Protein Adsorption Patterns and Analysis on IV Nanoemulsions—The Key Factor Determining the Organ Distribution

Abstract
:1. Introduction
- no pharmaceutical products on the market for the therapy of patients;
- despite the first successes in academic research, no “academically available” system is efficient/safe enough to be used for targeting in therapy.
- overcoming the recognition of the injected particles as being foreign and their subsequent clearance by the macrophages of the mononuclear phagocytic system (MPS), mainly uptake by liver and spleen macrophages (up to 90%–95% of the injected dose);
- and the lack of a sufficiently specific targeting moiety, which at the same time is not a too complex system to be realized over a foreseen period.
1.1. Previous Strategies for IV Targeting
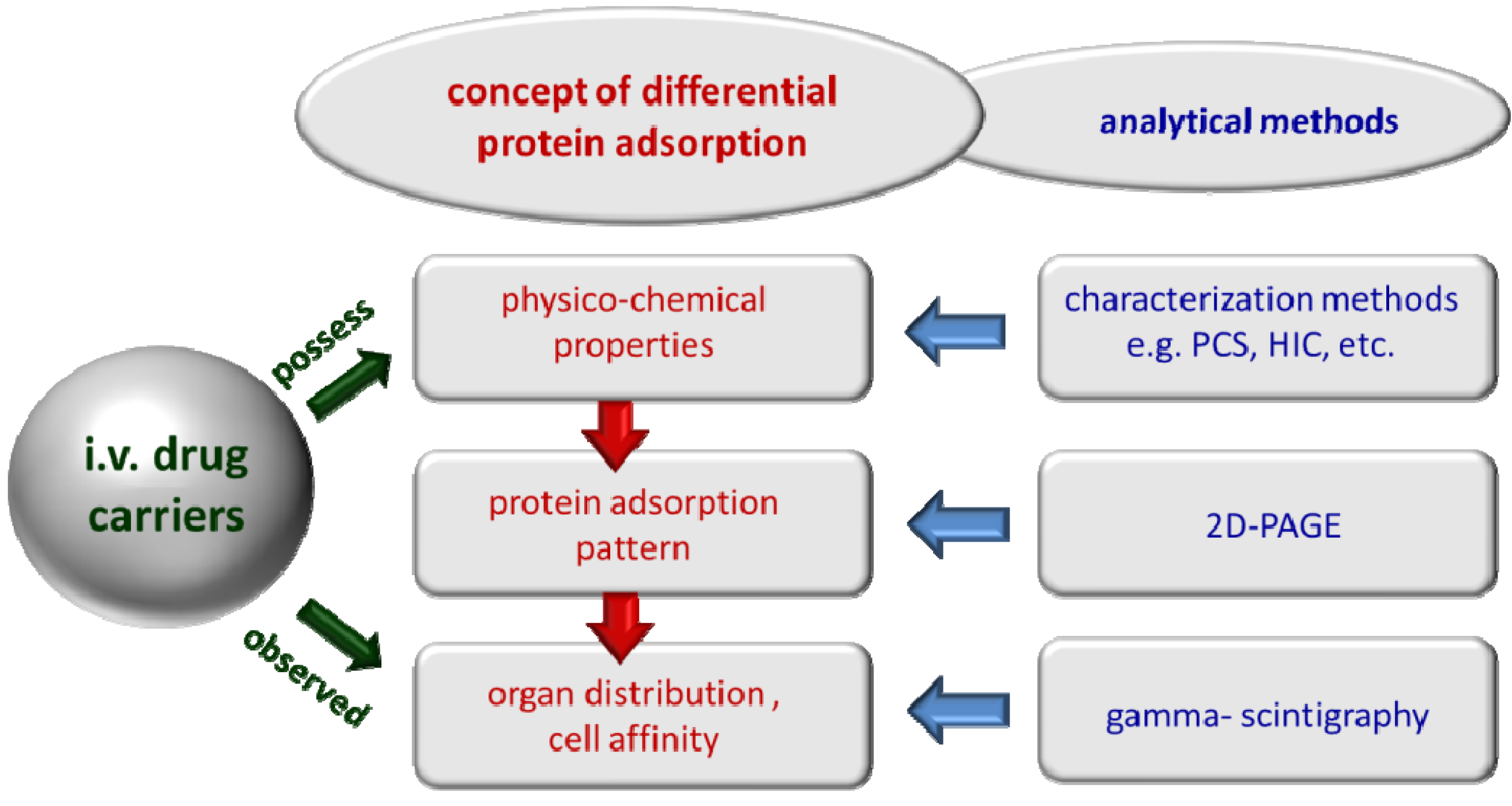
1.2. The Concept of Differential Protein Adsorption

1.3. Basic Considerations for Analysis of Protein Adsorption Patterns
- choice of incubation medium;
- species specific patterns;
- ratio of particle suspension to incubation medium;
- separation method;
- washing medium and number of washing steps;
- incubation time;
- number of samples/repetitions.
- incubating 2 mL of the emulsion in 6 mL citrate stabilized human plasma for 5 min at 37 °C;
- separating from excess plasma by centrifugation at 15,000g for 1h at 20 °C;
- washing three times with 0.05 M phosphate buffer, pH 7.4 and subsequent centrifugation (15,000g for 1h at 20 °C);
- solubilizing with sodium dodecyl sulfate (SDS) and dithioerythritol (DTE) for 5 min at 95 °C;
- cooling down to room temperature and incubation with a solution containing DTE (Dithioerythritol), CHAPS (3-(3-Cholamidopropyl)dimethylammonio-1-propansulfat), urea, Tris (Tris(hydroxymethyl)-aminomethan) and bromphenol blue, stirring and centrifuging 10–15 min at 12,000g;
- following steps according to the respective references given.
2. Protein Adsorption Patterns of Different IV Nanoemulsions
2.1. General Aspects
2.2. Comparison of Adsorption Patterns of Different Nanoemulsions and Influencing Parameters
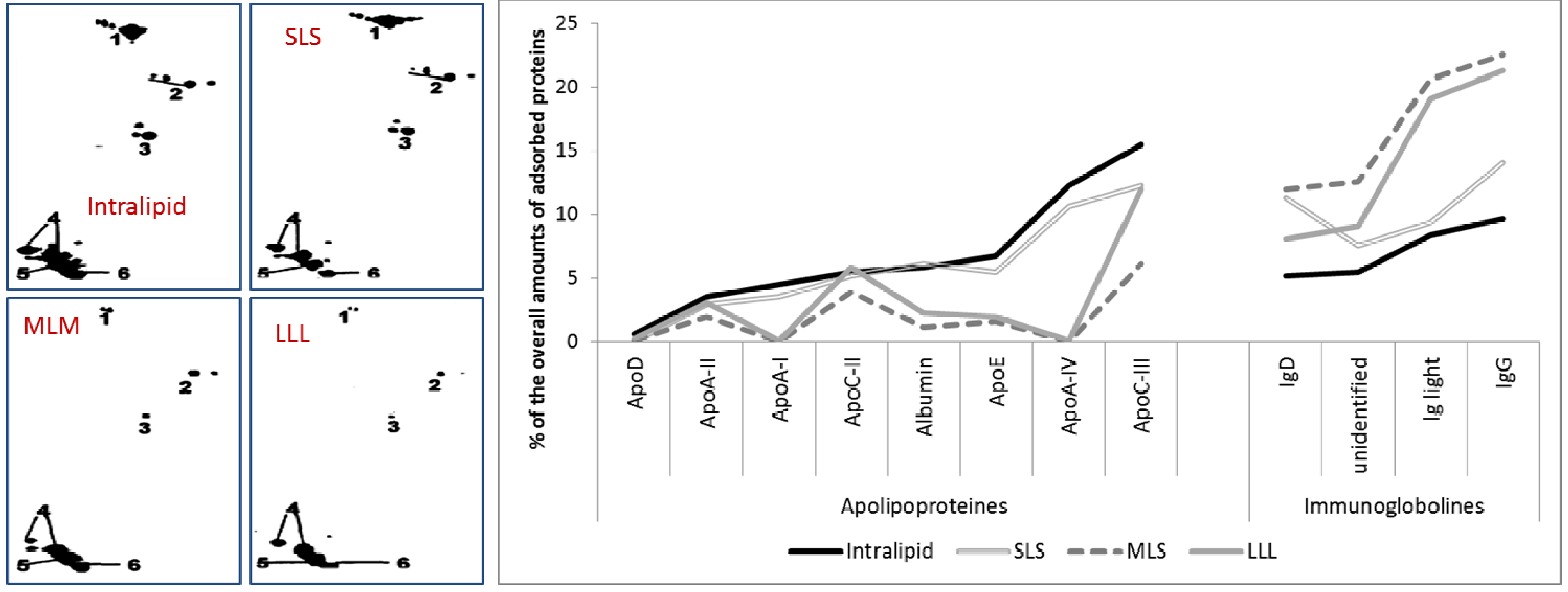
2.2.1. Adsorption Patterns of 20% Emulsions Stabilized with Lecithin from Different Manufactures
2.2.2. Adsorption Patterns of Different Oil Compositions: LCT vs. LCT/MCT

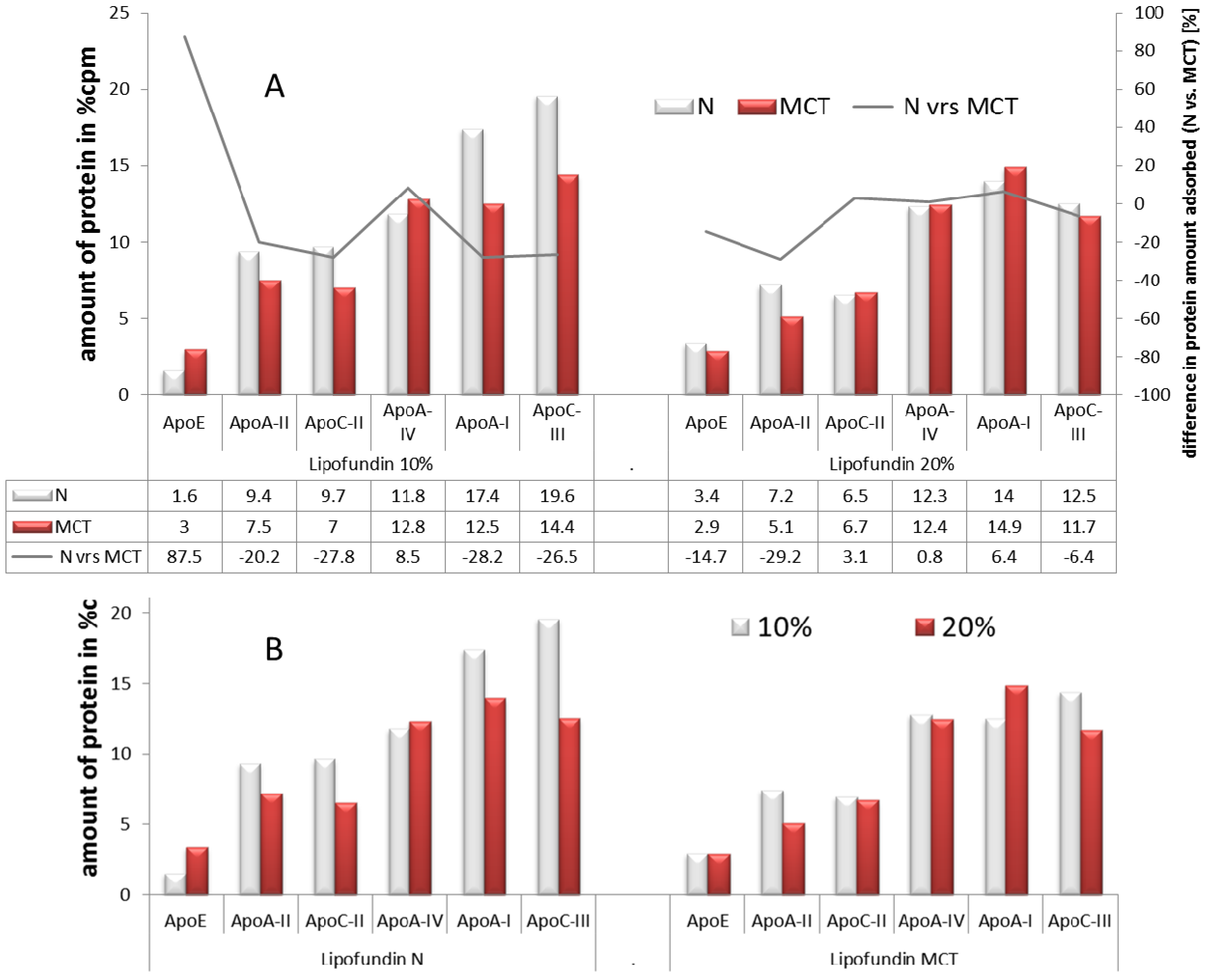
2.2.3. Adsorption Patterns of Different Oil Concentrations: 10% vs. 20%
2.2.4. Effect of Type of Oil Phase on Protein Adsorption
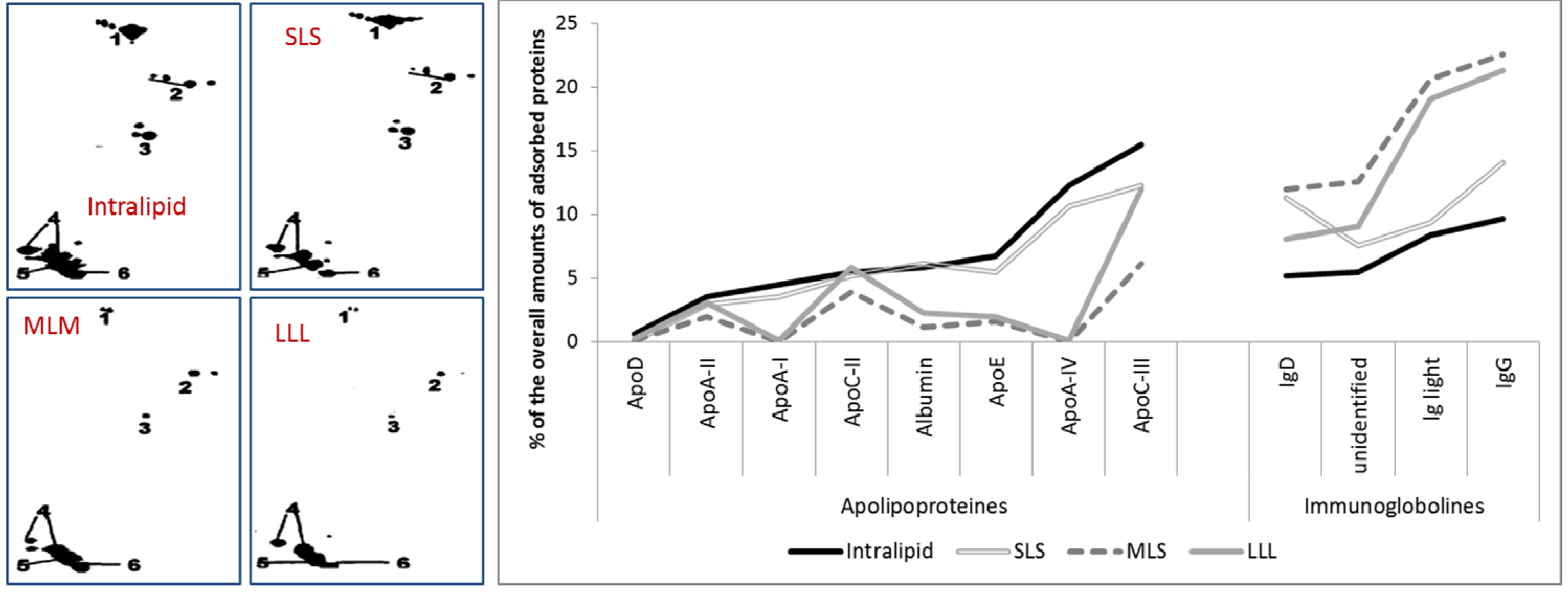
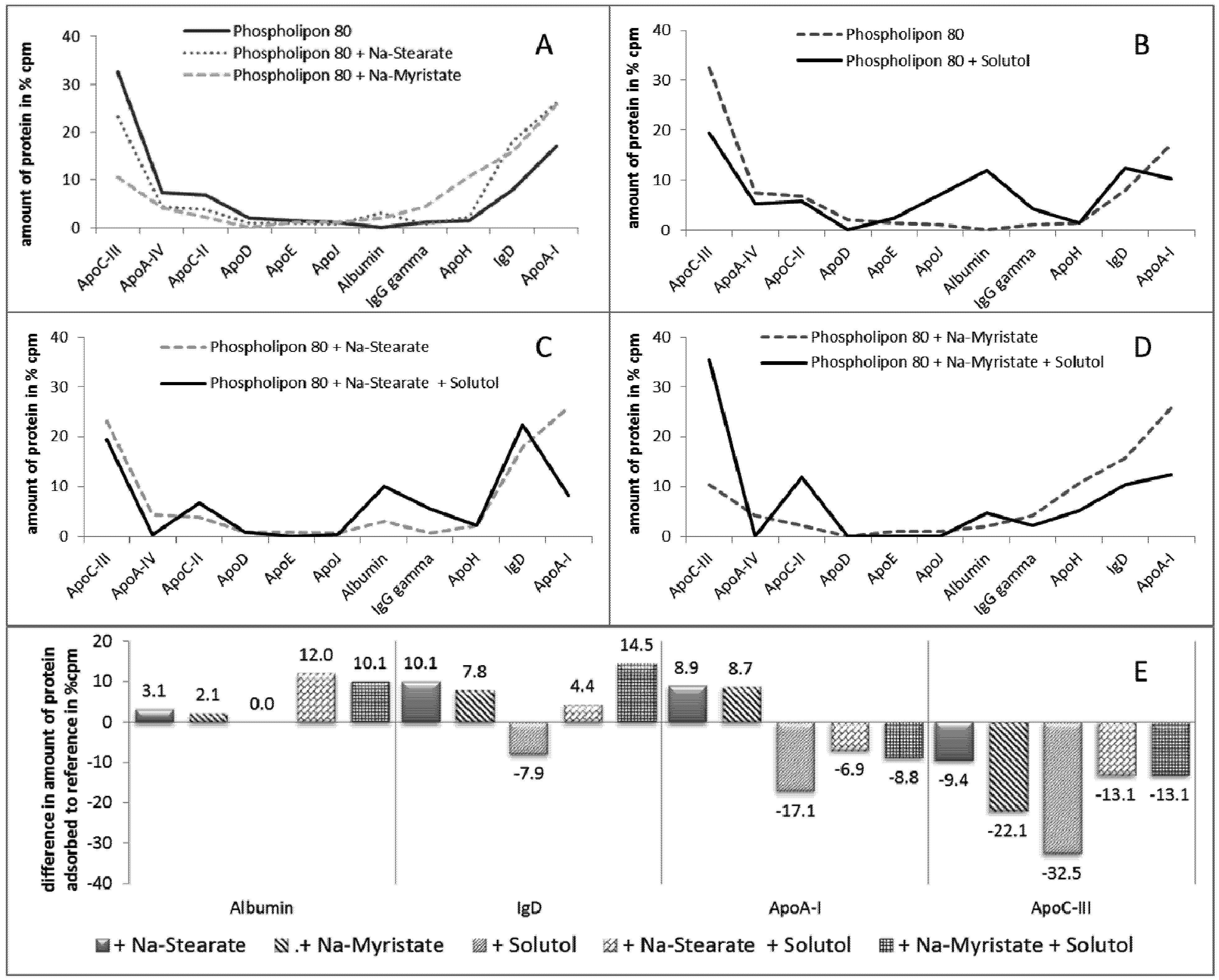
2.2.5. Effect of Stabilizer Composition on Adsorption Patterns

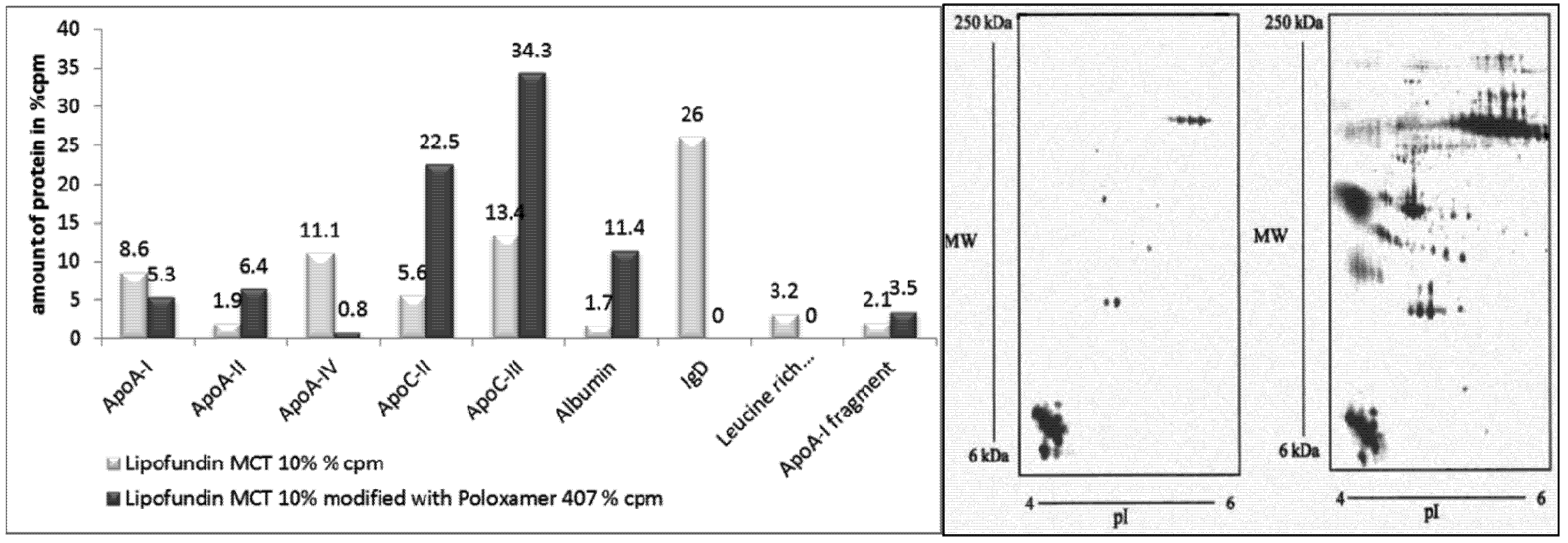
2.2.6. Surface Modification of Lipofundin Emulsions for Drug Targeting
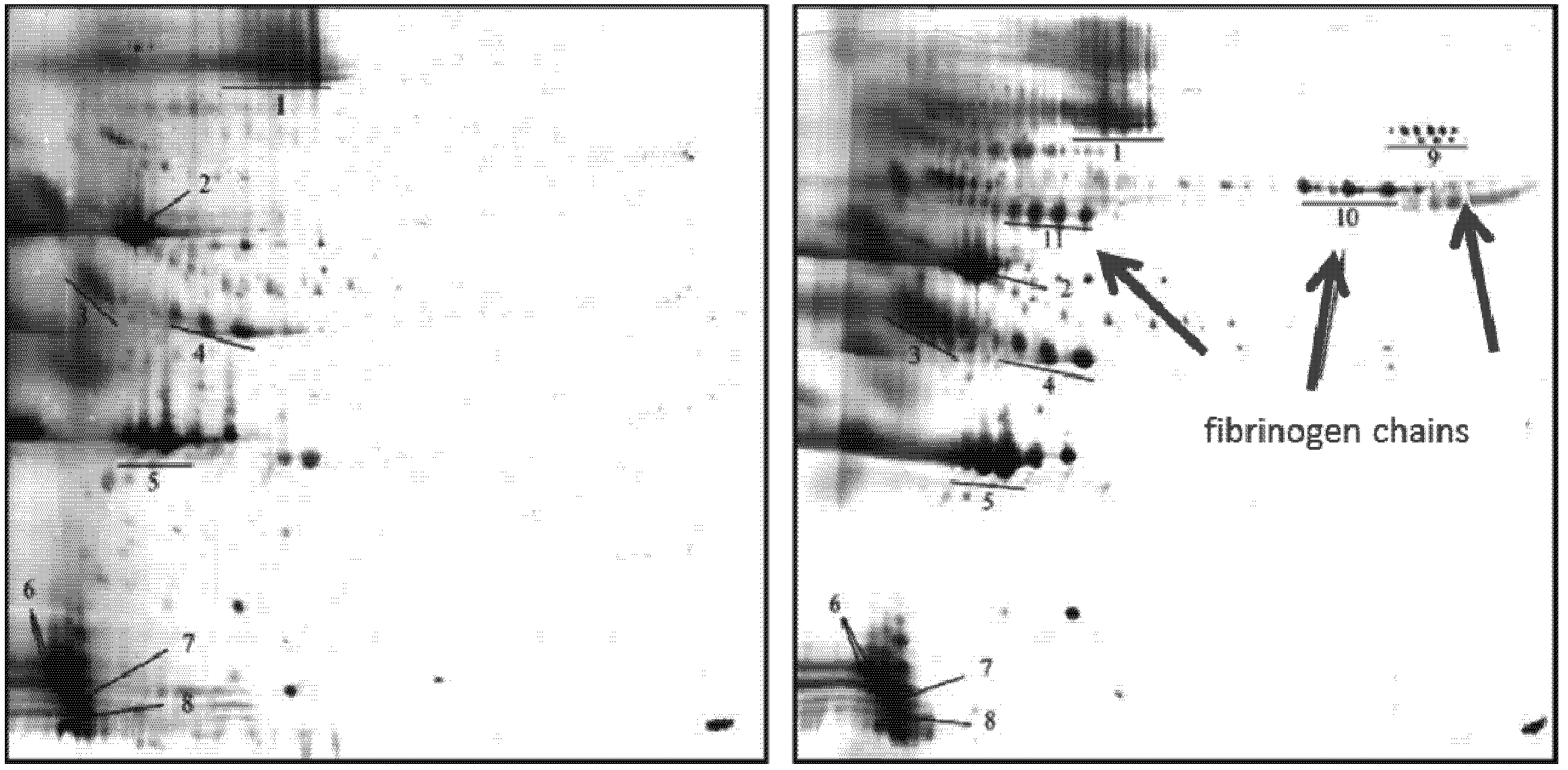
2.2.7. Influence of Surface Charge

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Formulations | Anionic emulsions prepared based on | Cationic emulsions prepared based on | Lipofundin MCT 10% | ||
|---|---|---|---|---|---|
| Oleic acid | deoxycholic acid | stearylamine | oleylamine | ||
| A: Physical characterization of the nanoemulsions | |||||
| particle size * (nm ± SD) | 205 ± 52 | 192 ± 48 | 183 ± 30 | 167 ± 50 | 250 ± 14 |
| zeta potential * (mV ± SD) | (−36.1) ± 3.6 | (−34.4) ± 0.8 | 47.8 ± 0.9 | 37.2 ± 2.4 | (−36.0) ± 0.3 |
| B: Volume percentage of detected plasma proteins after their adsorption onto particles | |||||
| Albumin | 0.0 | 2.9 | 2.7 | 2.5 | 2.0 |
| ApoA-I | 25.0 | 9.7 | 33.2 | 30.5 | 9.7 |
| ApoA-II | 2.5 | 6.8 | 6.1 | 4.8 | 9.2 |
| ApoA-IV | 4.6 | 36.9 | 6.7 | 4.5 | 10.5 |
| ApoC-II | 1.8 | 12.0 | 6.2 | 4.5 | 5.6 |
| ApoC-IIII | 4.5 | 14.4 | 8.6 | 7.9 | 10.6 |
| ApoE | 0.0 | 1.9 | 1.5 | 1.2 | 2.9 |
| ApoJ | 0.0 | 0.4 | 0.8 | 0.8 | 0.5 |
| Ig-gamma-chain | 2.2 | 0.0 | 0.9 | 1.2 | 8.1 |
| Ig-light chain | 6.3 | 1.3 | 1.5 | 3.0 | 0.0 |
| total | 46.9 | 86.3 | 68.2 | 60.9 | 59.1 |
2.2.8. Effects of Drug Incorporation
2.2.8.1. Effect of Amphotericin B

