
Real-Time Expression Analysis of Selected Anticarsia gemmatalis multiple nucleopolyhedrovirus Gene Promoters during Infection of Permissive, Semipermissive and Nonpermissive Cell Lines


Abstract
:1. Introduction
2. Materials and Methods
2.1. Insect Cells and Viruses
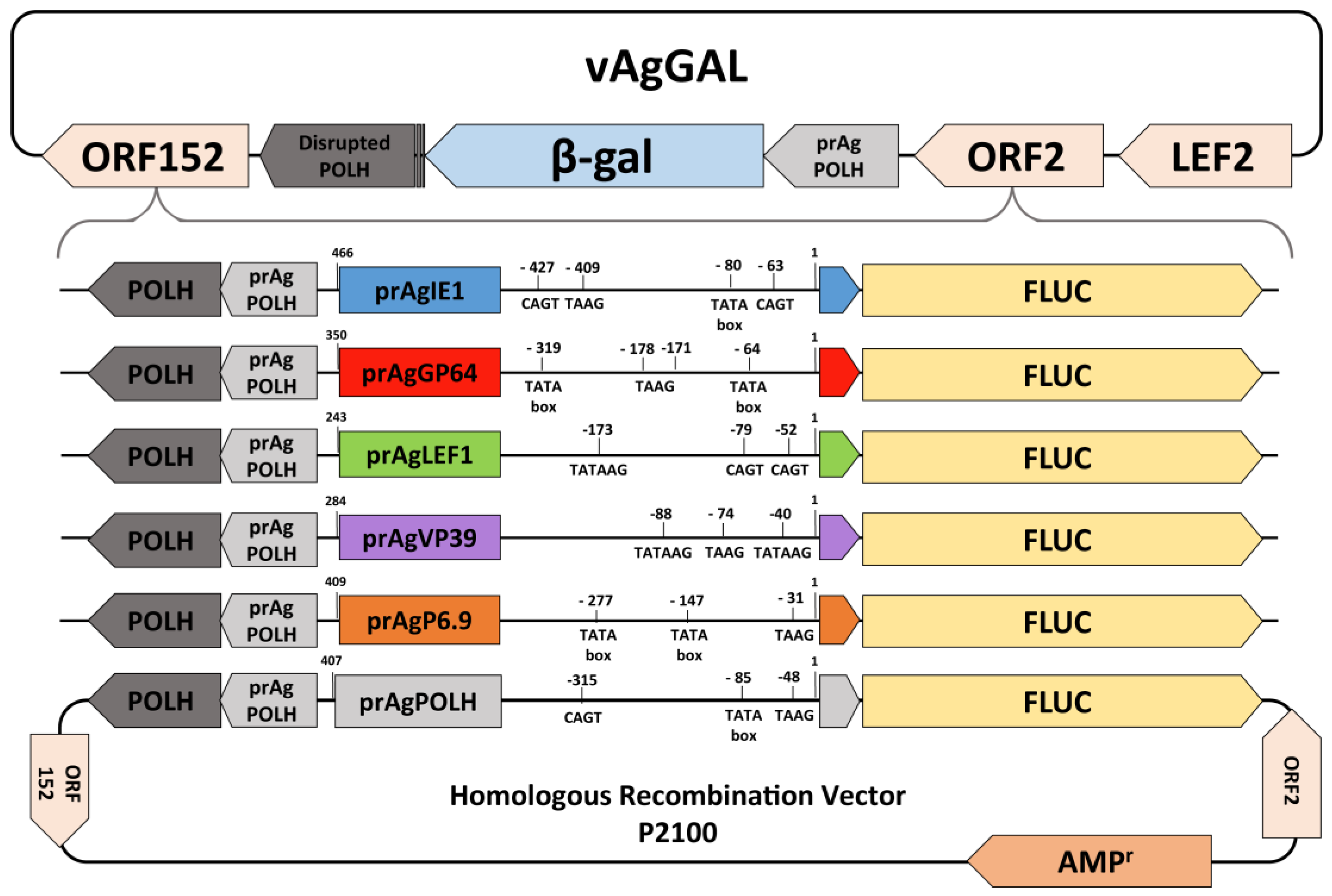
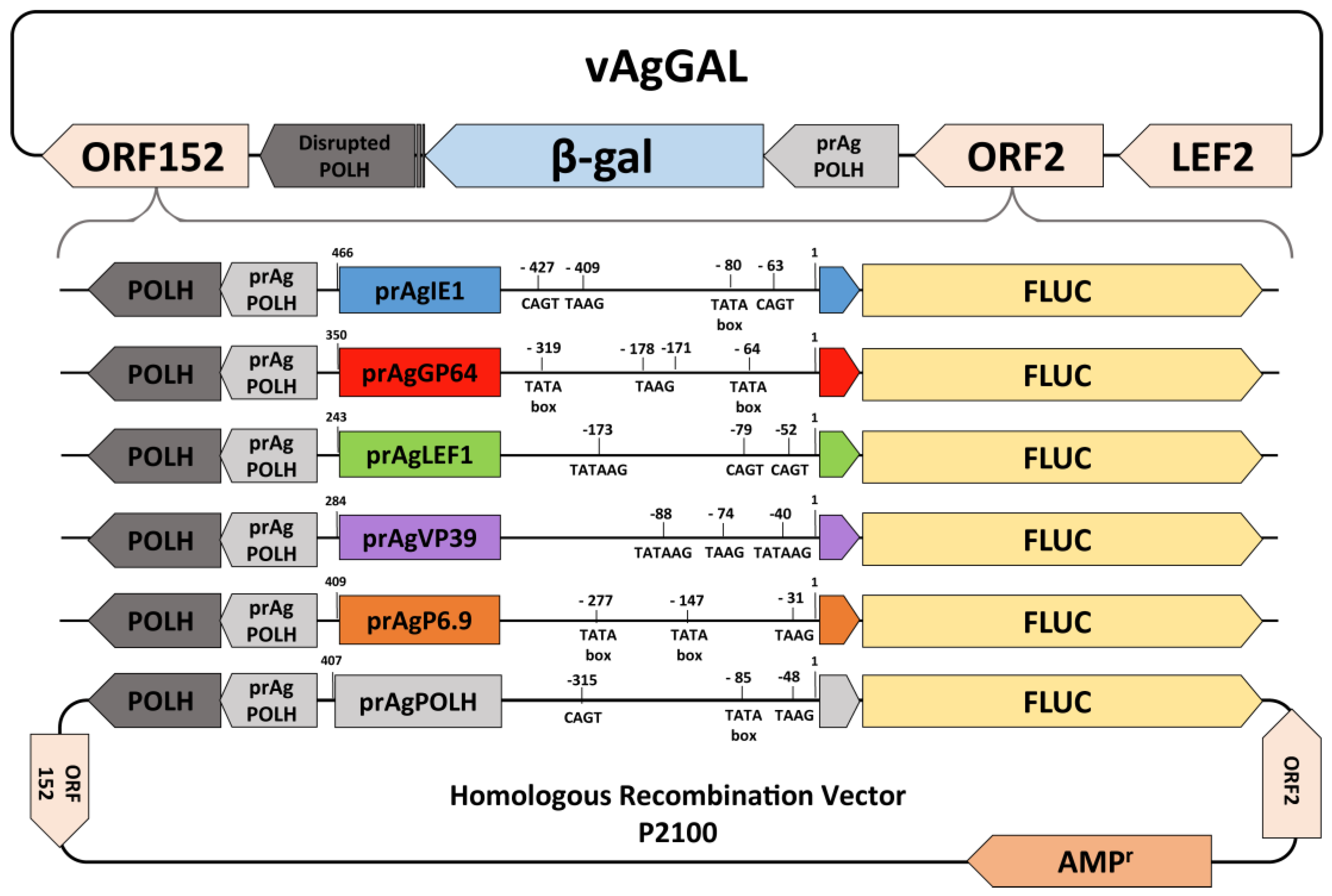
