Determination of the Pharmacokinetics and Oral Bioavailability of Salicylamine, a Potent γ-Ketoaldehyde Scavenger, by LC/MS/MS

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
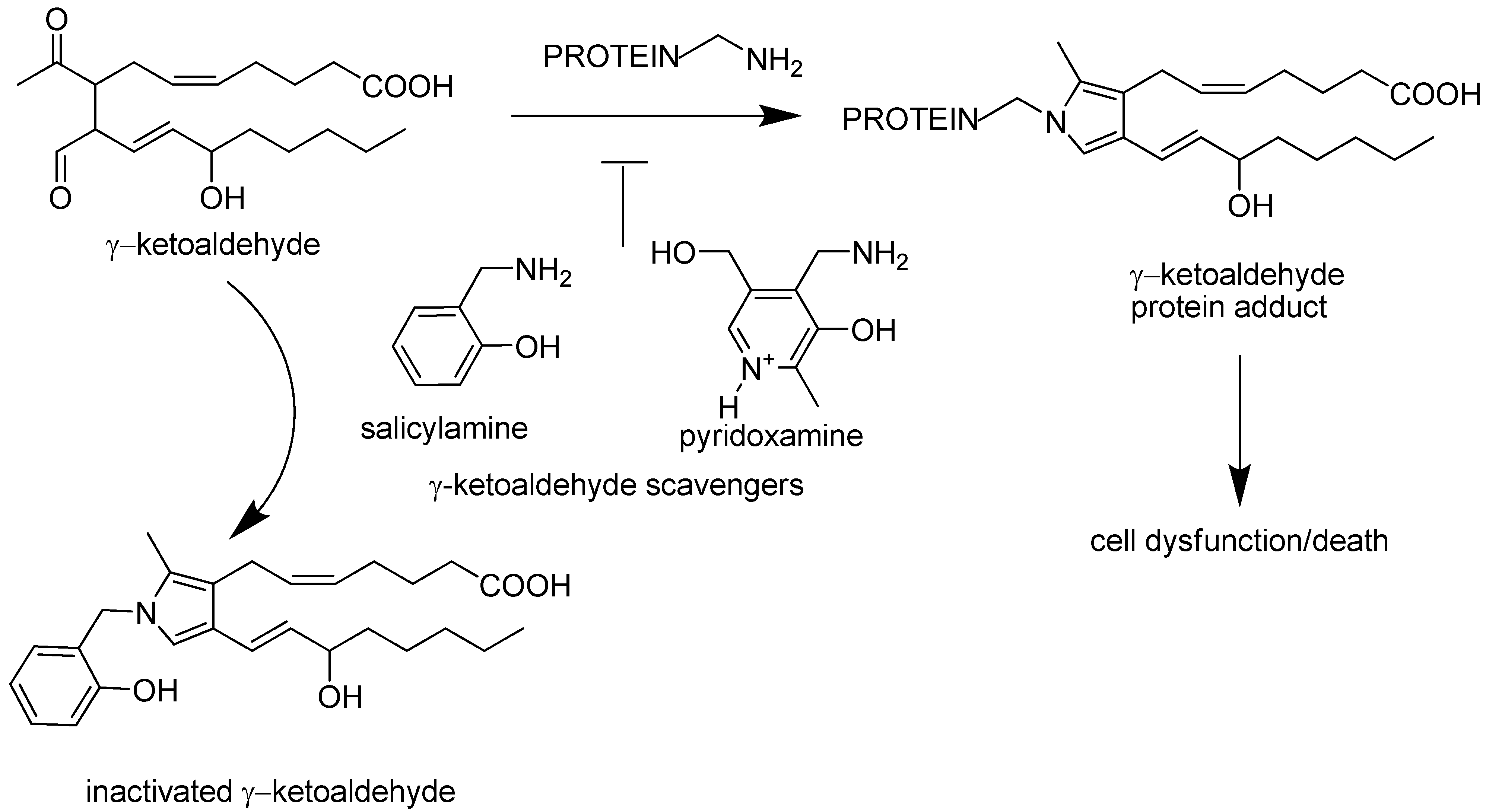