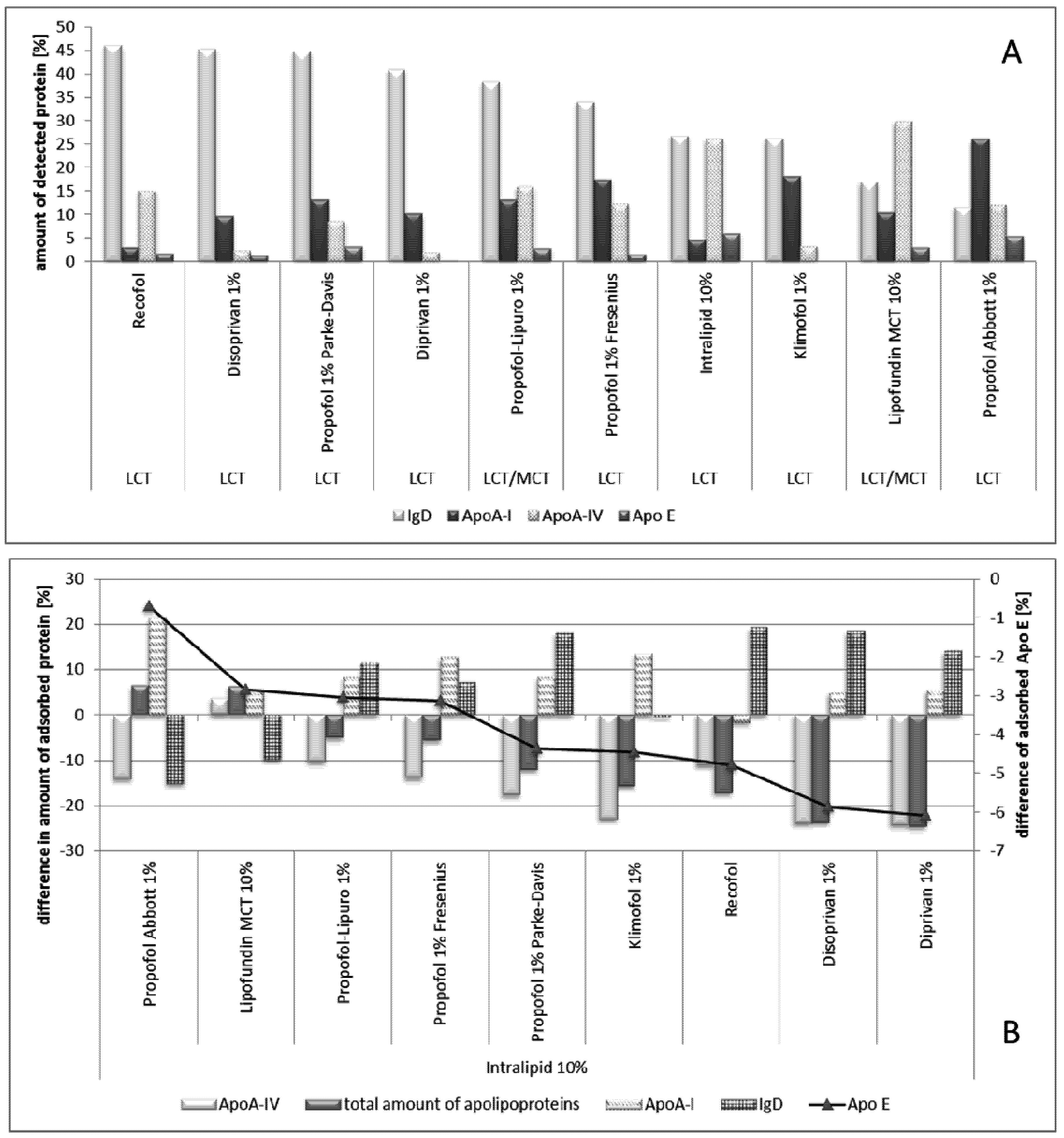
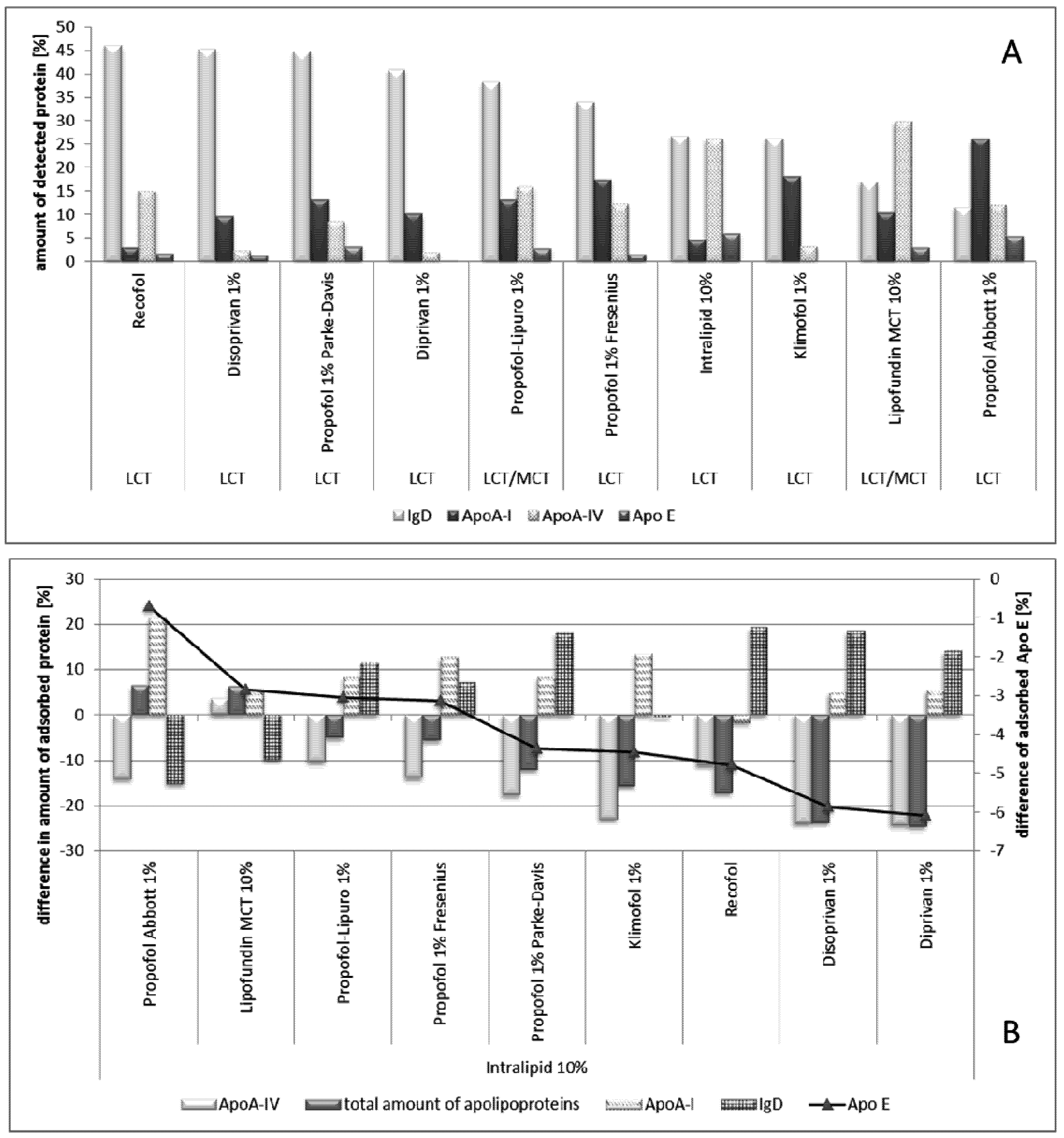
2.2.8.2. Effect of Propofol
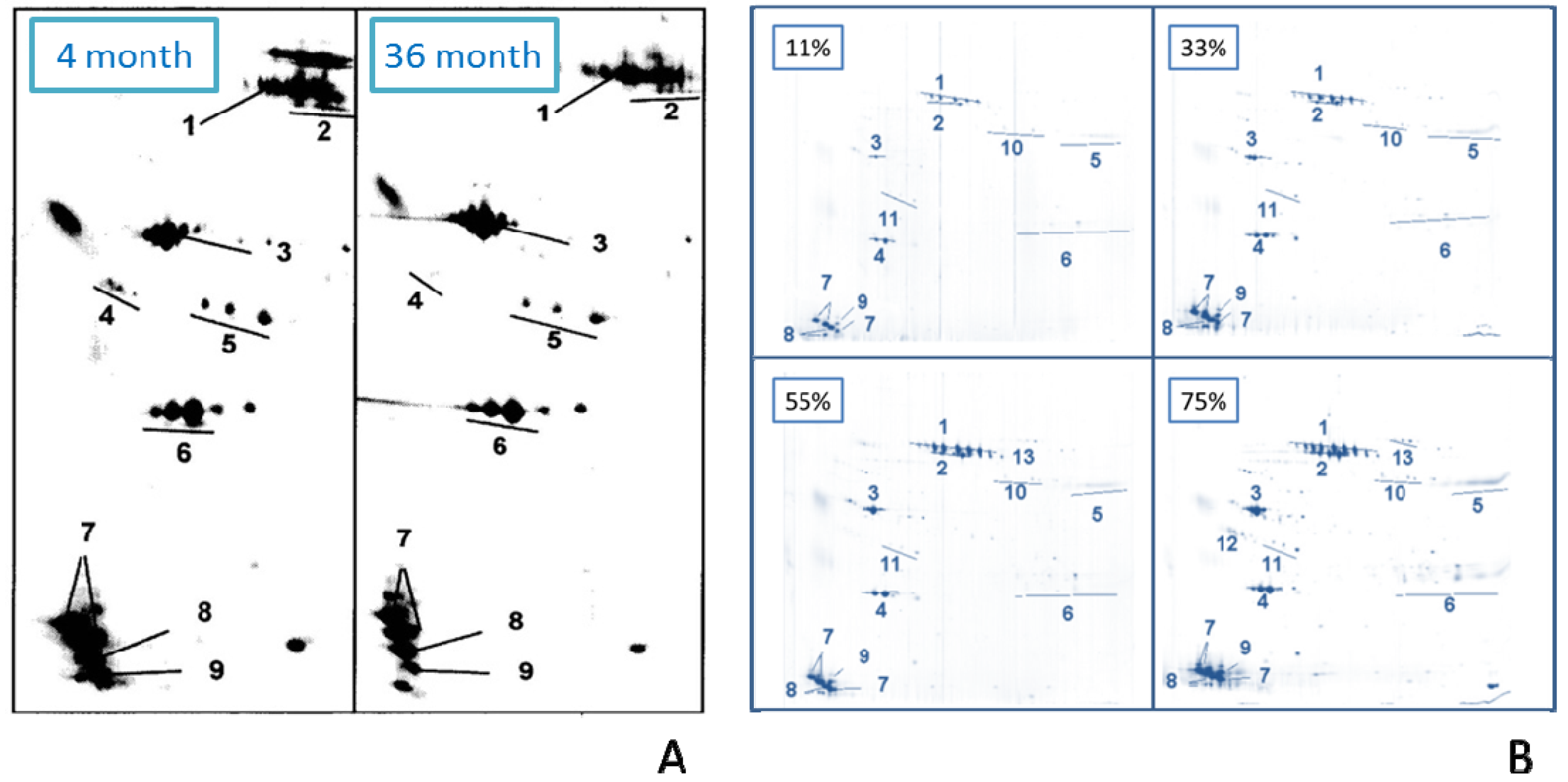
2.2.9. Effect of Age

2.2.10. Adsorption Kinetics (Effect of Incubation Time)
3. Conclusions
Acknowledgement
Conflict of Interest
References
- Ehrlich, P. Collected Studies on Immunity; John Wiley & Sons: New York, NY, USA, 1906. [Google Scholar]
- Kreuter, J. Nanoparticles—A historical perspective. Int. J. Pharm. 2007, 331, 1–10. [Google Scholar] [CrossRef]
- Torchilin, V.P. Liposomes as delivery agents for medical imaging. Mol. Med. Today 1996, 2, 242–249. [Google Scholar] [CrossRef]
- Lub-de Hooge, M.N.; Kosterink, J.G.; Perik, P.J.; Nijnuis, H.; Tran, L.; Bart, J.; Suurmeijer, A.J.; de Jong, S.; Jager, P.L.; de Vries, E.G. Preclinical characterisation of 111In-DTPA-trastuzumab. Br. J. Pharmacol. 2004, 143, 99–106. [Google Scholar] [CrossRef]
- Van Den Bossche, B.; van Christophe, D.W. Receptor imaging in oncology by means of nuclear medicine: Current status. J. Clin. Oncol. 2004, 22, 3593–3607. [Google Scholar] [CrossRef]
- Alyautdin, R.; Gothier, D.; Petrov, V.; Kharkevich, D.; Kreuter, J. Analgesic activity of the hexapeptide dalargin adsorbed on the surface of polysorbate 80-coated poly (butyl cyanoacrylate) nanoparticles. Eur. J. Pharm. Biopharm. 1995, 41, 44–48. [Google Scholar]
- Kreuter, J.; Alyautdin, R.N.; Kharkevich, D.A.; Ivanov, A.A. Passage of peptides through the blood-brain barrier with colloidal polymer particles (nanoparticles). Brain Res. 1995, 674, 171–174. [Google Scholar] [CrossRef]
- Kreuter, J.; Gelperina, S. Use of nanoparticles for cerebral cancer. Tumori 2008, 94, 271–277. [Google Scholar]
- Kreuter, J. Nanoparticulate systems for brain delivery of drugs. Adv. Drug. Deliv. Rev. 2001, 47, 65–81. [Google Scholar] [CrossRef]
- Kreuter, J. Influence of the surface properties on nanoparticle-mediated transport of drugs to the brain. J. Nanosci. Nanotechnol. 2004, 4, 484–488. [Google Scholar] [CrossRef]
- Senior, J.; Crawley, J.C.; Gregoriadis, G. Tissue distribution of liposomes exhibiting long half-lives in the circulation after intravenous injection. Biochim. Biophys. Acta 1985, 839, 1–8. [Google Scholar] [CrossRef]
- Davis, S.S. Colloids as drug-delivery systems. Pharm. Technol. 1981, 5, 71–88. [Google Scholar]
- Müller, R.H. Colloidal Carriers for Controlled Drug Delivery and Targeting—Modification, Characterization and in vivo Distribution; CRC Press: Boca Raton, FL, USA, 1991. [Google Scholar]
- Wilkins, D.J.; Myers, P.A. Studies on the relationship between the electrophoretic properties of colloids and their blood clearance and orrat, g.d.i.t. Br. J. Exp. Pathol. 1966, 47, 568–576. [Google Scholar]
- Schwendener, R.A.; Lagocki, P.A.; Rahman, Y.E. The effects of charge and size on the interaction of unilamellar liposomes with macrophages. Biochim. Biophys. Acta 1984, 772, 93–101. [Google Scholar] [CrossRef]
- Müller, R.H.; Davis, S.S.; Illum, L.; Mak, E. Particle charge and surface hydrophobicity of colloidal drug carriers. In Targeting of Drugs with Synthetic Systems; Gregoriadis, G., Senior, J., Poste, G., Eds.; Plenum Press: New York, NY, USA, 1986; pp. 239–263. [Google Scholar]
- Müller, R.H.; Davis, S.S.; Illum, L.; Mak, E. Surface Characterisation of Colloidal Drug Carriers Coated with Polymers; ACS: Washington, WA, USA, 1986; pp. 161–162. [Google Scholar]
- Mak, E. Determination of the Surface Hydrophobicity of Colloidal Drug Carriers using a Rose Bengal Binding Method. Acta Pharm. Technol. Int. J. Drug Formul. Biopharm. 1986, 32, 215–228. [Google Scholar]
- Lukowski, G.; Müller, R.H.; Müller, B.W.; Dittgen, M. Acrylic acid copolymer nanoparticles for drug delivery. I: Characterization of the surface properties relevant for in vivo organ distribution. Int. J. Pharm. 1992, 84, 23–31. [Google Scholar] [CrossRef]
- Krekeler, C.; Ziehr, H.; Klein, J. Physical methods for characterization of microbial surfaces. Experientia 1989, 45, 1047–1055. [Google Scholar] [CrossRef]