2.2. Recombinant Baculovirus Construction
2.3. Real-Time Luciferase Assay
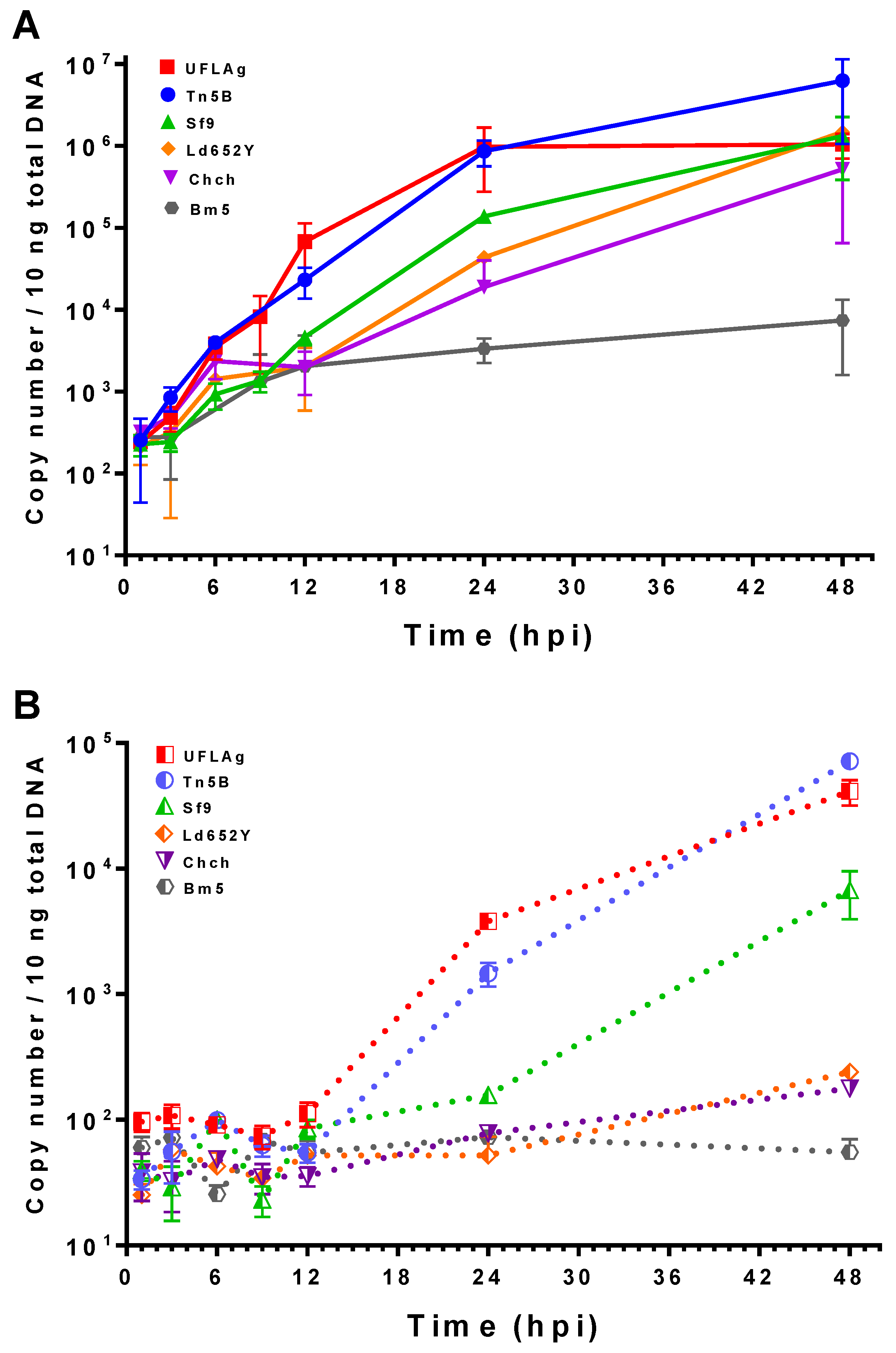
2.4. Viral DNA Quantification by Quantitative Polymerase Chain Reaction
2.5. Viability and Caspase Activity Measurements
3. Results
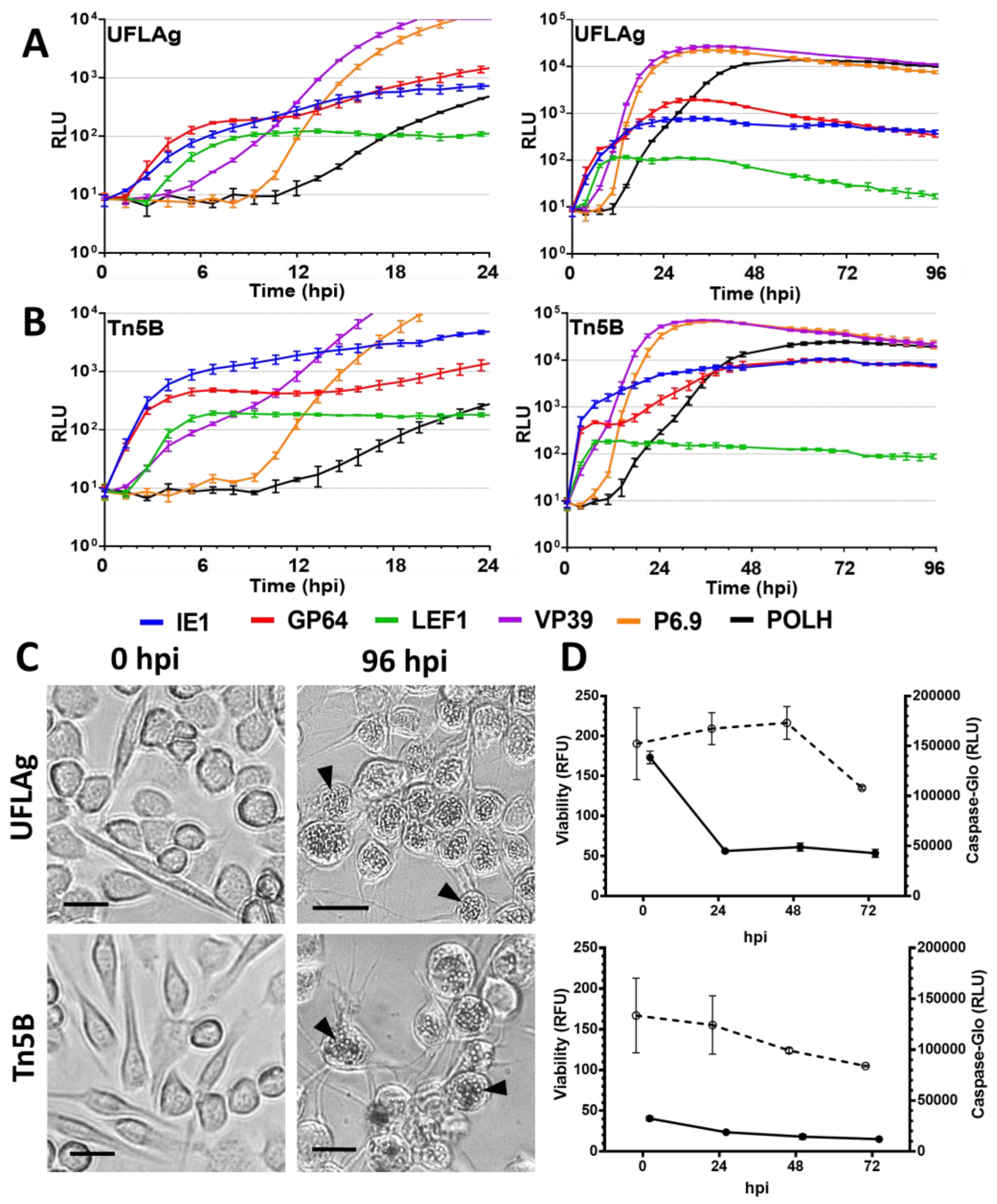
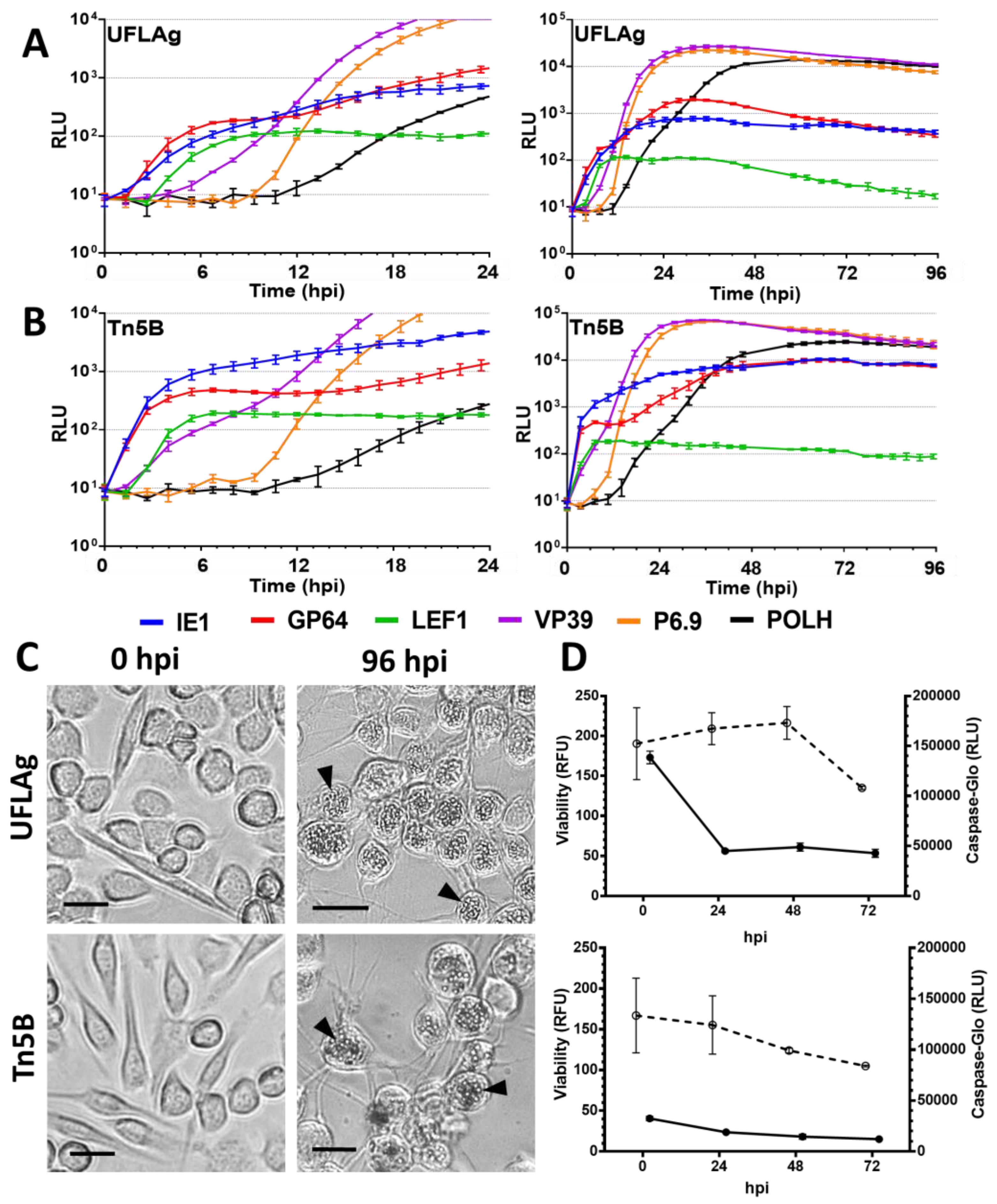
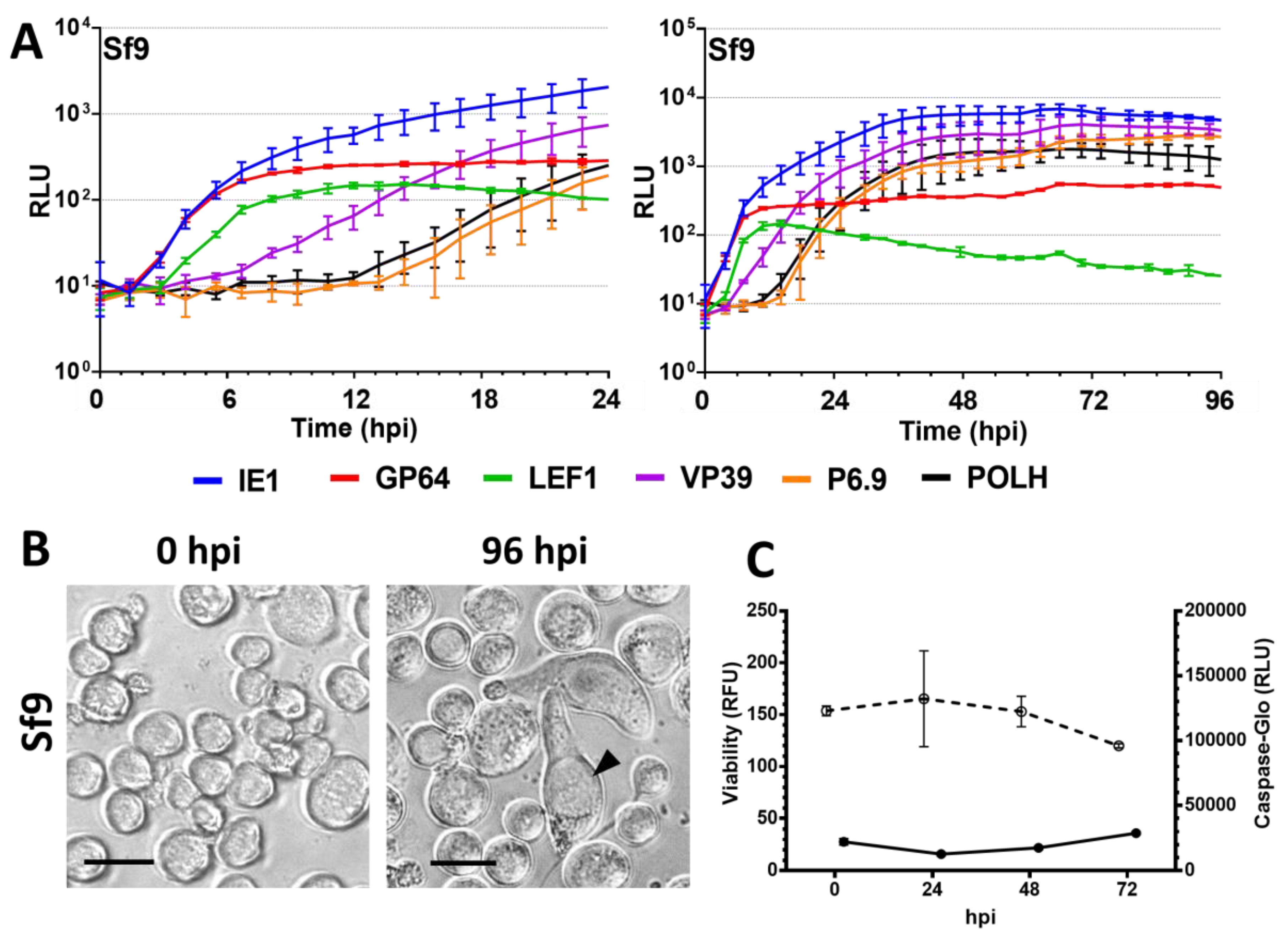