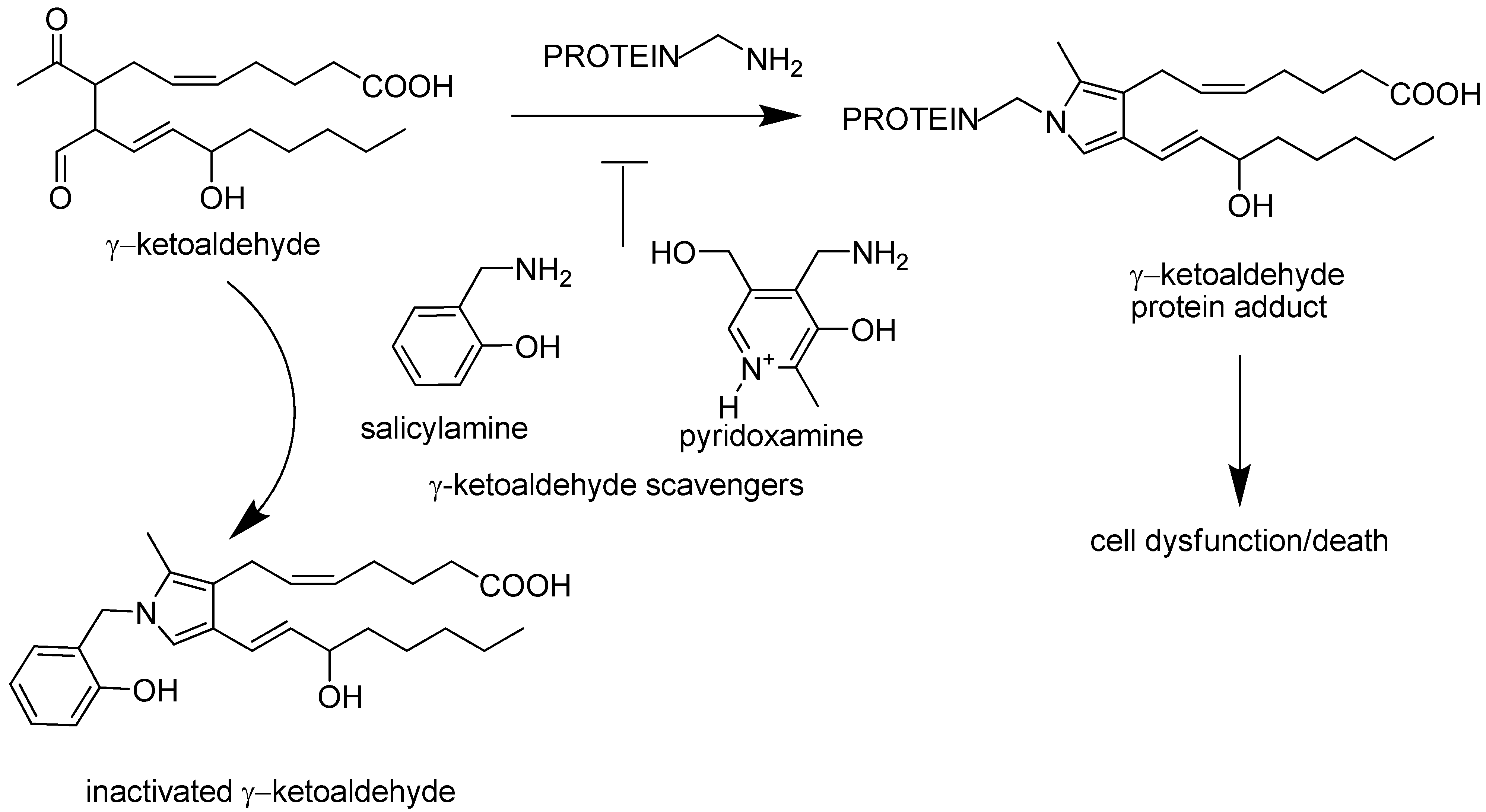
:1. Introduction
2. Experimental Section
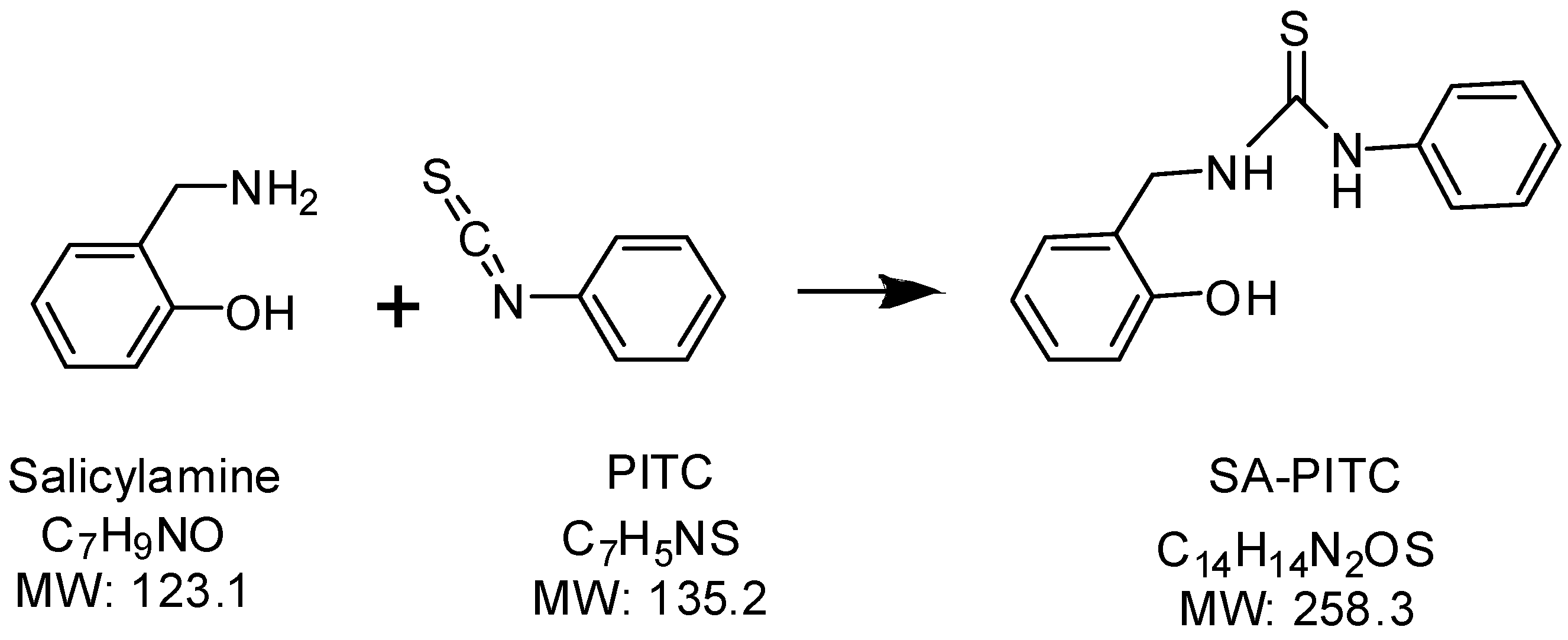
Synthesis of isotope-labeled salicylamine
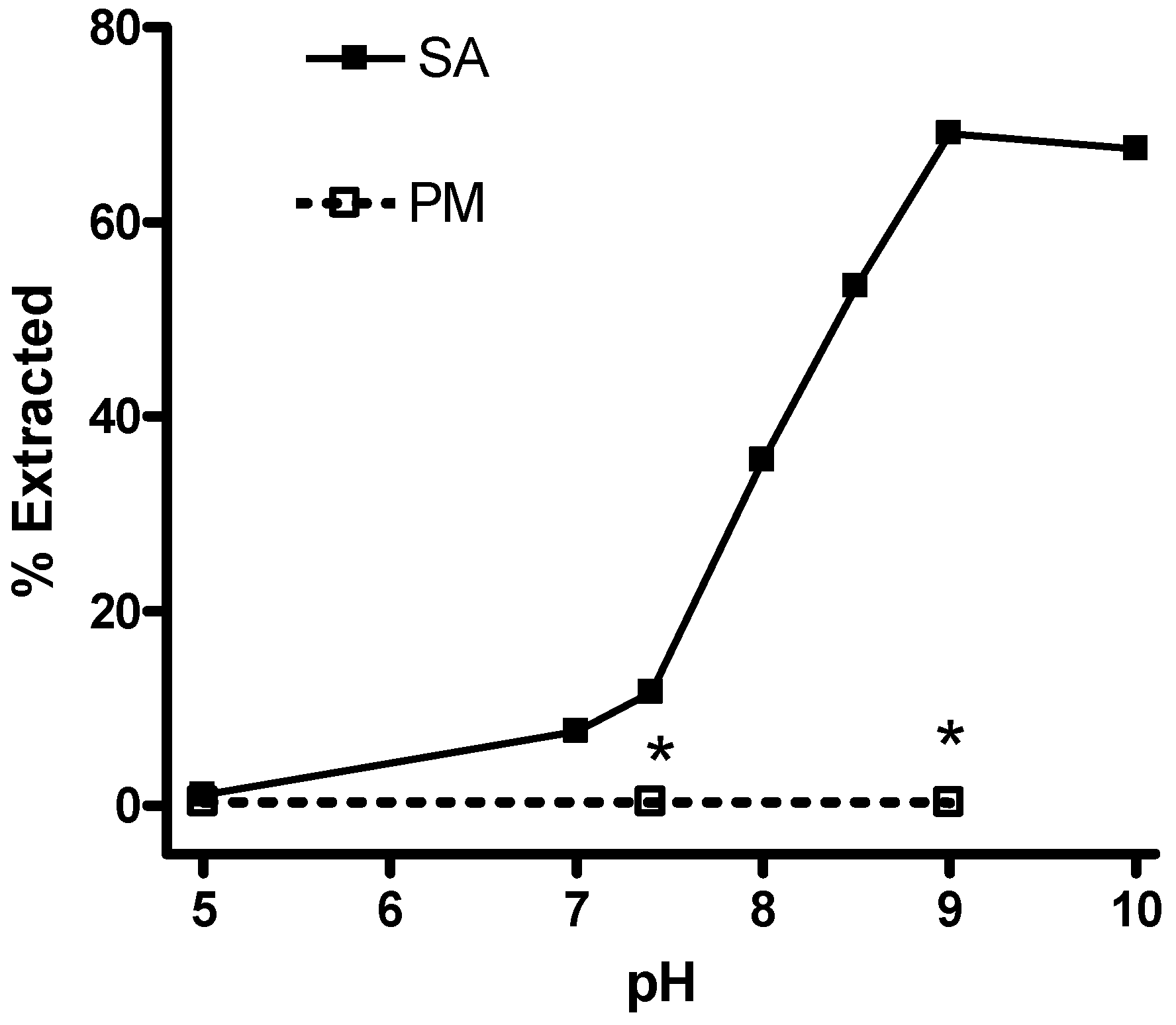