- Carstensen, H.; Müller, B.W.; Müller, R.H. Adsorption of ethoxylated surfactants on nanoparticles. I. Characterization by Hydrophobic Interaction Chromatography. Int. J. Pharm. 1991, 67, 29–37. [Google Scholar] [CrossRef]
- Wallis, K.H.; Müller, R.H. Determination of the surface hydrophobicity of colloidal dispersions by mini-hydrophobic interaction chromatography. Pharm. Ind. 1999, 55, 1124–1128. [Google Scholar]
- Illum, L.; Davis, S.S.; Müller, R.H.; Mak, E.; West, P. The organ distribution and circulation time of intravenously injected colloidal carriers sterically stabilized with a block copolymer-poloxamine 908. Life Sci. 1987, 40, 367–374. [Google Scholar] [CrossRef]
- Müller, R.H.; Lück, M.; Harnisch, S.; Thode, K. Intravenously Injected Particles; Surface Properties and Interaction with Blood Proteins—The Key Determining the Organ Distribution. In Scientific and Clinical Applications of Magnetic Carriers; Schütt, W., Teller, J., Häfeli, U., Zborowski, M., Eds.; Plenum Press: New York, NY, USA, 1997; pp. 135–148. [Google Scholar]
- Illum, L.; Davis, S.S. Targeting of colloidal particles to the bone marrow. Life Sci. 1987, 40, 1553–1560. [Google Scholar] [CrossRef]
- Juliano, R.L. Factors affecting the clearance kinetics and tissue distribution of liposomes, microspheres and emulsions. Adv. Drug Deliv. Rev. 1988, 2, 31–54. [Google Scholar] [CrossRef]
- Müller, R.H.; Heinemann, S. Surface Modelling of Microparticles as Parenteral Systems with High Tissue Affinity. In Bioadhesion-Possibilities and Future Trends; Gurny, R., Junginger, H.E., Eds.; Wissenschaftliche Verlagsgesellschaft: Stuttgart, Germany, 1989; pp. 202–214. [Google Scholar]
- Müller, R.H. Differential adsorption for the targeting of drug carriers. Acta Pharm. Technol. Int. J. Drug Formul. Biopharma. 1990, 36, 34. [Google Scholar]
- Davis, S.S.; Illum, L.; Moghimi, S.M.; Davis, M.C.; Porter, C.J.H.; Muir, I.S.; Brindley, A.; Christy, N.M.; Norman, M.E.; Williams, P.; et al. Microspheres for targeting drugs to specific body sites. J. Contr. Release 1993, 24, 157–163. [Google Scholar] [CrossRef]
- Armstrong, T.I.; Davies, M.C.; Illum, L. Human serum albumin as a probe for protein adsorption to nanoparticles: Relevance to biodistribution. J. Drug Target. 1997, 4, 389–398. [Google Scholar] [CrossRef]
- Basinska, T. Adsorption studies of human serum albumin, human gamma-globulins, and human fibrinogen on the surface of p(S/PGL) microsphere. J. Biomater. Sci. Polym. Ed. 2001, 12, 1359–1371. [Google Scholar] [CrossRef]
- Absolom, D.R.; Zingg, W.; Neumann, A.W. Protein adsorption to polymer particles: Role of surface properties. J. Biomed. Mat. Res. 1987, 21, 161–171. [Google Scholar] [CrossRef]
- Pieper, R.; Gatlin, C.L.; Makusky, A.J.; Russo, P.S.; Schatz, C.R.; Miller, S.S.; Su, Q.; McGrath, A.M.; Estock, M.A.; Parmar, P.P.; et al. The human serum proteome: display of nearly 3700 chromatographically separated protein spots on two-dimensional electrophoresis gels and identification of 325 distinct proteins. Proteomics 2003, 3, 1345–1364. [Google Scholar] [CrossRef]
- Paulke, B.R.; Möglich, P.; Knippel, E.; Budde, A.; Nitzsche, R.; Müller, R.H. Electrophoretic 3D-mobility profiles of latex particles with different surface groups. Langmuir 1995, 11, 70–74. [Google Scholar] [CrossRef]
- Lück, M.; Paulke, B.R.; Schröder, W.; Müller, R.H. Two-dimensional electrophoresis (2-DE) for the determination of plasma proteins adsorbed on model drug carriers. In Future Strategies for Drug Delivery with Particulate Systems; Diederichs, J.E., Müller, R.H., Eds.; CRC Press: Boca Raton, FL, USA, 1998; pp. 109–117. [Google Scholar]
- Müller, R.H.; Rühl, D.; Lück, M.; Paulke, B.R. Influence of fluorescent labelling of polystyrene particles on phagocytic uptake, surface hydrophobicity, and plasma protein adsorption. Pharm. Res. 1997, 14, 18–24. [Google Scholar] [CrossRef]
