Live Cell in Vitro and in Vivo Imaging Applications: Accelerating Drug Discovery

Abstract
: Dynamic regulation of specific molecular processes and cellular phenotypes in live cell systems reveal unique insights into cell fate and drug pharmacology that are not gained from traditional fixed endpoint assays. Recent advances in microscopic imaging platform technology combined with the development of novel optical biosensors and sophisticated image analysis solutions have increased the scope of live cell imaging applications in drug discovery. We highlight recent literature examples where live cell imaging has uncovered novel insight into biological mechanism or drug mode-of-action. We survey distinct types of optical biosensors and associated analytical methods for monitoring molecular dynamics, in vitro and in vivo. We describe the recent expansion of live cell imaging into automated target validation and drug screening activities through the development of dedicated brightfield and fluorescence kinetic imaging platforms. We provide specific examples of how temporal profiling of phenotypic response signatures using such kinetic imaging platforms can increase the value of in vitro high-content screening. Finally, we offer a prospective view of how further application and development of live cell imaging technology and reagents can accelerate preclinical lead optimization cycles and enhance the in vitro to in vivo translation of drug candidates.1. Introduction: Challenges in Preclinical Drug Discovery
The past two decades have witnessed significant scientific and technical advances in the fields of drug discovery and translational medicine. Despite increased knowledge of disease mechanism and higher levels of financial investments in drug discovery research and development (R&D) the number of novel medicines approved by regulatory agencies such as the US Food and Drug Administration (FDA) has been in steady decline, with 50% fewer new molecular entities (NME) approved in the past five years compared with the previous five year period [1]. The development of truly innovative medicines that provide superior efficacy and safety in clinical settings over existing standards of care represents a formidable challenge. High attrition rates of candidate drugs in late stage preclinical development or clinical trials due to toxicity and limited efficacy suggests current pre-clinical drug discovery strategies are sub-optimal and poorly predict clinical outcome. The conventional target-directed drug discovery approach, where candidate drugs are developed on the basis of potency and selectivity against a single nominated target complements high-throughput serendipitous screening strategies of large chemical libraries. However, high costs, protracted timelines, low success rates and impending patent expirations suggest that the target directed drug discovery approach, on its own, no longer represents an effective R&D strategy.
Alternative drug discovery strategies that offer faster, more cost-effective and more opportunistic drug development approaches for new chemical entities, existing drug portfolios, multi-targeted agents and drug combinations are urgently required. A critical requirement of any new drug discovery approach is to provide greater predictivity of clinical response over existing methods and thus more productive translation of preclinical findings to clinically effective therapy. Recent advances in next generation sequencing, quantitative proteomics, RNAi technology, systems biology approaches and quantitative in vitro and in vivo imaging all embrace the biological complexity of disease and offer alternative strategies for target selection, target validation, candidate drug profiling and patient stratification [2,3]. Quantitative in vitro and in vivo imaging represents a more holistic approach to evaluation of drug efficacy, providing an unbiased and thus more opportunistic assessment of drug response in complex biological systems. Real-time image based analysis of drug response upon target activity and pathophysiology in vitro and in vivo may accelerate drug development timelines, reduce costs, provide novel mechanistic insight into adaptive response and increased clinical predictivity when applied appropriately to relevant model systems. In this review article we describe the latest research tools and approaches that facilitate live cell imaging and which are advancing the field of dynamic in vitro and in vivo imaging applications in drug discovery.
2. Live Cell Imaging In Vitro
Optical microscopes developed in the early 17th century represent the original transformative scientific technology for detailed physiological analysis of tissue and cell biology. Traditionally, microscopic imaging in research has been applied subjectively through manual application to small numbers of experimental or clinical specimens. However, recent developments of fully automated fluorescent microscopic acquisition platforms and associated image analysis algorithms have enhanced the throughput and statistical robustness of quantitative imaging. These advances have facilitated the integration of quantitative microscopic imaging into the drug development process and created the new discipline of high content screening. High content microscopic platforms and image analysis tools have evolved rapidly over the last 5 years and become established to different levels of sophistication and application within the pharmaceutical and biotechnology industry [4,5].
The widespread adoption of high content screening platforms by the pharmaceutical and biotechnology industry indicates a new trend in incorporating complex biological endpoints into drug discovery. High content screening is typically placed as a secondary screening strategy to confirm the quality of hits, identified through high throughput biochemical screening. However, high content assays are increasingly being applied to primary screening approaches, and have been proposed as a strategy to reduce attrition at later stages in drug development by frontloading the evaluation of novel target classes, chemical libraries and biologic therapeutics in more physiological cell systems [6]. Several publications describe the utility of high content imaging and analysis for profiling drug mechanism and defining structure activity relationship based upon phenotypic endpoints suggesting that quantitative imaging may offer an alternative approach to target-directed drug discovery [7-10]. The impact of high content biology on increasing the predictivity of the drug discovery process and transforming R&D productivity, in terms of delivering more successful therapies into the clinic, remains to be determined and will largely be dictated by the quality of the underlying biological models being subjected to investigation.