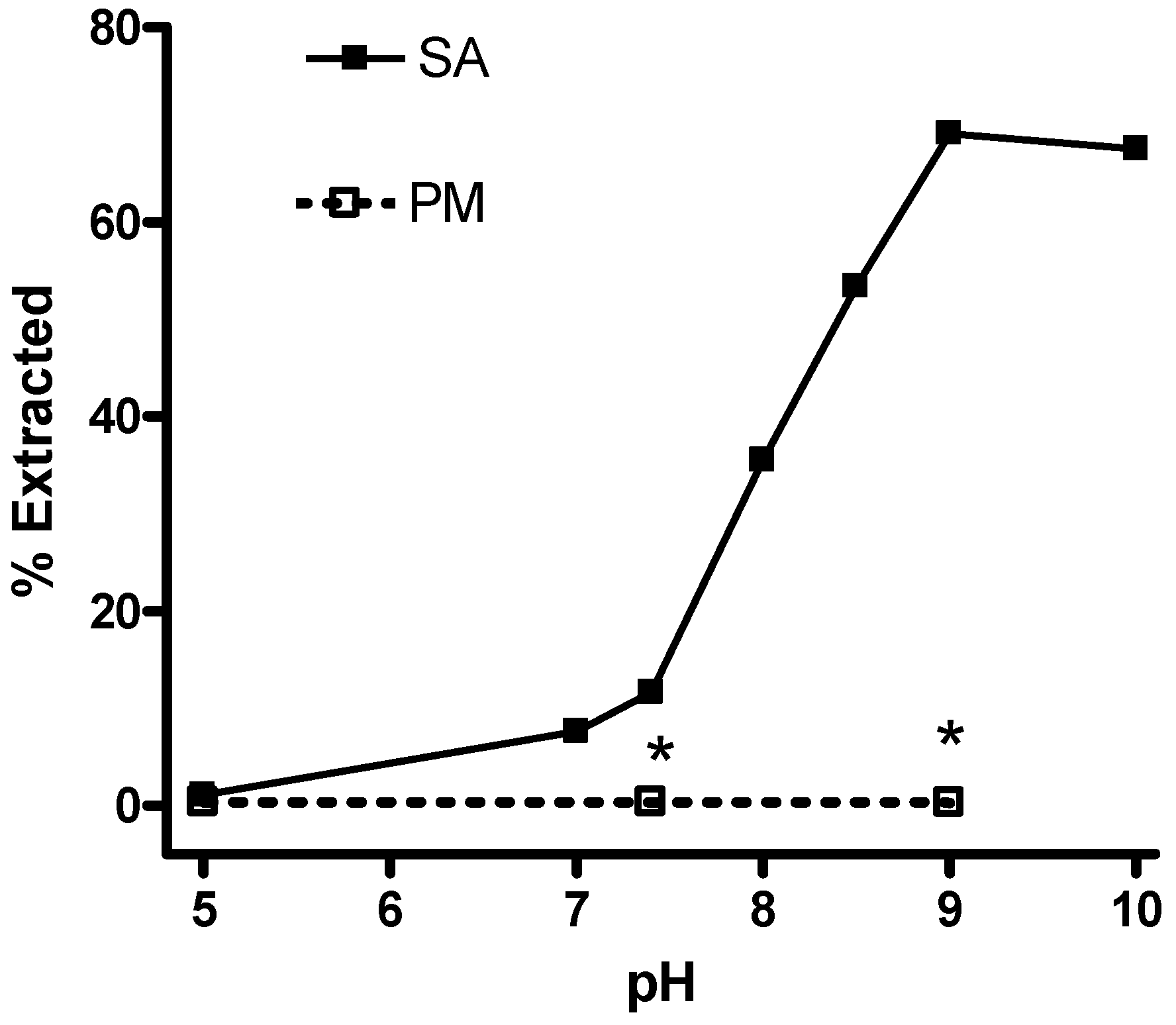
Measurement of salicylamine lipophilicity
Animals
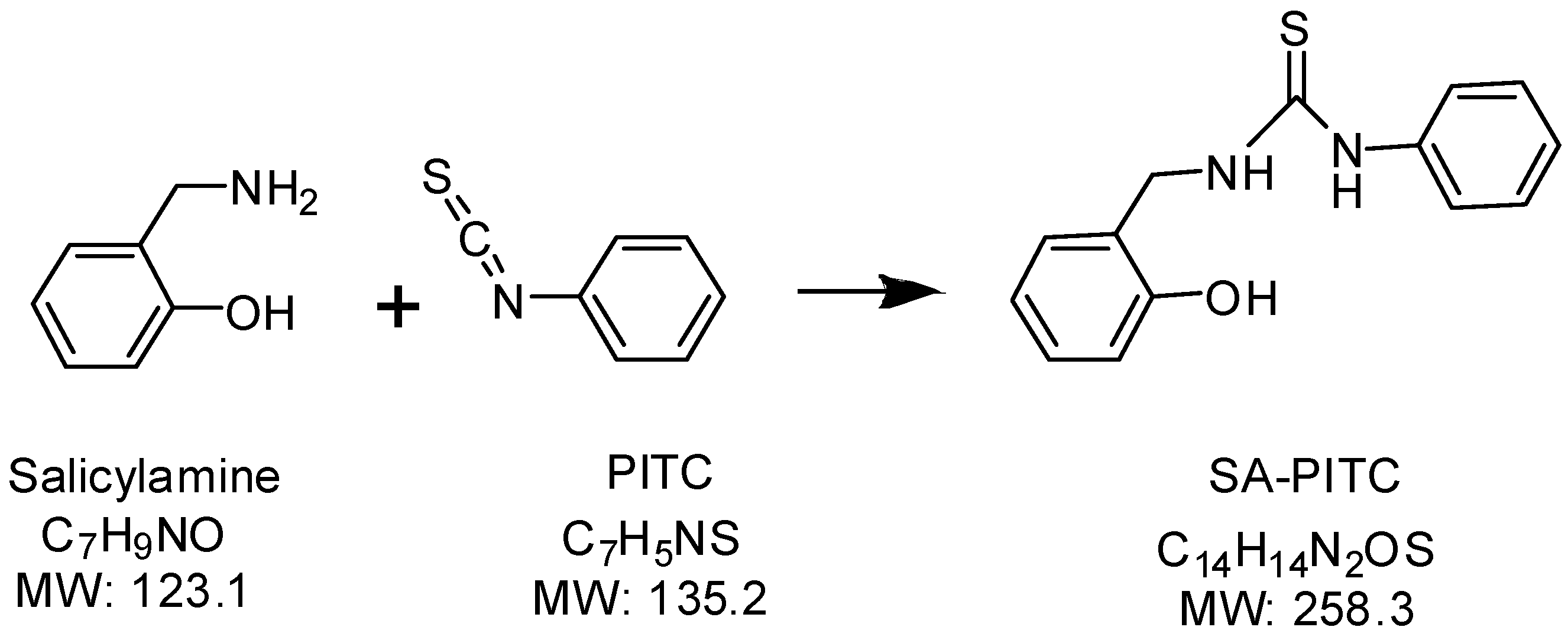
Salicylamine assay
Pharmacokinetic Analysis
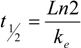
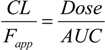