3.1. Permissive Cell Lines
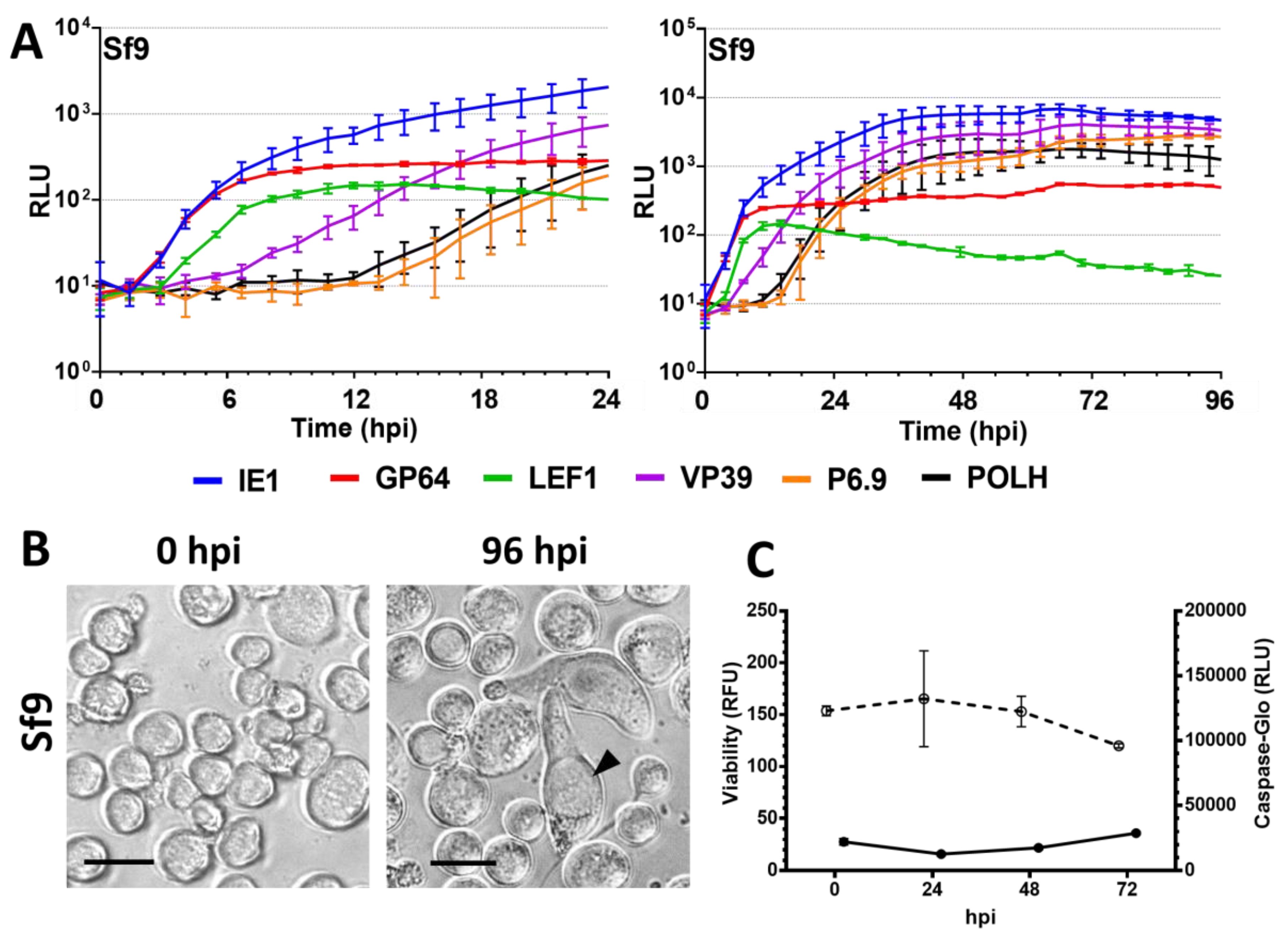
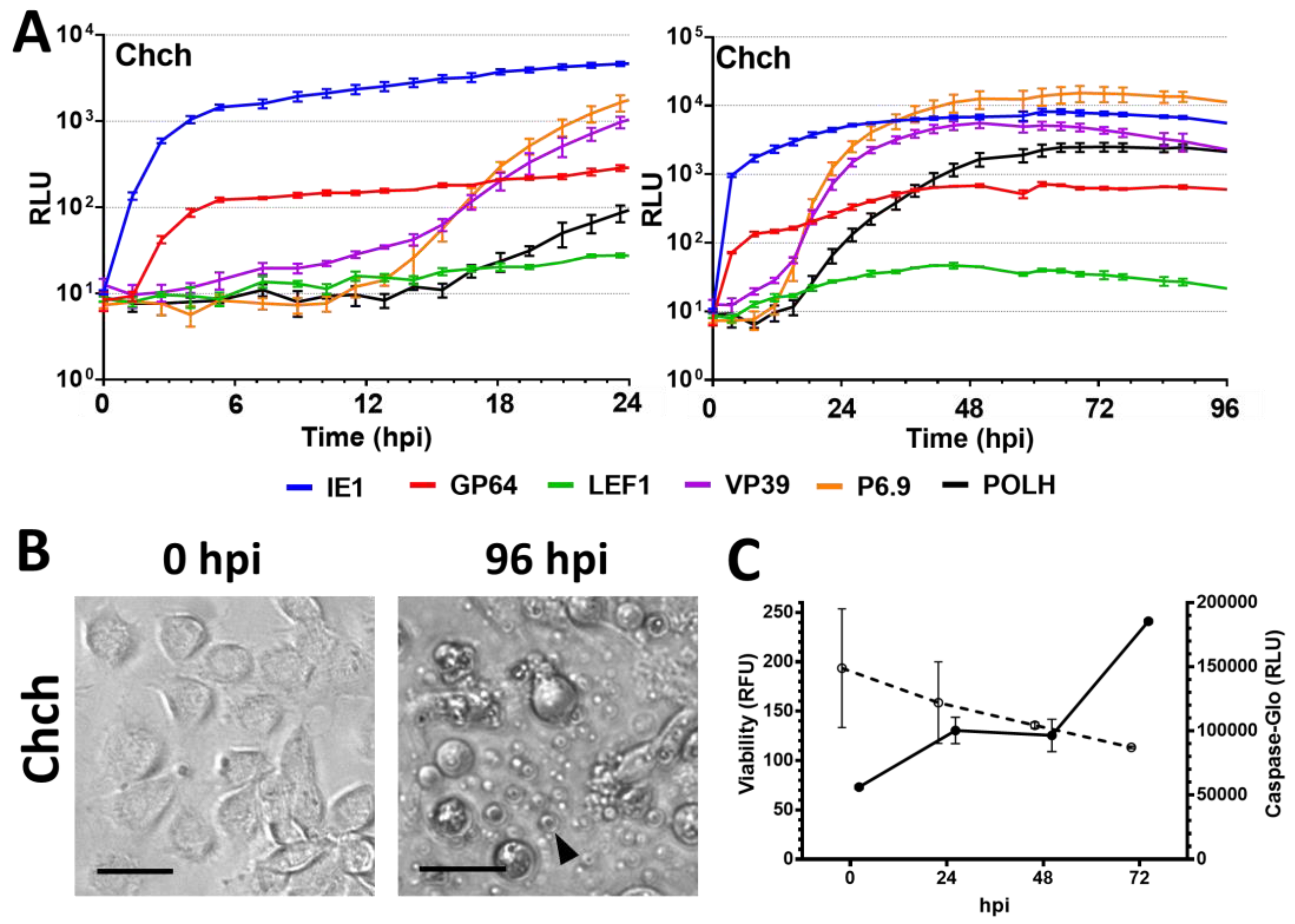
3.2. Semipermissive Cell Lines
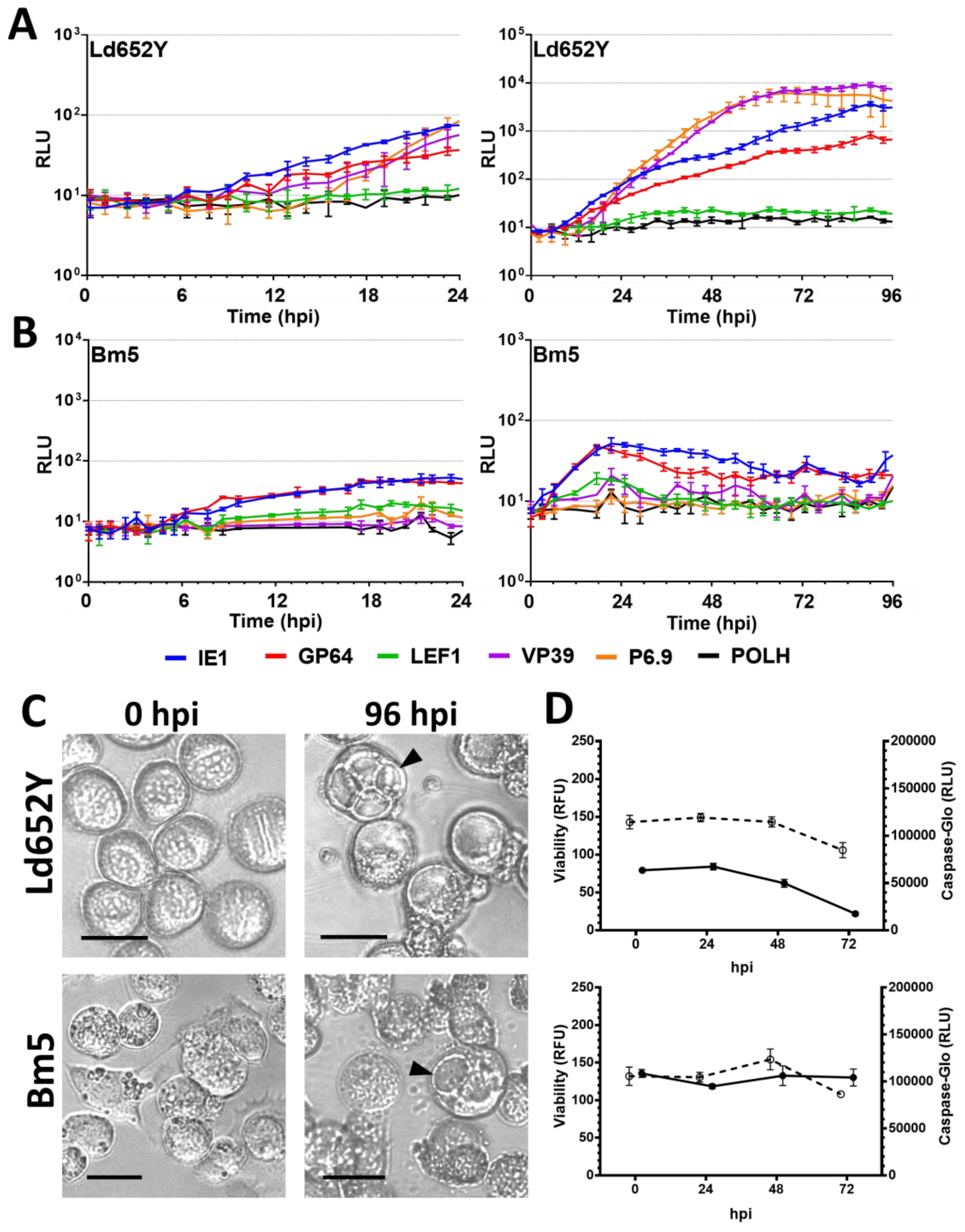
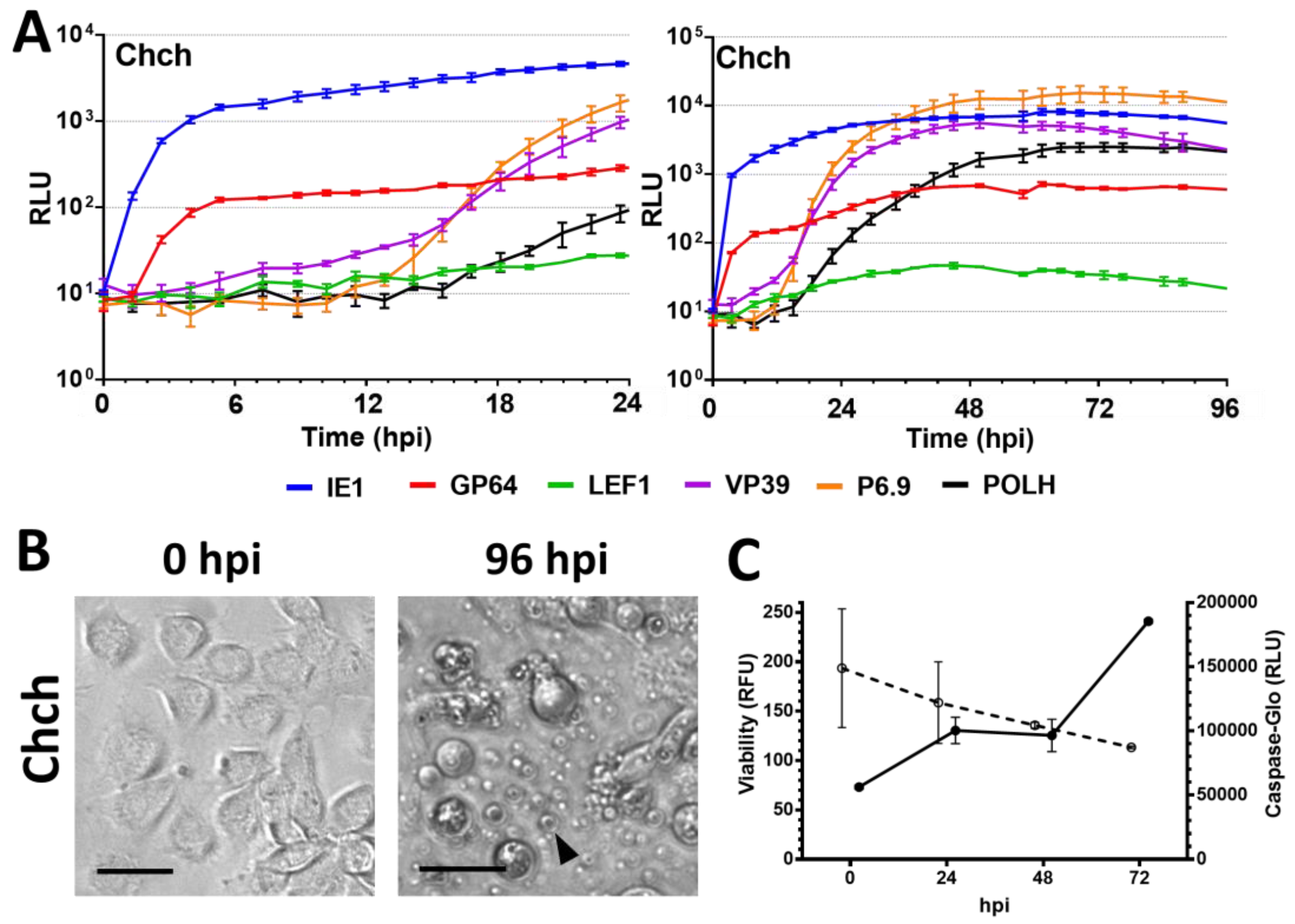
3.3. Nonpermissive Cell Lines
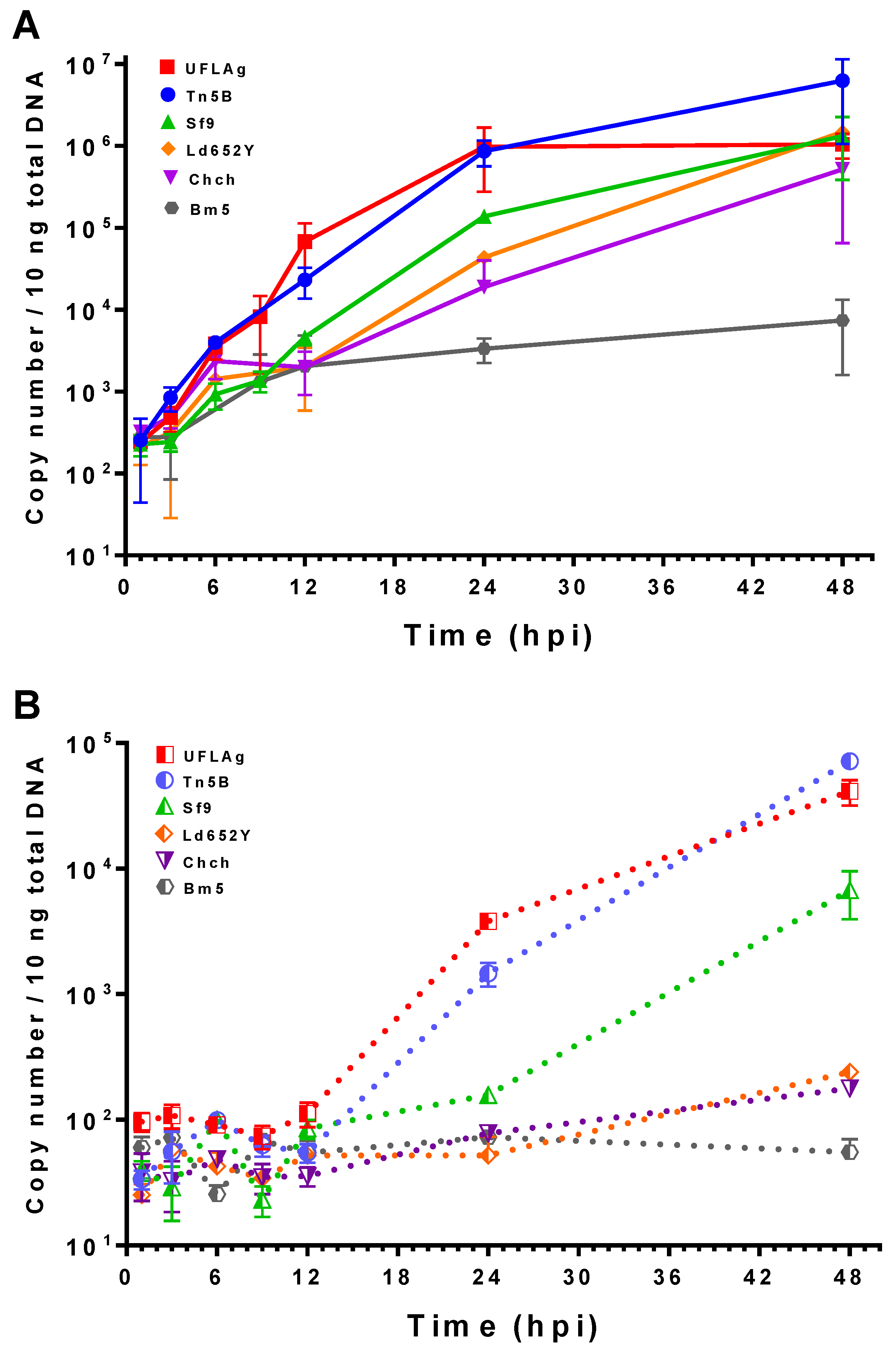
3.4. Viral DNA Quantification by qPCR
3.5. Viability and Caspase Activity Assay
4. Discussion
4.1. Methods for the Investigation of Baculovirus Promoter Activity
4.2. Temporal Expression Patterns of Individual Promoters and Correlations with Viral Replication in Permissive Cell Lines
4.3. The Semipermissive Cell Line Sf9 and Restriction of Very Late Promoter Expression
4.4. Viral Infection in Nonpermissive Cell Lines and the Effects on the Promoters
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Moscardi, F. Assessment of the application of baculoviruses for control of Lepidoptera. Annu. Rev. Entomol. 1999, 44, 257–289. [Google Scholar] [CrossRef] [PubMed]
- Rohrmann, G.F. Baculovirus Molecular Biology, 3rd ed.; National Center for Technology Information: Bethesda, MD, USA, 2013.