- Lück, M.; Schröder, W.; Harnisch, S.; Thode, K.; Blunk, T.; Paulke, B.R.; Kresse, M.; Müller, R.H. Identification of plasma proteins facilitated by enrichment on particulate surfaces: Analysis by two-dimensional electrophoresis and N-terminal microsequencing. Electrophoresis 1997, 18, 2961–2967. [Google Scholar] [CrossRef]
- Lück, M.; Paulke, B.R.; Schröder, W.; Blunk, T.; Müller, R.H. Analysis of plasma protein adsorption on polymeric nanoparticles with different surface characteristics. J. Biomed. Mat. Res. 1998, 39, 478–485. [Google Scholar] [CrossRef]
- Lück, M.; Schröder, W.; Paulke, B.R.; Blunk, T.; Müller, R.H. Complement activation by model drug carriers for intravenous application: Determination by two-dimensional electrophoresis. Biomaterials 1999, 20, 2063–2068. [Google Scholar] [CrossRef]
- Gessner, A.; Lieske, A.; Paulke, B.R.; Müller, R.H. Functional groups on polystyrene model nanoparticles: Influence on protein adsorption. J. Biomed. Mat. Res. 2003, 65, 319–326. [Google Scholar]
- Gessner, A.; Paulke, B.R.; Müller, R.H.; Göppert, T.M. Protein rejecting properties of PEG-grafted nanoparticles: Influence of PEG-chain length and surface density evaluated by two-dimensional electrophoresis and bicinchoninic acid (BCA)-proteinassay. Die Pharm. 2006, 61, 293–297. [Google Scholar]
- Gessner, A.; Waicz, R.; Lieske, A.; Paulke, B.; Mäder, K.; Müller, R.H. Nanoparticles with decreasing surface hydrophobicities: Influence on plasma protein adsorption. Int. J. Pharm. 2000, 196, 245–249. [Google Scholar] [CrossRef]
- Gessner, A.; Lieske, A.; Paulke, B.; Müller, R.H. Influence of surface charge density on protein adsorption on polymeric nanoparticles: Analysis by two-dimensional electrophoresis. Eur. J. Pharm. Biopharm. 2002, 54, 165–170. [Google Scholar] [CrossRef]
- Thode, K.; Lück, M.; Semmler, W.; Müller, R.H.; Kresse, M. Determination of plasma protein adsorption on magnetic iron oxides: Sample preparation. Pharm. Res. 1997, 14, 905–910. [Google Scholar] [CrossRef]
- Thode, K.; Lück, M.; Schröder, W.; Blunk, T.; Müller, R.H.; Kresse, M. The influence of the sample preparation on plasma protein adsorption patterns on polysaccharide-stabilized iron oxide particles and N-terminal microsequencing of unknown proteins. J. Drug Target. 1998, 5, 459–469. [Google Scholar] [CrossRef]
- Lind, K.; Kresse, M.; Müller, R.H. Comparison of protein adsorption patterns onto differently charged hydrophilic superparamagnetic iron oxide particles obtained in vitro and ex vivo. Electrophoresis 2001, 22, 3514–3521. [Google Scholar] [CrossRef]
- Lind, K.; Kresse, M.; Müller, R.H. Evaluation of desorption of proteins adsorbed to hydrophilic surfaces by two-dimensional electrophoresis. Proteomics 2001, 1, 1059–1066. [Google Scholar] [CrossRef]
- Petri, B.; Bootz, A.; Khalansky, A.; Hekmatara, T.; Müller, R.H.; Uhl, R.; Kreuter, J.; Gelperina, S. Chemotherapy of brain tumour using doxorubicin bound to surfactant-coated poly(butyl cyanoacrylate) nanoparticles: Revisiting the role of surfactants. J. Contr. Release 2007, 117, 51–58. [Google Scholar] [CrossRef]
- Kreuter, J.; Petrov, V.E.; Kharkevich, D.A.; Alyautdin, R.N. Influence of the type of surfactant on the analgesic effects induced by the peptide dalargin after its delivery across the blood-brain barrier using surfactant-coated nanoparticles. J. Contr. Release 1997, 49, 81–87. [Google Scholar] [CrossRef]
- Kreuter, J.; Shamenkov, D.; Petrov, V.; Ramge, P.; Cychutek, K.; Koch-Brandt, C.; Alyautdin, R. Apolipoprotein-mediated transport of nanoparticle-bound drugs across the blood-brain barrier. J. Drug Target. 2002, 10, 317–325. [Google Scholar] [CrossRef]
- Steiniger, S.C.; Kreuter, J.; Khalansky, A.S.; ISkidan, N.; Bobruskin, A.I.; Smirnova, Z.S.; Severin, S.E.; Uhl, R.; Kock, M.; Geiger, K.D.; et al. Chemotherapy of glioblastoma in rats using doxorubicin-loaded nanoparticles. Int. J. Cancer 2004, 109, 759–767. [Google Scholar] [CrossRef]