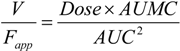
3. Results and Discussion
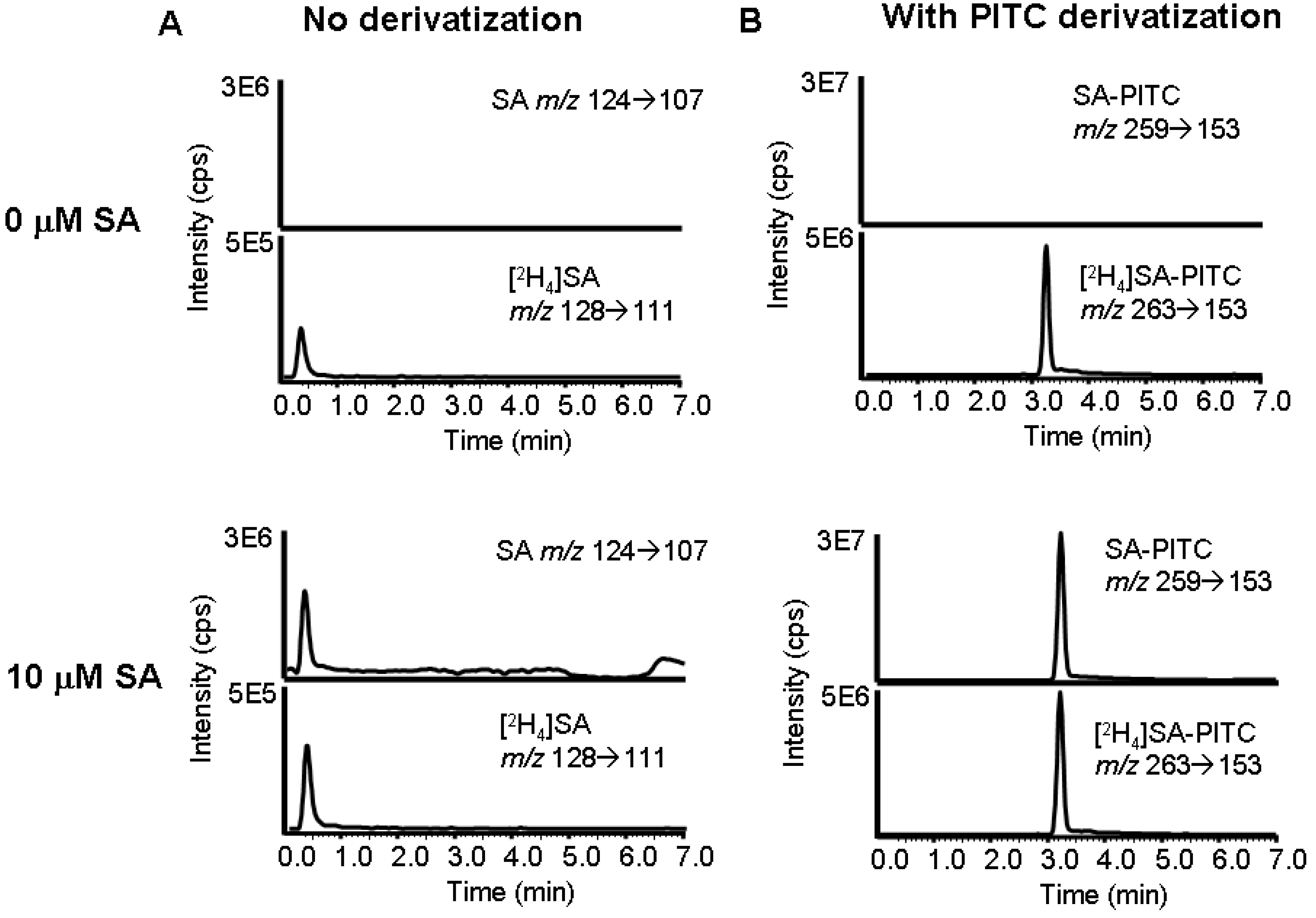
Development of method to assay SA in biological tissue
LC/MS/MS measurement of SA in plasma and tissue.
