
KSHV Entry and Trafficking in Target Cells—Hijacking of Cell Signal Pathways, Actin and Membrane Dynamics


Abstract
:1. Introduction
Kaposi’s Sarcoma-Associated Herpesvirus (KSHV) or Human Herpesvirus-8 (HHV-8)
2. KSHV Utilizes Diverse Endocytic Pathways for Entry into the Target Cells
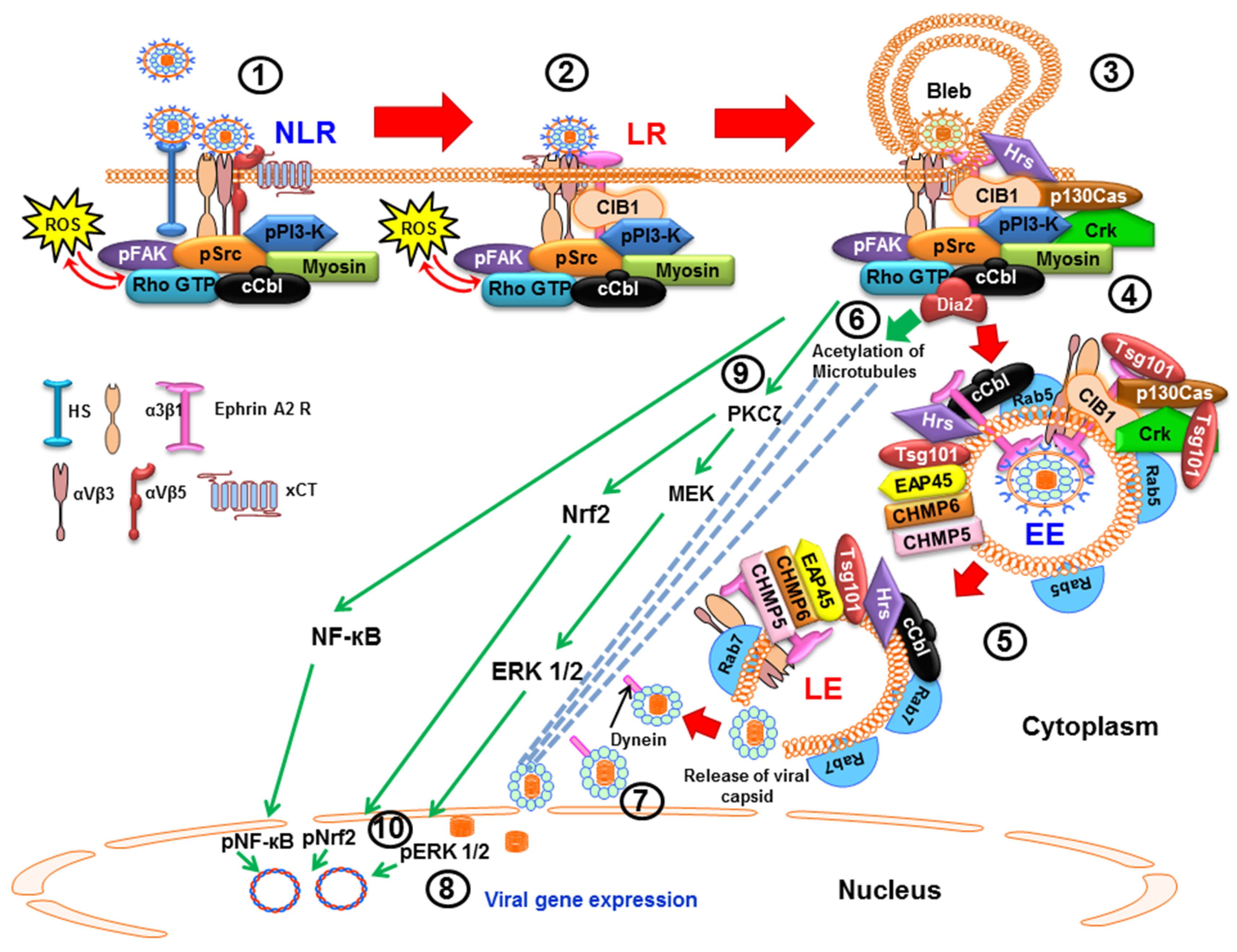
2.1. Macropinocytic Mode of Entry
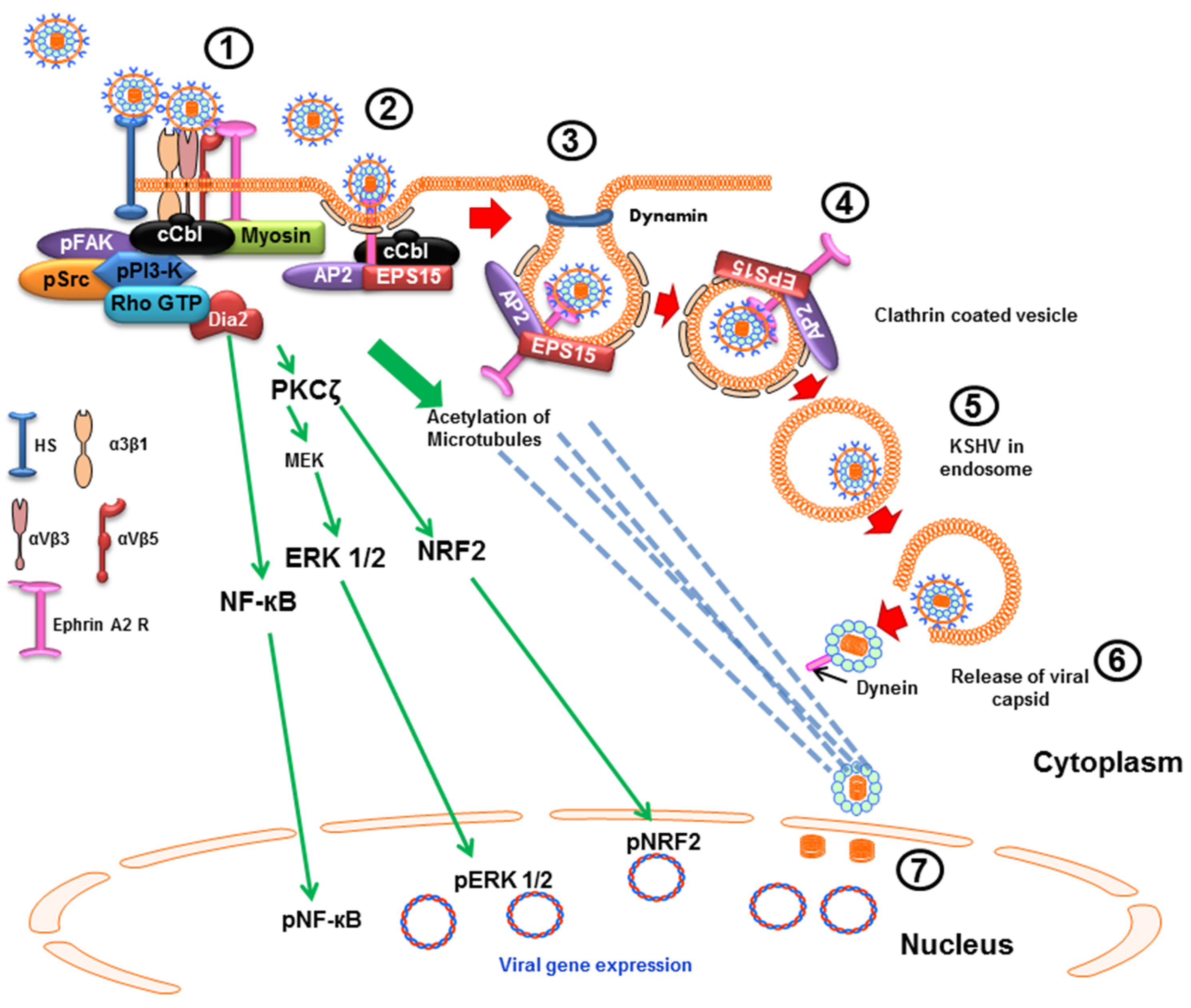
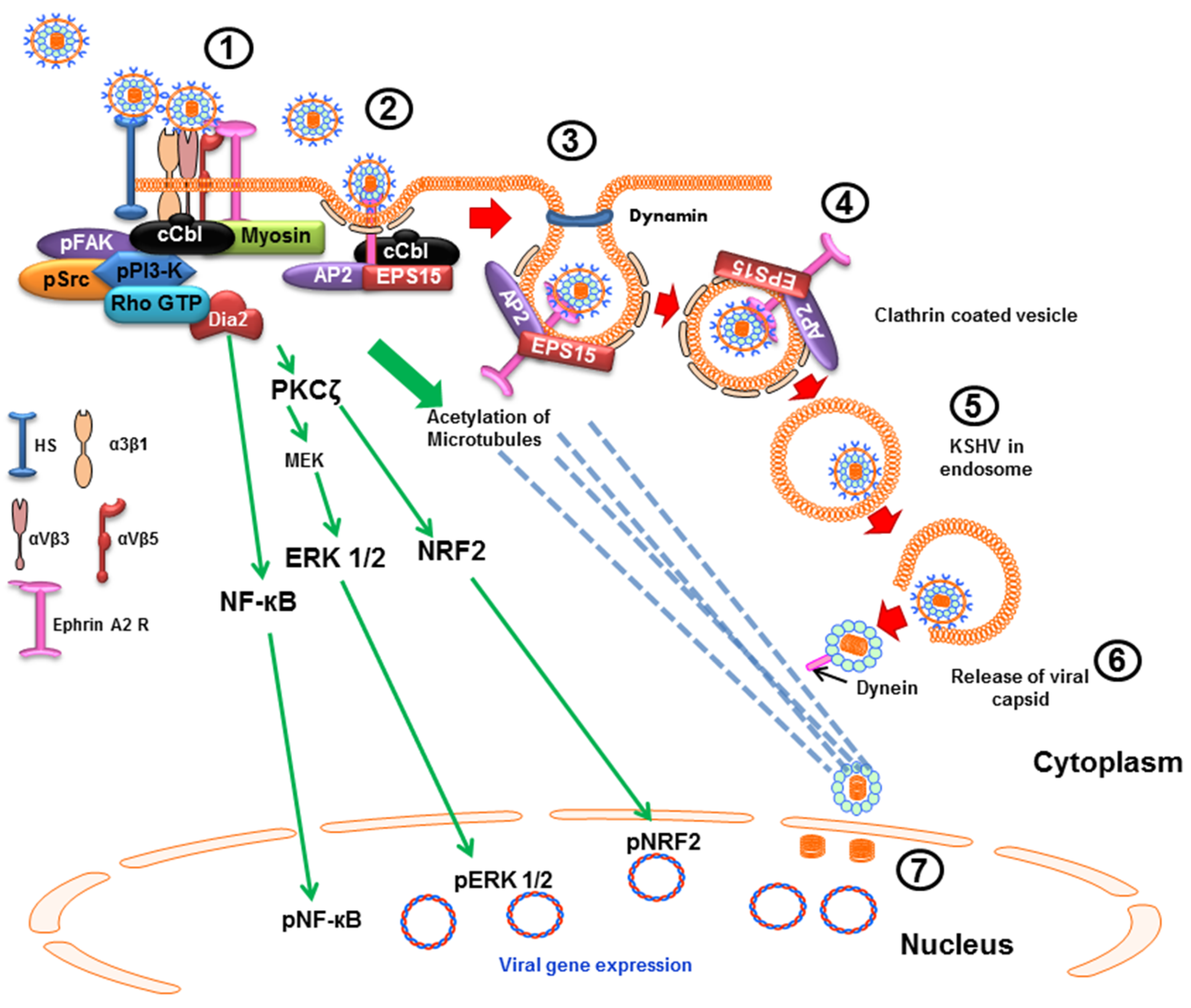
2.2. Clathrin-Mediated Endocytic Mode of Entry
3. KSHV Infection Is Initiated by Its Binding to Receptors on the Host Cell Membrane
3.1. Interactions with Cell Surface Proteins and Viral Entry Are Mediated by KSHV Envelope Glycoproteins
3.2. Heparan Sulfate (HS) Is Utilized by KSHV as the Initial Binding Receptor
3.3. Integrins on Endothelial, Fibroblast and Monocyte Cell Surfaces Play a Role as KSHV Entry Receptors
3.4. Role of xCT and DC-SIGN Molecules in KSHV Infection
3.5. Role of Ephrin Type-A Receptor 2 (EphA2R) in KSHV Infection
4. KSHV Interactions with Cell Surface Receptors Induce a Cascade of Host Preexisting Signal Pathways That Aid in Virus Entry and Trafficking
4.1. Early during Infection, KSHV Induces FAK, Src, PI3-K and Rho-GTPase to Facilitate Its Entry and Infection
4.2. Early during Infection, KSHV Induces c-Cbl, CIB1, EphA2R, Cas, and Crk to Facilitate Its Entry and Trafficking
5. Lipid Rafts (LR) of Infected Cells Regulate KSHV Entry and Trafficking
6. KSHV Induces Signal Pathways to Regulate Its Trafficking in the Cytoplasm Early during Infection
6.1. KSHV Infection Induces RhoA-GTPase to Modulate the Acetylation of Microtubules to Facilitate Intracellular Capsid Movement
6.2. KSHV Utilizes the ESCRT Complex Proteins for Its Entry and Trafficking