- Kreuter, J.; Ramge, P.; Petrov, V.; Hamm, S.; Gelperina, S.E.; Engelhardt, B.; Alyautdin, R.; von Briesen, H.; Begley, D.J. Direct evidence that polysorbate-80-coated poly (butylcyanoacrylate) nanoparticles deliver drugs to the CNS via specific mechanisms requiring prior binding of drug to the nanoparticles. Pharm. Res. 2003, 20, 409–416. [Google Scholar] [CrossRef]
- Gref, R.; Lück, M.; Quellec, P.; Marchand, M.; Dellacherie, E.; Harnisch, S.; Blunk, T.; Müller, R.H. "Stealth” corona-core nanoparticles surface modified by polyethylene glycol (PEG): Influences of the corona (PEG chain length and surface density) and of the core composition on phagocytic uptake and plasma protein adsorption. Colloids Surf. B Biointerfaces 2000, 18, 301–313. [Google Scholar] [CrossRef]
- Rouzes, C.; Gref, R.; Leonard, M.; Delgado, A.D.; Dellacherie, E. Surface modification of poly(lactic acid) nanospheres using hydrophobically modified dextrans as stabilizers in an o/w emulsion/evaporation technique. J. Biomed. Mat. Res. 2000, 50, 557–565. [Google Scholar] [CrossRef]
- Owens, D.E., III; Peppas, N.A. Opsonization, biodistribution, and pharmacokinetics of polymeric nanoparticles. Int. J. Pharm. 2006, 307, 93–102. [Google Scholar] [CrossRef]
- Meng, H.C.; Kuyama, T.; Thompson, S.W., II; Ferrell, J.F. Toxicity Testing of Fat Emulsions. I. Tolerance Study of Long-Term Intravenous Administration of Intralipid in Rats. Am. J. Clin. Nutr. 1965, 16, 29–36. [Google Scholar]
- Hochstrasser, D.F.; Funk, M.; Appel, R.D.; Pun, T.; James, R.W.; Hochstrasser, A.C.; Scherrer, J.R.; Pellegrini, C.; Müller, A.F. From biopsy to automatic diagnosis. Schweiz. Med. Wochenschr. 1990, 120, 1862–1866. [Google Scholar]
- Tissot, J.D.; Schneider, P.; James, R.W.; Daigneault, R.; Hochstrasser, D.F. High-resolution two-dimensional protein electrophoresis of pathological plasma/serum. Appl. Theor. Electrophor. 1991, 2, 7–12. [Google Scholar]
- Kim, H.R.; Andrieux, K.; Delomenie, C.; Chacun, H.; Appel, M.; Desmaele, D.; Taran, F.; Georgin, D.; Couvreur, P.; Taverna, M. Analysis of plasma protein adsorption onto PEGylated nanoparticles by complementary methods: 2-DE, CE and Protein Lab-on-chip system. Electrophoresis 2007, 28, 2252–2261. [Google Scholar]
- Liu, G.; Men, P.; Harris, P.L.; Rolston, R.K.; Perry, G.; Smith, M.A. Nanoparticle iron chelators: A new therapeutic approach in Alzheimer disease and other neurologic disorders associated with trace metal imbalance. Neurosci. Lett. 2006, 406, 189–193. [Google Scholar] [CrossRef]
- Dobrovolskaia, M.A.; Aggarwal, P.; Hall, J.B.; McNeil, S.E. Preclinical studies to understand nanoparticle interaction with the immune system and its potential effects on nanoparticle biodistribution. Mol. Pharmacol. 2008, 5, 487–495. [Google Scholar] [CrossRef]
- Dobrovolskaia, M.A.; McNeil, S.E. Immunological properties of engineered nanomaterials. Nat. Nanotechnol. 2007, 2, 469–478. [Google Scholar] [CrossRef]
- Leroux, J.C.; Gravel, P.; Balant, L.; Volet, B.; Anner, B.M.; Allemann, E.; Doelker, E.; Gurny, R. Internalization of poly(D,L-lactic acid) nanoparticles by isolated human leukocytes and analysis of plasma proteins adsorbed onto the particles. J. Biomed. Mat. Res. 1994, 28, 471–481. [Google Scholar] [CrossRef]
- Allemann, E.; Gravel, P.; Leroux, J.C.; Balant, L.; Gurny, R. Kinetics of blood component adsorption on poly(D,L-lactic acid) nanoparticles: Evidence of complement C3 component involvement. J. Biomed. Mat. Res. 1997, 37, 229–234. [Google Scholar] [CrossRef]
- Price, M.E.; Cornelius, R.M.; Brash, J.L. Protein adsorption to polyethylene glycol modified liposomes from fibrinogen solution and from plasma. Biochim. Biophys. Acta 2001, 1512, 191–205. [Google Scholar] [CrossRef]
- Cornelius, R.M.; Archambault, J.G.; Berry, L.; Chan, A.K.; Brash, J.L. Adsorption of proteins from infant and adult plasma to biomaterial surfaces. J. Biomed. Mat. Res. 2002, 60, 622–632. [Google Scholar] [CrossRef]