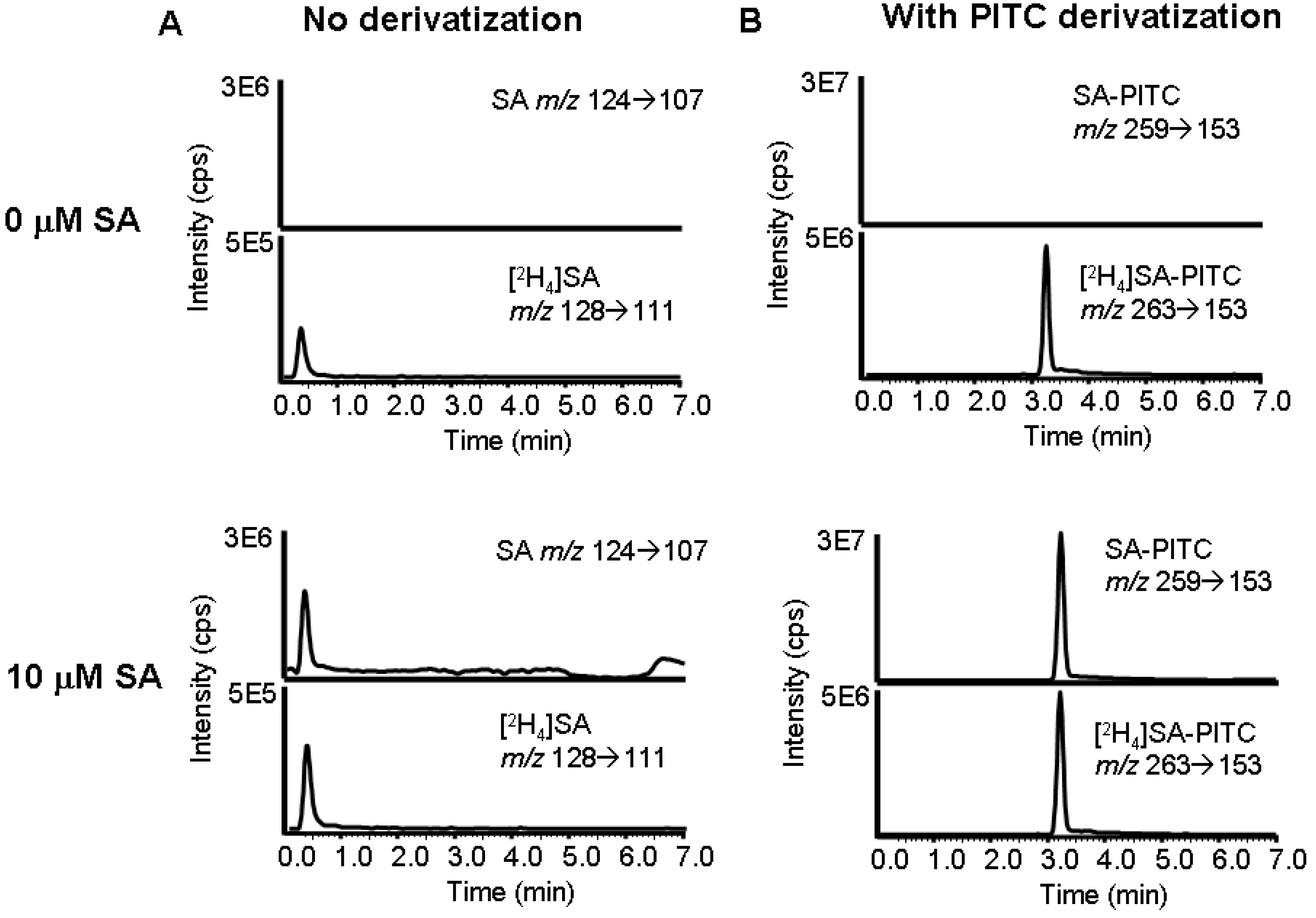
Validation of LC/MS/MS method to measure SA
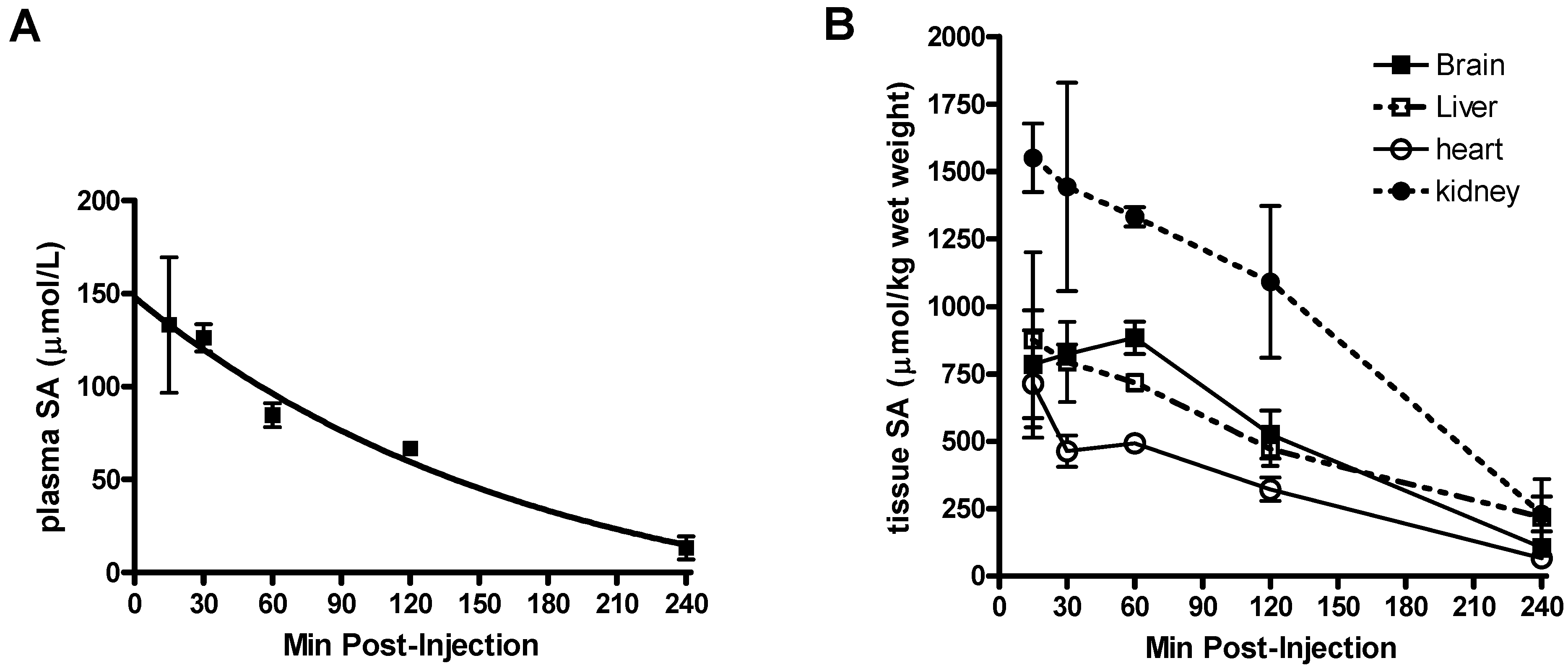
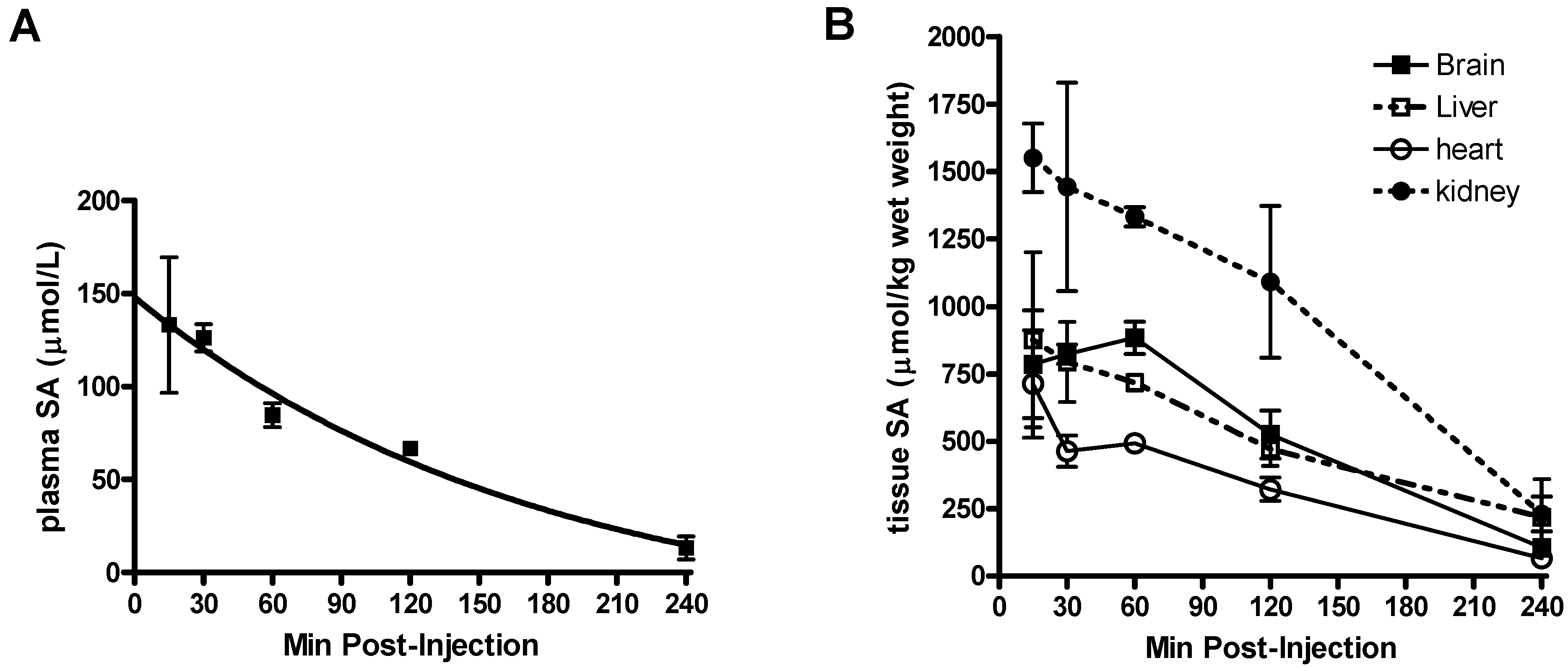
Pharmacokinetic parameters of SA in mice.
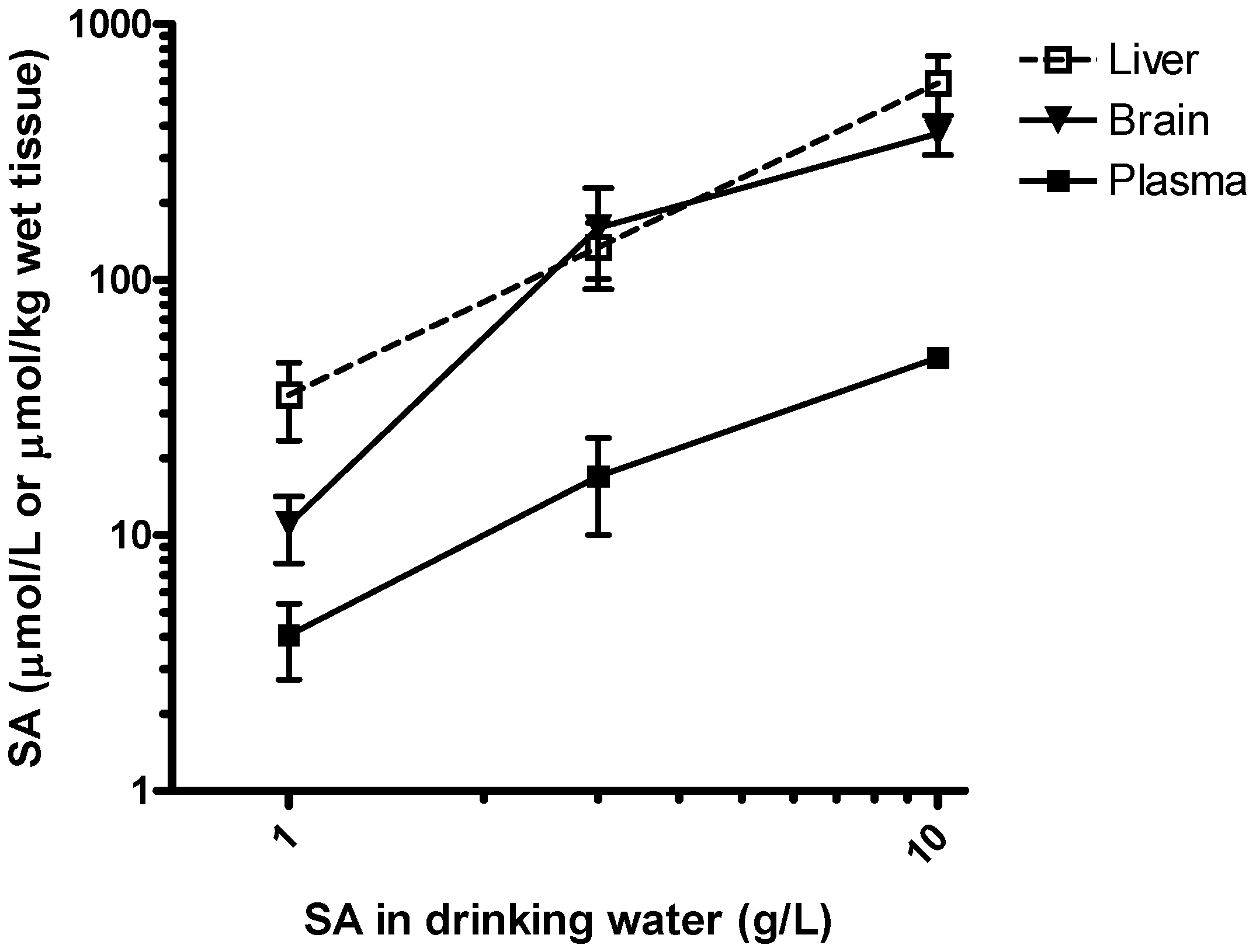
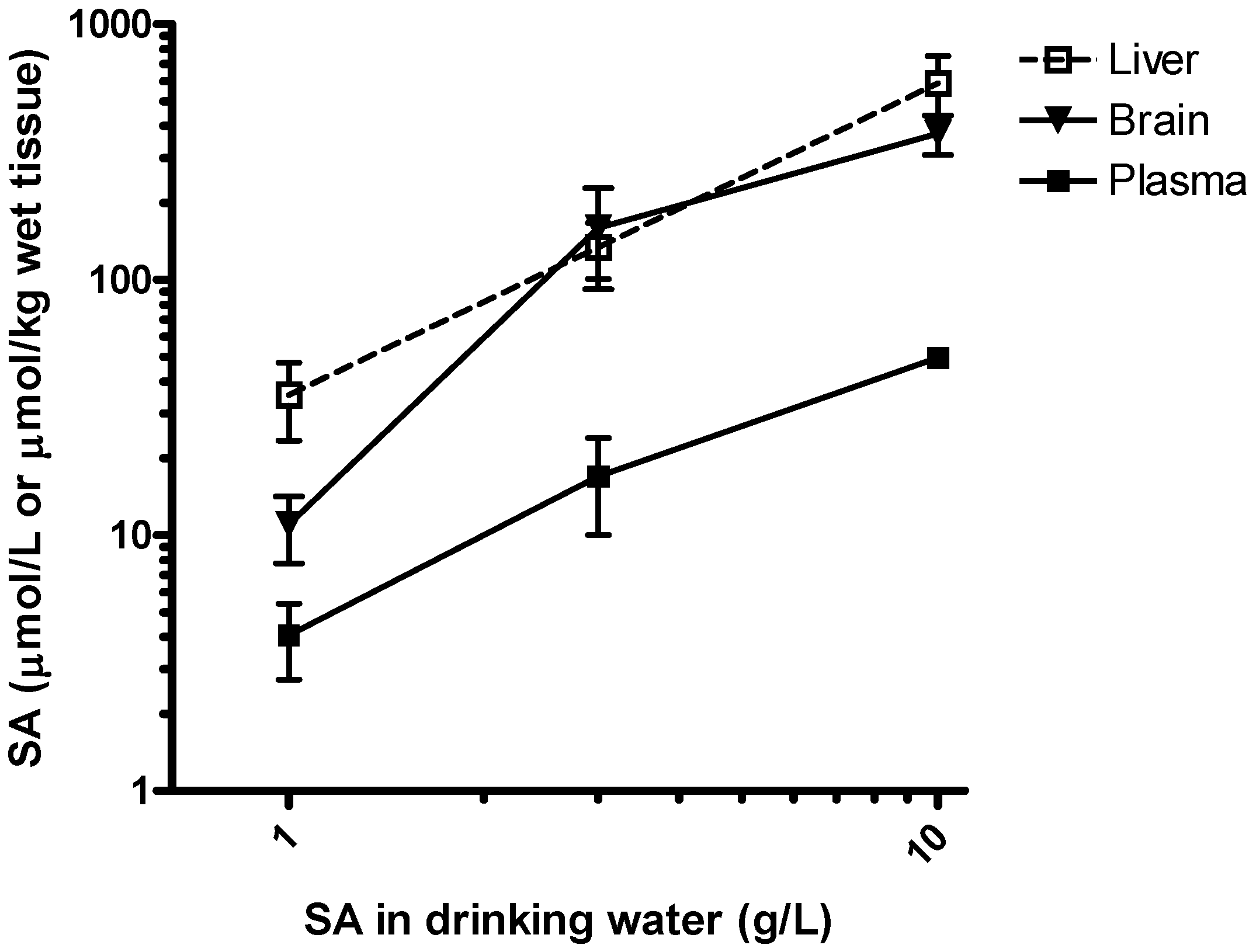
Feasibility of salicylamine administration in drinking water
4. Conclusions
Acknowledgments
References
- Salomon, R.G.; Miller, D.B.; Zagorski, M.G.; Coughlin, D.J. Solvent Induced Fragmentation of Prostaglandin Endoperoxides. New Aldehyde Products from PGH2 and Novel Intramolecular 1,2-Hydride Shift During Endoperoxide Fragmentation in Aqueous Solution. J. Am. Chem. Soc. 1984, 106, 6049–6060. [Google Scholar] [CrossRef]
- Brame, C.J.; Salomon, R.G.; Morrow, J.D.; Roberts, L.J., 2nd. Identification of extremely reactive gamma-ketoaldehydes (isolevuglandins) as products of the isoprostane pathway and characterization of their lysyl protein adducts. J. Biol. Chem. 1999, 274, 13139–13146. [Google Scholar] [CrossRef]
- Sullivan, C.B.; Matafonova, E.; Roberts, L.J., II; Amarnath, V.; Davies, S.S. Isoketals form cytotoxic phosphatidylethanolamine adducts in cells. J. Lipid Res. 2009, in press. [Google Scholar]
- Zagol-Ikapitte, I.; Masterson, T.S.; Amarnath, V.; Montine, T.J.; Andreasson, K.I.; Boutaud, O.; Oates, J. A. Prostaglandin H(2)-derived adducts of proteins correlate with Alzheimer's disease severity. J. Neurochem. 2005, 94, 1140–1145. [Google Scholar] [CrossRef]