- Miller, L. (Ed.) The Baculoviruses; Springer: New York, NY, USA, 1997. [Google Scholar]
- O’Reilly, D.R.; Miller, L.K.; Luckow, V.A. Baculovirus Expression Vectors: A Laboratory Manual; Oxford University Press: Oxford, NY, USA, 1993. [Google Scholar]
- Lauzon, H.A.M.; Jamieson, P.B.; Krell, P.J.; Arif, B.M. Gene organization and sequencing of the Choristoneura fumiferana defective nucleopolyhedrovirus genome. J. Gen. Virol. 2005, 86, 945–961. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.E.B.; Paula, D.P.; Almeida, G.F.; Ribeiro, Z.M.A.; Souza, M.L.; Inglis, P.W.; Ribeiro, B.M. Identification and sequence analysis of the Condylorrhiza vestigialis MNPV p74 gene. Virus Genes 2011, 43, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Jehle, J.A.; Blissard, G.W.; Bonning, B.C.; Cory, J.S.; Herniou, E.A.; Rohrmann, G.F.; Theilmann, D.A.; Thiem, S.M.; Vlak, J.M. On the classification and nomenclature of baculoviruses: A proposal for revision. Arch.Virol. 2006, 151, 1257–1266. [Google Scholar] [CrossRef] [PubMed]
- de Castro Oliveira, J.V.; Wolff, J.L.C.; Garcia-Maruniak, A.; Ribeiro, B.M.; de Castro, M.E.B.; de Souza, M.L.; Moscardi, F.; Maruniak, J.E.; de A Zanotto, P.M. Genome of the most widely used viral biopesticide: Anticarsia gemmatalis multiple nucleopolyhedrovirus. J. Gen. Virol. 2006, 87, 3233–3250. [Google Scholar] [CrossRef] [PubMed]
- Slack, J.M.; Ribeiro, B.M.; de Souza, M.L. The gp64 locus of Anticarsia gemmatalis multicapsid nucleopolyhedrovirus contains a 3′ repair exonuclease homologue and lacks v-cath and ChiA genes. J. Gen. Virol. 2004, 85, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Kaba, S.A.; Salcedo, A.M.; Wafula, P.O.; Vlak, J.M.; van Oers, M.M. Development of a chitinase and v-cathepsin negative bacmid for improved integrity of secreted recombinant proteins. J. Virol. Methods 2004, 122, 113–118. [Google Scholar] [CrossRef] [PubMed]
- Hitchman, R.B.; Possee, R.D.; Siaterli, E.; Richards, K.S.; Clayton, A.J.; Bird, L.E.; Owens, R.J.; Carpentier, D.C.J.; King, F.L.; Danquah, J.O.; et al. Improved expression of secreted and membrane-targeted proteins in insect cells. Biotechnol. Appl. Biochem. 2010, 56, 85–93. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.E.; Souza, M.L.; Araujo, S.; Bilimoria, S.L. Replication of Anticarsia gemmatalis nuclear polyhedrosis virus in four lepidopteran cell lines. J. Invertebr. Pathol. 1997, 69, 40–45. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.E.; Souza, M.L.; Bilimoria, S.L. Host-specific transcription of nucleopolyhedrovirus gene homologues in productive and abortive Anticarsia gemmatalis MNPV infections. Arch. Virol. 1999, 144, 1111–1121. [Google Scholar] [CrossRef] [PubMed]
- Castro, M.E.B.; Ribeiro, Z.M.A.; Souza, M.L. Infectivity of Anticarsia gemmatalis nucleopolyhedrovirus to different insect cell lines: Morphology, viral production, and protein synthesis. Biol. Control 2006, 36, 299–304. [Google Scholar] [CrossRef]
- Pombo, V.; Velloso, L.; Ribeiro, B.; Bao, S. Structural and ultrastructural changes during the infection of UFL-AG-286 cells with the baculovirus AgMNPV. J. Invertebr. Pathol. 1998, 72, 239–245. [Google Scholar] [CrossRef] [PubMed]
- Lynn, D.E. Comparative susceptibilities of twelve insect cell lines to infection by three baculoviruses. J. Invertebr. Pathol. 2003, 82, 129–131. [Google Scholar] [CrossRef]
- Sokal, N.; Nie, Y.; Willis, L.G.; Yamagishi, J.; Blissard, G.W.; Rheault, M.R.; Theilmann, D.A. Defining the roles of the baculovirus regulatory proteins IE0 and IE1 in genome replication and early gene transactivation. Virology 2014, 468–470, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Clem, R.J. Baculoviruses and apoptosis: A diversity of genes and responses. Curr. Drug Target 2007, 8, 1069–1074. [Google Scholar] [CrossRef]
- Carpes, M.P.; de Castro, M.E.B.; Soares, E.F.; Villela, A.G.; Pinedo, F.J.R.; Ribeiro, B.M. The inhibitor of apoptosis gene (iap-3) of Anticarsia gemmatalis multicapsid nucleopolyhedrovirus (AgMNPV) encodes a functional IAP. Arch. Virol. 2005, 150, 1549–1562. [Google Scholar] [CrossRef] [PubMed]
- Kool, M.; Ahrens, C.H.; Goldbach, R.W.; Rohrmann, G.F.; Vlak, J.M. Identification of genes involved in DNA replication of the Autographa californica baculovirus. Proc. Natl. Acad. Sci. USA 1994, 91, 11212–11216. [Google Scholar] [CrossRef]
- Guarino, L.A.; Xu, B.; Jin, J.; Dong, W. A virus-encoded RNA polymerase purified from baculovirus-infected cells. J. Virol. 1998, 72, 7985–7991. [Google Scholar] [PubMed]
- Beniya, H.; Funk, C.J.; Rohrmann, G.F.; Weaver, R.F. Purification of a virus-induced RNA polymerase from Autographa californica nuclear polyhedrosis virus-infected Spodoptera frugiperda cells that accurately initiates late and very late transcription in vitro. Virology 1996, 216, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Carson, D.D.; Guarino, L.A.; Summers, M.D. Functional mapping of an AcNPV immediately early gene which augments expression of the IE-1 trans-activated 39K gene. Virology 1988, 162, 444–451. [Google Scholar] [CrossRef]
- Guarino, L.A.; Smith, M. Regulation of delayed-early gene transcription by dual TATA boxes. J. Virol. 1992, 66, 3733–3739. [Google Scholar] [PubMed]
- Pullen, S.S.; Friesen, P.D. The CAGT motif functions as an initiator element during early transcription of the baculovirus transregulator IE-1. J. Virol. 1995, 69, 3575–3583. [Google Scholar] [PubMed]
- Chen, Y.R.; Zhong, S.; Fei, Z.; Hashimoto, Y.; Xiang, J.Z.; Zhang, S.; Blissard, G.W. The transcriptome of the baculovirus Autographa californica multiple nucleopolyhedrovirus in Trichoplusia ni cells. J. Virol. 2013, 87, 6391–6405. [Google Scholar] [CrossRef] [PubMed]
- Blissard, G.W.; Kogan, P.H.; Wei, R.; Rohrmann, G.F. A synthetic early promoter from a baculovirus: Roles of the TATA box and conserved start site CAGT sequence in basal levels of transcription. Virology 1992, 190, 783–793. [Google Scholar] [CrossRef]
- Wang, R.; Deng, F.; Hou, D.; Zhao, Y.; Guo, L.; Wang, H.; Hu, Z. Proteomics of the Autographa californica Nucleopolyhedrovirus Budded Virions. J. Virol. 2010, 84, 7233–7242. [Google Scholar] [CrossRef] [PubMed]
- Glocker, B.; Hoopes, R.R.; Hodges, L.; Rohrmann, G.F. In vitro transcription from baculovirus late gene promoters: Accurate mRNA initiation by nuclear extracts prepared from infected Spodoptera frugiperda cells. J. Virol. 1993, 67, 3771–3776. [Google Scholar] [PubMed]
- Todd, J.W.; Passarelli, A.L.; Miller, L.K. Eighteen baculovirus genes, including lef-11, p35, 39K, and p47, support late gene expression. J. Virol. 1995, 69, 968–974. [Google Scholar] [PubMed]
- Lu, A.; Miller, L.K. The roles of eighteen baculovirus late expression factor genes in transcription and DNA replication. J. Virol. 1995, 69, 975–982. [Google Scholar] [PubMed]
- Morris, T.D.; Miller, L.K. Mutational analysis of a baculovirus major late promoter. Gene 1994, 140, 147–153. [Google Scholar] [CrossRef]
- Xu, B.; Yoo, S.; Guarino, L.A. Differential transcription of baculovirus late and very late promoters: Fractionation of nuclear extracts by phosphocellulose chromatography. J. Virol. 1995, 69, 2912–2917. [Google Scholar] [PubMed]
- Sieburth, P.J.; Maruniak, J.E. Growth characteristics of a continuous cell line from the velvetbean caterpillar, Anticarsia gemmatalis Hübner (Lepidoptera: Noctuidae). In Vitro Cell. Dev. Biol. 1988, 24, 195–198. [Google Scholar] [CrossRef]
- Granados, R.R.; Guoxun, L.; Derksen, A.C.G.; McKenna, K.A. A new insect cell line from Trichoplusia ni (BTI-Tn-5B1-4) susceptible to Trichoplusia ni single enveloped nuclear polyhedrosis virus. J. Invertebr. Pathol. 1994, 64, 260–266. [Google Scholar] [CrossRef]
- Vaughn, J.L.; Goodwin, R.H.; Tompkins, G.J.; McCawley, P. The establishment of two cell lines from the insect Spodoptera frugiperda (Lepidoptera; Noctuidae). In Vitro 1977, 13, 213–217. [Google Scholar] [CrossRef] [PubMed]
