1. Introduction
The Picornaviridae family of viruses is a group of small, non-enveloped viruses that contain single-stranded, positive-polarity RNA genomes. Picornaviruses are significant pathogens of humans because they are widespread and capable of causing serious diseases such as poliomyelitis, hepatitis, meningitis, and encephalitis as well as less serious diseases including the common cold. The enteroviruses are a single genera within the picornavirus family, of which poliovirus is the type species. Due to the inherent limited protein coding capacity of their small RNA genomes, enteroviruses require the functions of cellular proteins to complete their infectious cycle. Because enteroviral replication is composed of a series of discrete steps that demand particular protein functions, there are dynamic changes to the composition of ribonucleoprotein (RNP) complexes throughout this cycle.
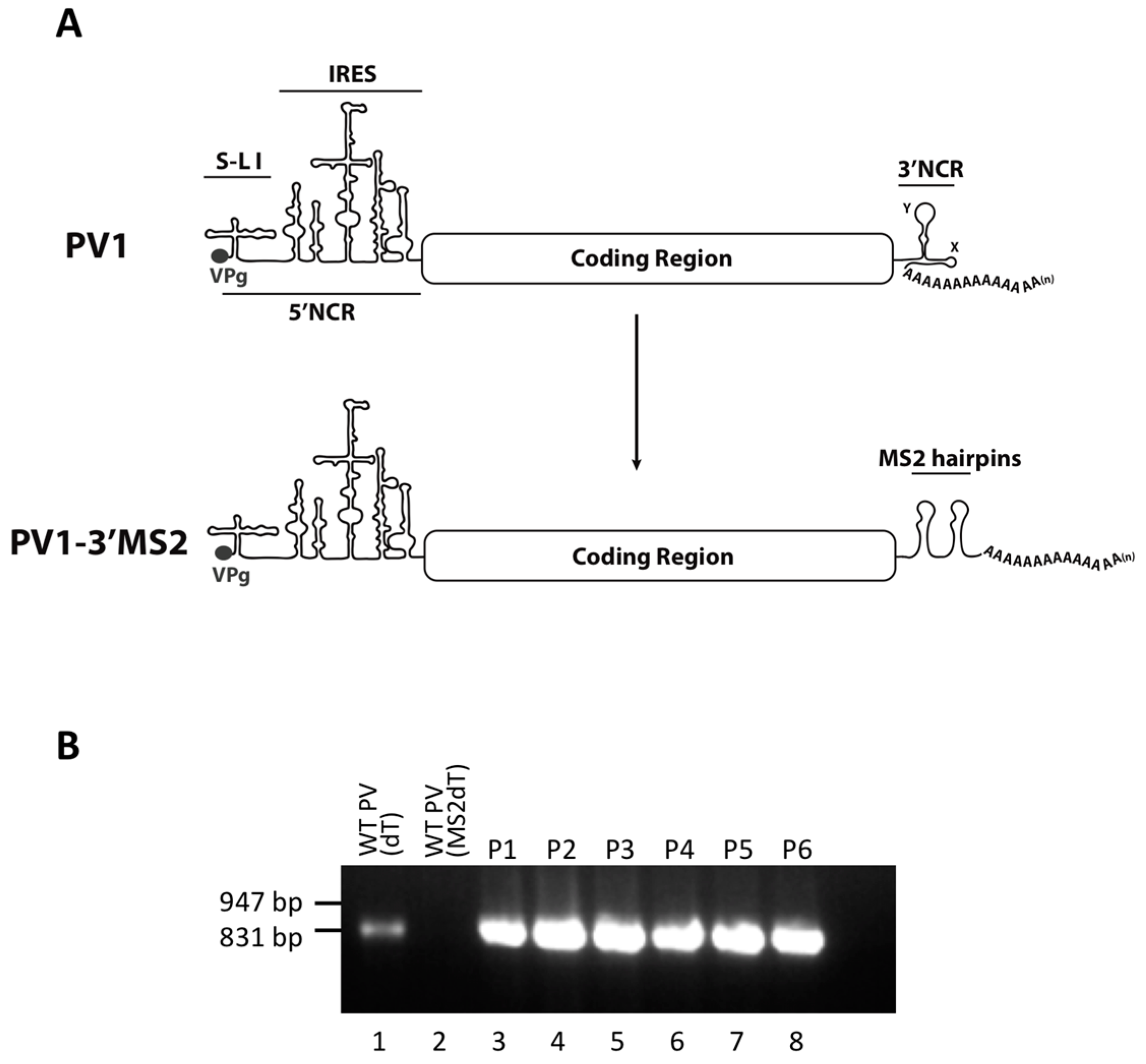
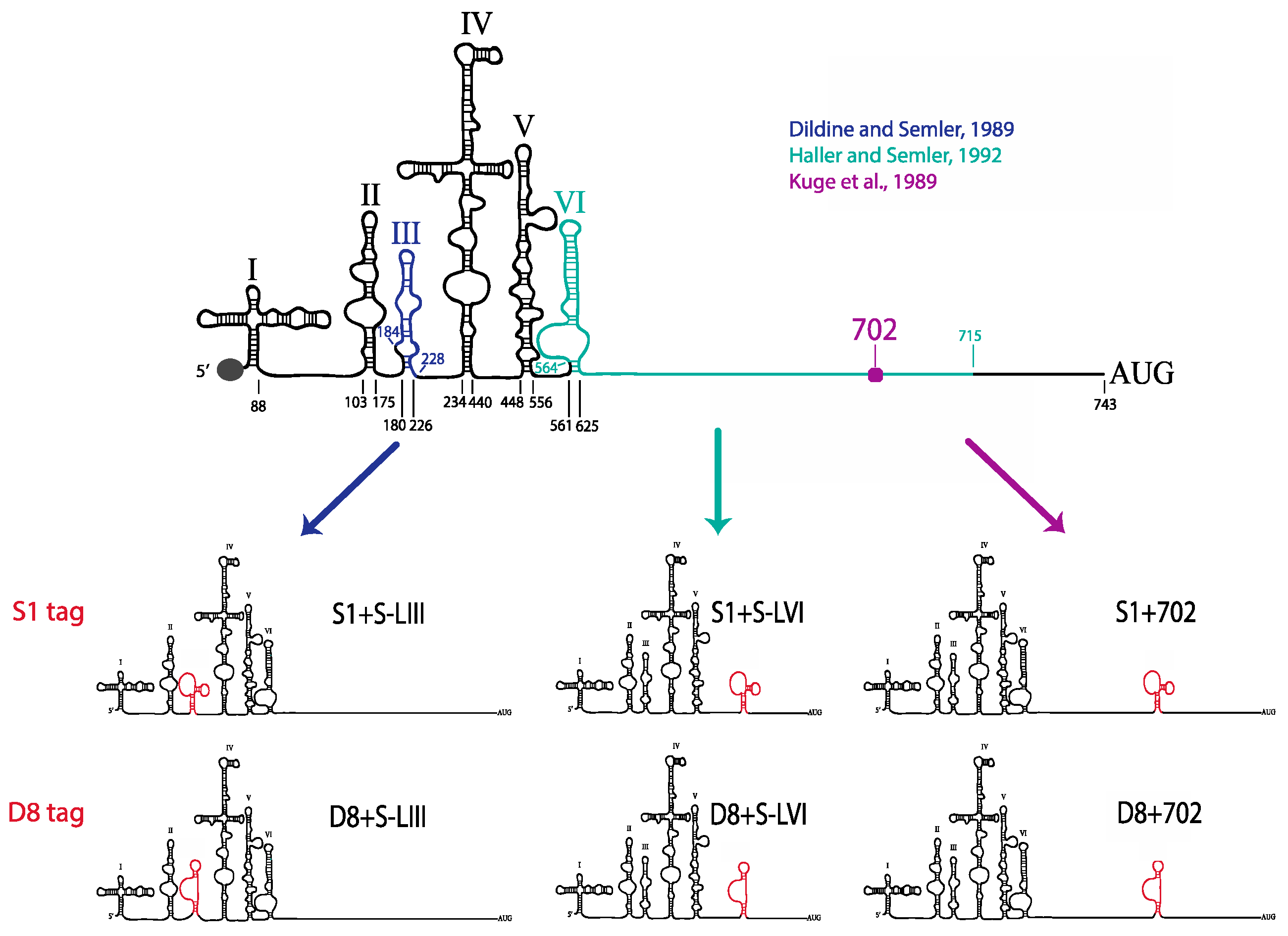
Much of what is known about the identity of cellular proteins that are usurped during the replication cycle of enteroviruses is a result of studies involving poliovirus. The poliovirus genome consists of a small viral protein (VPg) covalently linked to the RNA at the very 5′ terminus followed by a relatively long (742 nucleotide) and highly structured 5′ noncoding region (5′NCR). There are six stem-loop (S-L I-VI) structures within the 5′NCR, with the internal ribosome entry site (IRES) comprised of S-L II-VI. Downstream of the 5′NCR the poliovirus genome encodes a single open reading frame. The 3′ region of the genome contains the ~75 nucleotide 3′ noncoding region (3′NCR), made up of two predicted stem-loop structures called X and Y, and the genetically encoded poly(A) tract of ~60 nucleotides [
1]. The function of the 3′NCR is not clear, but the poly(A) tract is required for infectivity and is the putative binding site for the viral RNA-dependent RNA polymerase (3D
pol) during initiation of negative-sense RNA synthesis [
2,
3].
Following cellular entry and uncoating, the initial step in the replication cycle of poliovirus is the translation of the ~7500 nucleotide genomic RNA molecule in the cytoplasm of the infected cell. Unlike cellular mRNAs, the poliovirus genome lacks a 5′ 7-methylguanosine cap structure and relies on cap-independent, IRES-mediated translation resulting in the production of a single 250-kDa polyprotein. The polyprotein is proteolytically processed by viral proteinases to produce 11 mature proteins, as well as intermediate precursor proteins, which have distinct functions. In addition to generating the proteins required for viral RNA replication directly, translation of the viral genome also produces proteins that alter the infected cell to support conditions required for viral RNA synthesis. This includes induction of membranous structures that originate from the secretory pathway and/or autophagosomal pathways during infection [
4,
5,
6,
7,
8,
9,
10]. Viral RNA is synthesized in close association with these membranous structures induced during infection, and are together known as replication complexes [
11].
Once sufficient levels of viral proteins have been produced, the genomic RNA that was a template for translation is subsequently used as a template for the generation of viral RNA molecules. This involves a template usage switch that is dependent, in part, upon cleavage of the host-cell protein polypyrimidine tract-binding protein 1 (PTB1) and/or poly(rC)-binding protein 2 (PCBP2), by the viral proteinase 3CD [
12,
13]. Poliovirus RNA replication can be divided into two distinct stages: (i) the production of negative-sense RNA intermediates from genomic RNA templates and (ii) the synthesis of nascent genomic RNAs (positive-sense RNA molecules) from negative-sense templates. Due to the asymmetric nature of enterovirus RNA replication, the ratio of positive- to negative-sense RNAs within an infected cell is approximately 50:1 [
14,
15,
16]. Consequently, the majority of viral RNA in infected cells is single-stranded and positive-sense. Poliovirus RNA replication is dependent upon the formation of ribonucleoprotein (RNP) complexes that result from interactions between structured regions of the viral RNA molecule and proteins of host and viral origin. The RNA structures that nucleate various RNP complexes are found in the positive-sense RNA within in the 5′ NCR, the
cis-acting replication element (
cre) (a hairpin structure found within the coding region of 2C involved in the initiation of RNA synthesis), and the 3′NCR. RNP complexes function in stimulating viral RNA replication, through direct or indirect recruitment of 3D
pol, and are thought to be the determinants of 3D
pol template specificity.
The RNA structural element known as the cloverleaf or S-L I, located at the very 5′ terminus of poliovirus genomic RNA, is required for efficient synthesis of negative-sense viral RNA, by acting as a scaffold for protein associations [
14,
17,
18]. Poliovirus proteinase/polymerase precursor 3CD and host protein PCBP2 interact with the S-L I structure forming a ternary complex on the 5′ terminus of the template [
19,
20,
21]. On the opposite end of the same RNA molecule, poly(A)-binding protein 1 (PABP1) associates with the 3′ poly(A) tract. Because PABP1 has been shown to interact with both PCBP2 as well as 3CD, the current model of negative-sense RNA synthesis is based on the circularization of the genomic template through interaction of the proteins present in the ternary complex and PABP1 [
22]. End-to-end interaction is predicted to bring 3D
pol (perhaps in the form of 3CD that is bound to S-L I) into close proximity to the site of replication initiation at the 3′ terminus of the template RNA. Many features of this model have been verified
in vitro, and the specificity of 3D
pol for viral RNA templates containing these RNP complexes is bolstered by the requirement for translation in
cis,
i.e., a viral RNA template must be translated before being used for RNA replication. As a result, RNA templates enter the replication cycle already associated with various cellular proteins that function in the translation of the viral genome, and may also be required for efficient RNA replication [
23]. Because negative-sense RNA templates are not translated, they likely have a unique set of viral and host protein requirements compared to their genomic RNA counterparts. Previous studies have identified a cellular protein that promotes positive-sense RNA synthesis: heterogeneous nuclear ribonucleoprotein C1/C2 (hnRNP C1/C2). hnRNP C1/C2 interacts with RNA sequences that correspond to both the 5′ and 3′ terminal regions of negative-sense RNA intermediates (regions complementary to the 3′NCR and 5′NCR of genomic RNA, respectively) [
24,
25,
26]. The association of hnRNP C1/C2 with both termini of the negative-sense RNA molecule has been proposed to promote the end-to-end interaction of this RNA species through multimerization of hnRNP C1/C2 isoforms [
27]. Newly synthesized positive-sense RNA molecules are subsequently used as templates for translation, recycled back into the RNA replication cycle, or packaged into viral capsids producing progeny virions that are released from the cell.
Studies that have contributed to our understanding of enterovirus RNA replication have defined several proteins that interact directly with poliovirus RNA during the process of viral RNA production. However, previous work has relied heavily on in vitro assays and subgenomic poliovirus RNA constructs. To define the RNP complexes using full length genomic RNA in the context of infection, we have generated recombinant polioviruses containing RNA affinity tags within the noncoding regions of the genome. The use of polioviruses possessing stable, specifically isolatable genomes and replication intermediates could ultimately allow for strand-specific RNP complex characterization directly from infected cells, throughout the course of infection. Here we present a biological characterization of these recombinant viruses and demonstrate that exogenous sequence insertions can be stable in the poliovirus genome for multiple passages in cell culture while maintaining wild type-like growth kinetics. Our initial attempts to isolate these genetically-tagged viral RNAs and associated proteins from infected cells has been hampered by high levels of co-purification of nonspecific proteins and the limited binding efficiency between RNA affinity sequences and their respective matrices. Nonetheless, our results provide a foundation for the generation of enteroviruses that could eventually allow for a description of the dynamic changes in protein composition of viral RNP complexes that occur throughout the course of infection, and that reflect the distinct steps of the viral RNA replication cycle.