- Salomon, R.G.; Batyreva, E.; Kaur, K.; Sprecher, D.L.; Schreiber, M.J.; Crabb, J.W.; Penn, M.S.; DiCorletoe, A.M.; Hazen, S.L.; Podrez, E.A. Isolevuglandin-protein adducts in humans: products of free radical-induced lipid oxidation through the isoprostane pathway. Biochim. Biophys. Acta 2000, 1485, 225–235. [Google Scholar] [CrossRef]
- Fukuda, K.; Davies, S.S.; Nakajima, T.; Ong, B.H.; Kupershmidt, S.; Fessel, J.; Amarnath, V.; Anderson, M.E.; Boyden, P.A.; Viswanathan, P.C.; Roberts, L.J., 2nd; Balser, J.R. Oxidative mediated lipid peroxidation recapitulates proarrhythmic effects on cardiac sodium channels. Circ. Res. 2005, 97, 1262–1269. [Google Scholar] [CrossRef]
- Poliakov, E.; Brennan, M.L.; Macpherson, J.; Zhang, R.; Sha, W.; Narine, L.; Salomon, R.G.; Hazen, S.L. Isolevuglandins, a novel class of isoprostenoid derivatives, function as integrated sensors of oxidant stress and are generated by myeloperoxidase in vivo. Faseb J 2003, 17, 2209–2220. [Google Scholar] [CrossRef]
- Davies, S.S.; Talati, M.; Wang, X.; Mernaugh, R.L.; Amarnath, V.; Fessel, J.; Meyrick, B.O.; Sheller, J.; Roberts, L.J., 2nd. Localization of isoketal adducts in vivo using a single-chain antibody. Free Radic. Biol. Med. 2004, 36, 1163–1174. [Google Scholar] [CrossRef]
- Hoppe, G.; Subbanagounder, G.; O'Neil, J.; Salomon, R.G.; Hoff, H.F. Macrophage recognition of LDL modified by levuglandin E2, an oxidation product of arachidonic acid. Biochim. Biophys. Acta 1997, 1344, 1–5. [Google Scholar] [CrossRef]
- Schmidley, J.W.; Dadson, J.; Iyer, R.S.; Salomon, R.G. Brain tissue injury and blood-brain barrier opening induced by injection of LGE2 or PGE2. Prostaglandins Leukot. Essent. Fatty Acids 1992, 47, 105–110. [Google Scholar] [CrossRef]