7. Early during Infection, KSHV Induces the Signal Pathways to Regulate the Host Transcription Factors NF-κB, ERK1/2 and Nrf2 to Facilitate Infection
8. Nuclear Delivery of KSHV dsDNA Genome Induces Host Nuclear Innate Responses
8.1. Nuclear DNA Damage Response (DDR) Induction Early during KSHV Infection
8.2. Nuclear Innate Immune Response Induction Early during KSHV Infection
9. Conclusions
Acknowledgments
Conflicts of Interest
References
- Carter, J.B.; Saunders, V.A. Virology: Principles and Applications; John Wiley & Sons: Chichester, UK; Hoboken, NJ, USA, 2007; p. 358. [Google Scholar]
- Roizman, B.; Carmichael, L.E.; Deinhardt, F.; de The, G.; Nahmias, A.J.; Plowright, W.; Rapp, F.; Sheldrick, P.; Takahashi, M.; Wolf, K. Herpesviridae. Definition, provisional nomenclature, and taxonomy. The Herpesvirus Study Group, the International Committee on Taxonomy of Viruses. Intervirology 1981, 16, 201–217. [Google Scholar] [CrossRef] [PubMed]
- Plancoulaine, S.; Abel, L.; van Beveren, M.; Tregouet, D.A.; Joubert, M.; Tortevoye, P.; de The, G.; Gessain, A. Human herpesvirus 8 transmission from mother to child and between siblings in an endemic population. Lancet 2000, 356, 1062–1065. [Google Scholar] [CrossRef]
- Bourboulia, D.; Whitby, D.; Boshoff, C.; Newton, R.; Beral, V.; Carrara, H.; Lane, A.; Sitas, F. Serologic evidence for mother-to-child transmission of Kaposi sarcoma-associated herpesvirus infection. JAMA 1998, 280, 31–32. [Google Scholar] [CrossRef] [PubMed]
- Engels, E.A.; Atkinson, J.O.; Graubard, B.I.; McQuillan, G.M.; Gamache, C.; Mbisa, G.; Cohn, S.; Whitby, D.; Goedert, J.J. Risk factors for human herpesvirus 8 infection among adults in the United States and evidence for sexual transmission. J. Infect. Dis 2007, 196, 199–207. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.; Cesarman, E.; Pessin, M.S.; Lee, F.; Culpepper, J.; Knowles, D.M.; Moore, P.S. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 1994, 266, 1865–1869. [Google Scholar] [CrossRef] [PubMed]