2. Materials and Methods
2.1. Cell Culture and DNA Constructs
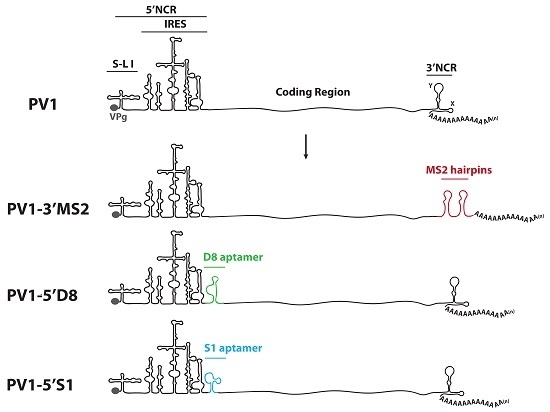
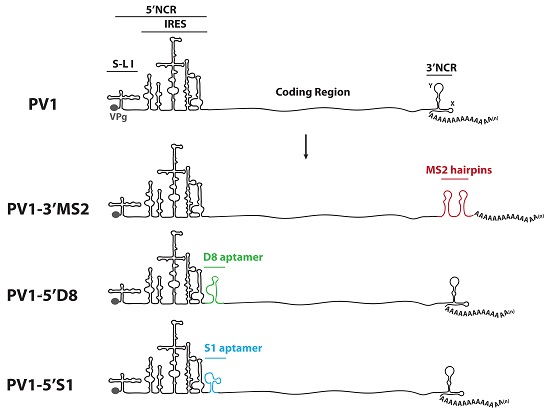
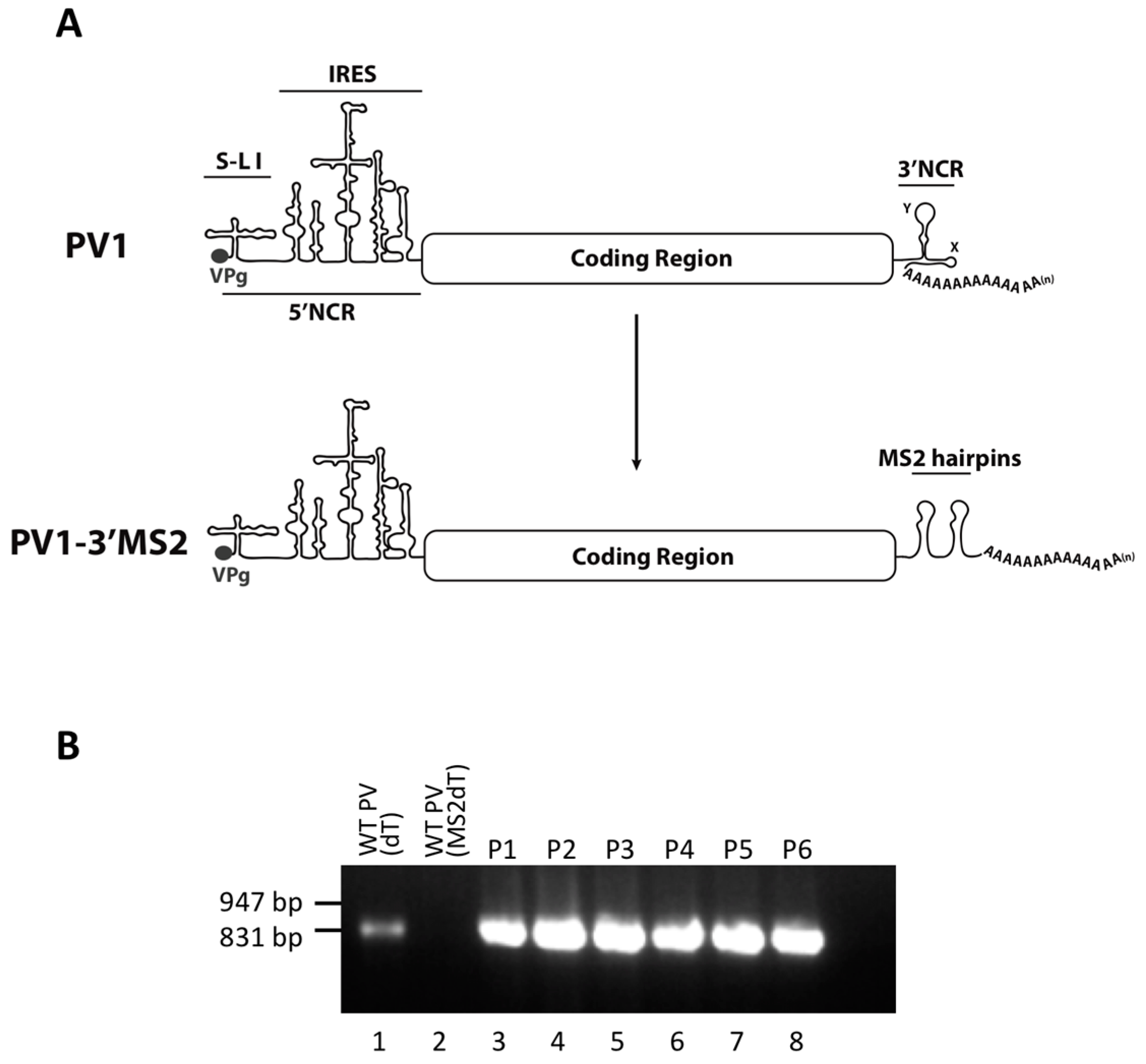
HeLa cells were grown as monolayers in Dulbecco’s Modified Eagle’s Medium (DMEM) or in suspension culture in Spinner Minimal Essential Medium (S-MEM), both supplemented with 8% newborn calf serum (NCS). To generate plasmids encoding MS2 tags in place of the 3′NCR, a HpaI site corresponding to nucleotide position 7378 in pT7PV1 [
28] was engineered into the pT7PV1(∆3′NCR) plasmid [
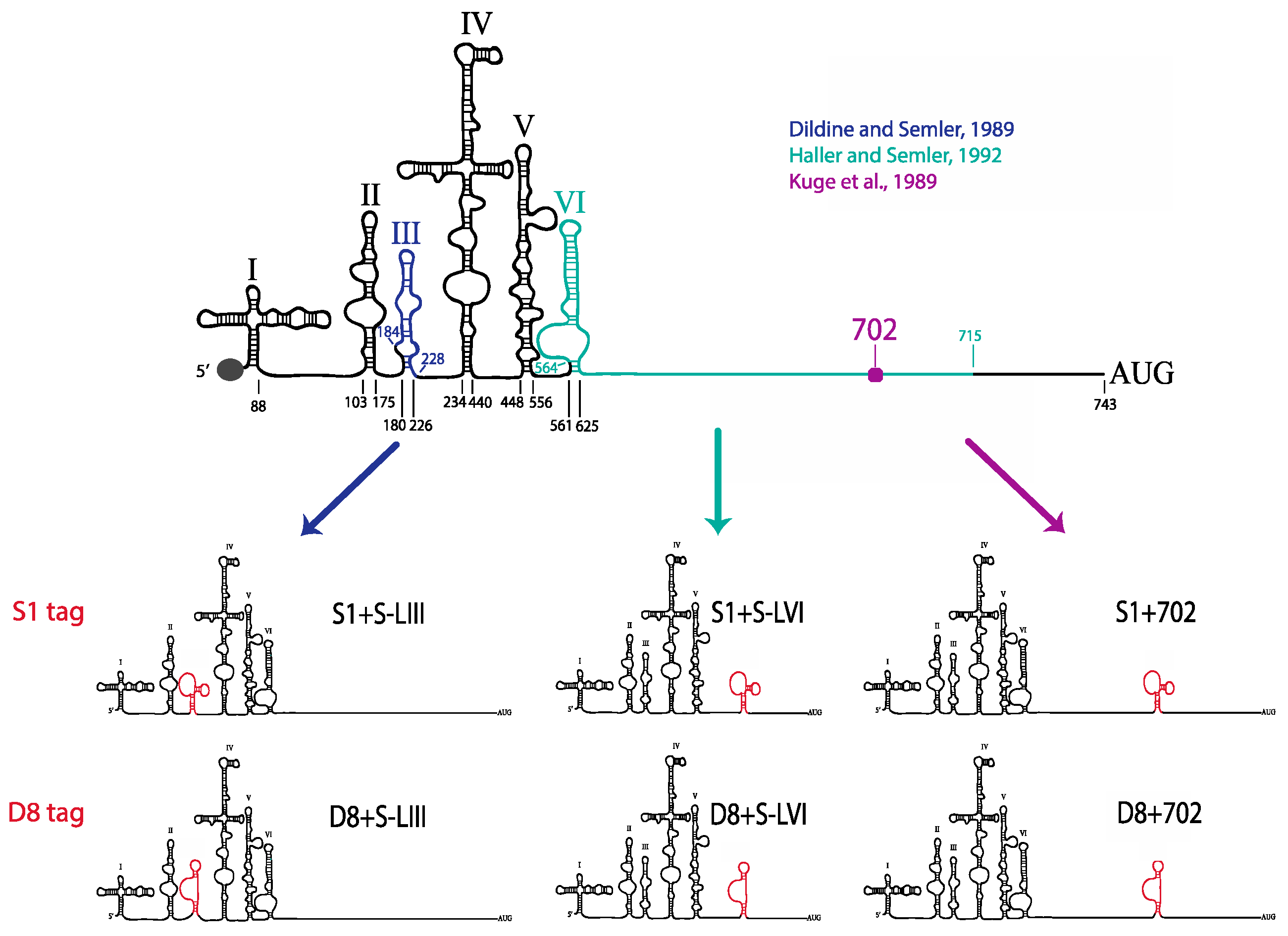
29]. Oligonucleotides corresponding to tandem MS2 hairpins were inserted into pT7PV1(∆3′NCR)HpaI at the HpaI site using blunt end ligation to generate pT7PV1-3′MS2 plasmid (MS2 stem-loop sequence obtained from SP73-βglobin-(MS2)4, kindly provided by Klemens Hertel (University of California, Irvine, CA, USA)). To generate plasmids encoding either the S1 or D8 aptamer tags in forward or reverse orientation, or the modified forms of these aptamers, within the 5′NCR of the poliovirus genome, three separate vectors were used. For constructs containing aptamer tag in place of S-L VI, the X585R plasmid [
30] was engineered to contain an XhoI site at nucleotide position 564 (X585RXhoI). For generating aptamer tags in place of S-L III, and at nucleotide position 702, pT7PV1 was digested with HincII and re-ligated to generate a subclone of this vector. XhoI sites were inserted flanking S-L III or at nucleotide position 702. These subclones, and X585RXhoI, were digested with XhoI then ligated with double stranded oligonucleotides corresponding to each one of the tag sequences containing XhoI recognition sites (
Table 1). Tagged subclones were then ligated into full-length poliovirus cDNA constructs.
Table 1.
Oligonucleotides corresponding to aptamer tag sequences incorporated into the 5′ noncoding region (5′NCR) of the poliovirus genome cDNA.
Table 1.
Oligonucleotides corresponding to aptamer tag sequences incorporated into the 5′ noncoding region (5′NCR) of the poliovirus genome cDNA.
| Affinity Tag | Oligonucleotide Sequence (5′–3′) |
|---|
| S1+ | top: TCGAACCGACCAGAATCATGCAAGTGCGTAAGATAGTCGCGGGCCGGG
bottom: TCGACCCGGCCCGCGACTATCTTACGCACTTGCATGATTCTGGTCGGT |
| S1− | top: TCGACCCGGCCCGCGACTATCTTACGCACTTGCATGATTCTGGTCGGT
bottom: TCGAACCGACCAGAATCATGCAAGTGCGTAAGATAGTCGCGGGCCGGG |
| S1m+ | top: TCGAAAGCGGCCGCCGACCAGAATCATGCAAGTGCGTAAGATAGTCGCGGGTCGGCGGCCGCTT
bottom: TCGAAAGCGGCCGCCGACCCGCGACTATCTTACGCACTTGCATGATTCTGGTCGGCGGCCGCTT |
| S1m− | top: TCGAAAGCGGCCGCCGACCCGCGACTATCTTACGCACTTGCATGATTCTGGTCGGCGGCCGCTT
bottom: TCGAAAGCGGCCGCCGACCAGAATCATGCAAGTGCGTAAGATAGTCGCGGGTCGGCGGCCGCTT |
| D8+ | top: TCGATCCGAGTAATTTACGTTTTGATACGGTTGCGGA
bottom: TCGATCCGCAACCGTATCAAAACGTAAATTACTCGGA |
| D8− | top: TCGATCCGCAACCGTATCAAAACGTAAATTACTCGGA
bottom: TCGATCCGAGTAATTTACGTTTTGATACGGTTGCGGA |
| D8m+ | top: TCGAAAGCGGCCTCCGAGTAATTTACGTTTTGATACGGTTGCGGAGGCCGCTT
bottom: TCGAAAGCGGCCTCCGCAACCGTATCAAAACGTAAATTACTCGGAGGCCGCTT |
| D8m− | top: TCGAAAGCGGCCTCCGCAACCGTATCAAAACGTAAATTACTCGGAGGCCGCTT
bottom: TCGAAAGCGGCCTCCGAGTAATTTACGTTTTGATACGGTTGCGGAGGCCGCTT |
2.2. RNA Constructs and Virus Stocks
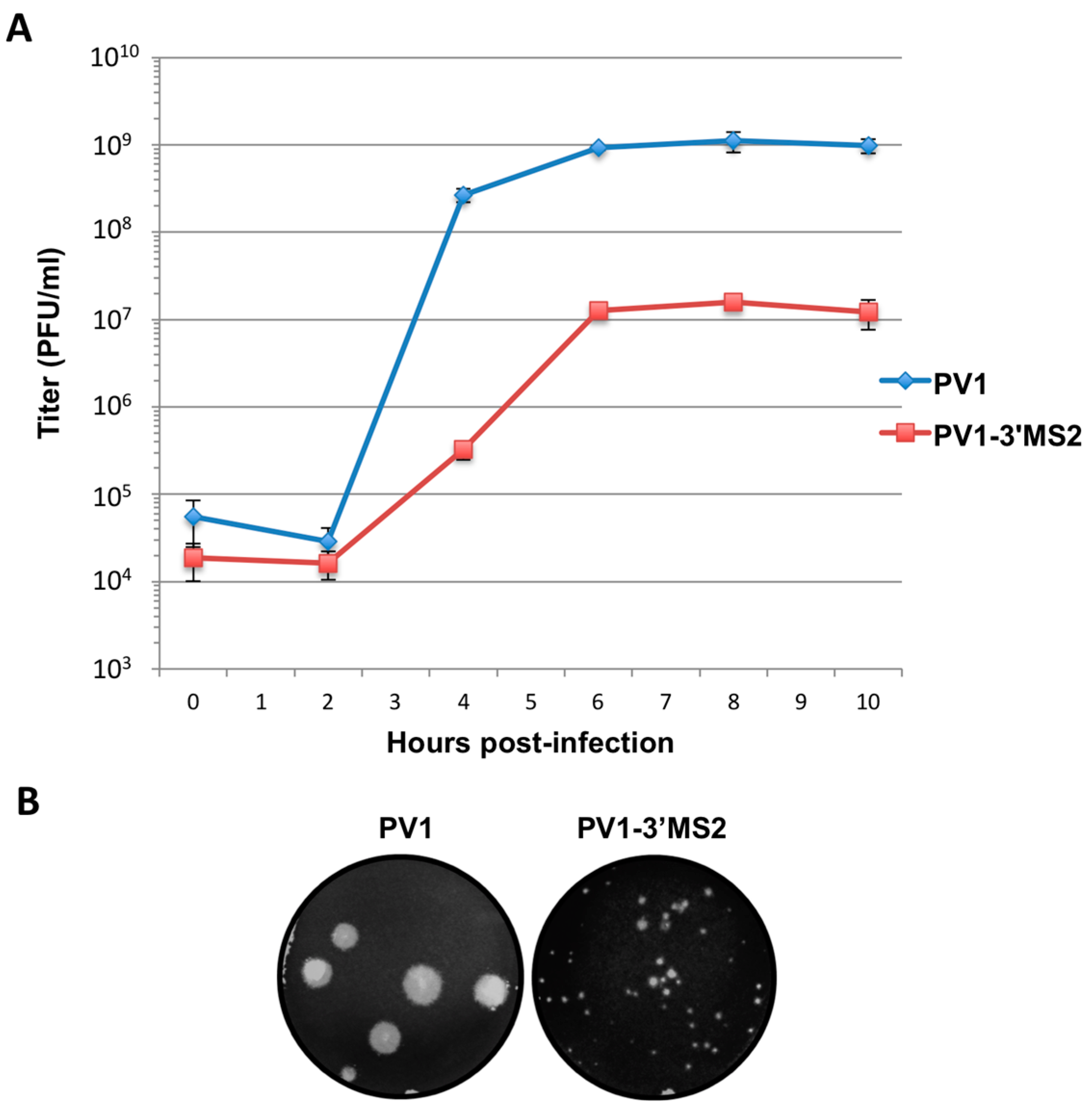