Typical high content cell-based screens employ fixed endpoint assays that provide a snapshot in time of a cellular response to pharmacological or molecular perturbation. In a similar manner to image-based immunohistochemical assessment of preclinical drug studies, such image based snapshots provide a reflection of pharmacological response at the time of sampling. Extension of image-based analysis to live cells, fresh tissue or live in vivo models in a time-resolved manner provides a more complete picture of dynamic biological processes through greater spatial and temporal understanding. Temporal analysis of drug response provides a number of unique advantages that enhance and complement fixed endpoint studies including: Quantification of transient phenotypic responses; Optimization of appropriate timepoints for endpoint studies; Interpretation of conflicting findings from endpoint studies that often arise from dynamic reversible processes that operate under precise temporal and spatial control; Characterization of adaptive responses; Determination of accurate scheduling and dosing regimes; Provision of additional data points of biological events collected over a time series, facilitating more robust quantitative analysis from less specimens. In the following sections of this article we shall review the development of the latest optical tools and enabling technology platforms, specifically optimized for live cell imaging applications in vitro and in vivo. We cite literature examples where live cell imaging has provided novel insight of pathophysiogical mechanism and drug response that cannot be extracted from fixed endpoint studies.
3. Live-cell Imaging in Vitro: Technical Advances and Novel Applications
3.1. Automated imaging platforms
Traditional time-lapse epifluorescent and confocal image acquisition microscopes provide detailed temporal and spatial analysis of cellular function. Throughput of such instruments is, however, restricted to one or a few samples per experiment, limiting their application within drug discovery. Most automated high content imaging microscopes can also be applied to live-cell kinetic studies where an environmental chamber is integrated onto the platform, however, throughput and analysis are once again rate-limited and environmental conditions are not optimal for long-term studies. Recent developments of more innovative microscopic configurations where the microscopic optical instrumentation is placed within a robust environmental chamber represent a significant technological advance in dynamic cell imaging. Such systems exemplified by IncuCyte™; Cell-IQ™ and Biostation CT™ incorporating software enabling remote control of, image acquisition, filter optic configurations and image analysis are optimized for long term kinetic studies across multiwell plates. This new generation of live-cell imaging microscopes increases the throughput and flexibility of examining dynamic cellular processes in response to multiple molecular or pharmacological intervention studies.
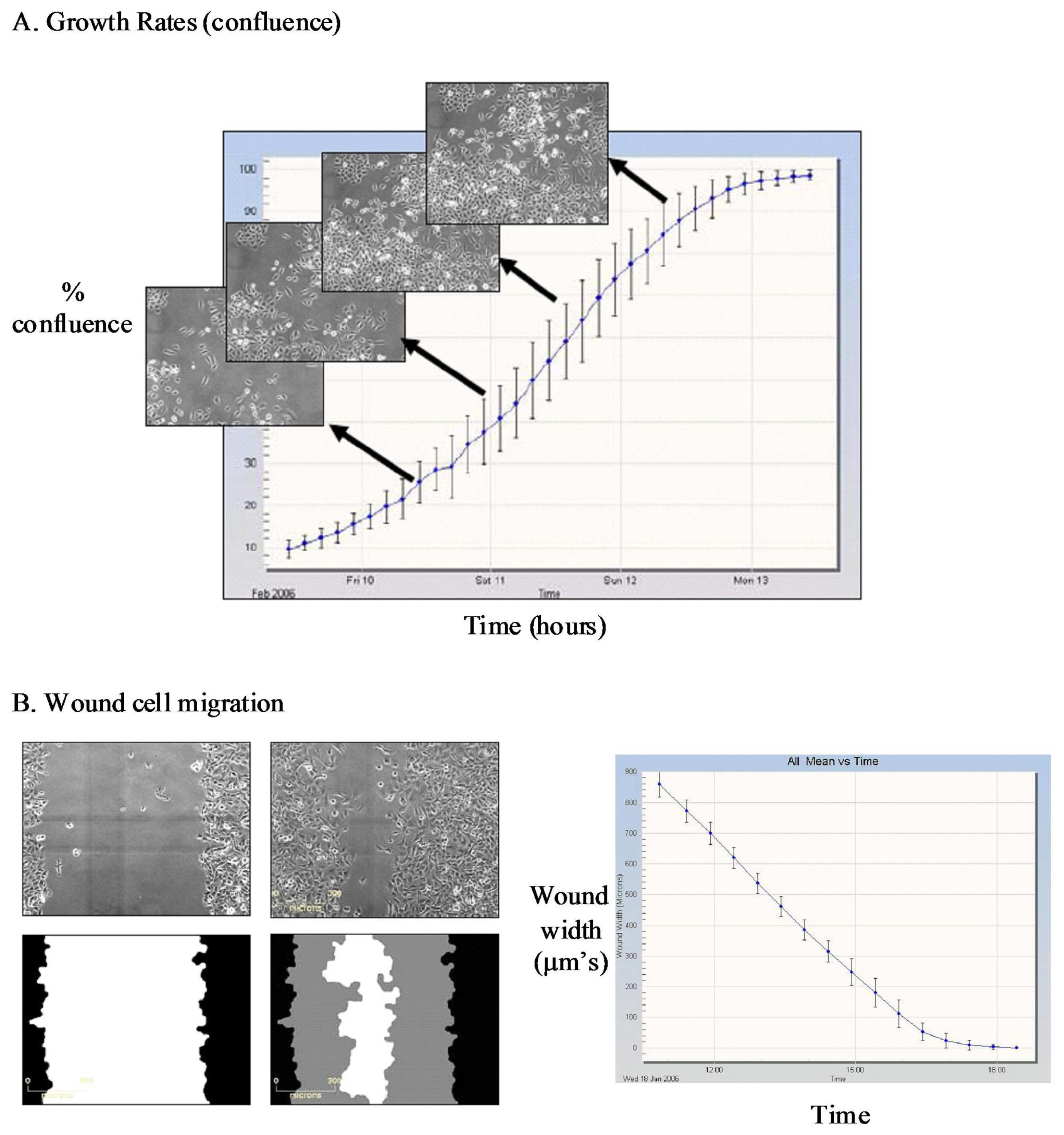
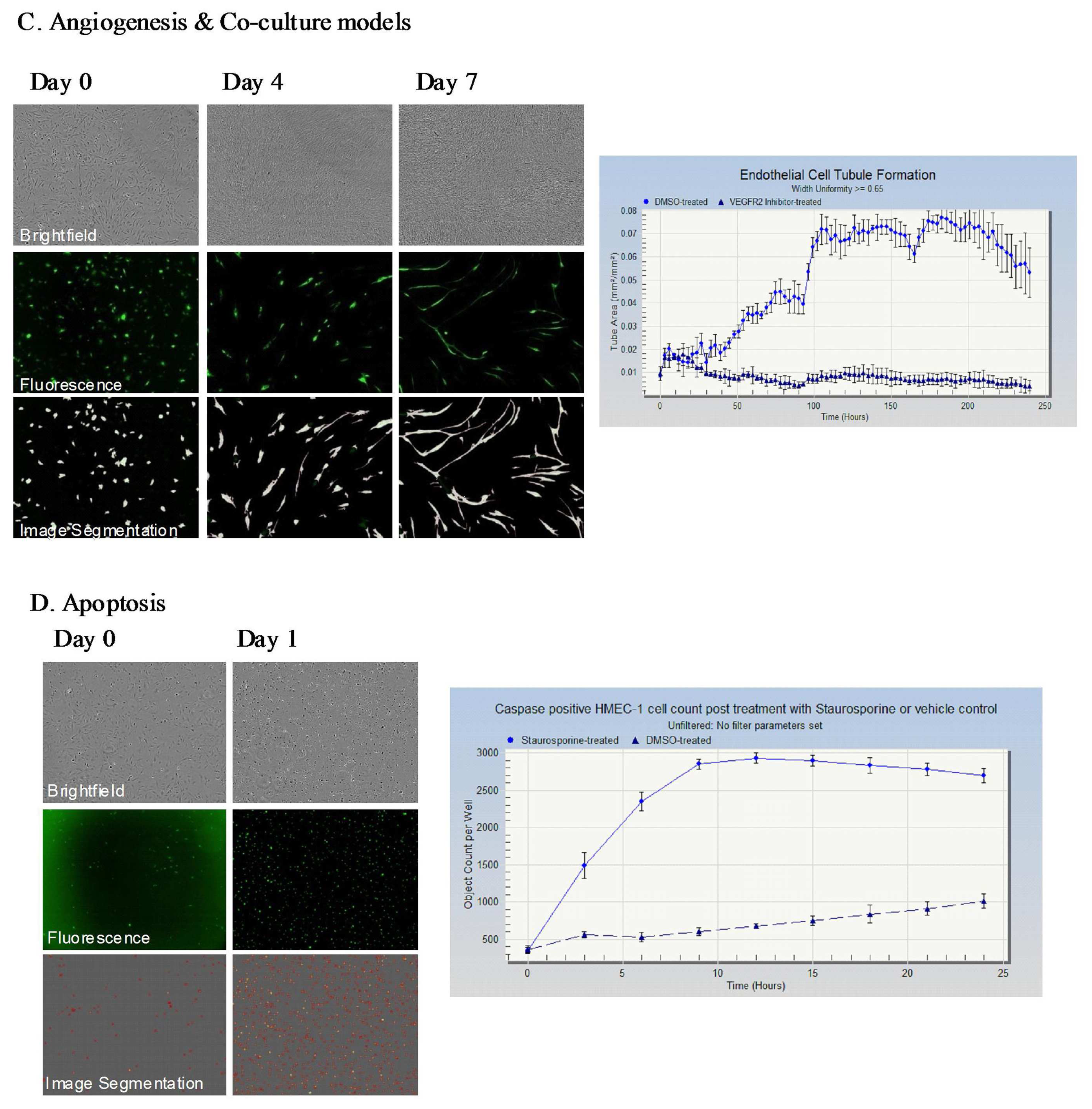
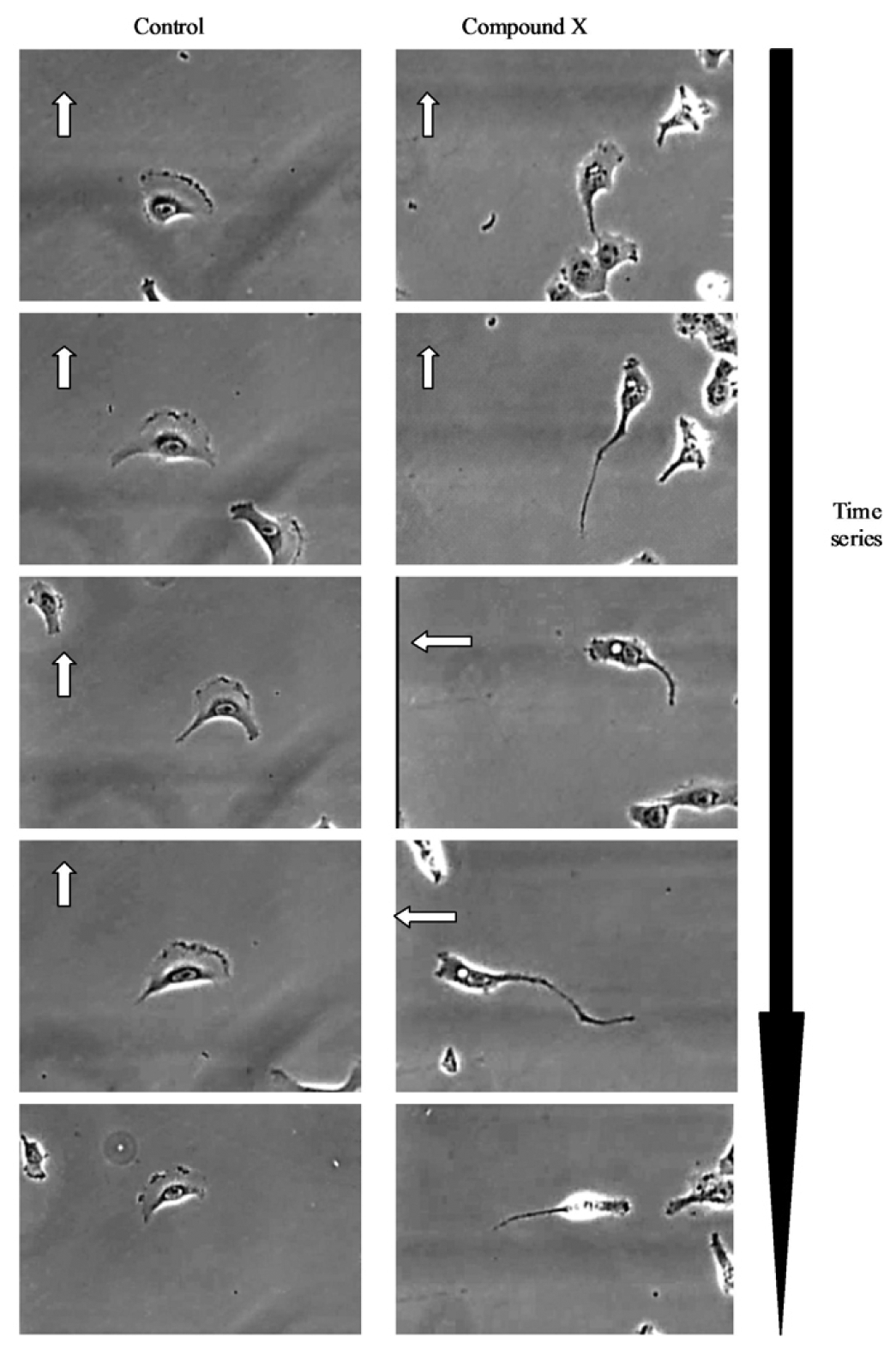
The IncuCyte-FLR™ is a compact, automated fluorescent and brightfield kinetic imaging platform that can be accommodated within a standard tissue culture incubator. The standard IncuCyte-FLR™ system can accommodate up to six 384-well plates to enable sequential monitoring of molecular and cell dynamics across multiple samples or drug treatment regimes in a single study. The IncuCyte™ platform also incorporates a suite of specific application modules comprising of image analysis algorithms and custom-designed consumables to facilitate automated monitoring of specific cellular processes such as cell growth, cell migration into a wounded monolayer, angiogenesis and apoptosis (Figure 1).
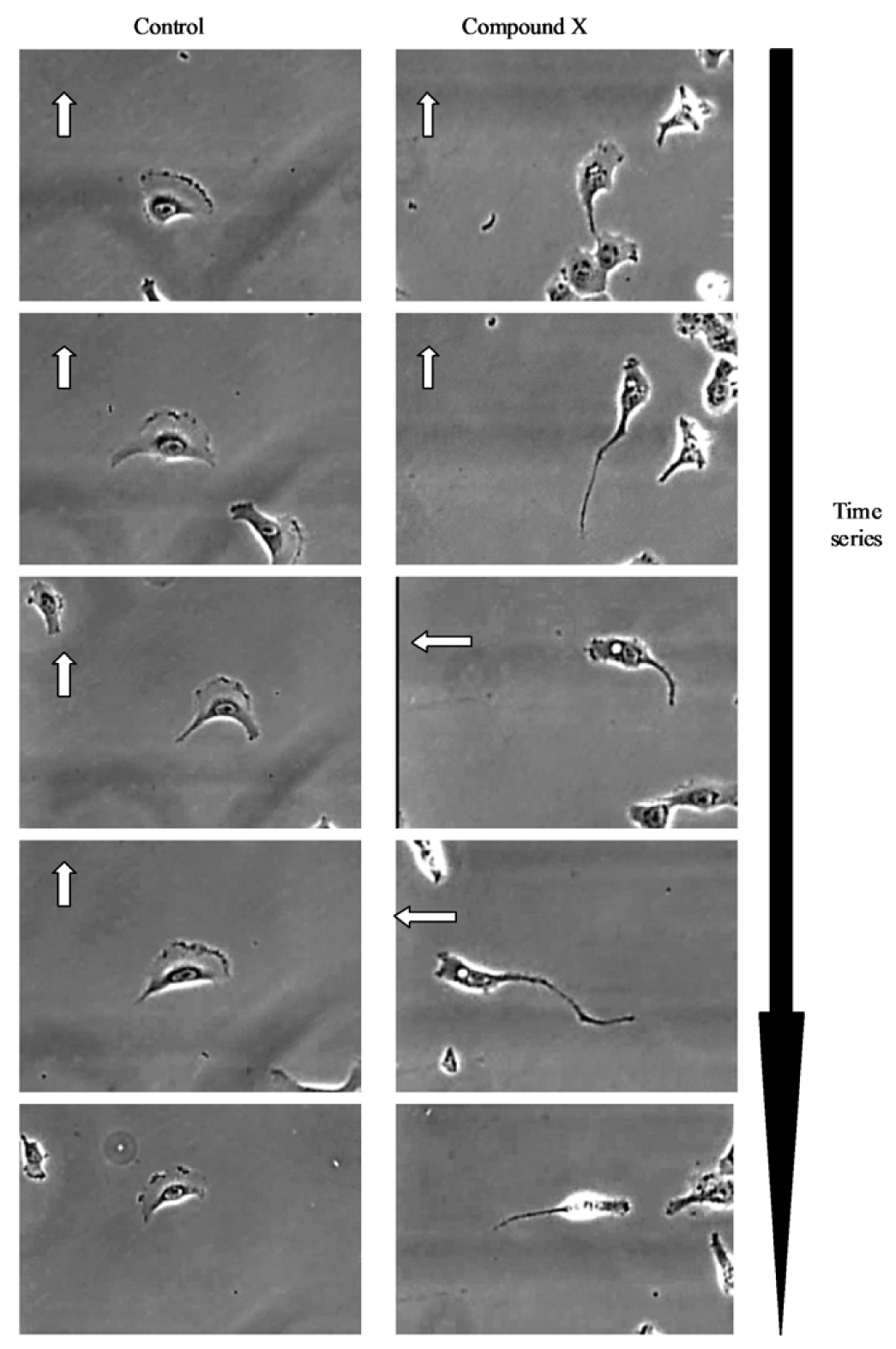
Further visual inspection and detailed analysis of kinetic responses reveal additional information upon cellular physiology. For example, long-term monitoring of cell migration with the IncuCyte™, clearly distinguishes protrusion of the leading edge, lamellipodia dynamics and uropod (rear) retraction (Figure 2). Such analysis of dynamic cellular events provides insightful mechanistic information that can assist with defining drug or target mechanism-of-action studies.
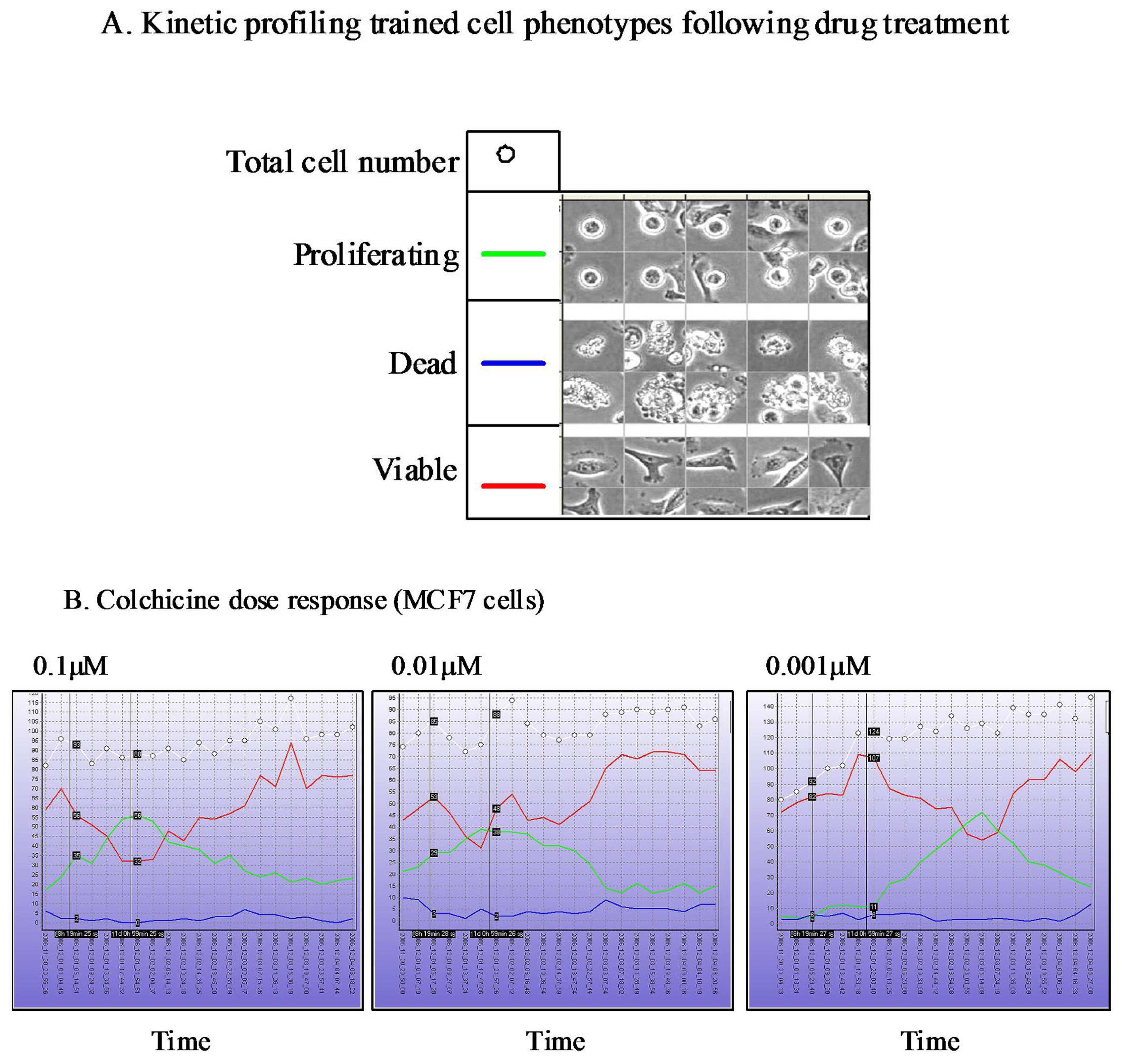
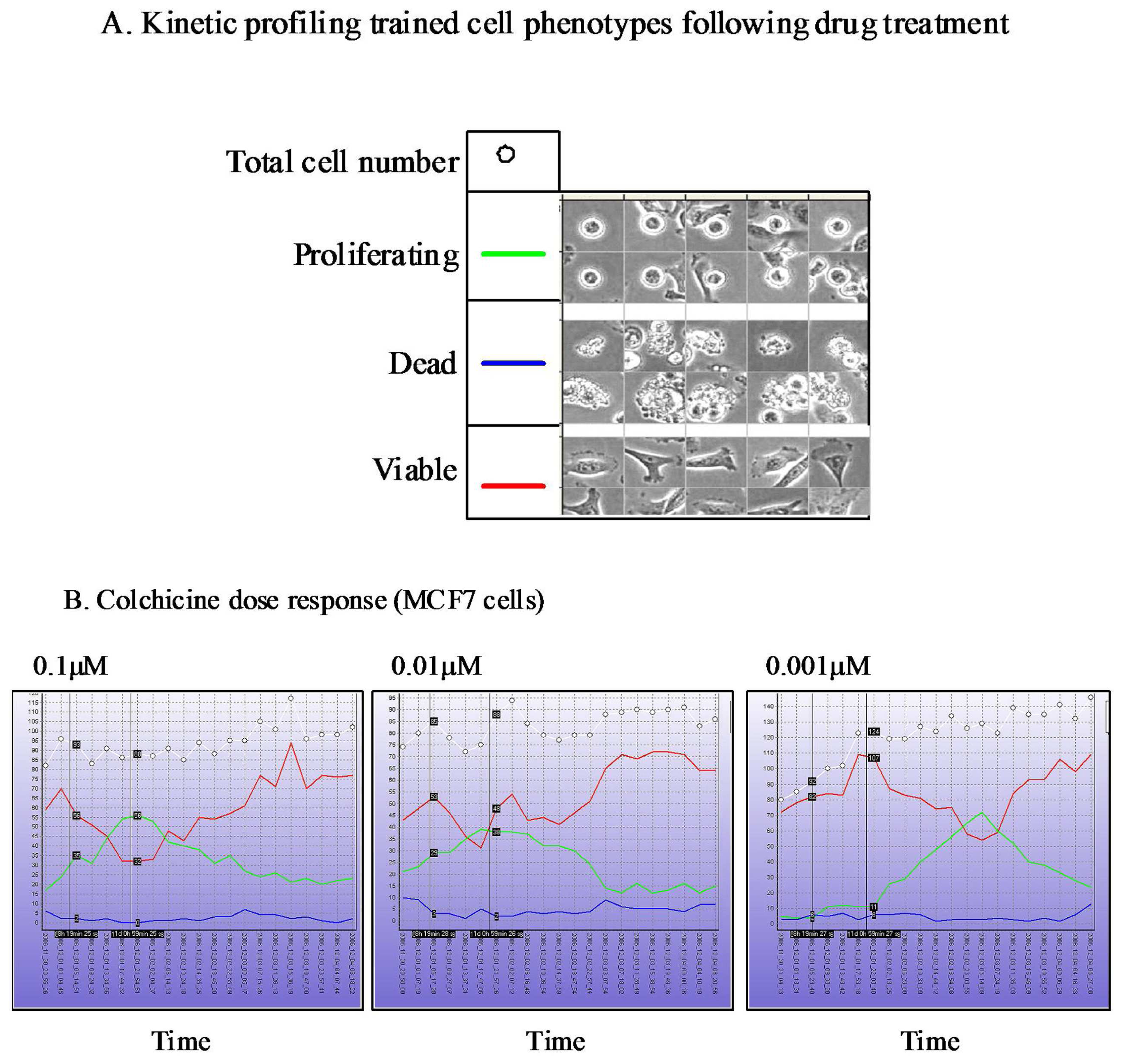
The Cell-IQ™ represents a fully integrated incubator, phase contrast and fluorescent image acquisition system linked to image analysis software based upon “machine vision” artificial intelligence approaches. The Cell-IQ™ acquisition software provides a flexible interface enabling fully automated or user defined image acquisition across 2D and 3D cell culture systems. The use of machine vision based training provides a flexible approach to monitoring proportions of distinct cellular phenotypes within complex samples such as co-culture models. In addition to classifying phenotypes other processes such as cell attachment, migration velocity, migration direction, neurite outgrowth, vesicle formation, angiogenesis and stem cell differentiation have all been recorded on the Cell-IQ™ platform. The Cell-IQ™ system enables robust kinetic profiling of phenotypic response following drug treatment including transient and long-term events (Figure 3). The temporal profile of phenotypic response and transient events often vary between cell type, drugs and drug doses (Figure 3). Thus, kinetic profiling provides robust phenotypic analysis of drug response and facilitates the selection of the most appropriate timepoints for standard high-content endpoint screens.