- Neipel, F.; Fleckenstein, B. The role of HHV-8 in Kaposi’s sarcoma. Semin. Cancer Biol. 1999, 9, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Cesarman, E.; Chang, Y.; Moore, P.S.; Said, J.W.; Knowles, D.M. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N. Engl. J. Med. 1995, 332, 1186–1191. [Google Scholar] [CrossRef] [PubMed]
- Gessain, A.; Sudaka, A.; Briere, J.; Fouchard, N.; Nicola, M.A.; Rio, B.; Arborio, M.; Troussard, X.; Audouin, J.; Diebold, J.; et al. Kaposi sarcoma-associated herpes-like virus (human herpesvirus type 8) DNA sequences in multicentric Castleman’s disease: Is there any relevant association in non-human immunodeficiency virus-infected patients? Blood 1996, 87, 414–416. [Google Scholar] [PubMed]
- Soulier, J.; Grollet, L.; Oksenhendler, E.; Cacoub, P.; Cazals-Hatem, D.; Babinet, P.; d’Agay, M.F.; Clauvel, J.P.; Raphael, M.; Degos, L.; et al. Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease. Blood 1995, 86, 1276–1280. [Google Scholar] [PubMed]
- Uldrick, T.S.; Wang, V.; O’Mahony, D.; Aleman, K.; Wyvill, K.M.; Marshall, V.; Steinberg, S.M.; Pittaluga, S.; Maric, I.; Whitby, D.; et al. An interleukin-6-related systemic inflammatory syndrome in patients co-infected with Kaposi sarcoma-associated herpesvirus and HIV but without Multicentric Castleman disease. Clin. Infect. Dis. 2010, 51, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Polizzotto, M.N.; Uldrick, T.S.; Wyvill, K.M.; Aleman, K.; Marshall, V.; Wang, V.; Whitby, D.; Pittaluga, S.; Jaffe, E.S.; Millo, C.; et