- Xu, F.; Lynn, D.E.; Roode, E.C.; Muñoz, D.; van Lent, J.W.M.; Vlak, J.M.; van Oers, M.M. Establishment of a cell line from Chrysodeixis chalcites permissive for Chrysodeixis chalcites and Trichoplusia ni nucleopolyhedrovirus. J. Invertebr. Pathol. 2010, 105, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, R.H.; Tompkins, G.J.; McCawley, P. Gypsy moth cell lines divergent in viral susceptibility. I. Culture and identification. In Vitro 1978, 14, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Grace, T.D. Establishment of a line of cells from the silkworm Bombyx mori. Nature 1967, 216, 613. [Google Scholar] [CrossRef] [PubMed]
- Johnson, D.W.; Maruniak, J.E. Physical Map of Anticarsia gemmatalis Nuclear Polyhedrosis Virus (AgMNPV-2) DNA. J. Gen. Virol. 1989, 70, 1877–1883. [Google Scholar] [CrossRef]
- Ribeiro, B.M.; Gatti, C.D.; Costa, M.H.; Moscardi, F.; Maruniak, J.E.; Possee, R.D.; Zanotto, P.M. Construction of a recombinant Anticarsia gemmatalis nucleopolyhedrovirus (AgMNPV-2D) harbouring the beta-galactosidase gene. Arch. Virol. 2001, 146, 1355–1367. [Google Scholar] [CrossRef] [PubMed]
- de Oliveira, V.C.; da Silva Morgado, F.; Ardisson-Araújo, D.M.P.; Resende, R.O.; Ribeiro, B.M. The silencing suppressor (NSs) protein of the plant virus Tomato spotted wilt virus enhances heterologous protein expression and baculovirus pathogenicity in cells and lepidopteran insects. Arch. Virol. 2015, 160, 2873–2879. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, B.A.; Tibúrcio, V.H.S.; Hallwass, M.; Paes, H.C.; Ribeiro, B.M.; Báo, S.N. Structural and ultrastructural alterations of Malpighian tubules of Anticarsia gemmatalis (Hübner) (Lepidoptera: Noctuidae) larvae infected with different Anticarsia gemmatalis multiple nucleopolyhedrovirus (AgMNPV) recombinant viruses. J. Invertebr. Pathol. 2008, 98, 7–19. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Russell, D.W. Molecular Cloning: A Laboratory Manual—Sambrook & Russel; Argentine, J., Ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2001; Volume 18. [Google Scholar]
- Reed, L.J.; Muench, H. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 1938, 27, 493–497. [Google Scholar] [CrossRef]
- Mukawa, S.; Goto, C. In vivo characterization of a group II nucleopolyhedrovirus isolated from Mamestra brassicae (Lepidoptera: Noctuidae) in Japan. J. Gen. Virol. 2006, 87, 1491–1500. [Google Scholar] [CrossRef] [PubMed]
- Kogan, P.H.; Blissard, G.W. A baculovirus gp64 early promoter is activated by host transcription factor binding to CACGTG and GATA elements. J. Virol. 1994, 68, 813–822. [Google Scholar] [PubMed]
- Glocker, B.; Hoopes, R.R.; Rohrmann, G.F. In vitro transactivation of baculovirus early genes by nuclear extracts from Autographa californica nuclear polyhedrosis virus-infected Spodoptera frugiperda cells. J. Virol. 1992, 66, 3476–3484. [Google Scholar] [PubMed]
- Thiem, S.M.; Miller, L.K. Differential gene expression mediated by late, very late and hybrid baculovirus promoters. Gene 1990, 91, 87–94. [Google Scholar] [CrossRef]
- Kogan, P.H.; Chen, X.; Blissard, G.W. Overlapping TATA-dependent and TATA-independent early promoter activities in the baculovirus gp64 envelope fusion protein gene. J. Virol. 1995, 69, 1452–1461. [Google Scholar] [PubMed]
- Morris, T.D.; Miller, L.K. Promoter influence on baculovirus-mediated gene expression in permissive and nonpermissive insect cell lines. J. Virol. 1992, 66, 7397–7405. [Google Scholar] [PubMed]
- Bleckmann, M.; Schürig, M.; Chen, F.-F.; Yen, Z.-Z.; Lindemann, N.; Meyer, S.; Spehr, J.; van den Heuvel, J. Identification of Essential Genetic Baculoviral Elements for Recombinant Protein Expression by Transactivation in Sf21 Insect Cells. PLoS ONE 2016, 11, e0149424. [Google Scholar] [CrossRef] [PubMed]
- Bleckmann, M.; Fritz, M.H.-Y.; Bhuju, S.; Jarek, M.; Schürig, M.; Geffers, R.; Benes, V.; Besir, H.; van den Heuvel, J. Genomic Analysis and Isolation of RNA Polymerase II Dependent Promoters from Spodoptera frugiperda. PLoS ONE 2015, 10, e0132898. [Google Scholar] [CrossRef] [PubMed]
- George, S.; Jauhar, A.M.; Mackenzie, J.; Kieβlich, S.; Aucoin, M.G. Temporal characterization of protein production levels from baculovirus vectors coding for GFP and RFP genes under non-conventional promoter control. Biotechnol. Bioeng. 2015, 112, 1822–1831. [Google Scholar] [CrossRef] [PubMed]
- Maier, T.; Güell, M.; Serrano, L. Correlation of mRNA and protein in complex biological samples. FEBS Lett. 2009, 583, 3966–3973. [Google Scholar] [CrossRef] [PubMed]
- Guarino, L.A.; Summers, M.D. Nucleotide sequence and temporal expression of a baculovirus regulatory gene. J. Virol. 1987, 61, 2091–2099. [Google Scholar] [PubMed]
- Kawasaki, Y.; Matsumoto, S.; Nagamine, T. Analysis of baculovirus IE1 in living cells: Dynamics and spatial relationships to viral structural proteins. J. Gen. Virol. 2004, 85, 3575–3583. [Google Scholar] [CrossRef] [PubMed]
- Nagamine, T.; Kawasaki, Y.; Iizuka, T.; Matsumoto, S. Focal distribution of baculovirus IE1 triggered by its binding to the hr DNA elements. J. Virol. 2005, 79, 39–46. [Google Scholar] [CrossRef] [PubMed]
- Granados, R.R.; Lawler, K.A. In vivo pathway of Autographa californica baculovirus invasion and infection. Virology 1981, 108, 297–308. [Google Scholar] [CrossRef]
- Blissard, G.W.; Rohrmann, G.F. Location, sequence, transcriptional mapping, and temporal expression of the gp64 envelope glycoprotein gene of the Orgyia pseudotsugata multicapsid nuclear polyhedrosis virus. Virology 1989, 170, 537–555. [Google Scholar] [CrossRef]
- Oomens, A.G.; Monsma, S.A.; Blissard, G.W. The baculovirus GP64 envelope fusion protein: Synthesis, oligomerization, and processing. Virology 1995, 209, 592–603. [Google Scholar] [CrossRef] [PubMed]
- Washburn, J.O.; Chan, E.Y.; Volkman, L.E.; Aumiller, J.J.; Jarvis, D.L. Early synthesis of budded virus envelope fusion protein GP64 enhances Autographa californica multicapsid nucleopolyhedrovirus virulence in orally infected Heliothis virescens. J. Virol. 2003, 77, 280–290. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, D.L.; Garcia, A. Biosynthesis and processing of the Autographa californica nuclear polyhedrosis virus gp64 protein. Virology 1994, 205, 300–313. [Google Scholar] [CrossRef] [PubMed]
- Passarelli, A.L.; Miller, L.K. Identification and characterization of lef-1, a baculovirus gene involved in late and very late gene expression. J. Virol. 1993, 67, 3481–3488. [Google Scholar] [PubMed]
- Todd, J.W.; Passarelli, A.L.; Lu, A.; Miller, L.K. Factors regulating baculovirus late and very late gene expression in transient-expression assays. J. Virol. 1996, 70, 2307–2317. [Google Scholar] [PubMed]
- Wilson, M.E.; Mainprize, T.H.; Friesen, P.D.; Miller, L.K. Location, transcription, and sequence of a baculovirus gene encoding a small arginine-rich polypeptide. J. Virol. 1987, 61, 661–666. [Google Scholar] [PubMed]
- Ishiyama, S.; Ikeda, M. High-level expression and improved folding of proteins by using the vp39 late promoter enhanced with homologous DNA regions. Biotechnol. Lett. 2010, 32, 1637–1647. [Google Scholar] [CrossRef] [PubMed]
- Possee, R.D.; Howard, S.C. Analysis of the polyhedrin gene promoter of the Autographa californica nuclear polyhedrosis virus. Nucleic Acids Res. 1987, 15, 10233–10248. [Google Scholar] [CrossRef] [PubMed]
- Rankin, C.; Ooi, B.G.; Miller, L.K. Eight base pairs encompassing the transcriptional start point are the major determinant for baculovirus polyhedrin gene expression. Gene 1988, 70, 39–49. [Google Scholar] [CrossRef]
- Thiem, S.M.; Miller, L.K. Identification, sequence, and transcriptional mapping of the major capsid protein gene of the baculovirus Autographa californica nuclear polyhedrosis virus. J. Virol. 1989, 63, 2008–2018. [Google Scholar] [PubMed]