The Nikon Biostation CT™ platform represents the first multi-objective fluorescent and phase contrast microscope integrated with automated plate handling robotics all within a cell-culture incubator. Such advances in kinetic imaging platforms have the potential to significantly increase the throughput and scope of live-cell studies by enabling more extensive and robust target identification, validation and secondary screening applications.
The examples provided here highlight the capabilities for new kinetic imaging technology to accurately record adapting transient responses that may also include dynamic flux through signaling pathways or temporal/spatial oscillations that are not readily detected by standard endpoints. The additional information provided by such temporal analysis is likely to provide further insight into drug or target mode-of-action and cross-talk between distinct pathways and mechanisms. The temporal analysis of phenotypic response also provides valuable insight into pharmacodynamic properties of candidate drugs. By relating the kinetics of pharmacodynamic phenotypic response to known in vivo pharmacokinetic properties of drug candidates shall facilitate more optimal scheduling and dosing strategies for in vivo studies. Thus, such technologies may drive the development of a new class of kinetic biomarkers that can be placed earlier in the drug discovery process to ensure more successful translation towards in vivo efficacy.
3.2. Multidimensional imaging
Multiphoton confocal microscopy offers a number of advantages over conventional fluorescent microscopy methods for live cell imaging such as greater depth penetration, better Z-resolution improving 3D reconstruction, less phototoxicity and photobleaching of samples. These advances facilitate the long term analysis of cell behavior in live-cell cultures and provide more sensitive and quantitative analysis of fluorescent reporters or labeled cells within more physiological 3D culture systems, thick tissue samples or live in vivo models. Cell migration assays traditionally used for monitoring the effect of drug treatments upon cell motility include Boyden chamber type transmigration or scratch wound endpoint assays [11,12]. While such assays provide a robust quantification of the number and relative proportions of migrating cells they provide limited mechanistic information on cell motility or drug response. Live cell imaging studies have provided additional mechanistic insight into heterogeneous mechanisms of cell motility and the effect of drug response upon such mechanisms in both 2D and 3D culture systems [13,14]. Multiphoton confocal analysis has further characterized several distinct modes of tumor invasion that occur in more physiological 3D environments and categorized these as, mesenchymal, amoeboid and collective [15-17]. Mesenchymal tumor invasion is characterized by single cell locomotion associated with integrin mediated adhesion and matrix metalloproteinase (MMP)–mediated remodeling of extracellular matrix (ECM) [15]. Amoeboid invasion is characterized by a spherical, single cell, locomotion phenotype that is independent of MMP activity [18]. Collective invasion is characterized by tumor cell locomotion as a multicellular sheet associated with both integrin mediated adhesion and MMP-mediated ECM remodeling. These dynamic imaging studies shed new light on the morphological and functional characteristics of multiple tumor invasion mechanisms. Plasticity between distinct tumor invasion mechanisms enables rapid adaption of tumor invasion in response to environmental factors including pharmacological intervention. Such mechanistic adaptation may explain conflicting pharmacological responses observed between distinct tumor cell models and recorded clinical resistance to potential anti-invasive therapies such as MMP inhibitors [19]. Further studies using live cell confocal microscopy uncover the influence that stromal cell types, within the tumor microenvironment, have upon tumor invasion [20]. MMP dependent invasion of fibroblasts through 3D ECM preparations creates tracks in the ECM environment that facilitates the collective invasion of squamous cell carcinoma cells through such tracks [20]. These live-cell imaging studies, therefore, uncover a protumorigenic role for stromal cell remodeling of ECM topology to create a more permissive microenvironment for tumor invasion. Through further discovery, phenotypic characterization and quantification of distinct tumor invasion mechanisms by live-cell imaging, drug discovery researchers can begin to tailor their experimental models appropriately to specific target classes or to recapitulate multiple drug resistance mechanisms to aid the evaluation of new pharmacological interventions that counteract such adaptive responses.
4. Advanced Imaging Technologies
4.1. Photactivation, photoswitching, photobleaching and photoinactivation
Serial site-directed amino acid mutation studies have created genetically engineered fluorescent proteins that are photoswitchable from dark to bright, or from one color to another following excitation at a specific wavelength [21]. Such photoactivatable probes can be used in live cell systems to track cell movement, monitor protein diffusion dynamics, protein stability and cell-substructure function [22-28]. Fluorescence recovery after photobleaching (FRAP) offers an alternative approach for monitoring protein diffusion and dynamic intracellular processes. FRAP analysis of GFP labeled integrin subunits at sites of cell-substrate adhesion has been applied as a quantitative measurement of the dynamic turnover of integrin associated adhesions, thus providing both spatial and temporal analysis of cell adhesion mechanisms not provided by traditional adhesion assays or endpoint measurements [29].
Photo-reactive proteins can also be exploited to positively manipulate protein activity. Recently, the photo-reactive domain of phototropin was fused to a constitutively active mutant of Rac1, causing sterical hindrance and impairing its interaction with downstream effectors [30]. Upon light illumination, the photo-reactive domain unwinds and allows the un-caging of the fused protein, leading to Rac1 activation. Using this approach light-guided timing and location of protein activation can be precisely regulated at sub-cellular resolution. This method has recently been used to show light-mediated guidance and control of cells movement both in vitro and in vivo [30,31]. Similarly, adaptations of light-gated proteins to control upstream regulators of the Rho family GTPases have recently been documented to control cell protrusion and migration [32]. Chromophore assisted light inactivation (CALI) is a specific technique for locally perturbing protein function through the generation of reactive oxygen species (ROS) that destroy specific proteins following excitation of an attached fluorophore. This approach provides a precise mechanism for inactivating protein targets with improved spatial and temporal control than provided by popular methods of genetic knockout or siRNA [33]. Using GFP as a CALI sensor, optical irradiation of GFP-α-actinin but not GFP-FAK (focal adhesion kinase) resulted in a loss of actin-fiber association with integrins. These are the first studies to demonstrate that FAK is not critical for stress fiber attachment to integrin mediated cellular adhesions [33]. The application of light-mediated activation/inactivation of other proteins both spatially and temporally in complex biological systems will provide a diverse platform in which to validate targets and investigate various drug regimes under appropriate biological context.
4.2. FRET and FLIM
Forster Resonance Energy Transfer (FRET) is a technique that monitors the transfer of energy from a donor to an acceptor fluorophore when donor-acceptor fluorescent pairs come into close proximity. FRET is performed following specific excitation of the donor flurorophore, and subsequent analysis of the fluorescent emission properties of the acceptor fluorphore. Alternatively, a change in the fluorescent decay rate of the donor can be used to measure FRET and changes in the fluorescent lifetime of the donor can be recorded by Fluorescent Lifetime Imaging Microscopy (FLIM) methods. The exponential decay rate of the fluorophore signal, rather than its intensity, is used to create the images in FLIM and can provide a more robust FRET readout due to less scattering of light or other artifacts associated with acceptor excitation/emission. Each fluorophore has unique decay rates, thus FLIM can also be used to distinguish between fluorophores in a multiplex assay. The exponential decay of fluorophores or autofluorescent biological structures can be altered by physiological events and thus label or label-free FLIM may provide an indirect readout of physiological modifications.
Both FRET and FLIM have been applied to monitor, protein-protein interactions, conformational changes in tertiary protein structure or proteolytic cleavage events in live cells [34]. Unlike ratiometric FRET, FLIM is independent of the donor concentration and therefore insensitive to bleaching. Specific FRET biosensors have been designed to monitor, in live cells, the activity of a number of distinct kinase (Src, FAK, EGFR), protease targets (calpain) [35-37] and intracellular signaling of calcium [38,39] amongst others [40]. The most common method used for quantification of FLIM reporters is Time Correlated Single Photon Counting (TCSPC). However, the application of longitudinal TCSPC FLIM to live cell applications is limited by long acquisition times particularly across large fields of view or multiple Z-planes. Thus, multiple acquisition of TCSPC FLIM in live cell systems is associated with a high risk of photobleaching and photodamage. A new innovation in FLIM technology is the development of, wide-field, time-gated, imaging using a gated optical intensifier (GOI) combined with a Nipkow disc confocal microscope that significantly reduces the time for FLIM data acquisition to 10 frames per second [41]. These advances have led to the development of high-speed FLIM systems suitable for high-content analysis of FLIM reporters in live cell and 3D model systems across multiple samples [42]. High-speed FLIM has been applied to monitor the interaction of Ras protein with Raf binding partners, a key event in the RAS-MAPK signaling cascade, in live-cell specimens in a format suitable for chemical or molecular screening [42]. Furthermore, such approaches have permitted a multiplexed FRET readout of the spatial and temporal resolution of both Ras activation and calcium flux in a single live cell assay [43]. Similarly, the simultaneous investigation of intensity based FRET probes within the same cells has recently been used to dissect the synchronized activity of individual RhoGTPases during coordinated cell migration [44]. Frequency domain based-FLIM has also been adapted to allow for fast FLIM in a 96-well high-throughput format for the in situ identification of tyrosine phosphorylation networks downstream of EGFR signaling [45]. The potential adaptation of these techniques for the simultaneous analysis of different probes within the same cell in a high throughput setting will enable the precise temporal and spatial monitoring of target activity and associated downstream or compensatory signaling pathways in response to a variety of drug screening regimes.
Lifetime imaging has recently been employed to compare the intrinsic lifetime of ECM proteins within spatially distinct regions of human tissue, such as the core or the periphery of a tumor [46]. Furthermore, this technique has been applied to evaluate invasive and non-invasive stromal tissue in fixed or freshly excised human cancer samples [47]. Lifetime imaging in this manner can provide vital intrinsic, contextual, and local environmental information on levels of hypoxia, NADH, the constitution of ECM components, and the overall metabolic status at specific areas within the tumor, such as detecting necrotic region within the tumor. This application of lifetime imaging to provide a histological profile which can document changes in the local tissue micro-environment upon therapeutic intervention will be of use in the assessment of drug delivery. Recently, lifetime imaging has been utilized in human tissue microarray (TMA) studies to understand not only the tumor status but the environment in which different tumor types prosper [48]. FLIM has also been used to obtain additional contrast from cells labeled with nuclear dyes (Figure 4) and properties of such dyes have been used to detect apoptosis [49] and identify changes in DNA/RNA content for the study of cellular proliferation, differentiation and cell cycle [50,51]. Lipid-targeting fluorescent probes (e.g. di-4-ANEPPDHQ) have also been used for FLIM applications in live cells to probe lipid order in the cellular membrane [42,52].
4.3. Fluorescent protein complementation
An alternative approach for monitoring protein-protein interactions dynamically in live cells is through the application of bimolecular fluorescent complementation (BiFC) assays [53]. BiFC monitors protein-protein interactions by expressing a split fragment of a fluorescent reporter protein on each individual protein binding partner. When the protein partners interact the split fluorescent reporter proteins complement together forming a functional fluorescent molecule that acts as a positive reporter for the protein-protein interaction. Engineered panels of cell lines expressing multiple fluorescent protein complementation pairs enables the dynamic profiling of multiple pathways in live cells [54].
The methods described above exploit the unique spectral properties of fluorescent molecules combined with the spatial and temporal resolution provided by microscopic imaging to enable detailed kinetic and spatial analysis of pathway events and cell physiology in live cell systems. These approaches provide additional insight and quantitative analysis of dynamic transient events that are not obtained from fixed endpoint studies. Specific applications of live cell imaging demonstrating added value over fixed endpoint studies include, cell migration, cell-cycle progression, endocytosis, autophagy, subcellular translocation, cell-cell interactions, intracellular calcium fluxes, oscillations in biochemical signaling and turnover rates of cell adhesions [55-58]. Extension of such live-cell analysis approaches to in vitro and in vivo preclinical drug discovery shall enable a more comprehensive temporal evaluation of drug response in complex biological systems. Integration of fluorescent reporter molecules and spectroscopic techniques with dedicated in vitro and in vivo live optical imaging systems shall further enhance the application of live-cell imaging into preclinical drug discovery.