al. Clinical features and outcomes of patients with symptomatic Kaposi Sarcoma Herpesvirus (KSHV)-associated inflammation: Prospective characterization of KSHV Inflammatory Cytokine Syndrome (KICS). Clin. Infect. Dis. 2016, 62, 730–738. [Google Scholar] [CrossRef] [PubMed]
- Renne, R.; Zhong, W.; Herndier, B.; McGrath, M.; Abbey, N.; Kedes, D.; Ganem, D. Lytic growth of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) in culture. Nat. Med. 1996, 2, 342–346. [Google Scholar] [CrossRef] [PubMed]
- Neipel, F.; Albrecht, J.C.; Fleckenstein, B. Human herpesvirus 8—The first human Rhadinovirus. J. Natl. Cancer Inst. Monogr. 1998, 23, 73–77. [Google Scholar] [CrossRef]
- Ganem, D. Fields Virology, 5th ed.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007; pp. 2875–2888. [Google Scholar]
- Chandran, B. Early events in Kaposi’s sarcoma-associated herpesvirus infection of target cells. J. Virol. 2010, 84, 2188–2199. [Google Scholar] [CrossRef] [PubMed]
- Cai, Q.; Verma, S.C.; Lu, J.; Robertson, E.S. Molecular biology of Kaposi’s sarcoma-associated herpesvirus and related oncogenesis. Adv. Virus Res. 2010, 78, 87–142. [Google Scholar] [PubMed]
- Dollery, S.J.; Santiago-Crespo, R.J.; Kardava, L.; Moir, S.; Berger, E.A. Efficient infection of a human B cell line with cell-free Kaposi’s sarcoma-associated herpesvirus. J. Virol. 2014, 88, 1748–1757. [Google Scholar] [CrossRef] [PubMed]
- Myoung, J.; Ganem, D. Active lytic infection of human primary tonsillar B cells by KSHV and its noncytolytic control by activated CD4+ T cells. J. Clin. Invest. 2011, 121, 1130–1140. [Google Scholar] [CrossRef] [PubMed]