- Bonning, B.C.; Roelvink, P.W.; Vlak, J.M.; Possee, R.D.; Hammock, B.D. Superior expression of juvenile hormone esterase and beta-galactosidase from the basic protein promoter of Autographa californica nuclear polyhedrosis virus compared to the p10 protein and polyhedrin promoters. J. Gen. Virol. 1994, 75 (Pt 7), 1551–1556. [Google Scholar] [CrossRef] [PubMed]
- Coulibaly, F.; Chiu, E.; Gutmann, S.; Rajendran, C.; Haebel, P.W.; Ikeda, K.; Mori, H.; Ward, V.K.; Schulze-Briese, C.; Metcalf, P. The atomic structure of baculovirus polyhedra reveals the independent emergence of infectious crystals in DNA and RNA viruses. Proc. Natl. Acad. Sci. USA 2009, 106, 22205–22210. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.S.; Chang, I.-S.; Huang, L.-W.; Chen, P.-C.; Wen, C.-C.; Liu, S.-C.; Chien, L.-C.; Lin, C.-Y.; Hsiung, C.A.; Juang, J.-L. Temporal transcription program of recombinant Autographa californica multiple nucleopolyhedrosis virus. J. Virol. 2006, 80, 8989–8999. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, J.V.; de Brito, A.F.; Braconi, C.T.; de Melo Freire, C.C.; Iamarino, A.; de Andrade Zanotto, P.M. Modularity and evolutionary constraints in a baculovirus gene regulatory network. BMC Syst. Biol. 2013, 7, 87. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.H.; Senthil Kumar, C.M.; Arif, B.M.; Krell, P.J.; Zhang, C.X.; Cheng, X.W. Cell-dependent production of polyhedra and virion occlusion of Autographa californica multiple nucleopolyhedrovirus fp25k mutants in vitro and in vivo. J. Gen. Virol. 2013, 94, 177–186. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.-H.; Hillman, C.C.; Zhang, C.-X.; Cheng, X.-W. Reduction of polyhedrin mRNA and protein expression levels in Sf9 and Hi5 cell lines, but not in Sf21 cells, infected with Autographa californica multiple nucleopolyhedrovirus fp25k mutants. J. Gen. Virol. 2013, 94, 166–176. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Thiem, S.M. Responses of insect cells to baculovirus infection: Protein synthesis shutdown and apoptosis. J. Virol. 1997, 71, 7866–7872. [Google Scholar] [PubMed]
- Thiem, S.M.; Du, X.; Quentin, M.E.; Berner, M.M. Identification of baculovirus gene that promotes Autographa californica nuclear polyhedrosis virus replication in a nonpermissive insect cell line. J. Virol. 1996, 70, 2221–2229. [Google Scholar] [PubMed]
- Morris, T.D.; Miller, L.K. Characterization of Productive and Non-productive ACMNPV Infection in Selected Insect Cell Lines. Virology 1993, 197, 339–348. [Google Scholar] [CrossRef] [PubMed]
- Du, X.; Thiem, S.M. Characterization of Host Range Factor 1 (hrf-1) Expression in Lymantria dispar M Nucleopolyhedrovirus- and Recombinant Autographa californica M Nucleopolyhedrovirus-Infected IPLB-Ld652Y Cells. Virology 1997, 227, 420–430. [Google Scholar] [CrossRef] [PubMed]
- Thiem, S.M.; Chejanovsky, N. The role of baculovirus apoptotic suppressors in AcMNPV-mediated translation arrest in Ld652Y cells. Virology 2004, 319, 292–305. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, B.; Burma, S.; Talwar, G.P.; Hasnain, S.E. Transcriptional regulation of cell line-dependent, baculovirus-mediated expression of foreign genes. DNA Cell Biol. 1995, 14, 7–14. [Google Scholar] [CrossRef] [PubMed]
- Argaud, O.; Lopez-Ferber, M.; Croizier, L.; Croizier, G. Two key mutations in the host-range specificity domain of the p143 gene of Autographa californica nucleopolyhedrovirus are required to kill Bombyx mori larvae. J. Gen. Virol. 1998, 79, 931–935. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target | Primers | Primer Sequence | AgMNPV Genome Coordinates | Amplicon Size (bp) |
|---|---|---|---|---|
| prIE1 | FWRHind | CCCCAAGCTTGAATTGTCGGTGAGCGTTGCGCGT | 121,218−121,652 | 466 |
| REV | GCTATGCACGCGCAATCCG | |||
| prGP64 | FWR | GGATCCATTTTGATGAAGGTCTT | 106,097–106,446 | 360 |
| REV | AGATCTTTGTTATGTCTTGTAGC | |||
| prLEF1 | FWR | GTTGCGGCTTGACCACGG | 14,848–15,104 | 260 |
| REV | GGATCCTTGTAGGGCGTCTA | |||
| prVP39 | FWRHind | AAGCAAGCTTTTTCGCGCCACACAAGCGGCACCAACG | 71,414–71,703 | 290 |
| REVXma | TTACCCGGGTTTGCTACAATGGACGACTTTGTGATT | |||
| prP6.9 | FWRHind | ACTGAAGCTTTCGCCAGCCCTGTGATGCGTTACG | 81,693–82,175 | 483 |
| REVXma | ATTACCCGGGAAGTGTTTTACAATGTAGCTTTAA | |||
| prPOLH | FWRHind | AAGCAAGCTTATTTGGAGTGTTTGTACGATT | 729–1149 | 421 |
| REVXma | ATTACCCGGGAGTTATAGCAAATTTTACTACAAAG | |||
| fluc | qPCRFWR | AAACGCTGGGCGTTAATCAG | - | 145 |
| qPCRREV | TCGTCCCAGTAAGCTATGTC | |||
| fluc | FLUCFWR | GATTTAGGTGACACTATAG | - | 1807 |
| FLUCREV | AAGGGATCCAGCTCGATTTATTCGACCTCGA |
| Cell Line | Virus | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IE1FLUC | GP64FLUC | LEF1FLUC | VP39FLUC | P6.9FLUC | POLHFLUC | |||||||
| Avg | SD | Avg | SD | Avg | SD | Avg | SD | Avg | SD | Avg | SD | |
| UFLAg | 1:58 | 0:13 | 2:03 | 0:18 | 3:42 | 0:05 | 5:35 | 0:39 | 10:10 | 0:08 | 12:49 | 0:13 |
| Tn5B | 0:27 | 0:05 | 0:32 | 0:02 | 2:15 | 0:10 | 2:03 | 0:12 | 9:08 | 0:28 | 13:17 | 0:48 |
| Sf9 | 2:03 | 0:13 | 2:03 | 0:04 | 3:51 | 0:03 | 6:39 | 0:11 | 15:08 | 1:36 | 16:53 | 0:55 |
| Ld652Y | 11:54 | 0:34 | 13:19 | 0:16 | - | - | 17:07 | 2:22 | 17:25 | 1:06 | - | - |
| Bm5 | 8:30 | 0:04 | 6:46 | 0:36 | - | - | - | - | - | - | - | - |
| Chch | 1:46 | 0:12 | 1:55 | 0:08 | 18:09 | 1:38 | 8:52 | 0:18 | 13:47 | 0:33 | 17:38 | 0:56 |
| Cell Line | Virus | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IE1FLUC | GP64FLUC | LEF1FLUC | VP39FLUC | P6.9FLUC | POLHFLUC | |||||||
| Avg | SD | Avg | SD | Avg | SD | Avg | SD | Avg | SD | Avg | SD | |
| UFLAg | 32:15 | 3:18 | 31:58 | 3:21 | 18:41 | 10:30 | 36:58 | 1:55 | 34:54 | 1:24 | 59:24 | 0:26 |
| Tn5B | 66:25 | 0:14 | 65:19 | 2:29 | 12:31 | 3:01 | 36:05 | 1:09 | 38:26 | 1:32 | 72:10 | 0:55 |
| Sf9 | 63:09 | 1:56 | 76:49 | 8:52 | 12:45 | 0:50 | 76:01 | 8:19 | 89:47 | 4:01 | 88:59 | 1:16 |
| Ld652Y | 109:18 | 2:24 | 110:59 | 0:16 | - | - | 90:30 | 0:41 | 74:45 | 11:20 | - | - |
| Bm5 | 57:33 | 30:20 | 17:57 | 1:14 | - | - | - | - | - | - | - | - |
| Chch | 63:22 | 1:40 | 52:39 | 6:51 | 46:13 | 3:57 | 49:57 | 0:54 | 70:01 | 2:59 | 77:27 | 10:59 |
| Cell Line | Virus | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| IE1FLUC | GP64FLUC | LEF1FLUC | VP39FLUC | P6.9FLUC | POLHFLUC | |||||||
| Avg | SD | Avg | SD | Avg | SD | Avg | SD | Avg | SD | Avg | SD | |
| UFLAg | 804 | 94 | 1998 | 91 | 130 | 5 | 27170 | 1512 | 22339 | 2278 | 13997 | 601 |
| Tn5B | 10768 | 533 | 10209 | 780 | 204 | 16 | 72005 | 1483 | 68130 | 2111 | 24660 | 1617 |
| Sf9 | 7087 | 1187 | 583 | 19 | 158 | 11 | 4210 | 1250 | 2875 | 255 | 387 | 61 |
| Ld652Y | 4652 | 625 | 1210 | 137 | 18 | 1 | 9562 | 883 | 6483 | 2329 | 17 | 0 |
| Bm5 | 69 | 28 | 50 | 3 | 22 | 2 | 19 | 1 | 21 | 3 | 18 | 1 |
| Chch | 8257 | 726 | 739 | 25 | 52 | 1 | 5651 | 786 | 15364 | 4115 | 2586 | 354 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Morgado, F.D.S.; Ardisson-Araújo, D.M.P.; Ribeiro, B.M. Real-Time Expression Analysis of Selected Anticarsia gemmatalis multiple nucleopolyhedrovirus Gene Promoters during Infection of Permissive, Semipermissive and Nonpermissive Cell Lines. Viruses 2017, 9, 132. https://doi.org/10.3390/v9060132
Morgado FDS, Ardisson-Araújo DMP, Ribeiro BM. Real-Time Expression Analysis of Selected Anticarsia gemmatalis multiple nucleopolyhedrovirus Gene Promoters during Infection of Permissive, Semipermissive and Nonpermissive Cell Lines. Viruses. 2017; 9(6):132. https://doi.org/10.3390/v9060132
Chicago/Turabian StyleMorgado, Fabricio Da Silva, Daniel Mendes Pereira Ardisson-Araújo, and Bergmann Morais Ribeiro. 2017. "Real-Time Expression Analysis of Selected Anticarsia gemmatalis multiple nucleopolyhedrovirus Gene Promoters during Infection of Permissive, Semipermissive and Nonpermissive Cell Lines" Viruses 9, no. 6: 132. https://doi.org/10.3390/v9060132