5. Live Cell Imaging In Vivo
Complimentary advances in advanced imaging technologies, near-infrared fluorescent NIRF imaging reagents, intravital microscopic methods and dedicated in vivo optical imaging systems have driven the application of fluorescent imaging into live animal models and fresh tissue specimens [59-61]. The development and commercial availability of a variety of near-infrared fluorophores and nanoparticles optimized for in vivo application (included in Table 1) has stimulated the development and adoption of optical in vivo imaging for preclinical drug discovery. Longitudinal in vivo imaging studies in live animals provide more detailed information from fewer animals at significant cost savings compared with traditional preclinical methods thus, representing an attractive approach for preclinical drug evaluation studies.
5.1. Non invasive in vivo imaging
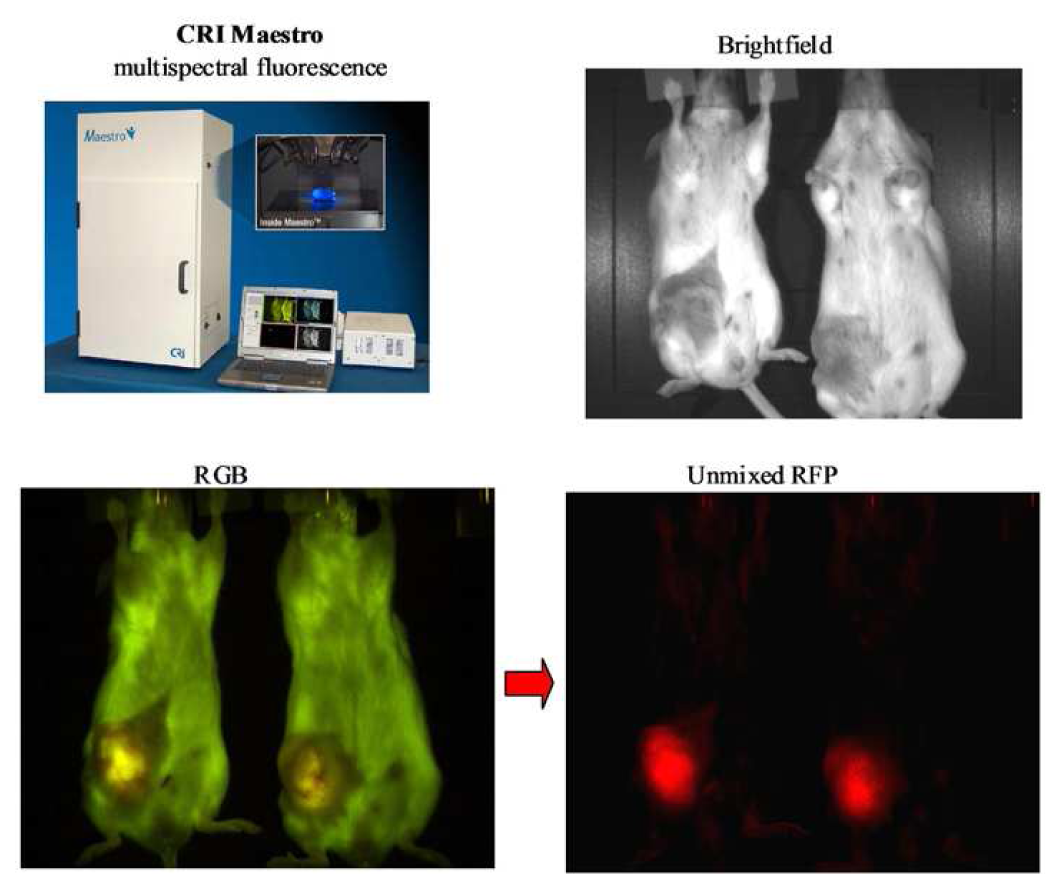
Recent advances in in vivo fluorescent imaging technologies include; multispectral imaging, tomographic reconstruction and high resolution intravital multiphoton confocal microscopy. Specific examples of dedicated fluorescent in vivo imaging systems offering non-invasive analysis of fluorescent reporters include the Maestro™ system from Cambridge Research & Instrumentation, Inc. (CRi). The Maestro™ platform incorporates a liquid crystal tunable filter and multispectral unmixing software to facilitate subtraction of background autofluorescence and multiplexing of multiple overlapping fluorophores in vivo. The increase in signal to noise associated with spectral unmixing can increase the sensitivity of analyzing distinct fluorescent signals in vivo when compared with standard monochrome reflectance imaging approaches [62] (Figure 5). The recent development of the Maestro dynamic™ system further facilitates the acquisition of temporal multispectral data enabling quantitative assessment of the biodistribution and pharmacokinetic properties of multiple probes in a sequential manner. Advances in the development of more sensitive charge coupled device (CCD) cameras linked to optimized photon amplification and counting technology such as that exemplified by the Photon Imager from Biospace Lab offer maximal sensitivity for both bioluminescent and fluorescent in vivo imaging. The intensified CCD technology within the Photon Imager amplifies all light emitted from the specimen by a sequential process of converting photons to electrons, electron amplification and conversion back to photons by a phosphorescent screen. As a direct result of this process the CCD camera in the Photon Imager is able to record and forward the optical signal emitted from in vivo samples at a rapid constant frame rate of 43 Hz facilitating real-time analysis of optical reporters. A key consideration for the detection and quantification of optical probes in vivo or in thick tissue samples is the scattering of light by biological tissue limiting quantitative assessment of signal strength, size and depth. Advances in fluorescent molecular tomography systems such as the FMT2500 LX, commercialized initially by VisEn Medical and subsequently by Perkin Elmer, provides a transmitted light excitation and emission configuration and associated software that compensates for the scattering of light through tissue. The FMT reconstruction software provides a more accurate assessment of the light emitting signal including robust quantification of size and depth of fluorescent reporters at any position in a mouse model [63]. The Ivis® spectrum and Ivis® Kinetic instruments from Caliper represent fully integrated non-invasive optical devices that facilitate both non invasive bioluminescent and fluorescent imaging in small animal models. The Ivis® systems are highly versatile platforms offering both spectral unmixing and single view 3D tomographic capabilities. The added sensitivity provided by a highly sensitive electron multiplying charge coupled device (EMCCD) camera on the Ivis®Kinetic system enables acquisition of bioluminescent or fluorescent reporter signals in milliseconds facilitating real-time analysis of biologically relevant events recorded by optical biosensors. The development and application of non-invasive in vivo optical imaging systems described above facilitate the longitudinal study of physiological responses, pharmacokinetics and pharmacodynamics in preclinical models without the need for sacrificing the animal. However, the application of non-invasive imaging is limited by the spatial resolution of images obtained and the availability of optimized probe sets.
5.2. High resolution intravital imaging
Advances in intravital confocal microscopy that provide cellular and subcellular resolution of biological events in vivo, more akin to in vitro high-content studies, provide a more adaptable platform for the development of live-cell in vivo imaging applications [61]. A number of recent technical developments have facilitated high resolution confocal microscopy in live in vivo models. The design of new lens configurations such as the microprobe lenses supplied by Olympus and miniaturized fiber optic fluorescent microscopic endoscopic (FME) devices facilitate physical access and functional analysis of deep tissue sites [64]. Such technology has been further developed to enable multidimensional fluorescent imaging in vivo with the introduction of fluorescent lifetime measurement capability on the Cellvizio endomicroscope platform [65].
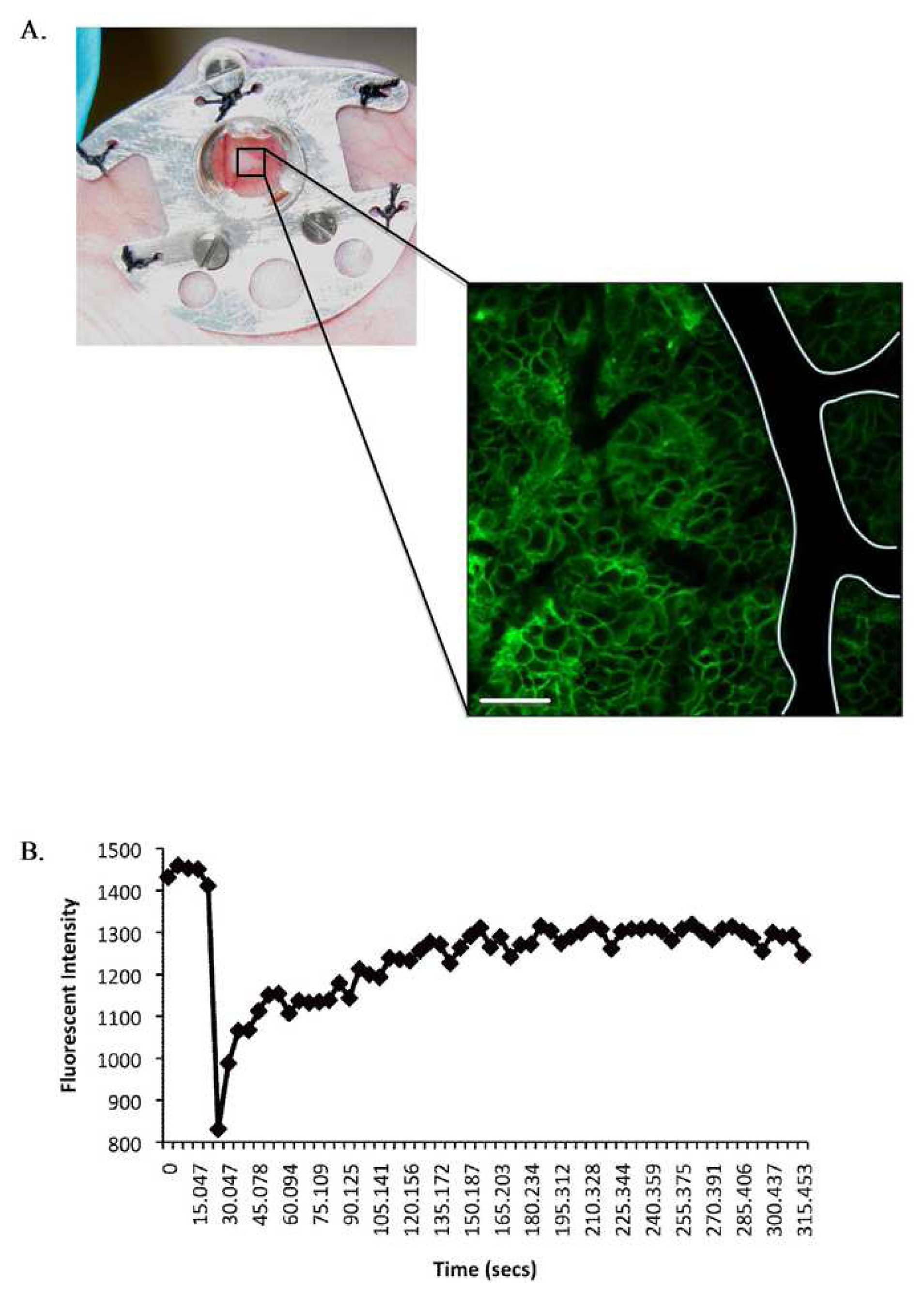
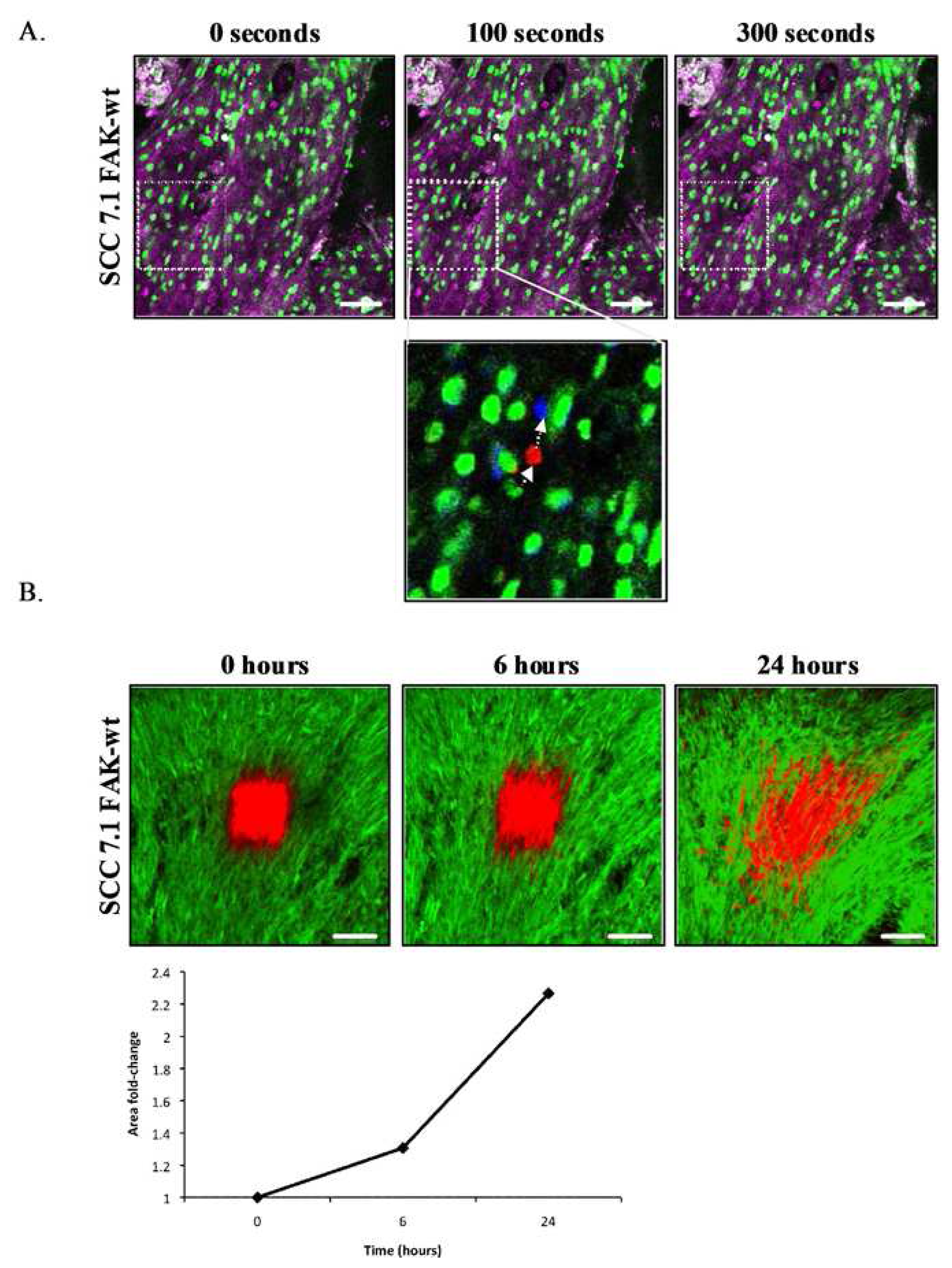
Implantation of tissue window devices such as optically clear glass coverslips in the skin enable long-term repeated high-resolution in vivo imaging without the elicitation of artifactual inflammatory responses associated with surgical trauma at time of image acquisition (Figure 6A). The design and application of tissue window devices for live in vivo imaging represents an increasing trend as demonstrated by a number of recent publications. Several studies describe the design of tissue window devices tailored to specific tissue sites under examination, including cranial windows [66], mammary windows [23] and dorsal skinfold windows [26,67]. Tissue window devices offer a number of advantages over alternative surgical exposure methods including the so called “skin flap” method including, sample stability, maintenance of tumor environment and repeated long term imaging in conjunction with recoverable anesthesia. Furthermore, in our experience the mechanical stability offered by the use of dorsal skinfold windows is of significant benefit when acquiring photobleaching/photoactivation time series. Two important considerations in the application of tissue window devices are the surgical methods of implantation to ensure minimal tissue damage and proper sterilization prior to use including anti-bacterial and anti-fungal treatment. Further optimization of the design and commercial availability of more cost effective and improved tissue window devices in association with approved surgical implantation protocols will provide a significant stimulus to the field of “live” in vivo imaging. Specific advantages of applying multiphoton confocal approaches together with intravital microscopy include, enhanced spatial resolution, and reduced image degradation due to light scattering. Greater depth penetration through tissue, improved optical efficiency, better z-registration for 3D reconstruction and less photodamage and phototoxicity enabling longer acquisition time and repeated imaging in live specimens. Multiphoton imaging can also provide additional detail regarding interactions with the surrounding extracellular tissue microenvironment provided by second harmonic signal generation which highlights non-centrosymmetric protein structures such as collagen and elastin.
