incubation. Incubations were initiated as follows: substrate and inhibitor solutions were prepared in Leibovitz medium to a concentration three times that of the final incubation. A blank (no inhibitor) solution was also prepared with the same DMSO concentration as the inhibitor solutions. Equal volumes (300 µL) of blank/inhibitor solution and cell suspension were mixed in a glass vial and allowed to pre-incubate for 15 min in water bath set to 100 rpm (linear shaking) at 37 °C. Experiments were started by addition of substrate solution (300 µL). The final DMSO content in all incubations was 0.15%
v/
v. At designated time points (15, 30, 45, 60, 120, and 180 s) a 100 µL aliquot of incubate was removed and dispensed into a microtube containing an upper layer of oil and a lower layer of cesium chloride. Upon rapid centrifugation (7000
g; 15 s) hepatocytes are passed through the oil layer separating them from the incubation medium. The microtubes were directly placed on dry ice before cutting (just below the oil/cesium chloride interface) of the frozen tube tips containing the cell pellets into a 96-well plate for sample processing as previously described [
11]. Incubations were performed in triplicate.
Samples were analyzed by LC–MS–MS: HTS PAL injector (CTC Analytics, Zwingen, Switzerland), HP 1100 LC binary pump (Agilent Technology, Waldbronn, Germany) and a Micromass Quattro Ultima mass spectrometer (Waters Corp., Milford, MA, USA). A mobile phase (A: 0.2% formic acid, 2% acetonitrile; B: 0.2% formic acid in acetonitrile) with a flow rate of 0.7 mL/min was used with a Sunniest C18 RP-aqua column (ChromaNik Technologies Inc, Osaka, Japan) for chromatography.
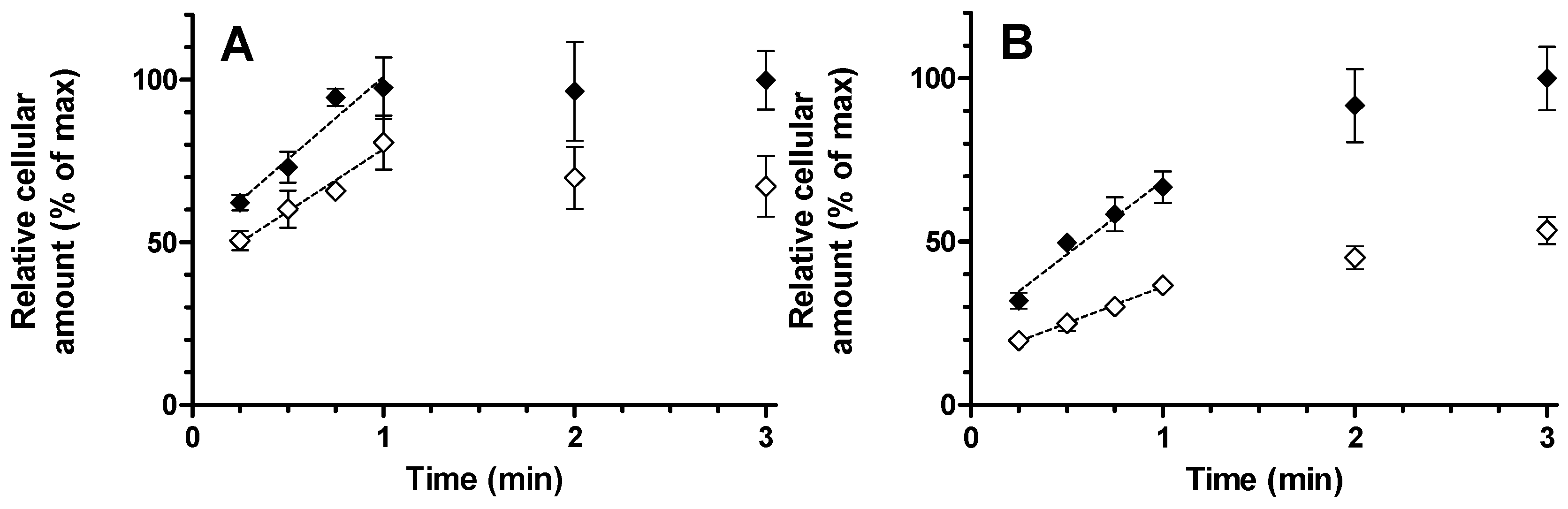
Relative rates of uptake were determined by linear regression. Data generated up to 60 s after substrate addition gave for montelukast as well as for E
13S profiles the optimal statistical confidence in slope determination and was, therefore, included in analysis. Significance of inhibition of uptake by rifampicin and erlotinib was calculated from a two-tailed linear regression
t-test (in-built functionality Prism 5, GraphPad Software, USA). Obtained
p-values were corrected for multiple comparisons using the Holm–Bonferroni method [
12]. Adjusted
p-values < 0.05 were considered as significant.