- Blunk, T.; Hochstrasser, D.F.; Sanchez, J.C.; Müller, B.W.; Müller, R.H. Colloidal carriers for intravenous drug targeting: Plasma protein adsorption patterns on surface-modified latex particles evaluated by two-dimensional polyacrylamide gel electrophoresis. Electrophoresis 1993, 14, 1382–1387. [Google Scholar] [CrossRef]
- Jahangir, R.; McCloskey, C.B.; Clung, W.G.M.; Labow, R.S.; Brash, J.L.; Santerre, J.P. The influence of protein adsorption and surface modifying macromolecules on the hydrolytic degradation of a poly(ether-urethane) by cholesterol esterase. Biomaterials 2003, 24, 121–130. [Google Scholar] [CrossRef]
- Archambault, J.G.; Brash, J.L. Protein resistant polyurethane surfaces by chemical grafting of PEO: Amino-terminated PEO as grafting reagent. Colloids Surf. B Biointerfaces 2004, 39, 9–16. [Google Scholar] [CrossRef]
- Unsworth, L.D.; Sheardown, H.; Brash, J.L. Protein resistance of surfaces prepared by sorption of end-thiolated poly(ethylene glycol) to gold: Effect of surface chain density. Langmuir 2005, 21, 1036–1041. [Google Scholar] [CrossRef]
- Leroux, J.-C.; Alleman, E.; de Jaeghere, F.; Doelker, E.; Gurny, R. Biodegradable nanoparticles: From sustained release formulations to improved site specific drug delivery. J. Contr. Release 1996, 39, 339–350. [Google Scholar] [CrossRef]
- Dobrovolskaia, M.A.; Guszcysski, T.; Specht, S.; McLeland, C.B. NCL Method ITA-4—Analysis of Nanoparticle Interaction with Plasma Proteins by 2D PAGE. Available online: http://ncl.cancer.gov/NCL_Method_ITA-4.pdf (accessed on 22 May 2011).
- Göppert, T.M.; Müller, R.H. Adsorption kinetics of plasma proteins on solid lipid nanoparticles for drug targeting. Int. J. Pharm. 2005, 302, 172–186. [Google Scholar] [CrossRef]
- Göppert, T.M.; Müller, R.H. Polysorbate-stabilized solid lipid nanoparticles as colloidal carriers for intravenous targeting of drugs to the brain: Comparison of plasma protein adsorption patterns. J. Drug Target. 2005, 13, 179–187. [Google Scholar] [CrossRef]
- Göppert, T.M.; Müller, R.H. Alternative sample preparation prior to two-dimensional electrophoresis protein analysis on solid lipid nanoparticles. Electrophoresis 2004, 25, 134–140. [Google Scholar] [CrossRef]
- Göppert, T.M.; Müller, R.H. Plasma protein adsorption of Tween 80- and poloxamer 188-stabilized solid lipid nanoparticles. J. Drug Target. 2003, 11, 225–231. [Google Scholar] [CrossRef]
- Müller, R.H.; Harnisch, S. Physico-chemical characterization of propofol-loaded emulsions and interaction with plasma proteins. Eur. J. Hosp. Pharm. 2000, 6, 24–31. [Google Scholar]
- Harnisch, S.; Müller, R.H. Plasma protein adsorption patterns on emulsions for parenteral administration: Establishment of a protocol for two-dimensional polyacrylamide electrophoresis. Electrophoresis 1998, 19, 349–354. [Google Scholar] [CrossRef]
- Harnisch, S.; Müller, R.H. Adsorption kinetics of plasma proteins on oil-in-water emulsions for parenteral nutrition. Eur. J. Pharm. Biopharm. 2000, 49, 41–46. [Google Scholar] [CrossRef]
- Schmidt, S.; Müller, R.H. Plasma protein adsorption patterns on surfaces of Amphotericin B-containing fat emulsions. Int. J. Pharm. 2003, 254, 3–5. [Google Scholar] [CrossRef]
- Blunk, T. Plasmaproteinadsorption auf kolloidalen Arzneistoffträgern. Ph.D. Thesis, Christian-Albrechts-Universität, Kiel, Germany, 1994. [Google Scholar]
- Gessner, A.; Paulke, B.R.; Müller, R.H. Analysis of plasma protein adsorption onto polystyrene particles by two-dimensional electrophoresis: Comparison of sample application and isoelectric focusing techniques. Electrophoresis 2000, 21, 2438–2442. [Google Scholar] [CrossRef]
- Diederichs, J.E. Plasma protein adsorption patterns on liposomes: Establishment of analytical procedure. Electrophoresis 1996, 17, 607–611. [Google Scholar] [CrossRef]
- Göppert, T.M. Plasma protein adsorption on parenterally administered colloidal drug carriers for crossing the blood brain barrier. Ph.D. Thesis, Freie Universität, Berlin, Germany, 2005. [Google Scholar]