Specialist microscopy systems that are optimized to facilitate the high resolution imaging of live animals are now reaching maturity. One example is the multiphoton multi-point scanning TriM-scope from LaVision Biotec. The design rational behind the system is to enhance the acquisition of optically sectioned imaging of thick samples at depths exceeding those possible via standard confocal microscopes. The TriM-scope achieves this by enabling customization of multiple imaging configurations tailored to the sample and experiment goals. This flexibility enables the scanner unit to couple into either an upright or inverted microscope body, accept the free space coupling of multiple excitation sources and multi-point beam scanning and detector types and locations optimized to the specimen being imaged.
The TriM-scope has two light input ports and the type of sources used are typically in the pulsed titanium sapphire (Ti:Sapphire) or optical parametric oscillators (OPO) class of laser. OPO cavities are pumped by the IR output from a Ti:Sapphire laser and which, through the choice of optical crystal, can produce excitation source light up to 1,400 nm. The generation of far-red shifted excitation has the obvious advantage of reduced scattering and absorption while passing through dense tissue, but also enables excitation of fluorescent probes in the NIR resulting in fluorescence that also suffers less scattering and absorption than the typically used GFPs. In addition to optimizing the delivery of excitation light, the TriM-scope design optimizes the detection of generated light through the placement of sensitive photo-multiplier tube (PMT) detectors as close to the sample as possible. By placing the PMT detectors directly behind the objective in both epi- or trans-illumination geometry the optical emission path length is significantly reduced when compared with standard confocal microscopes, thereby, maximizing sensitivity. However, if the experimental priority is the rate of acquisition, for example kinetic monitoring of drug responses, instead of depth penetration or sensitivity, then the option of multiplexing the excitation beam into a maximum of 64 separate beams allows the generation of multiple emission spots which can be imaged simultaneously using a CCD camera. Beam-splitting reduces the time to image a region of interest compared to imaging with a single beam, however, this approach is associated with a reduction in the power per excitation spot as it is distributed between the generated spot pattern, meaning that scattering will have a more apparent effect.
Research in several disease areas have benefited significantly from intravital confocal microscopy of preclinical models, most notably tumor biology. Pioneering intravital microscopy studies on tumor invasion in vivo have provided novel mechanistic insights with new pro-invasive roles being assigned to inflammatory macrophages and collagen fibres [68-70]. Recent studies exploiting the unique photoactivation and photobleaching properties of fluorescent probes have provided further mechanistic insight into specific biological mechanisms of disease and therapeutic response [26,27]. The application of optical windows in conjunction with localized photobleaching of GFP-labeled E-cadherin expressed at adherens junctions in engineered tumor cells has been used to monitor E-cadherin dynamics both in vitro and in vivo [27] (Figure 6). E-cadherin represents a family of transmembrane cell adhesion molecules that bind to the E-cadherin molecules on adjacent cells to mediate cell-cell adhesion. Thus, dynamics play a critical role in regulating inter-cellular adhesion between epithelial cells and many epithelial derived tumors. Experimental validation demonstrates that quantification of E-cadherin mobility into photobleached areas provides a preclinical measure of E-cadherin function and cell adhesion strength. E-cadherin mobility recorded, following exponential curve fitting, as the half-time of recovery provides a rapid and robust quantitative measure of E-cadherin function and inter-cellular adhesion in vivo [27] (Figure 6B). Photoactivation of a caged (non-fluorescent) plasma membrane targeting probe provides quantitative assessment of protein diffusion at the plasma membrane and in parallel with photobleaching can be used to quantify the proportion of the immobile fraction of protein stabilized at cell-cell junctions [27]. These approaches have been used to confirm the mechanism-of-action of small molecule kinase inhibitors [26,71].
The instantaneous readouts provided by such intravital imaging methods are in stark contrast to long-term metastasis models that can take up to several months to assess tumor invasion. Rapid image based evaluation of drug responses directly within the tumors of live animals facilitate more accurate scheduling and dosing, require fewer animals and accelerates the evaluation of preclinical response enabling the chemical optimization of lead compounds based on quantifiable in vivo biological endpoints. Interestingly, the dynamics of E-cadherin-GFP and plasma membrane diffusion rates in tumor cells implanted in vivo differ significantly between identical cell clones cultured on in vitro 2D culture substrates [27]. In addition, the observed effect of a licensed kinase inhibitor drug, dasatinib upon increasing the immobile fraction of E-cadherin in vivo was not observed when the cells where cultured on standard 2D plastic substrates. Critically, these studies demonstrate that 2D in vitro culture conditions do not accurately recapitulate the in vivo pharmacological mechanism-of-action of dasatinib and possibly other drugs, suggesting that 2D in vitro cell based models commonly used in high-content biology poorly predict in vivo drug response. The application of precise image-based monitoring of protein dynamics and physiological responses in vivo and in vitro may help calibrate the predictivity of in vitro model systems and ensure development of more predictive in vitro high content assays.
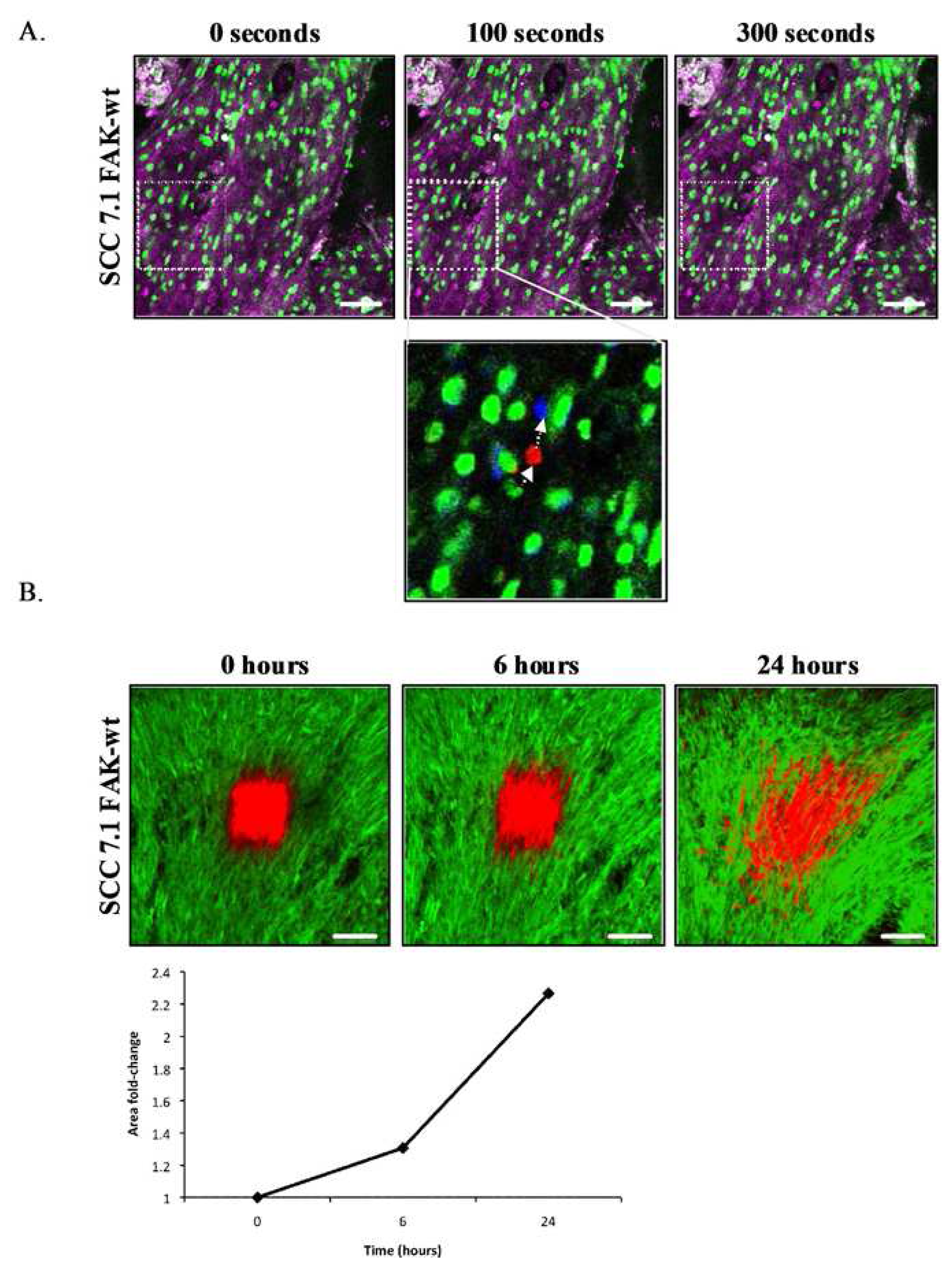
Non-invasive, repeated imaging of single cells expressing photoswitchable probes in real-time has recently been used to examine breast cancer motility and intravasation [23]. This approach has facilitated in the assessment of therapeutic intervention on other tumor types, including the brain, and has also been utilized to monitor the cross-linking and quantity of surrounding ECM components at different stages of drug treatment within the same animal [24,25,72]. Cell tracking algorithms have commonly been used to provide quantitative analysis of cell migration in vivo by following fluorescent labeled cells (Figure 7A), however the statistical robustness of such methods are limited by the low number of events recorded. Simultaneous photoactivation of a population of Dendra2 expressing cells followed by temporal analysis of distribution patterns of labeled tumor cell populations provides a more robust quantitative assessment of in vivo tumor migration [71] (Figure 7B).
The application of specific fluorescent reporters of protein activity in conjunction with intravital microscopy enables specific pathway analysis in vivo with high spatial and temporal dynamics. Recent studies using a Smad2-GFP fusion reporter that translocates to the nucleus upon TGFb signaling was used to monitor TGFb mediated signaling in migrating Mtln3E tumor cells transplanted in vivo [73]. Time-lapse analysis reveals TGFb signaling activity and smad2-GFP nuclear localization in single moving cells, whereas, smad2 reporter remained cytoplasmic in cells invading in a collective manner or in non-motile cells [73]. This data implies that reversible TGFb signaling can regulate the mechanism of breast cancer cell invasion in vivo and further suggests transient TGFb signaling is an important determinant of tumor metastasis. Such transient events would be difficult to ascertain from fixed endpoint studies which can often lead to conflicting hypotheses in the absence of temporal information.