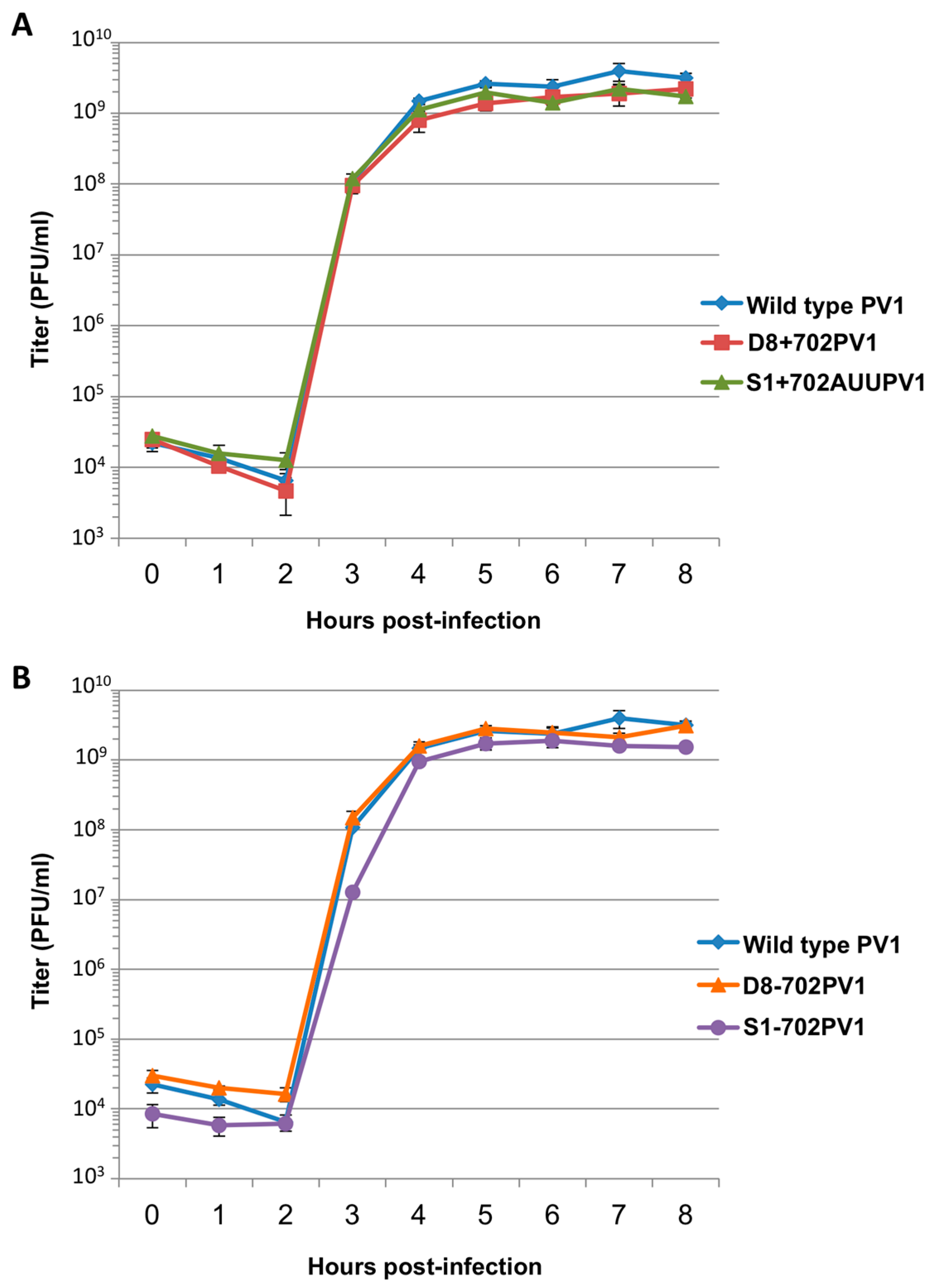
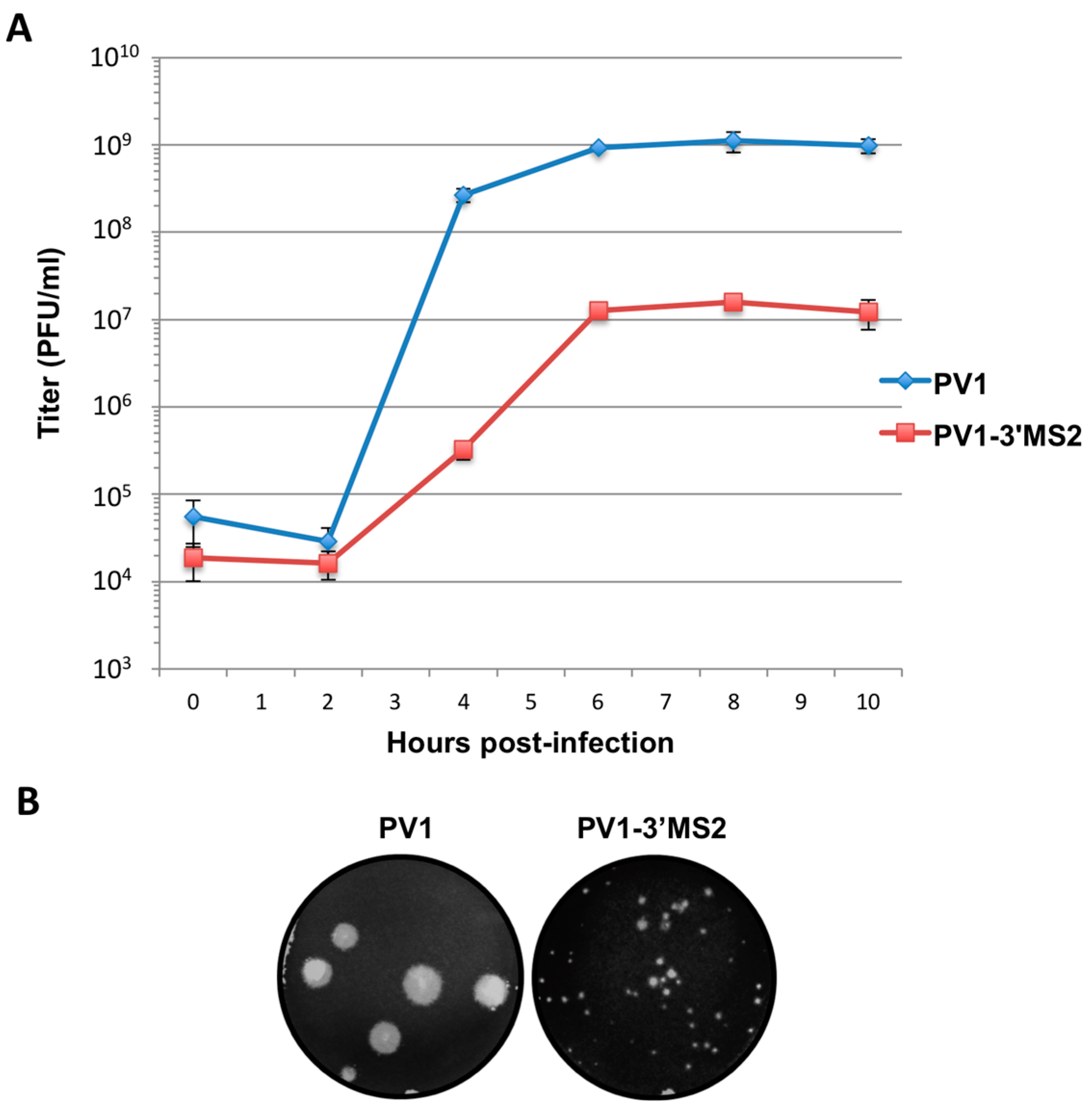
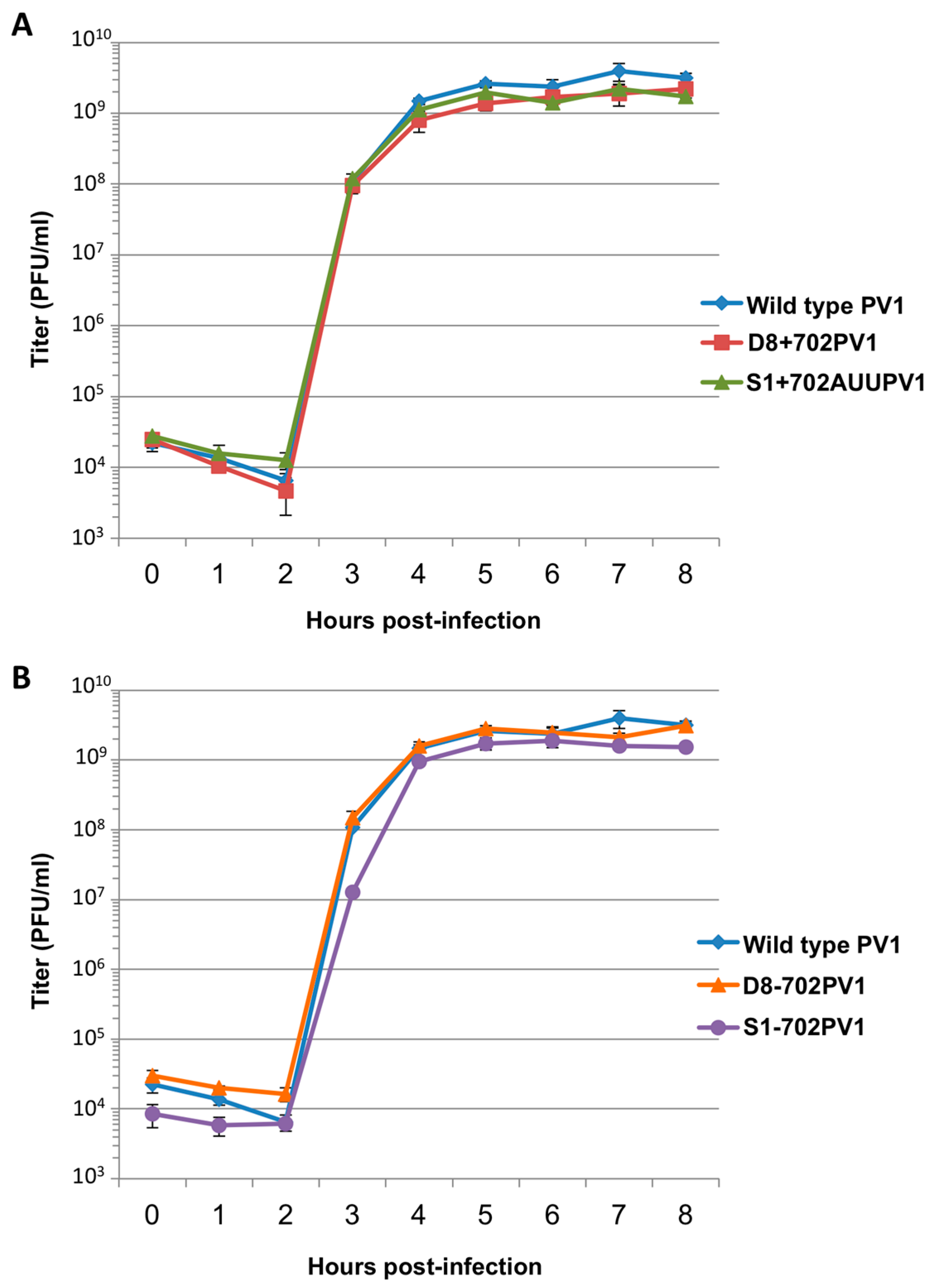
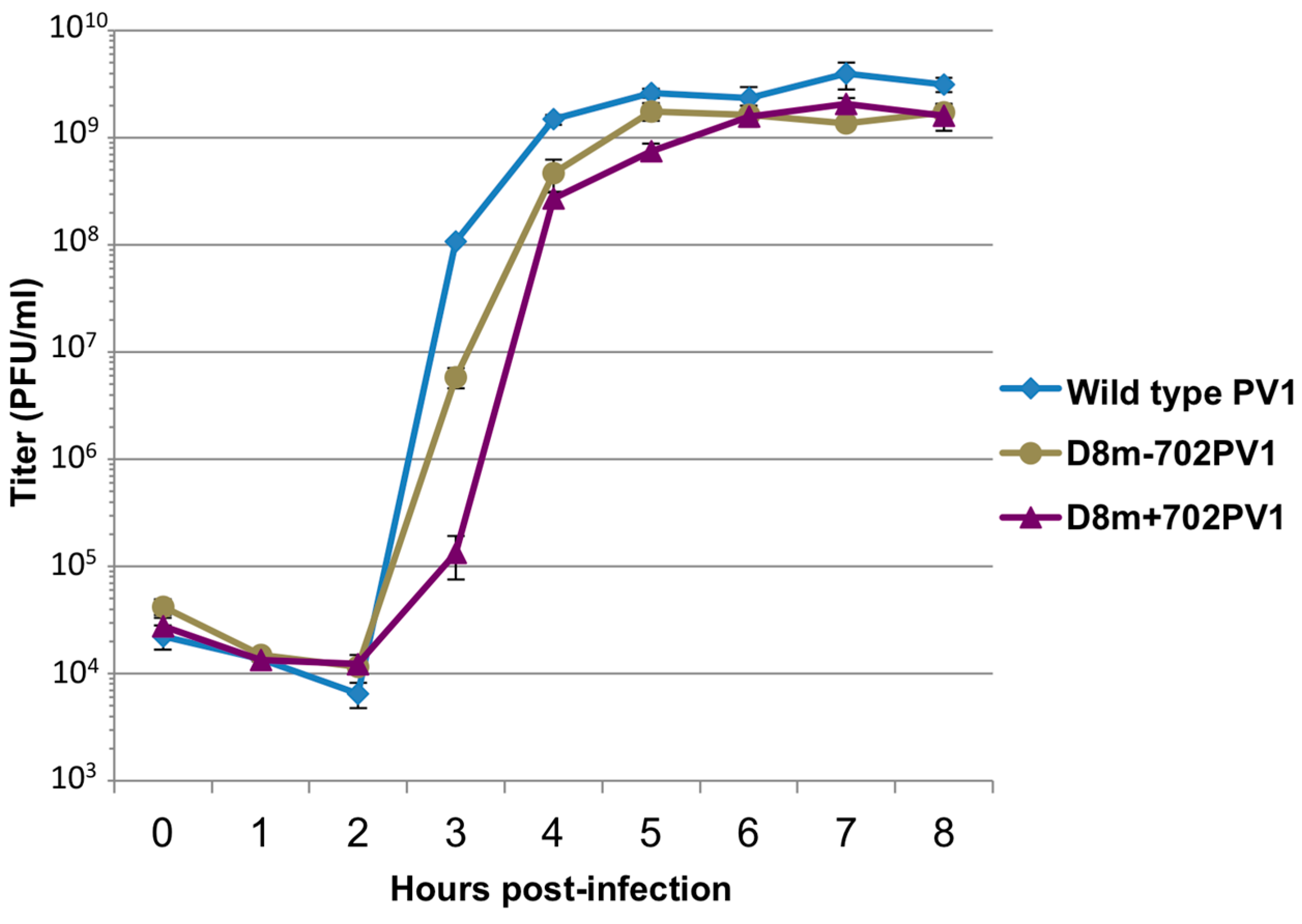
RNA corresponding to full-length PV1 harboring MS2 hairpins or aptamer tags within the 5′NCR was generated by in vitro transcription of plasmids linearized with EcoRI using the MEGAscript T7 transcription kit (Ambion, Waltham, MA). Following transcription, RNA was purified by phenol/chloroform extraction followed by two rounds of ethanol precipitation in the presence of 700 mM ammonium acetate or by using the RNeasy Mini kit (Qiagen, Hilden, Germany). For transfection of RNA into HeLa cells, 1 μg of transcribed RNA was incubated with TS buffer (137 mM NaCl, 4.4 mM KCl, 0.7 mM Na2HPO4, 0.5 mM MgCl2, 0.68 mM CaCl2, 25 mM Tris, pH 7.5) and 1 mg/mL DEAE-Dextran. Cells were washed twice with phosphate buffered saline (PBS) and 250 μL of Diethylaminoethyl(DEAE)-Dextran transfection mixture was added per 20 cm2 plate of HeLa cells. After 30 min incubation at room temperature, DMEM-8% NCS was added and cells were incubated at 37 °C and monitored for cytopathic effects (CPE). CPE was observed after 4 days for MS2-containing RNA transfections or after 1 day for RNAs containing aptamer tags in the 5′NCR. Following detection of CPE, cells and supernatant were collected, subjected to 3-5 freeze-thaw cycles, and used to infect HeLa cell monolayers with a semi-solid agar overlay (DMEM-6% NCS, 0.45% agarose). Single plaque isolates were recovered at 2 (aptamer-tagged RNA transfections) or 4 (MS2-tagged RNA transfections) days post-infection and used to infect fresh HeLa cell monolayers. The resulting virus was designated as passage 1. Virus was amplified by serial passage at a multiplicity of infection (MOI) of 20 in HeLa cell monolayers. Large-scale virus preparations of PV1-3’MS2 were generated for passages 3 and 4 using HeLa suspension cells (400 mL). Cells were pelleted, washed once with PBS, washed once with S-MEM, and infected at an MOI of 0.2 in 370 mL S-MEM. After 30 min adsorption at room temperature, NCS was added to 10% and infection was allowed to proceed at 37 °C for 11 h. Cells were pelleted at ~1500 relative centrifugal units (rcf), resuspended in 25 mL DMEM, and subjected to five freeze-thaw cycles to release virus. Large-scale preparations of D8+/D8−/S1+/S1−/D8m+/D8m−702PV1 were generated for passage 3 stocks using HeLa suspension cells (1 liter). Cells were pelleted, washed once with PBS, resuspended with S-MEM and infected at an MOI of 20. After 30 min adsorption at room temperature, NCS was added to 8% by volume and infection was allowed to proceed for 8 h. Cell cultures and fluids were subjected to three freeze-thaw cycles prior to centrifugation at ~1500 rcf. For single-cycle growth analysis, HeLa cell monolayers were washed twice with PBS and infected with wild type PV1, PV1-3′MS2, or D8+/D8−/S1+/S1−/D8m+/D8m−702PV1 at an MOI of 20 for 30 min at room temperature. Cells were washed twice in PBS, overlaid with DMEM-8% NCS, and incubated at 37 °C. At times indicated, cells and supernatant were collected, subjected to 3–5 freeze-thaw cycles to release virus, and virus yields were determined in HeLa cells by plaque assay.