- Murthi, K.K.; Salomon, R.G.; Sternlicht, H. Levuglandin E2 inhibits mitosis and microtubule assembly. Prostaglandins 1990, 39, 611–622. [Google Scholar]
- Boutaud, O.; Montine, T.J.; Chang, L.; Klein, W.L.; Oates, J.A. PGH2-derived levuglandin adducts increase the neurotoxicity of amyloid beta1-42. J. Neurochem. 2006, 96, 917–923. [Google Scholar]
- Brame, C.J.; Boutaud, O.; Davies, S.S.; Yang, T.; Oates, J.A.; Roden, D.; Roberts, L.J., 2nd. Modification of proteins by isoketal-containing oxidized phospholipids. J. Biol. Chem. 2004, 279, 13447–13451. [Google Scholar] [CrossRef]
- Davies, S.S.; Amarnath, V.; Montine, K.S.; Bernoud-Hubac, N.; Boutaud, O.; Montine, T.J.; Roberts, L.J., 2nd. Effects of reactive gamma-ketoaldehydes formed by the isoprostane pathway (isoketals) and cyclooxygenase pathway (levuglandins) on proteasome function. Faseb J 2002, 16, 715–717. [Google Scholar]
- Davies, S.S.; Brantley, E.J.; Voziyan, P.A.; Amarnath, V.; Zagol-Ikapitte, I.; Boutaud, O.; Hudson, B.G.; Oates, J.A.; Ii, L.J. Pyridoxamine Analogues Scavenge Lipid-Derived gamma-Ketoaldehydes and Protect against H(2)O(2)-Mediated Cytotoxicity. Biochemistry 2006, 45, 15756–15767. [Google Scholar] [CrossRef]
- Amarnath, V.; Amarnath, K.; Amarnath, K.; Davies, S.; Roberts, L.J., 2nd. Pyridoxamine: an extremely potent scavenger of 1,4-dicarbonyls. Chem. Res. Toxicol. 2004, 17, 410–415. [Google Scholar] [CrossRef]