- Thode, K. Specific contrast agents for magnetic resonance tomography: Physico-chemical characterisation and studies on plasma protein adsorption. PhD Thesis, Freie Universität, Berlin, Germany, 1996. [Google Scholar]
- Camner, P.; Lundborg, M.; Lastbom, L.; Gerde, P.; Gross, N.; Jarstrand, C. Experimental and calculated parameters on particle phagocytosis by alveolar macrophages. J. Appl. Physiol. 2002, 92, 2608–2616. [Google Scholar]
- Moghimi, S.M.; Muir, I.S.; Illum, L.; Davis, S.S.; Kolb-Bachofen, V. Coating particles with a block co-polymer (poloxamine-908) suppresses opsonization but permits the activity of dysopsonins in the serum. Biochim. Biophys. Acta 1993, 1179, 157–165. [Google Scholar] [CrossRef]
- Ogawara, K.; Furumoto, K.; Nagayama, S.; Minato, K.; Higaki, K.; Kai, T.; Kimura, T. Pre-coating with serum albumin reduces receptor-mediated hepatic disposition of polystyrene nanosphere: Implications for rational design of nanoparticles. J. Contr. Release 2004, 100, 451–455. [Google Scholar] [CrossRef]
- O’Mullane, J.E.; Artursson, P.; Tomlinson, E. Biopharmaceutics of microparticulate drug carriers. Ann. N. Y. Acad. Sci. 1987, 507, 120–140. [Google Scholar] [CrossRef]
- Liliemark, E.; Sjostrom, B.; Liliemark, J.; Peterson, C.; Kallberg, N.; Larsson, B.S. Targeting of teniposide to the mononuclear phagocytic system (MPS) by incorporation in liposomes and submicron lipid particles: An autoradiographic study in mice. Leuk. Lymphoma 1995, 18, 113–118. [Google Scholar] [CrossRef]
- Blunk, T.; Lück, M.; Calvör, A.; Hochstrasser, D.F.; Sanchez, J.C.; Müller, B.W.; Müller, R.H. Kinetics of Plasma Protein Adsorption on Model Particles for Controlled Drug Delivery and Drug Targeting. Eur. J. Pharma. Biopharm. 1996, 42, 262–268. [Google Scholar]
- Vroman, L.; Adams, A.L.; Fischer, G.C.; Munoz, P.C. Interaction of high molecular weight kininogen, factor XII, and fibrinogen in plasma at interfaces. Blood 1980, 55, 156–159. [Google Scholar]
- Vroman, L.; Adams, A.L. Adsorption of proteins out of plasma and solutions in narrow spaces. J. Colloid Interface Sci. 1986, 111, 391–402. [Google Scholar] [CrossRef]
- Rabilloud, T. Solubilization of proteins for electrophoretic analyses. Electrophoresis 1996, 17, 813–829. [Google Scholar] [CrossRef]
- Rabilloud, T.; Adessi, C.; Giraudel, A.; Lunardi, J. Improvement of the solubilization of proteins in two-dimensional electrophoresis with immobilized pH gradients. Electrophoresis 1997, 18, 307–316. [Google Scholar] [CrossRef] [Green Version]
- Benita, S.; Levy, M.Y. Submicron emulsions as colloidal drug carriers for intravenous administration: Comprehensive physicochemical characterization. J. Pharm. Sci. 1993, 82, 1069–1079. [Google Scholar] [CrossRef]
- Hallberg, D.; Holm, I.; Obel, A.L.; Schuberth, O.; Wretlind, A. Fat emulsion for complete intravenous nutrition. Postgrad. Med. 1967, 42, 149–152. [Google Scholar]
- Wretlind, A. Development of fat emulsions. J. Parenter. Enter. Nutr. 1981, 5, 230–235. [Google Scholar]
- Wretlind, A. Recollections of pioneers in nutrition: Landmarks in the development of parenteral nutrition. J. Am. Coll. Nutr. 1992, 11, 366–373. [Google Scholar]
- Davis, S.S.; Illum, L.; Müller, R.H.; Landry, F.; Wright, J.; Harper, G. The effect of infused fat emulsions on reticuloendothelial function in the rabbit. Clin. Nutr. 1990, 9, 260–265. [Google Scholar] [CrossRef]
- Davis, S.S.; Washington, C.; West, P.; Illum, L.; Liversidge, G.; Sternson, L.; Kirsh, B.R. Lipid emulsions as drug delivery systems. Ann. N. Y. Acad. Sci. 1987, 507, 75–88. [Google Scholar] [CrossRef]
- Davis, S.S.; Illum, L.; West, P.; Galloway, M. Studies on the fate of fat emulsions following intravenous administration to rabbits and the effect of added electrolyte. Clin. Nutr. 1987, 6, 13–19. [Google Scholar]
- Harnisch, S.; Müller, R.H. Analysis of adsorbed plasma proteins on commercial emulsions for parenteral nutrition. In Proceedings of International Symposium on Controlled Release and Bioactive Materials, Stockholm, Sweden, 1997.