2.3. RT-PCR Assays
For sequencing of viral RNA, HeLa cells were infected at an MOI of 5 for 6 h (PV1-3′MS2) or an MOI of 20 for 4 h (D8+/D8−/S1+/S1−/D8m+/D8m−702PV1) and total cellular RNA was extracted using TriReagent (Molecular Research Center, Inc., Cincinnati, OH, USA) or TRIzol (Invitrogen, Carlsbad, CA, USA). Reverse transcription was performed using avian myeloblastosis virus (AMV) reverse transcriptase (Life Sciences, Inc., St. Petersburg, FL, USA). PCR amplification was performed using PfuTurbo (Stratagene, San Diego, CA, USA) and PCR products were purified with QIAquick PCR purification kit (Qiagen) prior to sequencing (Laguna Scientific, Laguna Hills, CA, USA). RNA primers complementary to poliovirus positive-strand nucleotide 17 to 35, 771 to 798, 1534 to 1557, 2725 to 2742, 3510 to 3528, 4230 to 4248, 5719 to 5738, 6549 to 6572, and the reverse complements of each were used for PV1-3′MS2 RT-PCR and sequencing, while those listed in [
31] were used for D8+/D8−/S1+/S1−/D8m+/D8m−702PV1 and wild type poliovirus. For detection of MS2 hairpins and sequencing of the last 850 nucleotides of the viral RNA, MS2dT (5′-TTTTTTTTTGTTGACATGGGT-3′) or dT primer (5′-(T)
20-3′) and PV3CD6605 (5′-AAAAACCCAGGAGTGATAACAG-3′) primers were used. For detection of viral RNA after MS2 purification, 50% of purified RNA was used for reverse transcription (RT) reactions; 50% of the RT reaction was used in PCR amplification using previously described primers corresponding to the 3C coding region of poliovirus [
27]. For sequencing the 5′NCR of D8+/D8−/S1+/S1−/D8m+/D8m−702PV1 viruses, PV17+ (5′-GTTGTACCCACCCCAGAGG-3′) and PV895− (5V-CCTTGATGGGCTCGGTGAACTTG-3′) primers were used for RT-PCR.
2.4. Immunofluorescence Assays
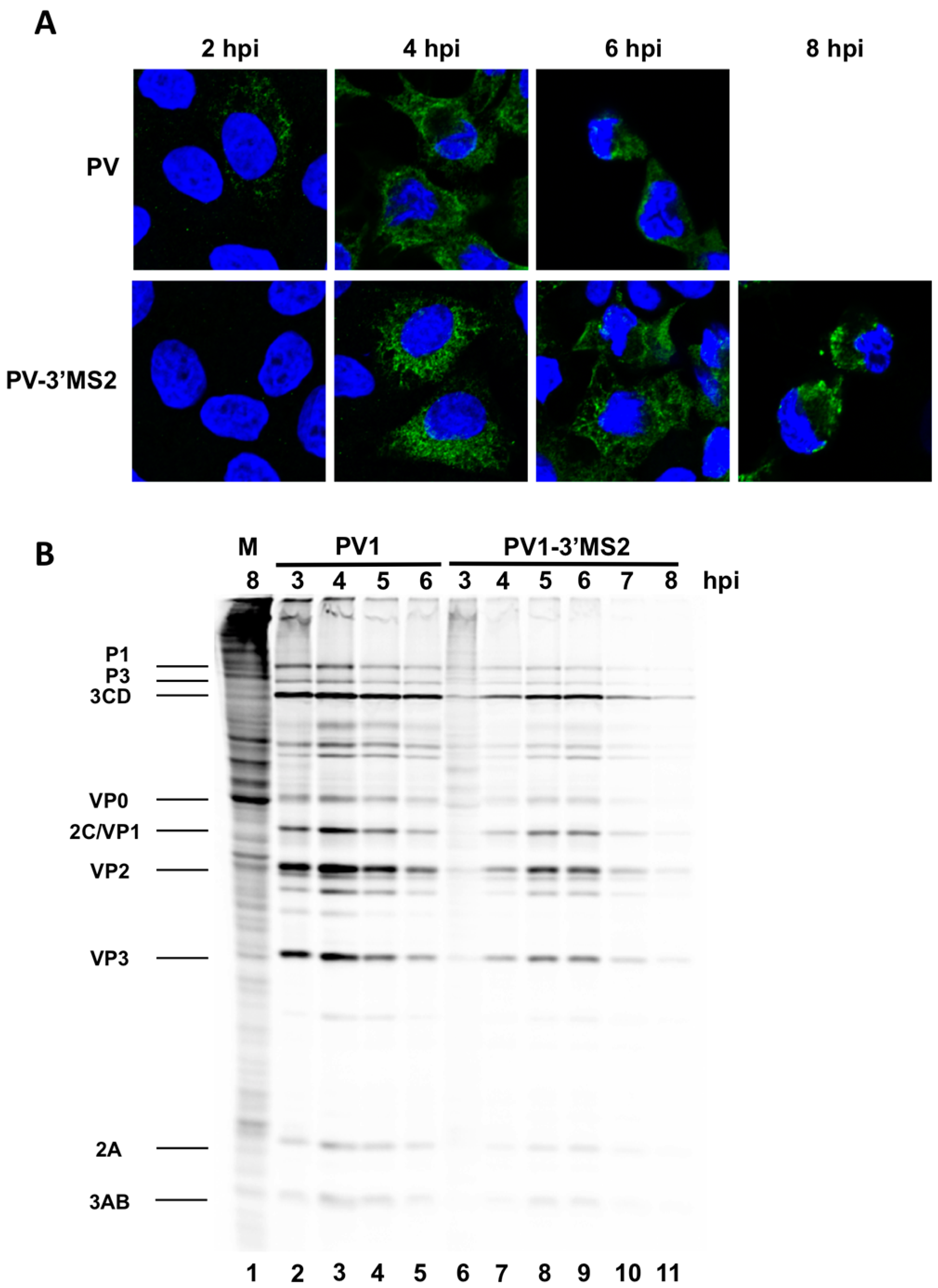
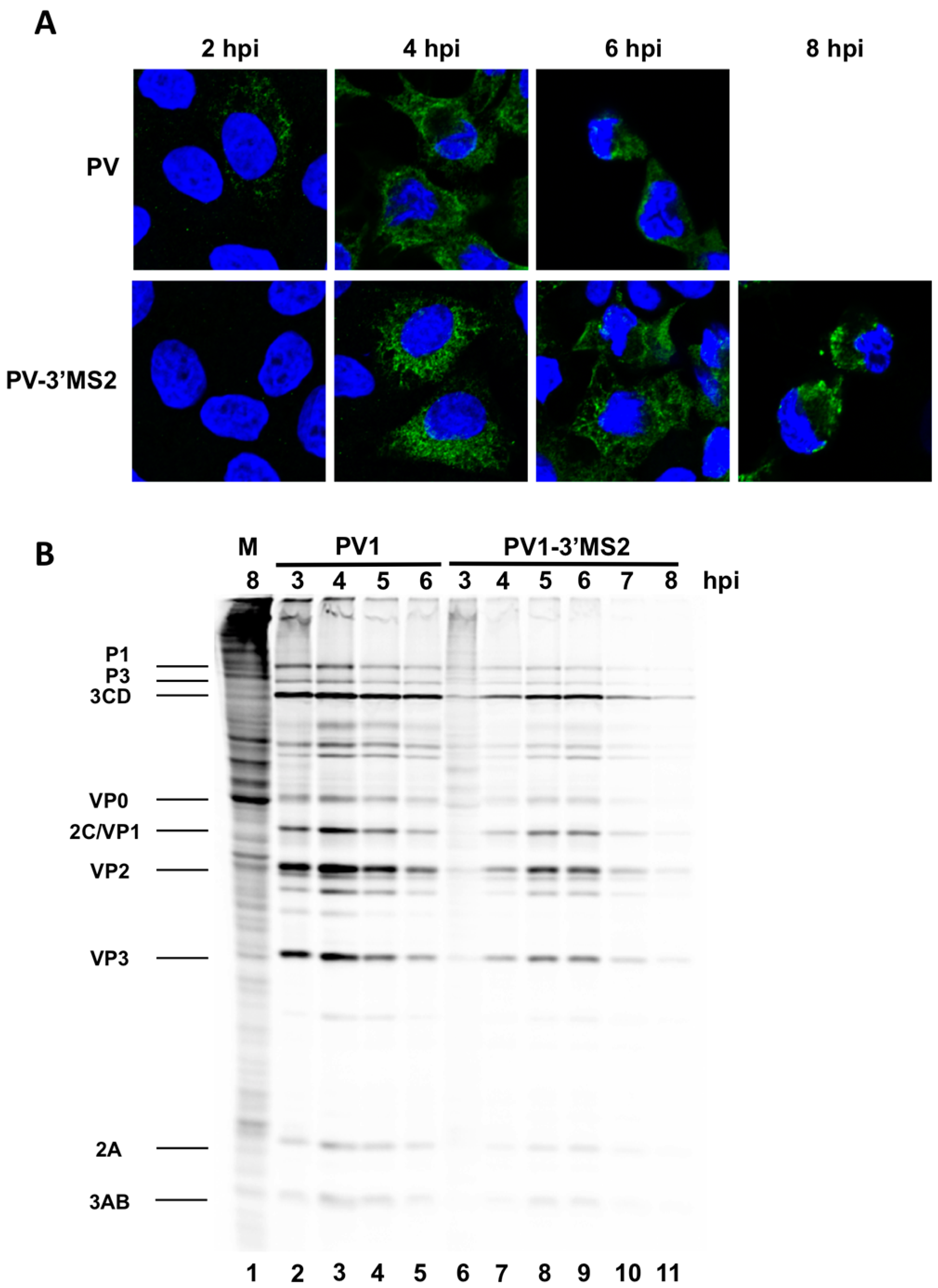
HeLa cells were seeded on coverslips then infected with PV1 or PV1-3′MS2 at an MOI of 20 and fixed with 3.7% formaldehyde for 15 min at indicated times post-infection. Cells were permeabilized with 0.5% NP-40 in PBS for 5 min, followed by three washes in 1% NCS-PBS. Cells were incubated with anti-3A monoclonal antibody (1:500) followed by Alexa Fluor 488 goat anti-rabbit IgG (Molecular Probes; 1:1000). To stain the nucleus, cells were incubated with 4′,6-diamidino-2-phenylindole (DAPI) for 10 min. Cells were washed after each incubation with 1% NCS-PBS. Proteins were visualized with a Zeiss LSM700 laser scanning confocal microscope and images were processed with Zen software (Zeiss, Germany). Poliovirus 3A antibody was generously provided by George Belov (University of Maryland, College Park, MD, USA).