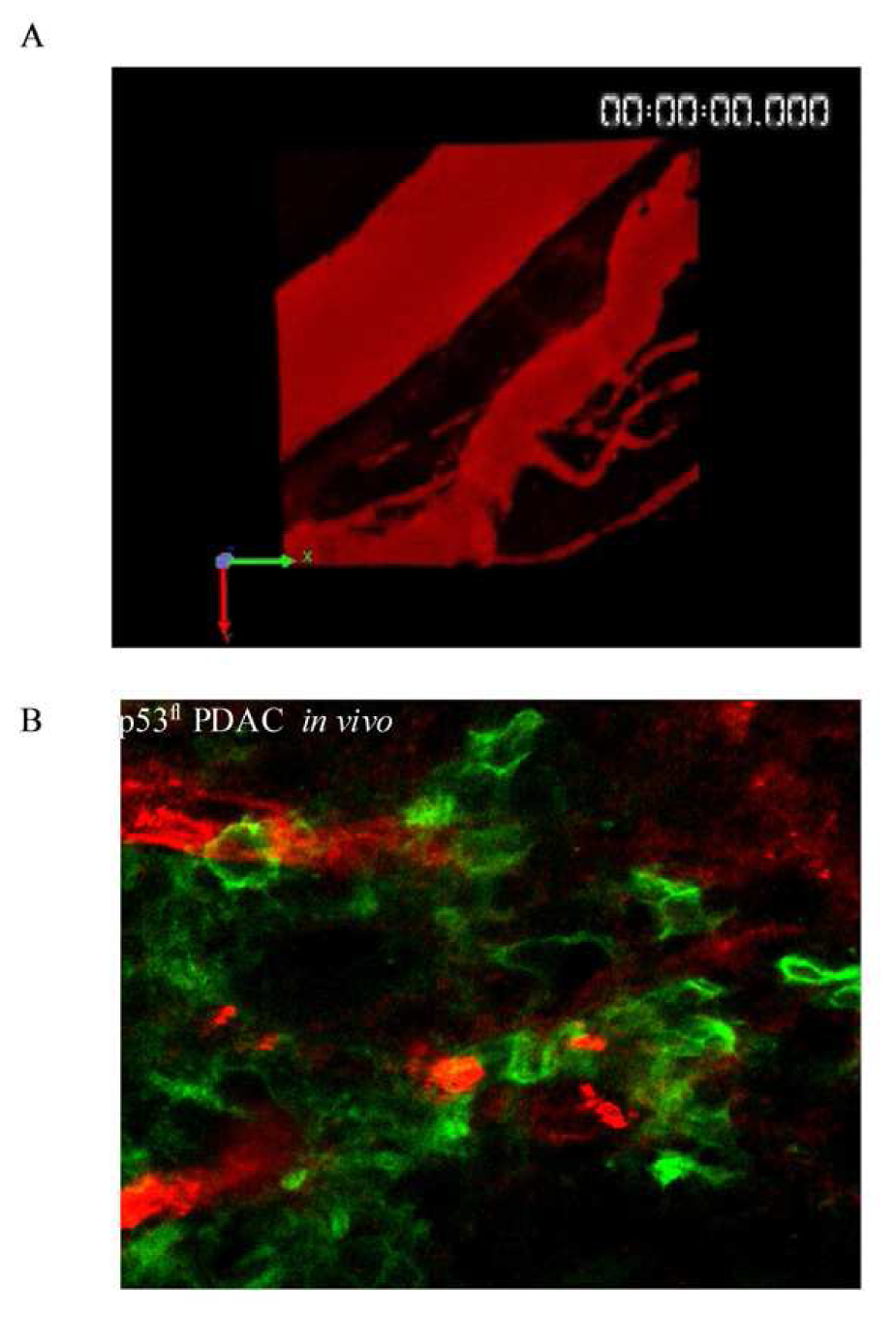
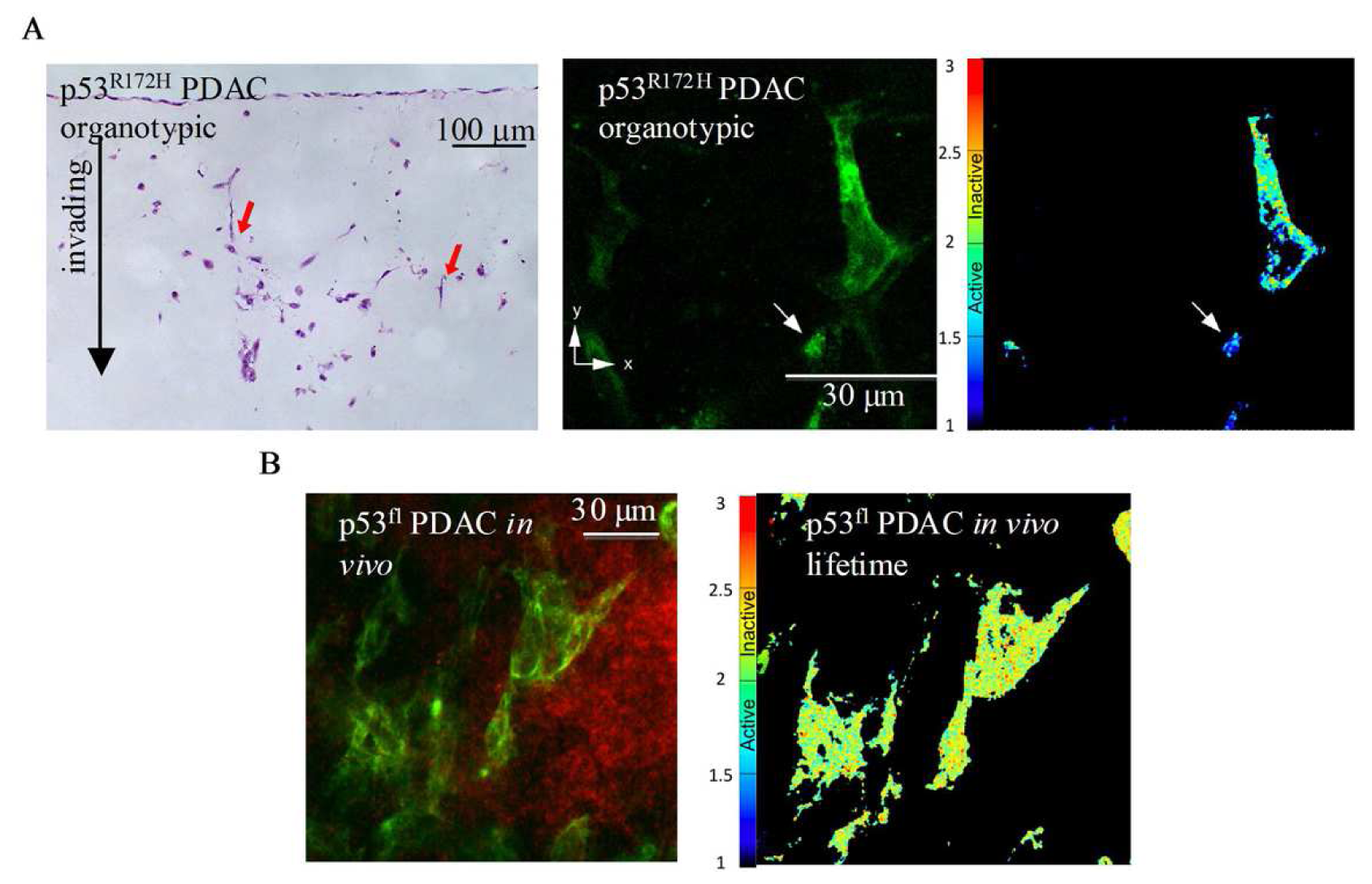
A recent study using multi-photon based FLIM-FRET demonstrated for the first time the capacity to apply FLIM to detect sub-cellular activation of protein species upon therapeutic intervention in live animals [74]. These studies demonstrated the presence of a distinct fraction of RhoA protein activity at the poles of invasive pancreactic ductal adenocarcinoma (PDAC) cells expressing mutant p53R172H protein [74]. Figure 8A displays images from organotypic cultures showing distinct regions of RhoA activity at the front of invading cells (Figure 8A middle and right panels). The preliminary finding that RhoA was spatially regulated during invasion was achieved using complimentary organotypic 3D matrices involving co-culture of pancreatic tumor cells on top of a fibrillar collagen I gel and embedded human fibroblasts (Figure 8A, left hand panel, H&E). This allowed the 3D manipulation and fluorescent lifetime imaging of RhoA activity during invasion to be investigated in a complex in vitro setting prior to in vivo examination of this biological process (see Figure 8A, middle and right hand panel). Importantly, this pool of RhoA activity at the front and rear of cells correlates with invasive potential in vivo and is absent in p53fl null PDAC cells in vivo (Figure 8B, [74]). Such detailed spatial analysis of protein activity within individual cells provided by in vivo FLIM enables the quantification of dynamic molecular events operating under precise subcellular control at a single cell level and thus intractable to standard biochemical techniques. The application of such technologies as drug response biomarkers or surrogate markers of drug activity provide precise quantification of biochemical signals and pharmacological response in vivo. The applications of these technologies display early promise as a new type of kinetic pharmacodynamic biomarker for preclinical drug evaluation studies. Analysis of kinetic biomarkers using a variety of distinct technologies including live cell imaging offers several advantages over static markers such as more sensitive quantification of flux through biochemical pathways and dynamic phenotypic response in integrated living systems [75]. Application of such methods to high quality preclinical models of disease, including genetically engineered humanized mouse models that utilize Cre-lox recombination technology to recapitulate the genetics and pathophysiology of human disease and incorporate fluorescent reporters, further enhance preclinical evaluation under appropriate biological context [76-79].
6. Biosensors
The discovery and application of natural fluorescent proteins, such as green fluorescent protein (GFP), as functional biosensors of protein activity and cellular contrast agents has revolutionized the study of protein function, location and cell activity in intact cells and living organisms [80,81]. The development of engineered cell lines stably incorporating fluorescent protein reporter molecules has contributed to the increased throughput and application of both fixed endpoint and live cell high content analysis screens in drug discovery [82-84].
Recent developments in the optimization of optical probe design specifically tailored towards live cell imaging applications are advancing both the variety of applications and quality of experimental investigation. Such advances include genetic mutagenesis of fluorescent proteins to give rise to distinct spectral properties facilitating multiplexing or specialized spectroscopic techniques such as photoactivation or photoswitching [21,28,85]. The discovery and development of more stable monomeric red and far-red fluorescent proteins minimizes perturbation of protein structure and function in living systems while increasing the penetration of fluorescent signals through deep tissue [85-87]. Chemical optimization of fluorescent dyes based on organic small molecules has produced fluorescent probe conjugates with more efficient spectral properties for live cell imaging applications [88-90]. Further chemical design of peptide scaffolds that act as substrates for specific enzymes and which incorporate fluorescent reporters of enzyme activity have further expanded the application of image-based biosensors in live cell and live in vivo models [91-93]. Advances in nanotechnology have created light emitting nanoparticles, such as quantum dots that possess superior spectral properties and greater flexibility as functional reporters [94]. Quantum dots are small nanocrystals which exhibit high extinction coefficients and quantum yields that make them brighter and more photostable than fluorescent proteins or typical fluorescent dyes. The broad excitation and tunable emission properties of quantum dots enables the design of nanoparticles with broad excitation but narrow emission properties within the near infrared fluorescent (NIRF) window of optimal tissue penetration. High photostability, superior brightness, flexible NIRF emission wavelengths and thus increased sensitivity exhibited by quantum dots are ideal properties for deep tissue imaging in vivo and long term sequential imaging in live specimens [95]. Figure 9B demonstrates GFP labeled pancreatic ductal adenocarcinoma (PDAC) cells in vivo (green) with bloodvessels labeled by intravenous injection of quantum dots in red, thus, providing contextual information and orientation to the in vivo location of tumor cells (Figure 9).
The advances in fluorescent probe design described above provide a number of advantages for long term study in live-cell in vitro, ex vivo or in vivo models, such as, enhanced tissue penetration, reduced spectral overlap with tissue autofluorescence, reduced risks of phototoxicity, photobleaching and perturbation upon protein function. The commercial availability of expanding portfolios of optical imaging probes (Table 1) has contributed to a dramatic increase in the number of live cell imaging investigations that can be routinely performed. Such advances in probe design have been complemented by advances in multiphoton confocal microscopy and automated kinetic imaging technology for visualizing fluorescent reporters in live systems. Thus, the mutual benefits of fluorescent probe design and platform technologies for detecting fluorescent reporters are synergistic and have been instrumental in driving the most significant advances in the field of optical imaging over the past decade.
7. Conclusions
The latest advances in live cell imaging technologies and functional fluorescent reporters have stimulated the development of a number of innovative live-cell imaging techniques providing detailed temporal profiling of cell behavior and dynamic molecular events in complex in vitro and in vivo model systems. High resolution in vitro and intravital in vivo live imaging applications are heralding a new era of “high-definition” drug response profiling. Profiling drug candidates across dynamic in vitro and in vivo live imaging assays can provide unique insights into therapeutic mode-of-action and robust quantification of transient responses. Fluorescent based biosensors of cell phenotype and target activity in vivo can act as early surrogate proof-of-mechanism or proof-of-principal kinetic biomarkers, thus facilitating precise pharmacodynamic and efficacy studies directly within diseased tissue in live animals. The precise temporal and spatial information on cellular and tissue phenotypes provided by live imaging approaches provides additional biological context and refined temporal information for identifying genomic or proteomic profiles of pathophysiology and or drug response. Live cell imaging applications in drug discovery are largely target-agnostic and encompass large areas of biological space and thus ideal for opportunistic drug reprofiling or drug combination studies. Live cell imaging is also an ideal companion technology for leveraging additional value from genome-wide siRNA screens and emerging stem cell differentiation studies. A key future prospect of live imaging in drug discovery applications is uncovering precise and novel phenotypic information from complex in vitro, ex-vivo and in vivo systems. Thus, live-cell imaging may provide new insight into old drugs and old model systems. A critical issue for current drug discovery strategies is determining whether image-based endpoints provide greater predictive power of clinical response outcomes over standard preclinical measurements. Retrospective studies performed on approved drugs and clinical trial failures may help identify which image-based endpoints, biomarkers and experimental models are predictive of clinical outcome. It is likely that a multiparametric approach encompassing several image-based endpoints and a suite of models systems will deliver the optimal predictions of drug efficacy or at least facilitate clinical positioning, dosing strategies and thus more effective clinical trial designs.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Probe | Application | Vendor |
|---|---|---|
| Premo™ FUCCI Cell cycle sensor | Cell-cycle | Invitrogen |
| Premo™Autophagy Sensor (LC3B-RFP or GFP) | Autophagy | Invitrogen |
| Premo™Calcium Sensor | Ca2+ signaling | Invitrogen |
| CellLight™Histon2BGFP | Nuclear morphology | Invitrogen |
| CellLight™Actin/Tubulin GFP/RFP | Cytoskeletal dynamics | Invitrogen |
| CellLight™Talin GFP/RFP | Adhesion dynamics | Invitrogen |
| Human EGFR live cell fluorescent biosensor assay | EGFR signaling dynamics | Sigma |
| CompoZr cellular reporter cell lines beta-actin, tubulin alpha-1B chain, LamininB1, HMGA1 | Cytoskeletal dynamics | Sigma |
| Lysotracker & LysoSensor | Lysosomes | Invitrogen |
| TMRE | Mitochondria function | Invitrogen |
| pHrodo™Indicators | Phagocytosis & Endocytosis | Invitrogen |
| NucView | Apoptosis (caspase activity) | Biotium |
| MitoView633 | Mitochondrial function | Biotium |
| Sulforhodamine 101-annexin V apoptosis or TexasRedTM-annexinV | Apoptosis | Biotium |
| CellBrite Cytoplasmic membrane labeling kits (Neuro-DiO) | Cell Tracking | Biotium |
| DiO/DPA FRET pair | Membrane potential | Biotium |
| Cell Trace™ Violet | Cell proliferation | Invitrogen |
| MMPSense | MMP activity | PerkinElmer |
| ProSense | Cathespin B, L, S, plasmin activity | PerkinElmer |
| OsteoSense | Bone turnover | PerkinElmer |
| AnnexinVivo | Apoptosis | PerkinElmer |
| Angiosense & Angiospark(pegylated-nanoparticles) | Vascular Labeling | PerkinElmer |
| IntegriSense | Angiogenesis/tumor cell metastasis | Perkin Elmer |
| ReninSense | Renin activity | Perkin Elmer |
| Neutrophil Elastase | Neutrophil Elastase activity | Perkin Elmer |
| CatK | Cathepsin K proteinase activity | Perkin Elmer |
| CatB | Cathepsin B protease activity | Perkin Elmer |
| Qtracker quantum dots | Cell/vascular labeling | Invitrogen |
| FM 4-64 | Membrane label | Invitrogen |
| Acrivlavin | cell label/mask | Sigma |
| Carbocyanine dye DiI, DiO | Membrane label | Invitrogen |
| Fluorescent lectins | Bloodvessel endothelium | Invitrogen |
| Fluorescent dextran | Vascular labeling | Invitrogen |
Acknowledgements
The authors wish to acknowledge all of the laboratory staff and scientific researchers at the Advanced Science and Technology Laboratory, AstraZeneca, Beatson Insititute for Cancer Research and Edinburgh Cancer Research Centre for their support in conducting live imaging microscopy studies over many years. We also wish to acknowledge Professor Paul French and colleagues at Imperial College for their support in conducting in vitro FLIM studies as part of a funded UK Technology Strategy Board Award (CHBT/007/00030, EP/C54269X, in partnership with GE Healthcare, GSK, Kentech Insturments Ltd and Perkin Elmer). In addition we wish to thank the sales and support staff from the various imaging vendors cited throughout the manuscript for their support and insight into live imaging technologies.