- Nakajima, T.; Davies, S.S.; Matafonova, E.; Potet, F.; Amarnath, V.; Tallman, K.A.; Serwa, R.A.; Porter, N.A.; Balser, J.R.; Kupershmidt, S.; Roberts, L.J., II. Selective gamma-ketoaldehyde scavengers protect NaV1.5 from oxidant-induced inactivation. J. Mol. Cell. Cardiol. 2009, in press. [Google Scholar]
- Reany, O.; Gunnlaugsson, T.; Parker, D. J. Chem. Soc. Perkin Trans. 2000, 2, 1819–1831.
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an Open Access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zagol-Ikapitte, I.; Matafonova, E.; Amarnath, V.; Bodine, C.L.; Boutaud, O.; Tirona, R.G.; Oates, J.A.; Roberts II, L.J.; Davies, S.S. Determination of the Pharmacokinetics and Oral Bioavailability of Salicylamine, a Potent γ-Ketoaldehyde Scavenger, by LC/MS/MS. Pharmaceutics 2010, 2, 18-29. https://doi.org/10.3390/pharmaceutics2010018
Zagol-Ikapitte I, Matafonova E, Amarnath V, Bodine CL, Boutaud O, Tirona RG, Oates JA, Roberts II LJ, Davies SS. Determination of the Pharmacokinetics and Oral Bioavailability of Salicylamine, a Potent γ-Ketoaldehyde Scavenger, by LC/MS/MS. Pharmaceutics. 2010; 2(1):18-29. https://doi.org/10.3390/pharmaceutics2010018
Chicago/Turabian StyleZagol-Ikapitte, Irene, Elena Matafonova, Venkataraman Amarnath, Christopher L. Bodine, Olivier Boutaud, Rommel G. Tirona, John A. Oates, L. Jackson Roberts II, and Sean S. Davies. 2010. "Determination of the Pharmacokinetics and Oral Bioavailability of Salicylamine, a Potent γ-Ketoaldehyde Scavenger, by LC/MS/MS" Pharmaceutics 2, no. 1: 18-29. https://doi.org/10.3390/pharmaceutics2010018