2.5. Translation of Viral Proteins During Infection and in Vitro
To radiolabel viral translation products in infected cells, HeLa cell monolayers were infected with PV1 or PV1-3′MS2 at an MOI of 10. Following a 30 min adsorption, DMEM without methionine (MP)-8% NCS was added to cells. Cells were pulsed with 20 μCi
35S-methionine (PerkinElmer, Waltham, MA, USA) 1 h prior to collection. Cells were washed twice with PBS, harvested, and resuspended in 2X Laemmli sample buffer (LSB). Crude lysates (25% of total) were resolved on a 12.5% polyacrylamide, SDS-containing gel. For
in vitro translation assays, HeLa cell S10 cytoplasmic extracts were generated as described elsewhere [
32]. HeLa S10 (60% of total volume) was incubated with 250 or 500 ng of
in vitro transcribed RNA constructs corresponding to aptamer-tagged viral genomes or poliovirus virion RNA (vRNA),
35S-methionine (PerkinElmer), and all-four buffer (1 mM ATP, 0.25 mM GTP, 0.25 mM UTP, 0.25 mM CTP, 60 mM potassium acetate, 30 mM creatine phosphate, 0.4 mg/mL creatine kinase, 15.5 mM HEPES-KOH [pH 7.4]). Translation was allowed to proceed at 30 °C for 5–6 h. 2X LSB was added to an equal volume of translation reaction and boiled for 3 min. Samples were then subjected to electrophoresis on an SDS-containing 12.5% polyacrylamide gel. Proteins were visualized by autoradiography following fluorography. Quantity One software (Bio-Rad Laboratories, Hercules, CA, USA) was used to quantify VP3 band intensity of
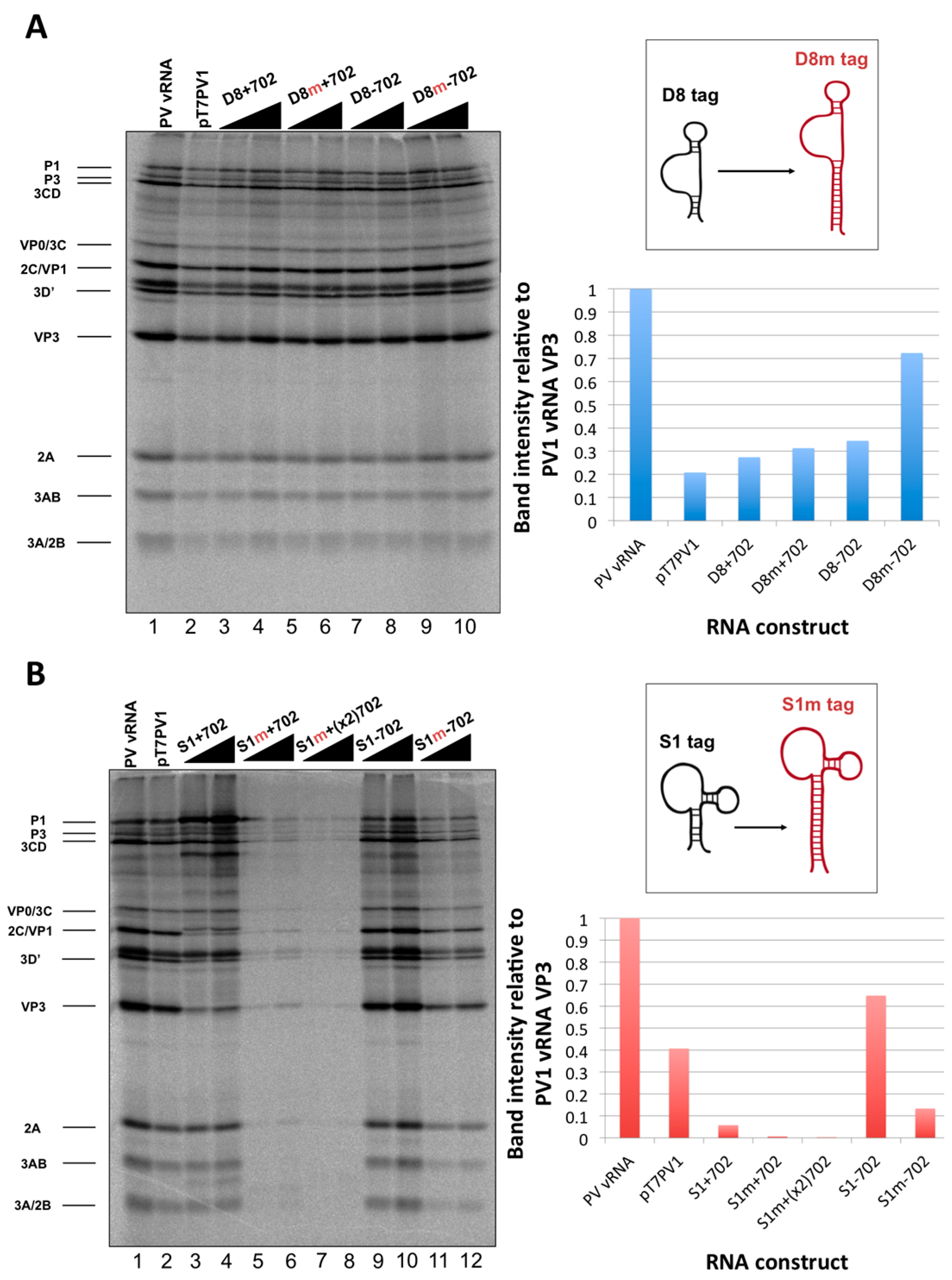
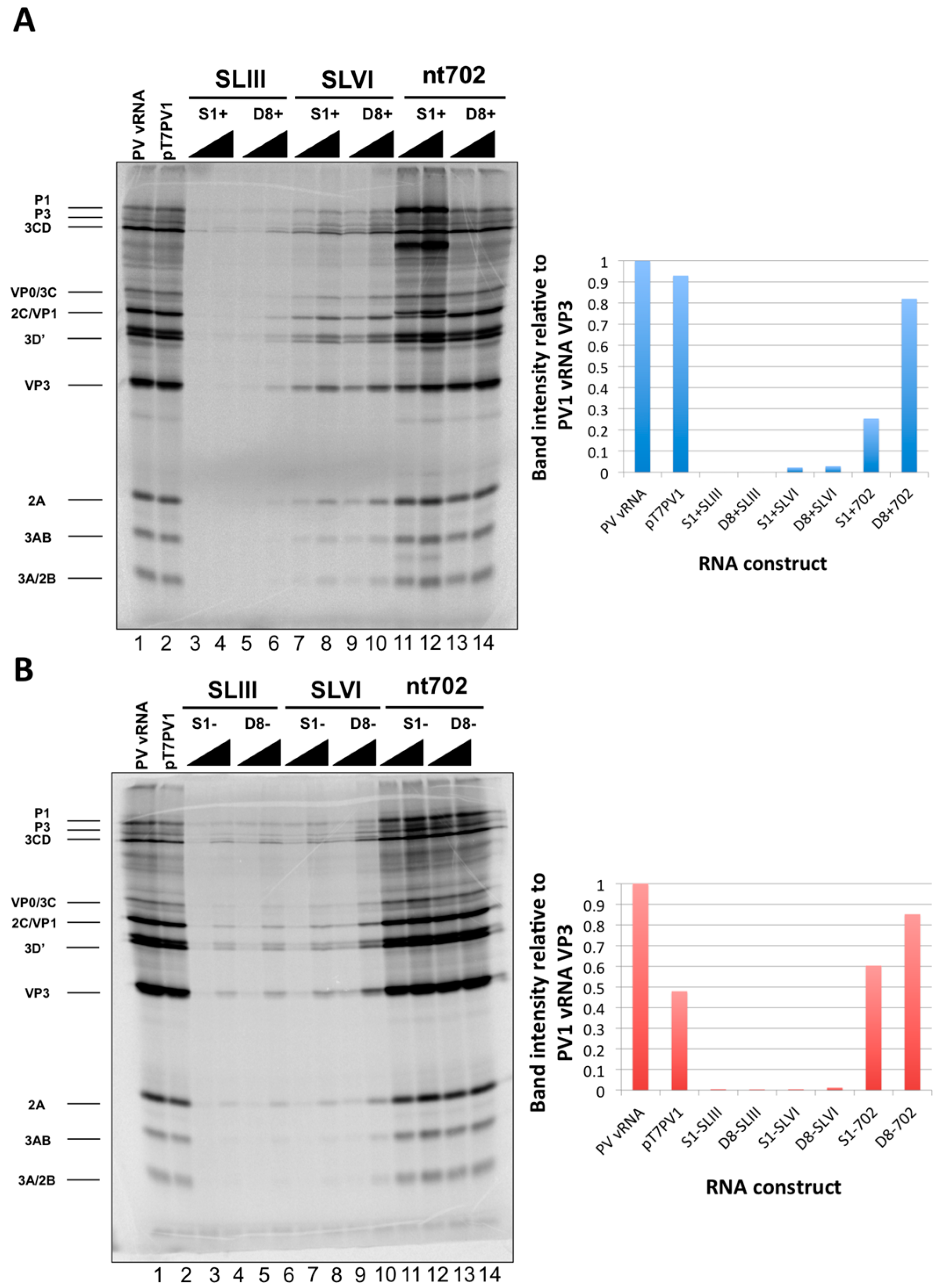
in vitro translation reactions that contained 250 ng of RNA relative to the band intensity of the poliovirus vRNA translation.
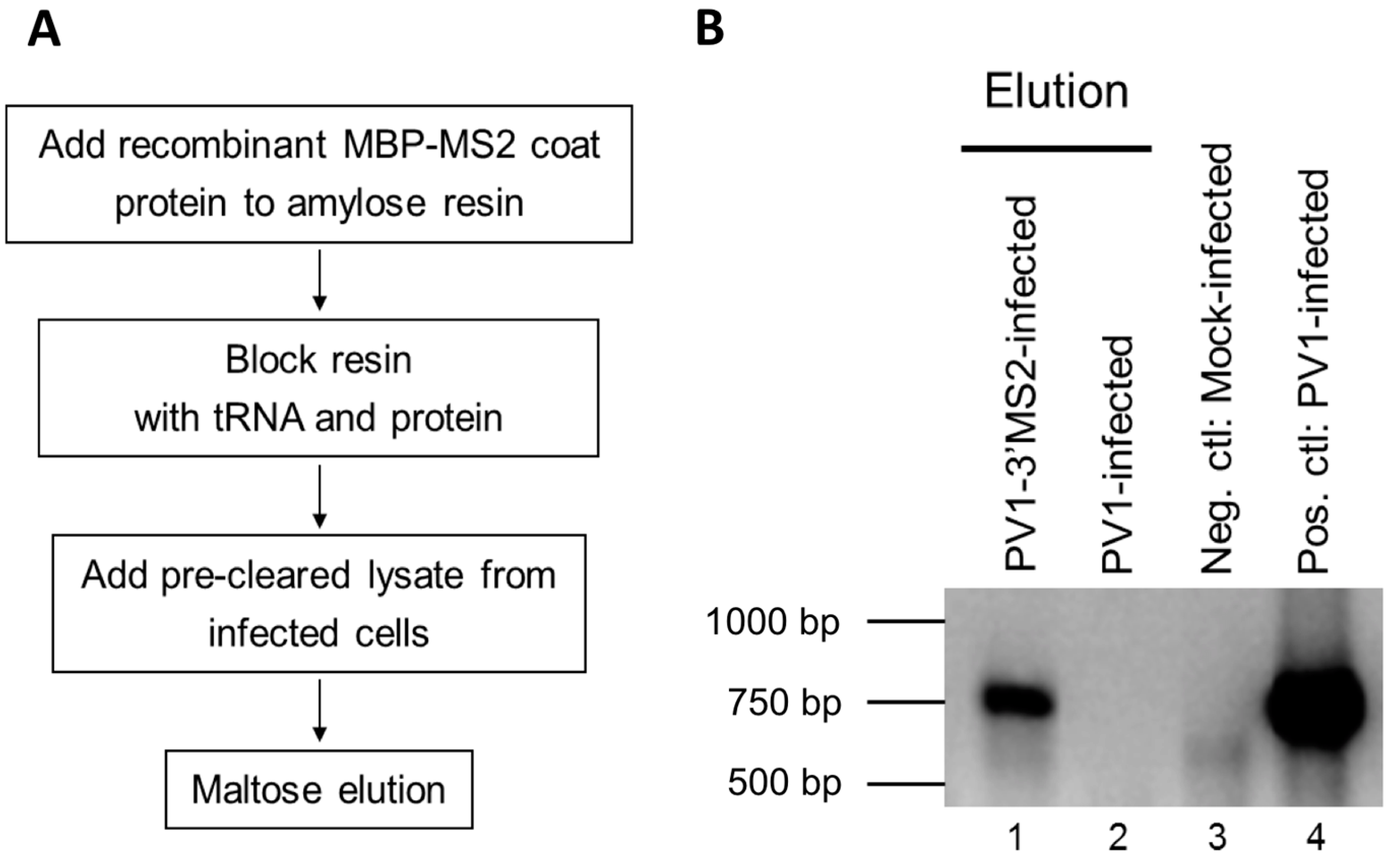
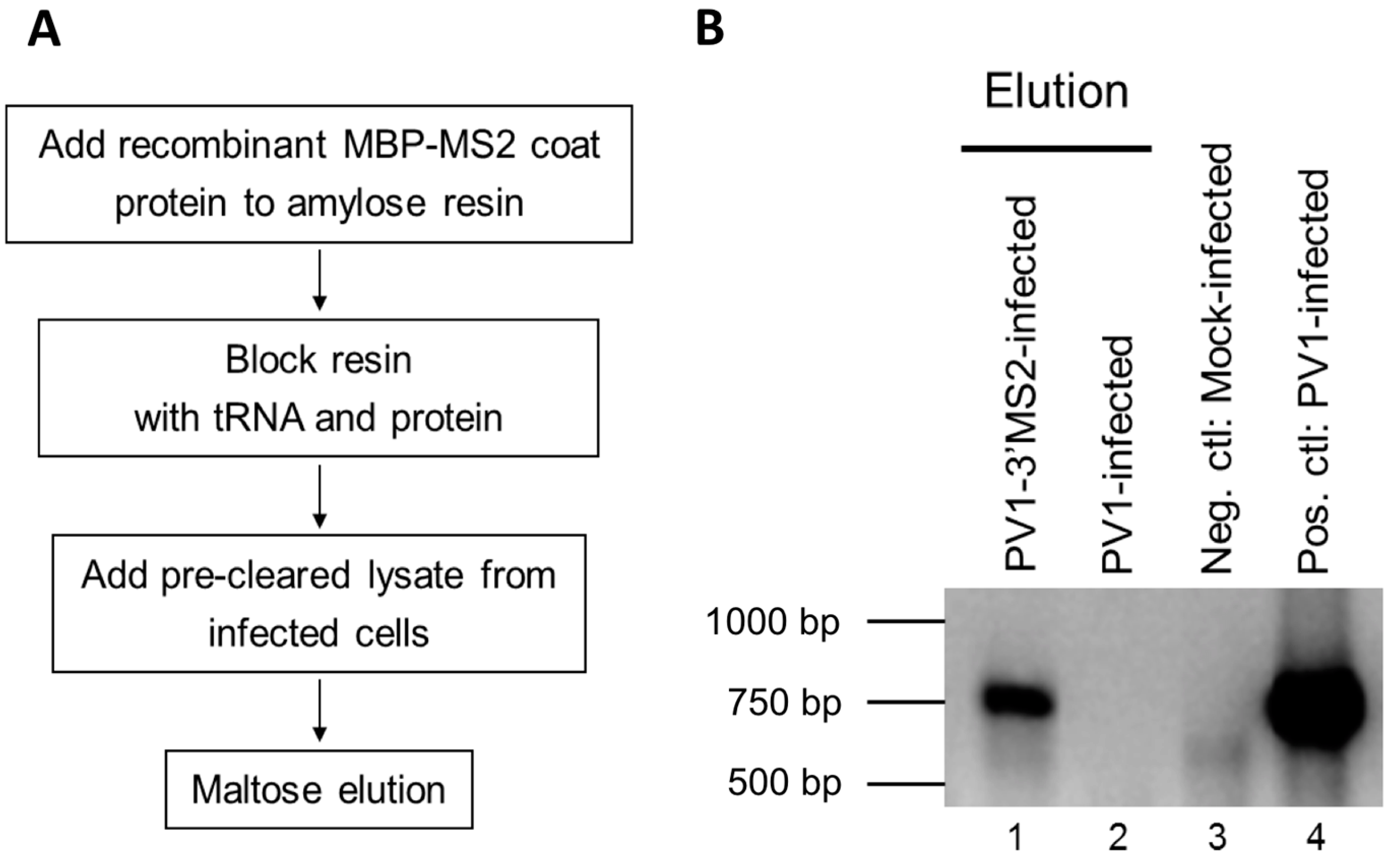
2.6. Purification of Recombinant MBP-MS2 Coat Protein
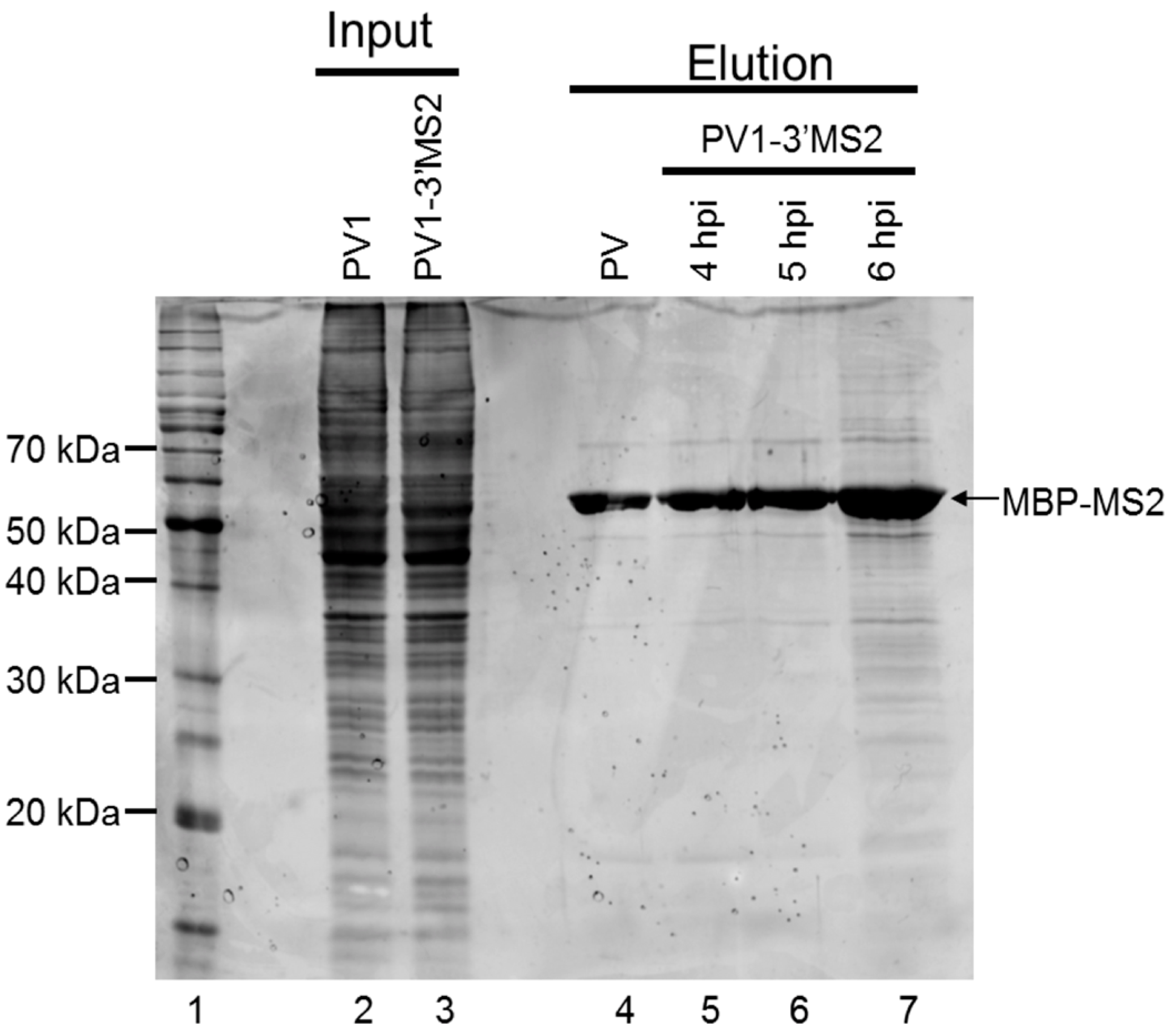
E. coli expressing MBP-MS2 was a gift from Yongsheng Shi (University of California, Irvine, CA, USA). Cells were grown at 37 °C until OD600 reached 0.4, and protein expression was induced with 0.5 mM isopropyl β-d-thiogalactoside (IPTG). After 3 h incubation at 37 °C, cells were pelleted, resuspended in lysis buffer (20 mM Tris HCl, pH 7.5, 200 mM NaCl, 0.5 mM PMSF), and lysed by sonication on ice 3 times at 30-second intervals. Lysates were cleared by centrifugation at ~7500 rcf for 10 min at 4 °C. The supernatant was added to washed amylose resin (NEB). Samples were incubated overnight at 4 °C with micrococcal nuclease and 5 mM CaCl2. The amylose resin was washed 3 times with lysis buffer, and MBP-MS2 was eluted with 5 mM Na2PO4, pH 7. Eluted protein was added to washed heparin agarose (Sigma-Aldrich, St. Louis, MO, USA) and incubated overnight at 4 °C. The heparin agarose was washed once with 5 mM Na2PO4, pH 7 and protein was eluted with buffer D-10% glycerol (20 mM HEPES, 100 mM KCl, 1 mM MgCl2, 0.2 mM EDTA) for 15 min at 4 °C. Protein concentration was determined by Bradford assay (Bio-Rad Laboratories).