References and Notes
- Paul, S.M.; Mytelka, D.S.; Dunwiddie, C.T.; Persinger, C.C.; Munos, B.H.; Lindborg, S.R.; Schacht, A.L. How to improve R&D productivity: The pharmaceutical industry's grand challenge. Nat. Rev. Drug Discov. 2010, 9, 203–214. [Google Scholar]
- Sams-Dodd, F. Target-based drug discovery: Is something wrong? Drug Discov. Today 2005, 10, 139–147. [Google Scholar]
- Butcher, E.C. Can cell systems biology rescue drug discovery? Nat. Rev. Drug Discov. 2005, 4, 461–467. [Google Scholar]
- Zock, J.M. Applications of high content screening in life science research. Comb. Chem. High Throughput Scr. 2009, 12, 870–876. [Google Scholar]
- Bickle, M. The beautiful cell: High-content screening in drug discovery. Anal. Bioanal. Chem. 2010, 398, 219–226. [Google Scholar]
- Bickle, M. High-content screening: A new primary screening tool? IDrugs 2008, 11, 822–826. [Google Scholar]
- Perlman, Z.E.; Slack, M.D.; Feng, Y.; Mitchison, T.J.; Wu, L.F.; Altschuler, S.J. Multidimensional drug profiling by automated microscopy. Science 2004, 306, 1194–1198. [Google Scholar]
- Tanaka, M.; Bateman, R.; Rauh, D.; Vaisberg, E.; Ramachandani, S.; Zhang, C.; Hansen, K.C.; Burlingame, A.L.; Trautman, J.K.; Shokat, K.M.; Adams, C.L. An unbiased cell morphology-based screen for new, biologically active small molecules. PLoS Biol. 2005, 3, e128. [Google Scholar]
- Young, D.W.; Bender, A.; Hoyt, J.; McWhinnie, E.; Chirn, G.W.; Tao, C.Y.; Tallarico, J.A.; Labow, M.; Jenkins, J.L.; Mitchison, T.J.; Feng, Y. Integrating high-content screening and ligand-target prediction to identify mechanism of action. Nat. Chem. Biol. 2008, 4, 59–68. [Google Scholar]
- Caie, P.D.; Walls, R.E.; Ingleston-Orme, A.; Daya, S.; Houslay, T.; Eagle, R.; Roberts, M.E.; Carragher, N.O. High-content phenotypic profiling of drug response signatures across distinct cancer cells. Mol. Cancer Ther. 2010, 9, 1913–1926. [Google Scholar]
- Hassoun, A.T.; Erdelyi, F.; Szabo, G.; Davis, M.I. A rapid screening method for population-specific neuronal motogens, substrates and associated signaling pathways. J. Neurosci. Methods 2007, 166, 178–194. [Google Scholar]
- Yarrow, J.C.; Perlman, Z.E.; Westwood, N.J.; Mitchison, T.J. A high-throughput cell migration assay using scratch wound healing, a comparison of image-based readout methods. BMC Biotechnol. 2004, 4, 21. [Google Scholar]
- Huttenlocher, A.; Palecek, S.P.; Lu, Q.; Zhang, W.; Mellgren, R.L.; Lauffenburger, D.A.; Ginsberg, M.H.; Horwitz, A.F. Regulation of cell migration by the calcium-dependent protease calpain. J. Biol. Chem. 1997, 272, 32719–32722. [Google Scholar]
- Friedl, P.; Wolf, K. Plasticity of cell migration: A multiscale tuning model. J. Cell Biol. 2010, 188, 11–19. [Google Scholar]
- Wolf, K.; Mazo, I.; Leung, H.; Engelke, K.; von Andrian, U.H.; Deryugina, E.I.; Strongin, A.Y.; Brocker, E.B.; Friedl, P. Compensation mechanism in tumor cell migration: Mesenchymal-amoeboid transition after blocking of pericellular proteolysis. J. Cell Biol. 2003, 160, 267–277. [Google Scholar]
- Sahai, E.; Marshall, C.J. Differing modes of tumour cell invasion have distinct requirements for Rho/ROCK signalling and extracellular proteolysis. Nat. Cell Biol. 2003, 5, 711–719. [Google Scholar]
- Wolf, K.; Friedl, P. Molecular mechanisms of cancer cell invasion and plasticity. Br. J. Dermatol. 2006, 154 (Suppl. 1), 11–15. [Google Scholar]
- Wyckoff, J.B.; Pinner, S.E.; Gschmeissner, S.; Condeelis, J.S.; Sahai, E. ROCK- and myosin-dependent matrix deformation enables protease-independent tumor-cell invasion in vivo. Curr. Biol. 2006, 16, 1515–1523. [Google Scholar]
- Carragher, N.O. Profiling distinct mechanisms of tumour invasion for drug discovery: Imaging adhesion, signalling and matrix turnover. Clin. Exp. Metastasis 2009, 26, 381–397. [Google Scholar]
- Gaggioli, C.; Hooper, S.; Hidalgo-Carcedo, C.; Grosse, R.; Marshall, J.F.; Harrington, K.; Sahai, E. Fibroblast-led collective invasion of carcinoma cells with differing roles for RhoGTPases in leading and following cells. Nat. Cell Biol. 2007, 9, 1392–1400. [Google Scholar]
- Lukyanov, K.A.; Chudakov, D.M.; Lukyanov, S.; Verkhusha, V.V. Innovation: Photoactivatable fluorescent proteins. Nat. Rev. Mol. Cell Biol. 2005, 6, 885–891. [Google Scholar]
- Gurskaya, N.G.; Verkhusha, V.V.; Shcheglov, A.S.; Staroverov, D.B.; Chepurnykh, T.V.; Fradkov, A.F.; Lukyanov, S.; Lukyanov, K.A. Engineering of a monomeric green-to-red photoactivatable fluorescent protein induced by blue light. Nat. Biotechnol. 2006, 24, 461–465. [Google Scholar]
- Kedrin, D.; Gligorijevic, B.; Wyckoff, J.; Verkhusha, V.V.; Condeelis, J.; Segall, J.E.; van Rheenen, J. Intravital imaging of metastatic behavior through a mammary imaging window. Nat. Methods 2008, 5, 1019–1021. [Google Scholar]
- Brown, E.; McKee, T.; diTomaso, E.; Pluen, A.; Seed, B.; Boucher, Y.; Jain, R.K. Dynamic imaging of collagen and its modulation in tumors in vivo using second-harmonic generation. Nat. Med. 2003, 9, 796–800. [Google Scholar]
- Perentes, J.Y.; McKee, T.D.; Ley, C.D.; Mathiew, H.; Dawson, M.; Padera, T.P.; Munn, L.L.; Jain, R.K.; Boucher, Y. In vivo imaging of extracellular matrix remodeling by tumor-associated fibroblasts. Nat. Methods 2009, 6, 143–145. [Google Scholar]
- Canel, M.; Serrels, A.; Anderson, K.I.; Frame, M.C.; Brunton, V.G. Use of photoactivation and photobleaching to monitor the dynamic regulation of E-cadherin at the plasma membrane. Cell Adh. Migr. 2010, 4, 491–501. [Google Scholar]
- Serrels, A.; Timpson, P.; Canel, M.; Schwarz, J.P.; Carragher, N.O.; Frame, M.C.; Brunton, V.G.; Anderson, K.I. Real-time study of E-cadherin and membrane dynamics in living animals: Implications for disease modeling and drug development. Cancer Res. 2009, 69, 2714–2719. [Google Scholar]
- Welman, A.; Serrels, A.; Brunton, V.G.; Ditzel, M.; Frame, M.C. Two-color photoactivatable probe for selective tracking of proteins and cells. J. Biol. Chem. 2010, 285, 11607–11616. [Google Scholar]
- Ballestrem, C.; Geiger, B. Application of microscope-based FRET to study molecular interactions in focal adhesions of live cells. Methods Mol. Biol. 2005, 294, 321–334. [Google Scholar]
- Wu, Y.I.; Frey, D.; Lungu, O.I.; Jaehrig, A.; Schlichting, I.; Kuhlman, B.; Hahn, K.M. A genetically encoded photoactivatable Rac controls the motility of living cells. Nature 2009, 461, 104–108. [Google Scholar]
- Wang, X.; He, L.; Wu, Y.I.; Hahn, K.M.; Montell, D.J. Light-mediated activation reveals a key role for Rac in collective guidance of cell movement in vivo. Nat. Cell Biol. 2010, 12, 591–597. [Google Scholar]
- Levskaya, A.; Weiner, O.D.; Lim, W.A.; Voigt, C.A. Spatiotemporal control of cell signalling using a light-switchable protein interaction. Nature 2009, 461, 997–1001. [Google Scholar]
- Rajfur, Z.; Roy, P.; Otey, C.; Romer, L.; Jacobson, K. Dissecting the link between stress fibres and focal adhesions by CALI with EGFP fusion proteins. Nat. Cell Biol. 2002, 4, 286–293. [Google Scholar]
- Wallrabe, H.; Periasamy, A. Imaging protein molecules using FRET and FLIM microscopy. Curr. Opin. Biotechnol. 2005, 16, 19–27. [Google Scholar]
- Sato, M.; Ozawa, T.; Inukai, K.; Asano, T.; Umezawa, Y. Fluorescent indicators for imaging protein phosphorylation in single living cells. Nat. Biotechnol. 2002, 20, 287–294. [Google Scholar]
- Ting, A.Y.; Kain, K.H.; Klemke, R.L.; Tsien, R.Y. Genetically encoded fluorescent reporters of protein tyrosine kinase activities in living cells. Proc. Natl. Acad. Sci. USA 2001, 98, 15003–15008. [Google Scholar]
- Vanderklish, P.W.; Krushel, L.A.; Holst, B.H.; Gally, J.A.; Crossin, K.L.; Edelman, G.M. Marking synaptic activity in dendritic spines with a calpain substrate exhibiting fluorescence resonance energy transfer. Proc. Natl. Acad. Sci. USA 2000, 97, 2253–2258. [Google Scholar]
- Miyawaki, A.; Llopis, J.; Heim, R.; McCaffery, J.M.; Adams, J.A.; Ikura, M.; Tsien, R.Y. Fluorescent indicators for Ca2+ based on green fluorescent proteins and calmodulin. Nature 1997, 388, 882–887. [Google Scholar]
- Mank, M.; Reiff, D.F.; Heim, N.; Friedrich, M.W.; Borst, A.; Griesbeck, O. A FRET-based calcium biosensor with fast signal kinetics and high fluorescence change. Biophys. J. 2006, 90, 1790–1796. [Google Scholar]
- Li, I.T.; Chiang, J.J.; Truong, K. FRET evidence that an isoform of caspase-7 binds but does not cleave its substrate. In Proceedings of IEEE Engineering in Medicine and Biology Society, NewYork, NY, USA, 30 August-3 September 2006.