4. Conclusions
Previously, it has been reported that OATP2B1 may be important in the human intestinal absorption of montelukast [
5]. These conclusions were drawn from
in vitro Caco-2 data in which the relationship between permeation rate and montelukast concentration was interpreted as being indicative of transporter mediated transport that could be described by Michaelis–Menten kinetics. Only the single concentration of 170 µM montelukast deviated from the linear relationship between rate and concentration, this data point being associated with the greatest variance, possibly due to limitations in the aqueous solubility of montelukast being in the 10–50 µM range [
13]. Vectorial transport of montelukast through MDCKII cells transfected with OATP2B1 was shown by the authors to be higher (1.4-fold) compared to control MDCKII cells. Although the result was calculated to be significant, the assay had a relatively small “window” even for the positive control E
13S (only 2.8-fold higher cell uptake in OATP2B1 cells compared to control). Additionally, the cell uptake was shown to be inhibited 27% by 125 µM sulfobromophthalein. In this context it is noteworthy that when E
13S has been previously used as a substrate of OATP2B1, an IC
50 of 2 µM was reported for sulfobromophthalein [
14]. Other investigators have subsequently challenged the conclusions of Mougey and co-workers [
7]. Chu
et al. provided MDCKII (control and OATP2B1-expressing) cellular uptake data that showed no significant difference in the amount of montelukast taken into control and OATP2B1 cells when incubated at 1 µM, pH 6.0. The same finding was observed at incubation pH 7.4, but in both cell lines the amount of drug taken up was approximately 1.5-fold lower compared to incubations performed at pH 6.0. A similar experiment was performed using 1 µM E
13S and the rate of uptake was approximately 10-fold higher in the OATP2B1 cells compared to control cells. Thus, an assay with a relatively large window for the positive control compound contradicted the findings of the earlier publication. Furthermore, cellular accumulation data for 1 µM E
13S and 3 µM montelukast presented by Chu
et al., concluded that E
13S accumulated to a greater extent in MDCKII OATP2B1 cells when added to the basolateral side than the apical side, but that this was not true for montelukast. However, higher montelukast accumulation in the control cells after apical administration, compared to basolateral administration, together with only a two-fold difference between basolateral and apical administration for E
13S, reduces the clarity of these results.