2.7. MBP-MS2 RNA Affinity Purification
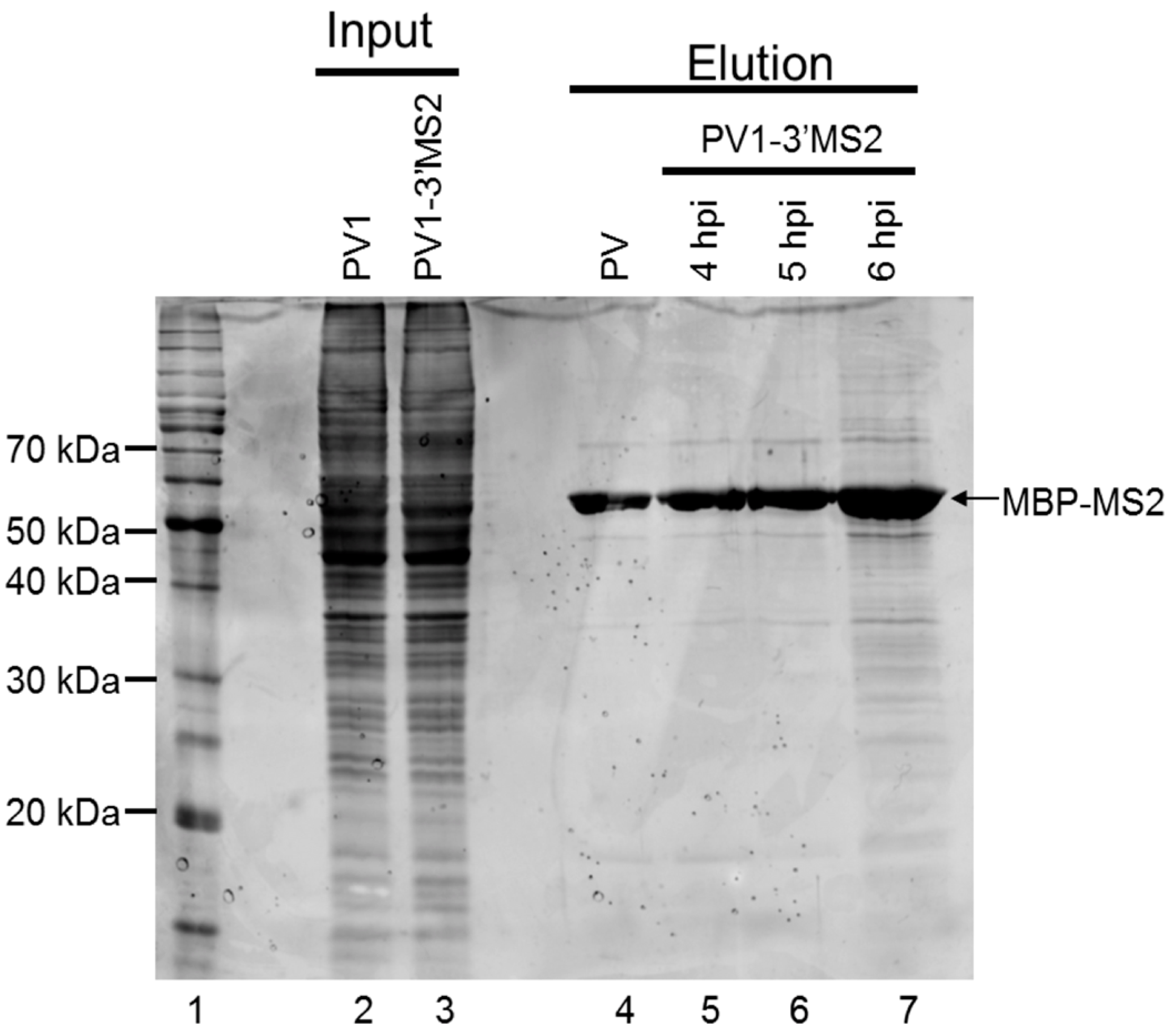
To generate lysate for MBP-MS2 RNA affinity, HeLa cell monolayers were infected with PV1-3′MS2 or wild type PV1 at an MOI of 5. After 30 min adsorption at room temperature, DMEM-8% NCS was added and cells were incubated at 37 °C for 4 h (wild type PV1) or 4 to 6 h (PV1-3′MS2). At indicated times post-infection, cells were washed twice with warm PBS, followed by cross linking with 0.4% formaldehyde in PBS for 10 min at room temperature with shaking. Formaldehyde was quenched with 0.25 M glycine for 5 min at room temperature. Cells were washed twice with ice-cold PBS, scraped, and pelleted at ~1000 rcf for 5 min at 4 °C. Cells were lysed in 100 μL NP-40 lysis buffer (50 mM Tris-HCl, pH 7.5, 5 mM EDTA, 150 mM NaCl, 1% NP-40) per 100 cm2 plate of HeLa cells for 25 min on ice. Cell debris was pelleted, supernatant was collected, and protein concentration was determined via Bradford assay.
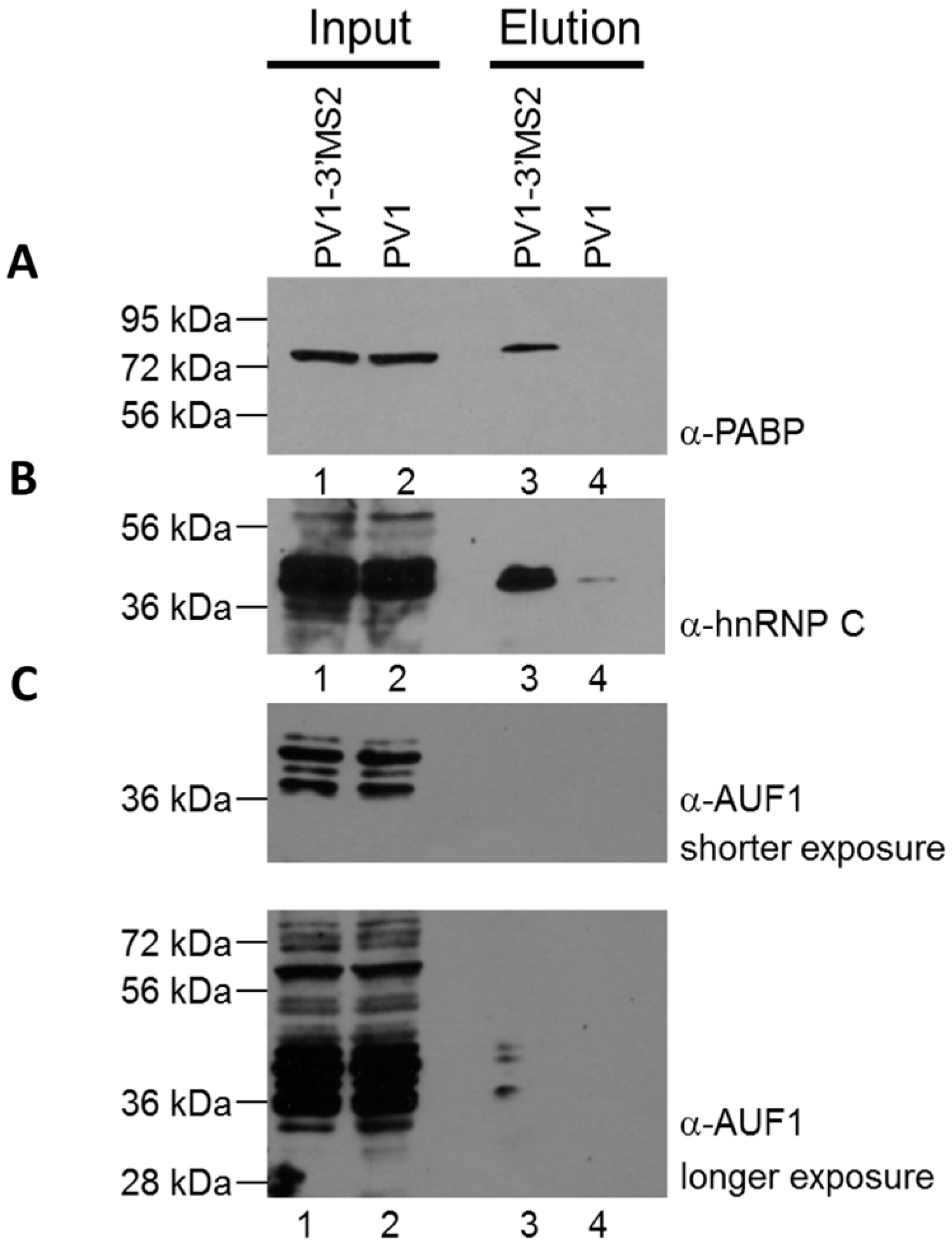
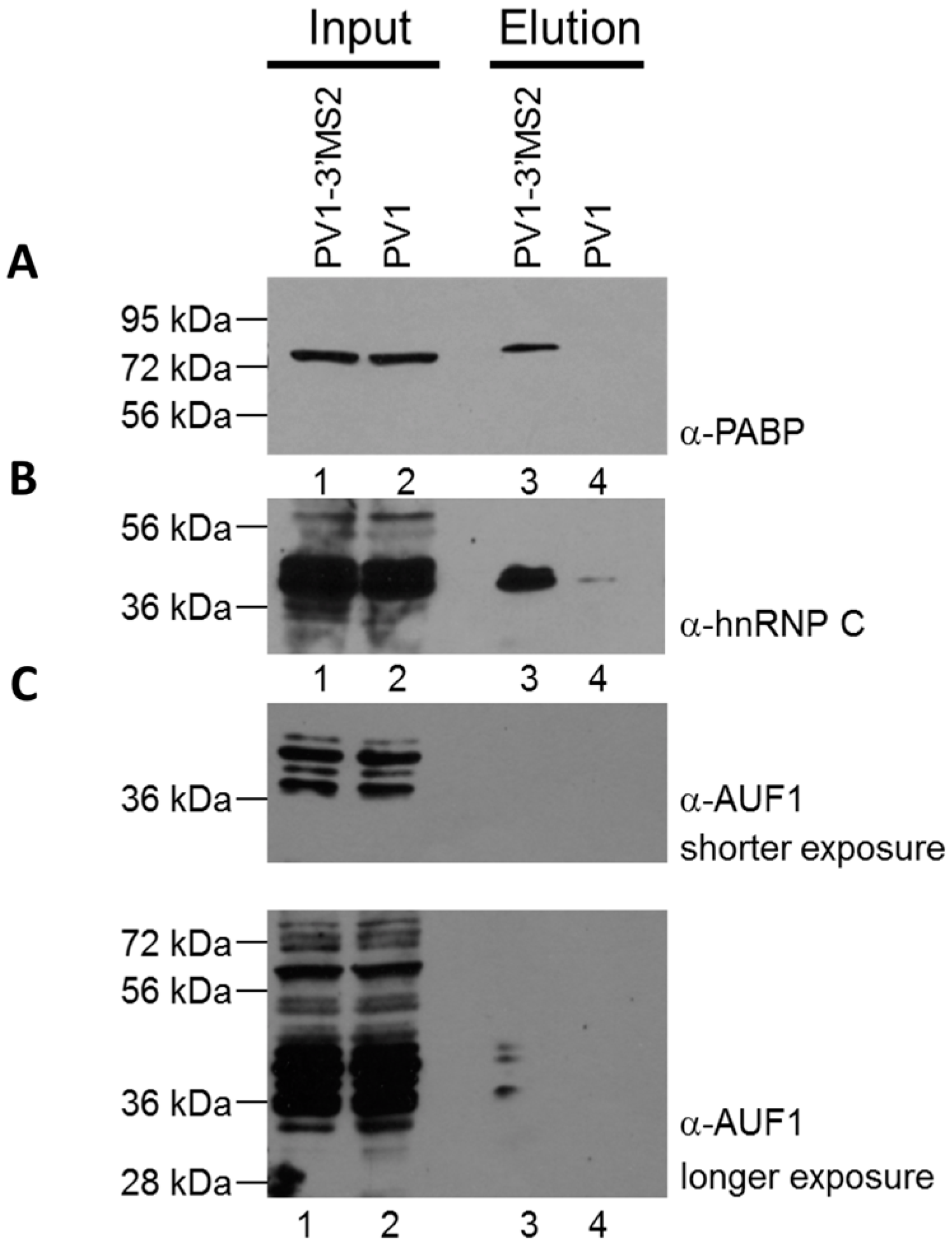
RNA affinity resin was generated by incubating 40 μg recombinant MBP-MS2 protein per 10 μL washed magnetic amylose resin (NEB) in buffer B (20 mM Tris HCl, pH 7.5, 100 mM KCl, 2.5 mM MgCl2, 2 mM DTT, 0.5 mM Pefabloc SC (Roche, Basel, Switzerland), 20 U/mL RNasin (Promega, Madison, WI, USA)) for 1 h with rotation at 4 °C. Unbound MBP-MS2 was removed, and the resin was blocked in buffer B with 1 μg BSA and 1 μg tRNA per μL magnetic amylose resin. After 1 h at 4 °C, the resin was washed 3 times in buffer B and then incubated with lysates from infected cells. NP-40 lysates from wild type PV1 or PV1-3′MS2 infected cells were pre-blocked with equal volume amylose resin for 1 h at 4 °C prior to incubation with MBP-MS2 amylose (1 μL resin per 10 μg lysate). Samples were incubated for 4 h at 4 °C with rotation. The resin was washed 3 times with buffer B and complexes were eluted with 30 mM maltose in buffer B for 30 min at 4 °C. Purified samples were either heated for 1 h at 70 °C for mass spectrometry analysis or LSB was added to samples which were then resolved by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Gels were analyzed by SYPRO Ruby staining (Lonza, Basel, Switzerland) or proteins were transferred to a PVDF membrane and analyzed by Western blot. For Western blot analysis, antibodies directed against PABP (Santa Cruz Biotechnology, Inc., Dallas, TX, USA), hnRNP C1 + C2 (Abcam, Cambridge, MA, USA), or AUF1 (Millipore, Temecula, CA, USA) were used. For mass spectrometry analysis, eluted samples were digested with trypsin and subjected to nano-liquid chromatography-tandem mass spectrometry (nanoLC-MS/MS). For purification of eluted RNA, 10% of final elution volume was subjected to phenol/chloroform extraction and ethanol precipitation.