- Hochstrasser, D.F.; Harrington, M.G.; Hochstrasser, A.C.; Miller, M.J.; Merril, C.R. Methods for increasing the resolution of two-dimensional protein electrophoresis. Anal. Biochem. 1988, 173, 424–435. [Google Scholar]
- Schmidt, S. Parenteral o/w emulsions: Drug incorporation and interaction with plasma proteins. Ph.D. Thesis, Freie Universität Berlin, Berlin, Germany, 2002. [Google Scholar]
- Gogos, C.A.; Kalfarentzos, F. Total parenteral nutrition and immune system activity: A review. Nutrition 1995, 11, 339–344. [Google Scholar]
- Eckart, J.; Adolph, M.; van der Mühlen, U.; Naab, V. Fat emulsions containing medium chain triglycerides in parenteral nutrition of intensive care patients. J. Parenter. Enteral Nutr. 1980, 4, 360–366. [Google Scholar]
- Hedeman, H.; Brondsted, H.; Mullertz, A.; Frokjaer, S. Fat emulsions based on structured lipids (1,3-specific triglycerides): An investigation of the in vivo fate. Pharm. Res. 1996, 13, 725–728. [Google Scholar] [CrossRef]
- Harnisch, S.; Blunk, T.; Müller, R.H. The Kinetics of Plasma Protein Adsorption on Oil-in-Water Emulsions. In Proceedings of Electrophorese Forum’97, Strassburg, France, 1997; p. 59.
- Harnisch, S.; Buszello, K.; Müller, B.W.; Müller, R.H. Influence of the Composition of Oil in Water Emulsions on the Pattern of Adsorbed Plasma Proteins. In Proceedings of 2nd World Meeting on Pharmaceutics, Biopharmaceutics and Pharmaceutical Technology, Paris, France, 1998.
- Jeppson, R.; Rössner, S. The influence of emulsifying agents and of lipid soluble drugs on the fractional removal rate of lipid emulsions from the blood stream of the rabbit. Acta Pharmacol. Toxicol. 1975, 37, 134–144. [Google Scholar] [CrossRef]
- Yang, S.C.; Benita, S. Enhanced adsorption and drug targeting by positively charged submicron emulsions. Drug Dev. Res. 2000, 50, 476–486. [Google Scholar] [CrossRef]
- Klang, S.H.; Parnas, M.; Benita, S. Emulsions as drug carriers-possibilities, limitations, and future perspectives. In Emulsions and Nanosuspensions for the Formulation of Poorly Soluble Drugs; Müller, R.H., Benita, S., Böhm, H.L., Eds.; Medpharm: Stuttgart, Germany, 1998; pp. 31–65. [Google Scholar]
- Korner, D.; Benita, S.; Albrecht, G.; Baszkin, A. Surface properties of mixed phospholipid–stearylamine monolayers and their interaction with a non-ionic surfactant (poloxamer). Colloids Surf. B Biointerfaces 1994, 3, 101–109. [Google Scholar] [CrossRef]
- Tamilvanan, S.; Schmidt, S.; Müller, R.H.; Benita, S. In vitro adsorption of plasma proteins onto the surface (charges) modified-submicron emulsions for intravenous administration. Eur. J. Pharm. Biopharm. 2005, 59, 1–7. [Google Scholar] [CrossRef]
- Chyle, M.; Chyle, P. Affecting the immunity response with deoxycholic acid. Sb Lek. 1982, 84, 212–218. [Google Scholar]
- Chyle, M.; Chyle, P. Deoxycholic acid in the therapy of herpes labialis (author’s transl). Cas. Lek. Cesk. 1975, 114, 1226–1229. [Google Scholar]
- Fassati, P.; Chyle, P.; Fassati, M.; Chyle, M. Deoxycholic acid in the therapy of liver cirrhosis (author’s transl). Cas. Lek. Cesk. 1975, 114, 1222–1226. [Google Scholar]
- Müller, R.H. Dispersions for the formulation of slightly or poorly soluble agents. DE10036871.9, 2001.
- Müller, R.H.; Schmidt, S.; Buttle, I.; Akkar, A.; Schmitt, J.; Bromer, S. SolEmuls-novel technology for the formulation of IV emulsions with poorly soluble drugs. Int. J. Pharm. 2004, 269, 293–302. [Google Scholar] [CrossRef]
- Janknegt, R.; de Marie, S.; Bakker-Woudenberg, I.A.; Crommelin, D.J. Liposomal and lipid formulations of amphotericin B. Clinical pharmacokinetics. Clin. Pharmacokinet. 1992, 23, 279–291. [Google Scholar] [CrossRef]
- Edwards, J.E.; Brouwer, K.R.; McNamara, P.J. GF120918, a P-glycoprotein modulator, increases the concentration of unbound amprenavir in the central nervous system in rats. Antimicrob. Agents Chemother. 2002, 46, 2284–2286. [Google Scholar] [CrossRef]
- Kambara, T.; Inada, T.; Kubo, K.; Shingu, K. Propofol suppresses prostaglandin E (2) production in human peripheral monocytes. Immunopharmacol. Immunotoxicol. 2009, 31, 117–126. [Google Scholar] [CrossRef]
- Washington, C.; Davis, S.S. Ageing effects in parenteral fat emulsions: the role of fatty acid. Int. J. Pharm. 1987, 39, 33–37. [Google Scholar] [CrossRef]
- Schmidt, S.; Gessner, A.; Müller, R.H. Comparison of plasma protein adsorption patterns on emulsions for parenteral nutrition dependent to their age. In Proceedings of International Symposium on Controlled Release and Bioactive Materials, San Diego, CA, USA, 2011.
- Schmidt, S.; Müller, R.H. Biological long-term stability of i. v. emulsions: Plasma protein adsorption on propofol-loaded emulsions. In Proceedings of 4th World Meeting on Pharmaceutics, Biopharmaceutics and Pharmaceutical Technology, Florence, Italy, 2002.
- Müller, R.H.; Lück, M.; Kreuter, J. Arzneistoffträgerpartikel für die gewebsspezifische Arzneistoffapplikation. DE19745950A1 1997.
Supplementary Files
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Keck, C.M.; Jansch, M.; Müller, R.H. Protein Adsorption Patterns and Analysis on IV Nanoemulsions—The Key Factor Determining the Organ Distribution. Pharmaceutics 2013, 5, 36-68. https://doi.org/10.3390/pharmaceutics5010036
Keck CM, Jansch M, Müller RH. Protein Adsorption Patterns and Analysis on IV Nanoemulsions—The Key Factor Determining the Organ Distribution. Pharmaceutics. 2013; 5(1):36-68. https://doi.org/10.3390/pharmaceutics5010036
Chicago/Turabian StyleKeck, Cornelia M., Mirko Jansch, and Rainer H. Müller. 2013. "Protein Adsorption Patterns and Analysis on IV Nanoemulsions—The Key Factor Determining the Organ Distribution" Pharmaceutics 5, no. 1: 36-68. https://doi.org/10.3390/pharmaceutics5010036