- Grant, D.M.; McGinty, J.; McGhee, E.J.; Bunney, T.D.; Owen, D.M.; Talbot, C.B.; Zhang, W.; Kumar, S.; Munro, I.; Lanigan, P.M.; Kennedy, G.T.; Dunsby, C.; Magee, A.I.; Courtney, P.; Katan, M.; Neil, M.A.; French, P.M. High speed optically sectioned fluorescence lifetime imaging permits study of live cell signaling events. Opt. Express 2007, 15, 15656–15673. [Google Scholar]
- Talbot, C.B.; McGinty, J.; Grant, D.M.; McGhee, E.J.; Owen, D.M.; Zhang, W.; Bunney, T.D.; Munro, I.; Isherwood, B.; Eagle, R.; Hargreaves, A.; Dunsby, C.; Neil, M.A.; French, P.M. High speed unsupervised fluorescence lifetime imaging confocal multiwell plate reader for high content analysis. J. Biophotonics 2008, 1, 514–521. [Google Scholar]
- Grant, D.M.; Zhang, W.; McGhee, E.J.; Bunney, T.D.; Talbot, C.B.; Kumar, S.; Munro, I.; Dunsby, C.; Neil, M.A.; Katan, M.; French, P.M. Multiplexed FRET to image multiple signaling events in live cells. Biophys. J. 2008, 95, L69–L71. [Google Scholar]
- Machacek, M.; Hodgson, L.; Welch, C.; Elliott, H.; Pertz, O.; Nalbant, P.; Abell, A.; Johnson, G.L.; Hahn, K.M.; Danuser, G. Coordination of Rho GTPase activities during cell protrusion. Nature 2009, 461, 99–103. [Google Scholar]
- Grecco, H.E.; Roda-Navarro, P.; Girod, A.; Hou, J.; Frahm, T.; Truxius, D.C.; Pepperkok, R.; Squire, A.; Bastiaens, P.I. In situ analysis of tyrosine phosphorylation networks by FLIM on cell arrays. Nat. Methods 2010, 7, 467–472. [Google Scholar]
- Elson, D.; Requejo-Isidro, J.; Munro, I.; Reavell, F.; Siegel, J.; Suhling, K.; Tadrous, P.; Benninger, R.; Lanigan, P.; McGinty, J.; Talbot, C.; Treanor, B.; Webb, S.; Sandison, A.; Wallace, A.; Davis, D.; Lever, J.; Neil, M.; Phillips, D.; Stamp, G.; French, P. Time-domain fluorescence lifetime imaging applied to biological tissue. Photochem. Photobiol. Sci. 2004, 3, 795–801. [Google Scholar]
- Galletly, N.P.; McGinty, J.; Dunsby, C.; Teixeira, F.; Requejo-Isidro, J.; Munro, I.; Elson, D.S.; Neil, M.A.; Chu, A.C.; French, P.M.; Stamp, G.W. Fluorescence lifetime imaging distinguishes basal cell carcinoma from surrounding uninvolved skin. Br. J. Dermatol. 2008, 159, 152–161. [Google Scholar]
- Kelleher, M.T.; Fruhwirth, G.; Patel, G.; Ofo, E.; Festy, F.; Barber, P.R.; Ameer-Beg, S.M.; Vojnovic, B.; Gillett, C.; Coolen, A.; Keri, G.; Ellis, P.A.; Ng, T. The potential of optical proteomic technologies to individualize prognosis and guide rational treatment for cancer patients. Target Oncol. 2009, 4, 235–252. [Google Scholar]
- van Zandvoort, M.A.; de Grauw, C.J.; Gerritsen, H.C.; Broers, J.L.; oude Egbrink, M.G.; Ramaekers, F.C.; Slaaf, D.W. Discrimination of DNA and RNA in cells by a vital fluorescent probe: Lifetime imaging of SYTO13 in healthy and apoptotic cells. Cytometry 2002, 47, 226–235. [Google Scholar]
- Cui, H.H.; Valdez, J.G.; Steinkamp, J.A.; Crissman, H.A. Fluorescence lifetime-based discrimination and quantification of cellular DNA and RNA with phase-sensitive flow cytometry. Cytometry A 2003, 52, 46–55. [Google Scholar]
- Murata, S.; Herman, P.; Lakowicz, J.R. Texture analysis of fluorescence lifetime images of AT- and GC-rich regions in nuclei. J. Histochem. Cytochem. 2001, 49, 1443–1451. [Google Scholar]
- Owen, D.M.; Lanigan, P.M.; Dunsby, C.; Munro, I.; Grant, D.; Neil, M.A.; French, P.M.; Magee, A.I. Fluorescence lifetime imaging provides enhanced contrast when imaging the phase-sensitive dye di-4-ANEPPDHQ in model membranes and live cells. Biophys. J. 2006, 90, L80–82. [Google Scholar]
- Magliery, T.J.; Regan, L. Reassembled GFP: Detecting protein-protein interactions and protein expression patterns. Methods Biochem. Anal. 2006, 47, 391–405. [Google Scholar]
- Michnick, S.W.; MacDonald, M.L.; Westwick, J.K. Chemical genetic strategies to delineate MAP kinase signaling pathways using protein-fragment complementation assays (PCA). Methods 2006, 40, 287–293. [Google Scholar]
- Ainscow, E.K.; Rutter, G.A. Glucose-stimulated oscillations in free cytosolic ATP concentration imaged in single islet beta-cells: Evidence for a Ca2+-dependent mechanism. Diabetes 2002, 51 (Suppl. 1), S162–170. [Google Scholar]
- Webb, D.J.; Donais, K.; Whitmore, L.A.; Thomas, S.M.; Turner, C.E.; Parsons, J.T.; Horwitz, A.F. FAK-Src signalling through paxillin, ERK and MLCK regulates adhesion disassembly. Nat. Cell Biol. 2004, 6, 154–161. [Google Scholar]
- Nelson, D.E.; Ihekwaba, A.E.; Elliott, M.; Johnson, J.R.; Gibney, C.A.; Foreman, B.E.; Nelson, G.; See, V.; Horton, C.A.; Spiller, D.G.; Edwards, S.W.; McDowell, H.P.; Unitt, J.F.; Sullivan, E.; Grimley, R.; Benson, N.; Broomhead, D.; Kell, D.B.; White, M.R. Oscillations in NF-kappaB signaling control the dynamics of gene expression. Science 2004, 306, 704–708. [Google Scholar]
- Bains, M.; Heidenreich, K.A. Live-cell imaging of autophagy induction and autophagosome-lysosome fusion in primary cultured neurons. Methods Enzymol. 2009, 453, 145–158. [Google Scholar]
- Rubin, M.A.; Zerkowski, M.P.; Camp, R.L.; Kuefer, R.; Hofer, M.D.; Chinnaiyan, A.M.; Rimm, D.L. Quantitative determination of expression of the prostate cancer protein alpha-methylacyl-CoA racemase using automated quantitative analysis (AQUA): A novel paradigm for automated and continuous biomarker measurements. Am. J. Pathol. 2004, 164, 831–840. [Google Scholar]
- Weigert, R.; Sramkova, M.; Parente, L.; Amornphimoltham, P.; Masedunskas, A. Intravital microscopy: A novel tool to study cell biology in living animals. Histochem. Cell Biol. 2010, 133, 481–491. [Google Scholar]
- Beerling, E.; Ritsma, L.; Vrisekoop, N.; Derksen, P.W.; van Rheenen, J. Intravital microscopy: New insights into metastasis of tumors. J. Cell Sci. 2011, 124, 299–310. [Google Scholar]
- Levenson, R.M.; Mansfield, J.R. Multispectral imaging in biology and medicine: Slices of life. Cytometry A 2006, 69, 748–758. [Google Scholar]
- Ntziachristos, V.; Bremer, C.; Weissleder, R. Fluorescence imaging with near-infrared light: New technological advances that enable in vivo molecular imaging. Eur. Radiol. 2003, 13, 195–208. [Google Scholar]
- Bullen, A. Microscopic imaging techniques for drug discovery. Nat. Rev. Drug Discov. 2008, 7, 54–67. [Google Scholar]
- Kennedy, G.T.; Manning, H.B.; Elson, D.S.; Neil, M.A.; Stamp, G.W.; Viellerobe, B.; Lacombe, F.; Dunsby, C.; French, P.M. A fluorescence lifetime imaging scanning confocal endomicroscope. J. Biophotonics 2010, 3, 103–107. [Google Scholar]
- Brown, E.; Munn, L.L.; Fukumura, D.; Jain, R.K. In vivo imaging of tumors. Cold Spring Harb. Protoc. 2010, 2010. pdb prot5452. [Google Scholar]
- Makale, M. Intravital imaging and cell invasion. Methods Enzymol. 2007, 426, 375–401. [Google Scholar]
- Condeelis, J.; Segall, J.E. Intravital imaging of cell movement in tumours. Nat. Rev. Cancer 2003, 3, 921–930. [Google Scholar]
- Xue, C.; Wyckoff, J.; Liang, F.; Sidani, M.; Violini, S.; Tsai, K.L.; Zhang, Z.Y.; Sahai, E.; Condeelis, J.; Segall, J.E. Epidermal growth factor receptor overexpression results in increased tumor cell motility in vivo coordinately with enhanced intravasation and metastasis. Cancer Res. 2006, 66, 192–197. [Google Scholar]
- Yamaguchi, H.; Wyckoff, J.; Condeelis, J. Cell migration in tumors. Curr. Opin. Cell Biol. 2005, 17, 559–564. [Google Scholar]
- Canel, M.; Serrels, A.; Miller, D.; Timpson, P.; Serrels, B.; Frame, M.C.; Brunton, V.G. Quantitative in vivo imaging of the effects of inhibiting integrin signaling via Src and FAK on cancer cell movement: Effects on E-cadherin dynamics. Cancer Res. 2010, 70, 9413–9422. [Google Scholar]
- Canel, M.; Serrels, A.; Miller, D.; Timpson, P.; Serrels, B.; Frame, M.C.; Brunton, V.G. Quantitative in vivo imaging of the effects of inhibiting integrin signaling via Src and FAK on cancer cell movement: Effects on E-cadherin dynamics. Cancer Res. 2010, 70, 9413–9422. [Google Scholar]
- Giampieri, S.; Manning, C.; Hooper, S.; Jones, L.; Hill, C.S.; Sahai, E. Localized and reversible TGFbeta signalling switches breast cancer cells from cohesive to single cell motility. Nat. Cell Biol. 2009, 11, 1287–1296. [Google Scholar]
- Timpson, P.; McGhee, E.J.; Morton, J.P.; von Kriegsheim, A.; Schwarz, J.P.; Karim, S.A.; Doyle, B.; Quinn, J.A.; Carragher, N.O.; Edward, M.; Olson, M.F.; Frame, M.C.; Brunton, V.G.; Sansom, O.J.; Anderson, K.I. Spatial Regulation of RhoA Activity during Pancreatic Cancer Cell Invasion Driven by Mutant p53. Cancer Res. 2011, 71, 747–757. [Google Scholar]
- Turner, S.M.; Hellerstein, M.K. Emerging applications of kinetic biomarkers in preclinical and clinical drug development. Curr. Opin. Drug Discov. Devel. 2005, 8, 115–126. [Google Scholar]
- Morton, J.P.; Timpson, P.; Karim, S.A.; Ridgway, R.A.; Athineos, D.; Doyle, B.; Jamieson, N.B.; Oien, K.A.; Lowy, A.M.; Brunton, V.G.; Frame, M.C.; Evans, T.R.; Sansom, O.J. Mutant p53 drives metastasis and overcomes growth arrest/senescence in pancreatic cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 246–251. [Google Scholar]
- Ahmad, I.; Morton, J.P.; Singh, L.B.; Radulescu, S.M.; Ridgway, R.A.; Patel, S.; Woodgett, J.; Winton, D.J.; Taketo, M.M.; Wu, X.R.; Leung, H.Y.; Sansom, O.J. beta-Catenin activation synergizes with PTEN loss to cause bladder cancer formation. Oncogene 2011, 30, 178–189. [Google Scholar]
- Cole, A.M.; Ridgway, R.A.; Derkits, S.E.; Parry, L.; Barker, N.; Clevers, H.; Clarke, A.R.; Sansom, O.J. p21 loss blocks senescence following Apc loss and provokes tumourigenesis in the renal but not the intestinal epithelium. EMBO Mol. Med. 2010, 2, 472–486. [Google Scholar]
- Doyle, B.; Morton, J.P.; Delaney, D.W.; Ridgway, R.A.; Wilkins, J.A.; Sansom, O.J. p53 mutation and loss have different effects on tumourigenesis in a novel mouse model of pleomorphic rhabdomyosarcoma. J. Pathol. 2010, 222, 129–137. [Google Scholar]
- Zhang, J.; Campbell, R.E.; Ting, A.Y.; Tsien, R.Y. Creating new fluorescent probes for cell biology. Nat. Rev. Mol. Cell Biol. 2002, 3, 906–918. [Google Scholar]
- Giepmans, B.N.; Adams, S.R.; Ellisman, M.H.; Tsien, R.Y. The fluorescent toolbox for assessing protein location and function. Science 2006, 312, 217–224. [Google Scholar]
- Haney, S.A.; LaPan, P.; Pan, J.; Zhang, J. High-content screening moves to the front of the line. Drug Discov. Today 2006, 11, 889–894. [Google Scholar]
- Lundholt, B.K.; Heydorn, A.; Bjorn, S.P.; Praestegaard, M. A simple cell-based HTS assay system to screen for inhibitors of p53-Hdm2 protein-protein interactions. Assay Drug Dev. Technol. 2006, 4, 679–688. [Google Scholar]
- Heydorn, A.; Lundholt, B.K.; Praestegaard, M.; Pagliaro, L. Protein translocation assays: Key tools for accessing new biological information with high-throughput microscopy. Methods Enzymol. 2006, 414, 513–530. [Google Scholar]
- Shcherbo, D.; Merzlyak, E.M.; Chepurnykh, T.V.; Fradkov, A.F.; Ermakova, G.V.; Solovieva, E.A.; Lukyanov, K.A.; Bogdanova, E.A.; Zaraisky, A.G.; Lukyanov, S.; Chudakov, D.M. Bright far-red fluorescent protein for whole-body imaging. Nat. Methods 2007, 4, 741–746. [Google Scholar]
- Campbell, R.E.; Tour, O.; Palmer, A.E.; Steinbach, P.A.; Baird, G.S.; Zacharias, D.A.; Tsien, R.Y. A monomeric red fluorescent protein. Proc. Natl. Acad. Sci. USA 2002, 99, 7877–7882. [Google Scholar]
- Shu, X.; Royant, A.; Lin, M.Z.; Aguilera, T.A.; Lev-Ram, V.; Steinbach, P.A.; Tsien, R.Y. Mammalian expression of infrared fluorescent proteins engineered from a bacterial phytochrome. Science 2009, 324, 804–807. [Google Scholar]
- Panchuk-Voloshina, N.; Haugland, R.P.; Bishop-Stewart, J.; Bhalgat, M.K.; Millard, P.J.; Mao, F.; Leung, W.Y. Alexa dyes, a series of new fluorescent dyes that yield exceptionally bright, photostable conjugates. J. Histochem. Cytochem. 1999, 47, 1179–1188. [Google Scholar]
- Escobedo, J.O.; Rusin, O.; Lim, S.; Strongin, R.M. NIR dyes for bioimaging applications. Curr. Opin. Chem. Biol. 2010, 14, 64–70. [Google Scholar]
- Klohs, J.; Wunder, A.; Licha, K. Near-infrared fluorescent probes for imaging vascular pathophysiology. Basic Res. Cardiol. 2008, 103, 144–151. [Google Scholar]
- Kasili, P.M.; Song, J.M.; Vo-Dinh, T. Optical sensor for the detection of caspase-9 activity in a single cell. J. Am. Chem. Soc. 2004, 126, 2799–2806. [Google Scholar]
- Tung, C.H.; Mahmood, U.; Bredow, S.; Weissleder, R. In vivo imaging of proteolytic enzyme activity using a novel molecular reporter. Cancer Res. 2000, 60, 4953–4958. [Google Scholar]
- Bremer, C.; Tung, C.H.; Weissleder, R. In vivo molecular target assessment of matrix metalloproteinase inhibition. Nat. Med. 2001, 7, 743–748. [Google Scholar]
- Bruchez, M.P. Turning all the lights on: Quantum dots in cellular assays. Curr. Opin. Chem. Biol. 2005, 9, 533–537. [Google Scholar]
- Bentolila, L.A.; Ebenstein, Y.; Weiss, S. Quantum dots for in vivo small-animal imaging. J. Nucl. Med. 2009, 50, 493–496. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Isherwood, B.; Timpson, P.; McGhee, E.J.; Anderson, K.I.; Canel, M.; Serrels, A.; Brunton, V.G.; Carragher, N.O. Live Cell in Vitro and in Vivo Imaging Applications: Accelerating Drug Discovery. Pharmaceutics 2011, 3, 141-170. https://doi.org/10.3390/pharmaceutics3020141
Isherwood B, Timpson P, McGhee EJ, Anderson KI, Canel M, Serrels A, Brunton VG, Carragher NO. Live Cell in Vitro and in Vivo Imaging Applications: Accelerating Drug Discovery. Pharmaceutics. 2011; 3(2):141-170. https://doi.org/10.3390/pharmaceutics3020141
Chicago/Turabian StyleIsherwood, Beverley, Paul Timpson, Ewan J McGhee, Kurt I Anderson, Marta Canel, Alan Serrels, Valerie G Brunton, and Neil O Carragher. 2011. "Live Cell in Vitro and in Vivo Imaging Applications: Accelerating Drug Discovery" Pharmaceutics 3, no. 2: 141-170. https://doi.org/10.3390/pharmaceutics3020141