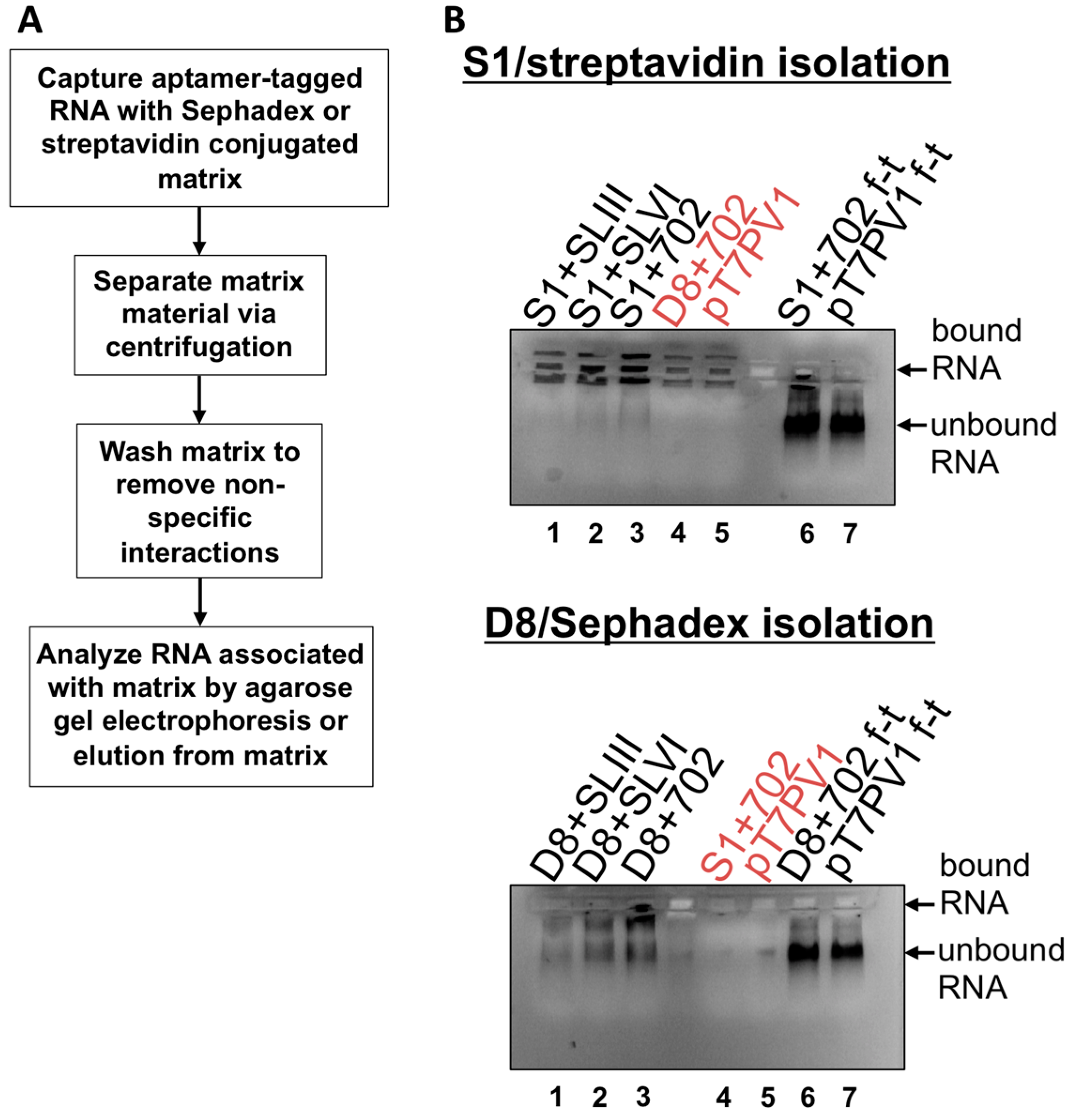
2.8. S1 and D8 Aptamer Affinity Purification
RNA affinity purification utilizing S1 and D8 aptamers was performed as described in [
33,
34].
In vitro transcribed RNA (11 μg) was renatured in TE buffer (10 mM Tris-HCl pH 7.5, 1 mM EDTA) by heating at 56 °C for 5 min, 37 °C for 10 min, and incubating at room temperature for 15 min. Streptavidin-agarose (SigmaAldrich,) was prepared by washing 10 times in lysis buffer (50 mM HEPES pH 7.4, 10 mM MgCl
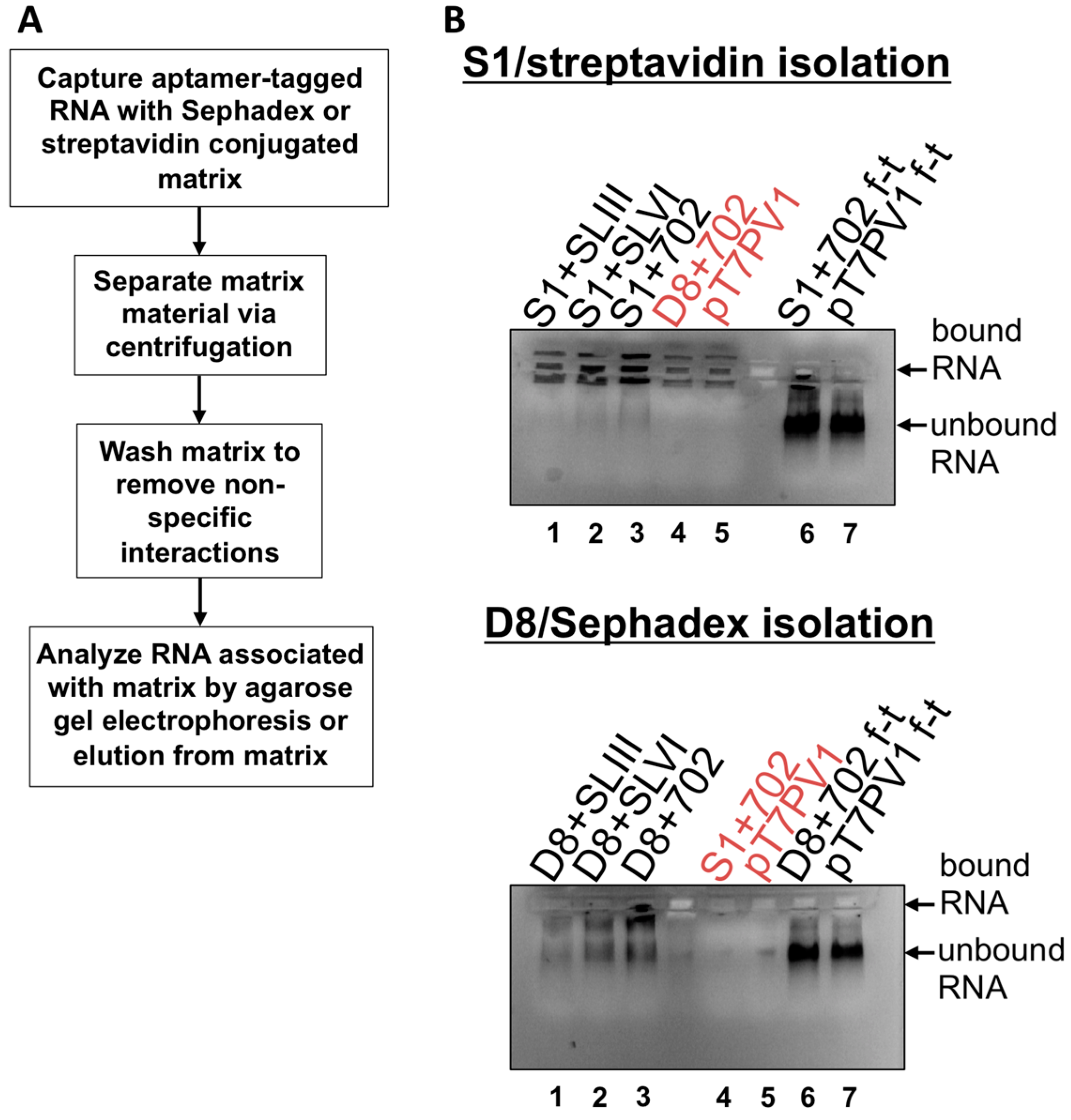
2, 100 mM NaCl, 1 mM DTT, 0.1% Triton X-100, 10% glycerol), Complete protease inhibitors (Roche) and resuspended to 50% slurry in lysis buffer. Sephadex matrix was prepared by swelling 0.5 g Sephadex G-200 (Sigma-Aldrich) in 40 mL lysis buffer overnight at room temperature. The Sephadex was then washed 3 times with lysis buffer and resuspended to 50% slurry. RNA (1 μg) was subjected to electrophoresis on a 1% agarose gel in Tris/Borate/EDTA (TBE) buffer to confirm an intact, homogenous RNA population. The remaining 10 μg of RNA was combined with 100 μL prepared streptavidin beads or Sephadex G-200 and 500 μL of lysis buffer. Samples were allowed to rotate at 4 °C for 4 h then subjected to centrifugation at ~25 rcf for 1 min to separate matrix, and supernatant was removed. Matrices were washed 5 times with 500 μL of lysis buffer, with rotation at 4 °C for 10 min. The matrix slurry was loaded directly onto a 1% TBE agarose gel containing ethidium bromide and subjected to electrophoresis to visualize RNA associated with matrix. As a comparison, the supernatant from the initial matrix separation (
i.e., the flow-through) was also subjected to electrophoresis on the agarose gel. Elution of aptamer-tagged RNA was also carried-out by transferring matrix material slurry, following wash steps, to Ultrafree-MC HV centrifugal filter units (0.45 μm pore size (Millipore)) with 10 mM biotin (Sigma-Aldrich) or 50 mg/mL dextran (average molecular weight 9000–11,000 Da (Sigma-Aldrich)) in lysis buffer. Filter units were subjected to rotation at 4 °C for 1.5 h then subjected to centrifugation at ~7500 rcf for 2.5 min.
Isolation of recombinant viral RNA from infected cells was carried-out as described above, but the streptavidin matrix was first blocked with 10 μg avidin from egg white (Sigma-Aldrich). To generate lysates from infected cells, two 150 mm plates of HeLa cell monolayers were infected with wild type PV1, D8+702PV1, or S1+702PV1 at an MOI of 20 following two washes with PBS. After 30 min adsorption, DMEM-8% NCS was added to cells which were then placed in 37 °C incubator for 4 h. Formaldehyde cross-linking was incorporated where indicated as described in [
35]: infected cells were washed twice with PBS then cross-linked with 0.2%–1% formaldehyde in PBS for 10 min at room temperature with shaking. Formaldehyde was quenched with 0.25 M glycine for 5 min at room temperature. Infected cells or infected and cross-linked cells were collected by scraping and pelleted at ~1500 rcf for 5 min, followed by washing twice with ice cold PBS. Pellets were resuspended in lysis buffer containing 20 U/μL RNasin (Promega). Cells were lysed by three rounds of sonication for 10 s followed by incubation on ice for 2 min. Lysates were pre-cleared by centrifugation and supernatant was incorporated into the isolation procedure.
4. Discussion
In an effort to isolate intact enteroviral RNA replication complexes from infected cells at different times following infection and to identify the novel cellular components of these complexes that correspond to the discrete steps of the RNA replication process, we have generated recombinant polioviruses containing genetically encoded RNA affinity tags.
Despite promising results related to the co-isolation of host proteins known to be involved in the replication cycle of poliovirus through MS2 affinity purification of viral RNA, the recombinant PV1-3′MS2 proved to be inadequate for the identification of novel host proteins involved in poliovirus RNA replication. This was in large part due to the sensitivity of mass spectrometry analysis, as significant co-isolation of proteins from negative control purifications (i.e., from wild type poliovirus infections) resulted in poor signal-to-noise ratios.