Here we performed experiments in an attempt to describe montelukast time- and concentration-dependence for HEK293 OATP2B1 cellular uptake. A novel human OATP2B1 expressing HEK293 cell line was used, created at Uppsala University [
8]. This cell line used was validated with the OATP substrate E
13S, shown to have a
Km value of 7.5 µM, in excellent agreement with previously observed values [
15,
16]. Drug transport was measured using HEK-OATP2B1 and HEK-mock (empty vector) cells at pH 6.5, consistent with the luminal pH of the small intestine and also because the transporter is reported to have a higher substrate transport capacity at acidic pH [
17]. Since high passive uptake could potentially still mask an active component, the experiment was repeated at 1 µM and the higher pH of 7.4, in order to reduce the passive permeability through decreasing the percentage of the un-ionized drug. Large differences in E
13S uptake were observed when comparing the OATP2B1- and mock-transfected HEK cell lines at both pH 6.5 and 7.4. This gave an advantageous window for defining test compounds as substrates of the drug transporter. In contrast to E
13S, montelukast uptake at pH 6.5 was very similar in the OATP2B1 and mock cell lines over a range of concentrations. Altering the pH in the incubation from 6.5 to 7.4 did not affect the OATP2B1/mock ratio substantially for E
13S. We, therefore, conclude that montelukast is not pronouncedly transported by OATP2B1 due to high inherent passive permeability and given the low variability in observed human exposure there is no reason to consider that the transporter is important in driving human intestinal absorption [
3,
18]. This data therefore supports a recent clinical study indicating that OATP2B1 polymorphisms do not affect the pharmacokinetics of montelukast [
19].
It is interesting to note that montelukast has been shown to be a relatively potent inhibitor of OATP1B1, 1B3, and 2B1 [
20,
21]. It is also worthy of note that the drug has been postulated to be actively transported into human hepatocytes
in vitro, although the mechanism of the uptake has, to our knowledge, never been investigated [
22]. Drugs that interact with OATP1B1 are typically acids and are most likely to be moderately to highly lipophilic with log
P values in the range 3.5 to 5.5 [
23]. Montelukast is more lipophilic than this, having a log
P of over 7. Whilst acidic drugs that are highly bound to plasma proteins typically have low steady state volume of distribution (
Vss) values in the order of 0.1 to 0.3 L/kg, for those whose hepatic disposition is influenced by OATP1B1/3 transport, the
Vss is likely to be larger [
24,
25]. Montelukast does not obviously fit as having OATP1B1 driven disposition, having a
Vss of approximately 0.15 L/kg [
4]. In the studies described in this manuscript we used HEK293 OATP1B1 and the corresponding HEK-mock (empty vector) cells in an attempt to clarify the involvement of the transport protein in the human pharmacokinetics of montelukast. The cell line was validated using E
217βG as a standard OATP1B1 substrate since
Km and
Vmax values of 4.1 µM and 697 pmol/mg/min were in line with previously published data [
9]. As with the OATP2B1 cell incubations, it was not possible to differentiate between the OATP1B1 and mock cells in terms of montelukast cellular uptake. Consequently, no concentration-dependent uptake experiments were performed, since the results indicate high passive transport and no detectable OATP involvement. Substantiating data was also produced indicating that active transport is at best a minor permeability component for the uptake of montelukast into primary human hepatocytes: Rifampicin was used at a concentration of 10 µM in order to facilitate approximately 80% inhibition of OATP1B1 and OATP1B3 activity [
8] but no significant inhibition on the assayed initial montelukast uptake rate was observed. In contrast, the initial E
13S uptake rate was reduced by 51%. The back-extrapolated
Y-intercept of the plots represents instantaneous non-specific drug binding which was understandably higher for montelukast than E
13S due to the higher lipophilicity. Taking into account this intercept, the uptake rate between 15 and 60 s appears a good measure of the true initial rate (
Figure 4). It is interesting to note that at the 1 min time point, the E
13S intracellular concentration was less than 70% of the concentration achieved by 3 min, whereas montelukast uptake was complete after 1 min. This more rapid cellular uptake may be more suggestive of a drug concentration-driven passive process. Nevertheless, the total amount of montelukast entering the cell was reduced in the presence of rifampicin and it cannot be ruled out that the active process plays a minor part in the overall hepatic permeability of the drug. Since 10 µM rifampicin should cause only minimal inhibition of OATP2B1 activity, 4 µM erlotinib was used to selectively inhibit this drug transporter [
8]. Erlotinib had no impact on the uptake of either compound, which most likely indicates a low OATP2B1 activity in the hepatocytes used, since E
13S is an OATP2B1 substrate.
In summary, it appears unlikely that OATPs are involved in the human absorption of montelukast or distribution into hepatocytes.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}