Due to the lack of specificity we observed during isolations of viral RNP complexes from cells infected with PV1-3′MS2, we turned to alternate affinity tags that could increase the specificity of viral RNA isolations. In addition to adjusting affinity sequences from MS2 hairpins to aptamers, we also explored alternative genomic locations to the 3′NCR for insertion of these purification tags. The replication defect of polioviruses lacking the 3’NCR of the genome, including PV1-3′MS2, is thought to be a result of the absence of the complementary 5′ terminal region within the negative-sense intermediate RNA. This region, although not strictly required for viral replication, has been proposed to be a binding site for hnRNP C1/C2, allowing for efficient positive-sense RNA synthesis to take place [
27]. Therefore, this recombinant virus was not ideal for identifying other proteins that may bind the 3′NCR of the poliovirus genome. Moreover, the coding region of the poliovirus genome is obviously restricted in its ability to tolerate exogenous nucleic acid insertions throughout its length due to the potential of altered viral protein production from disruptions to translation reading frame and/or irregular polyprotein processing. While there are examples of viable, recombinant polioviruses containing nucleic acid sequence insertions within the coding region of genomes to generate fusion or tagged viral proteins, these genomes are only quasi-stable [
55,
56]. To overcome the replication defect observed with PV1-3′MS2, the inherent deficiency this recombinant virus might offer in identifying proteins that interact with the genomic 3′NCR or negative-strand intermediate 5′ terminal region, and to promote the generation of a stable genome that could allow for growth kinetics that more closely matched wild type poliovirus, we focused on the 5′NCR to identify potential aptamer tag sequence insertion sites. While interference with regulatory RNA regions within the 5′NCR was a concern, there are several regions within the 5′NCR of the poliovirus genome that have previously been shown to tolerate sequence alterations with limited effects to viral replication processes. Nucleic acid sequences generated via systematic evolution of ligands by exponential enrichment (SELEX) to bind with high affinity to streptavidin (S1) or Sephadex (D8) were selected due to their short length and demonstrated potential for RNP complex isolation [
42,
43,
44,
45,
46,
47,
48,
49,
50,
51]. We tested three separate sites within the 5′NCR of the poliovirus genome: S-L III, S-L VI, and nucleotide position 702 for their capacity to tolerate aptamer insertions while maintaining biological activity. Making use of these regions, we generated poliovirus RNA constructs containing either of the two aptamer tags within the 5′NCR, in either a forward or reverse orientation to allow for aptamer sequences to be present in viral genomic RNAs or replication intermediate negative-strand RNAs and to permit strand-specific isolation from infected cells.
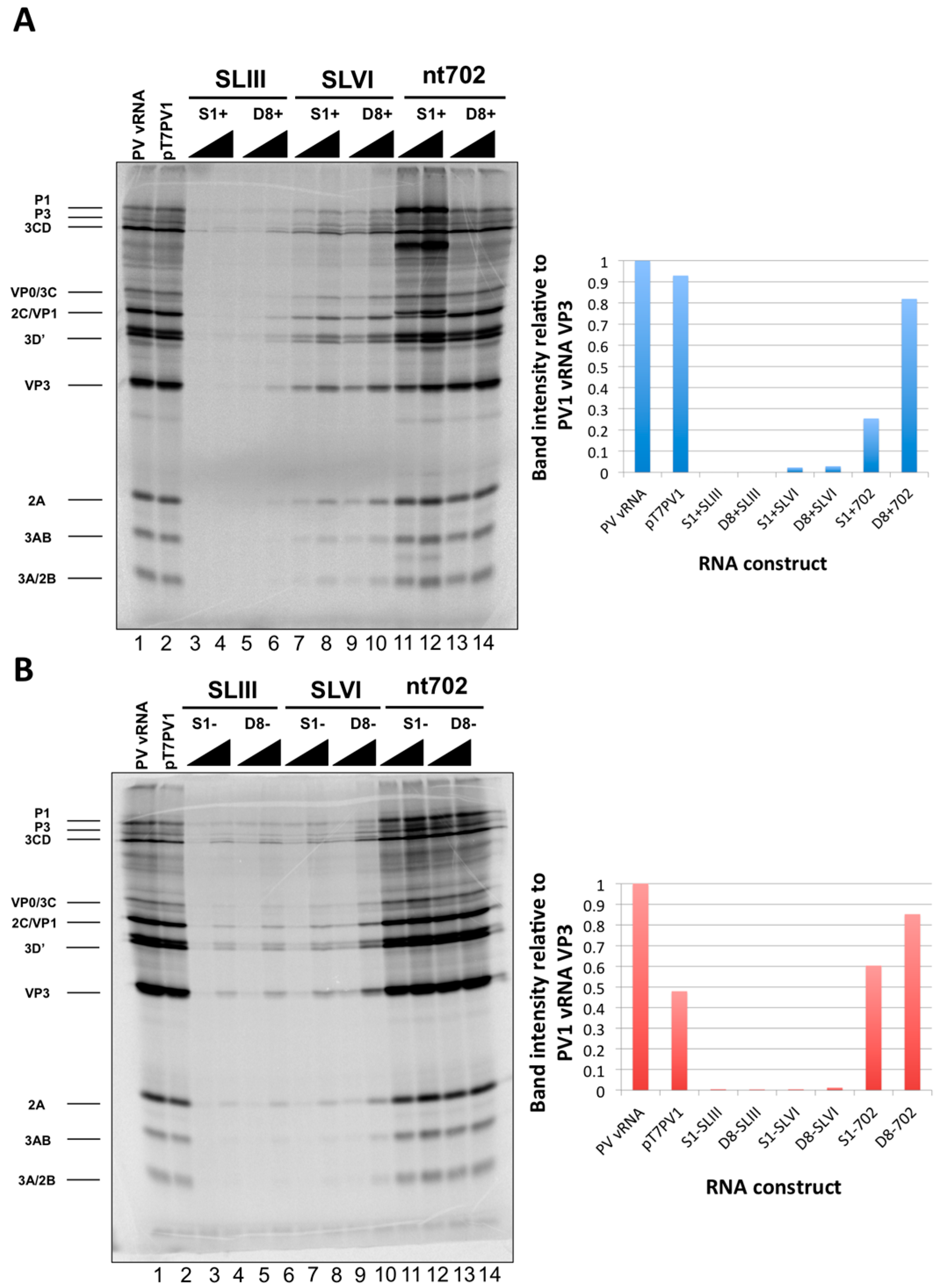
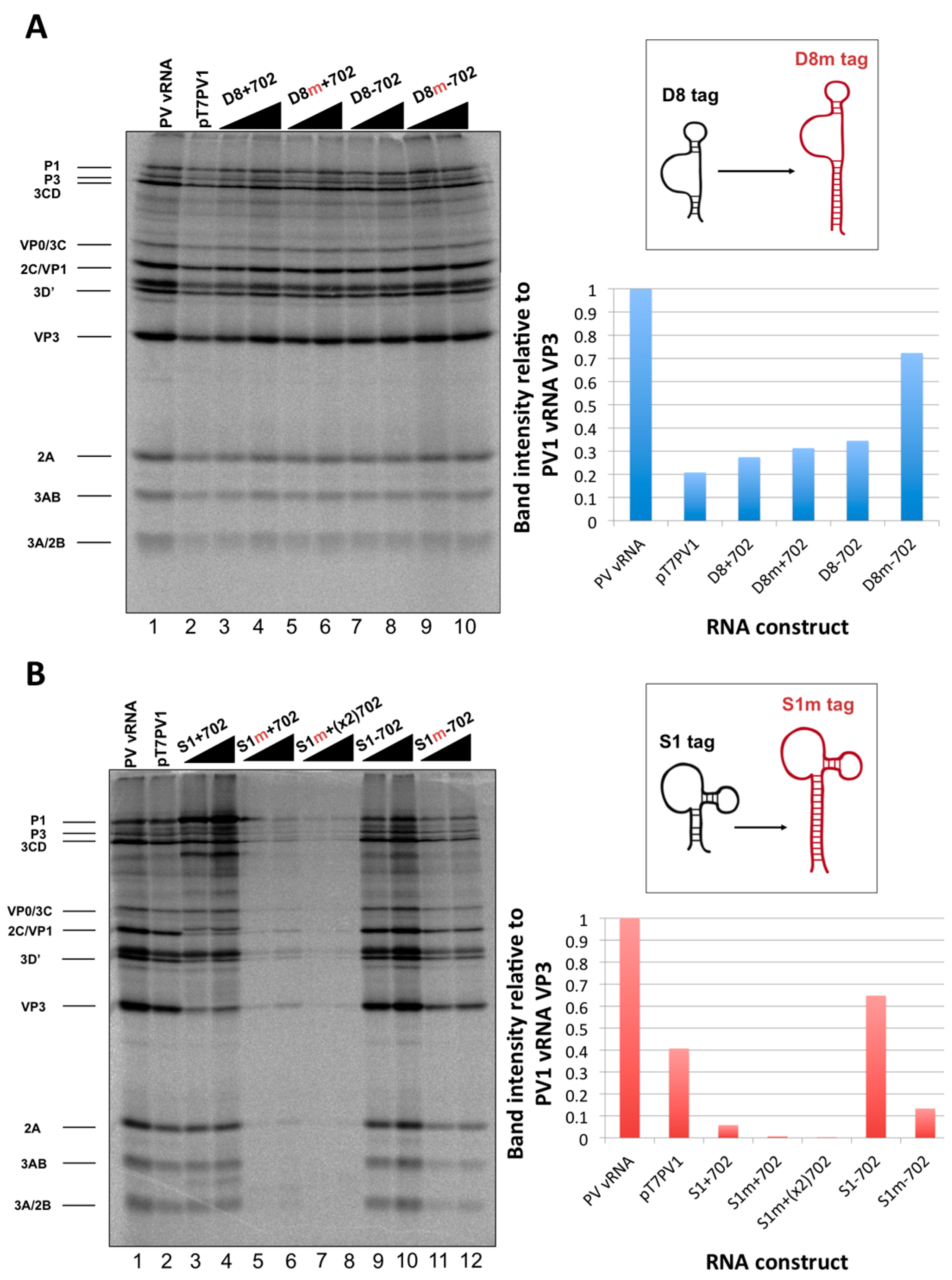
We initiated the RNP complex isolation study focused on the minimal, consensus sequences of the D8 and S1 aptamer tags with the rationale that their short length would be amenable to the production of recombinant virus containing a stable, RNA affinity-tagged genome. However, we observed insufficient RNA isolation efficiency associated with the S1 and D8 RNA affinity tags, and a loss of all specificity associated with these tags upon formaldehyde treatment. The minimal nature of these tags, particularly in the context of the relatively long poliovirus genomic RNA, likely limits the amount of affinity-tagged RNA that can be isolated. Our results agreed with the recent finding that the binding efficiency of the minimal S1 aptamer offers almost no specificity of isolation relative to an untagged RNA construct [
34]. The authors of this latter study presented modified aptamer structures and tandem conformations that allow for up to 15-fold increases in binding efficiencies. However, the best binding efficiencies reported correspond to the inclusion of multiple tags of increased nucleotide length, likely preventing the generation of stable, recombinant poliovirus genomes containing these tags. In an attempt to increase the binding efficiency of the aptamer tags within the poliovirus 5′NCR, while maintaining the stability of the tags through multiple passages, we increased the stem length of both the D8 and S1 by 16 nucleotides to promote the accessibility of RNA affinity sequence to matrix. However, increasing the stem length of the aptamer tags also proved to be insufficient for isolating poliovirus RNP complexes from infected cells.
Aside from the identification of the 3’NCR and nucleotide position 702 of the poliovirus genome as amenable sites for the insertion of RNA affinity tags, the generation of recombinant viruses containing these tags, while not yet optimized for RNP complex isolation, revealed insights into the biology of the poliovirus genome. The Mahoney strain of the poliovirus 1 genome contains eight AUG triplets upstream of the authentic translation initiation site at nucleotide position 743, within the 5′NCR. Kuge and colleagues have previously demonstrated that insertions into the poliovirus genome that contain an AUG triplet resulted in base substitutions to the AUG triplet alone, with no other alterations to the insertion sequence [
54]. Our results partially support this finding, in that the AUG triplet present in the S1 aptamer sequence was not tolerated in any of the viral isolates that were sequenced, with single nucleotide changes to this triplet sequence present in all cases. However, the sequence coding for the presence of the S1 aptamer within negative-sense RNA (S1−) also contains an AUG triplet (when present in the genomic RNA molecule) that is out-of-frame with authentic AUG start site. Interestingly, no alterations to protein production levels or polyprotein processing were observed upon
in vitro translation of this construct compared to wild type RNA sequence. Whether this can be explained by the presence of a UAA stop codon in-frame with this AUG triplet remains to be seen. Other investigations have suggested that the translational efficiency of picornaviruses depends on the particular spacing between a polypyrimidine tract at nucleotide 558 of the 5′NCR and one of the cryptic AUG triplets at nucleotide position 586, as well as whether AUG triplets are present within structured or unstructured elements [
57,
58,
59]. The function of the multiple AUG sequences upstream of the authentic translation initiation start site remains uncertain, but our work suggests that an additional AUG inserted into the poliovirus genome downstream of nucleotide position 586 is only selected against when this triplet is in-frame with the start codon, even when this AUG is within the structured region of an aptamer.
Our work also indicates that very short oligonucleotide sequences may not be competent for stringent isolation of the RNA when they occur within the context of a long, structured viral genome. Although the region between S-L VI and the translation start site of the poliovirus genome (including nucleotide position 702) is often considered more-or-less unstructured, recent global RNA structure analysis has suggested that this region of the genome may in fact contain higher-order structures [
60]. Incorporating aptamer tags within a structured RNA will impact the native structure of that region, and may impact the way in which the inserted sequences are able to make intramolecular contacts, possibly affecting proper secondary structure formation of the tags themselves. It is also possible that RNA affinity tags incorporated into the 5′NCR of the poliovirus genome are sterically blocked from interacting with affinity matrices due to the highly structured nature of this region. Furthermore, the fact that this region interacts with many different protein species, which promote both viral translation and RNA replication, could lead to further masking of RNA affinity tags, thus blocking these RNA sequences from interacting with the matrix. MS2 coat protein RNA affinity tags within the 3′NCR of the poliovirus genome should be sufficiently separated from RNA structural elements within the genome, which may aid in the isolation of the tagged RNA, but our work suggests the MS2 RNA affinity purification scheme itself allows for untenable levels of non-specific protein co-isolation. Adding complexity is the fact that poliovirus RNA is at least partially duplexed during RNA replication, obstructing affinity matrix/tag interactions and further complicating isolation schemes.
The minimal S1 tag has recently been shown be highly inefficient in regard to its ability to retain RNA on the relevant affinity matrix, as untagged, negative-control RNAs are retained with similar efficiency [
34]. Leppek and Stoecklin have demonstrated increased isolation efficiencies with the S1 aptamer by increasing both the stem length of this aptamer as well as the number of tags incorporated into the RNA of interest. Although it would no doubt be advantageous to incorporate multiple aptamer tags in series (and/or at the apex of a stem-loop structure) we were limited in our choice of sequence length and insertion site, due to the constraint that recombinant viral genomes containing RNA affinity tags must recapitulate the biological activities of the wild type poliovirus genome. The fact that there is a maximum size threshold for exogenous sequence insertions into the poliovirus genome before biological activity is affected was evidenced by cloning two modified streptavidin-binding aptamers into the nucleotide 702 region. This construct, S1m+(x2)702, produced the lowest levels of viral proteins of all constructs that were assayed by
in vitro translation (
Figure 11). Recent work has demonstrated the isolation of poliovirus RNA through infection of a cell line stably expressing uracil phosphoribosyltransferase followed by UV cross-linking and oligonucleotide-directed poly(A) isolation, suggesting that isolation of poliovirus RNA may be more tractable when using complementary oligonucleotide sequences rather than RNA affinity tags [
61].


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}