- Bechtel, J.T.; Liang, Y.; Hvidding, J.; Ganem, D. Host range of Kaposi’s sarcoma-associated herpesvirus in cultured cells. J. Virol. 2003, 77, 6474–6481. [Google Scholar] [CrossRef] [PubMed]
- Jarousse, N.; Chandran, B.; Coscoy, L. Lack of heparan sulfate expression in B-cell lines: Implications for Kaposi’s sarcoma-associated herpesvirus and murine gammaherpesvirus 68 infections. J. Virol. 2008, 82, 12591–12597. [Google Scholar] [CrossRef] [PubMed]
- Inoue, N.; Winter, J.; Lal, R.B.; Offermann, M.K.; Koyano, S. Characterization of entry mechanisms of human herpesvirus 8 by using an Rta-dependent reporter cell line. J. Virol. 2003, 77, 8147–8152. [Google Scholar] [CrossRef] [PubMed]
- Rappocciolo, G.; Hensler, H.R.; Jais, M.; Reinhart, T.A.; Pegu, A.; Jenkins, F.J.; Rinaldo, C.R. Human herpesvirus 8 infects and replicates in primary cultures of activated B lymphocytes through DC-SIGN. J. Virol. 2008, 82, 4793–4806. [Google Scholar] [CrossRef] [PubMed]
- Raghu, H.; Sharma-Walia, N.; Veettil, M.V.; Sadagopan, S.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus utilizes an actin polymerization-dependent macropinocytic pathway to enter human dermal microvascular endothelial and human umbilical vein endothelial cells. J. Virol. 2009, 83, 4895–4911. [Google Scholar] [CrossRef] [PubMed]
- Akula, S.M.; Naranatt, P.P.; Walia, N.S.; Wang, F.Z.; Fegley, B.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) infection of human fibroblast cells occurs through endocytosis. J. Virol. 2003, 77, 7978–7990. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Tang, Y.; Kuo, Y.L.; Liu, B.Y.; Xu, C.J.; Giam, C.Z. Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 transcriptional activator Rta is an oligomeric DNA-binding protein that interacts with tandem arrays of phased A/T-trinucleotide motifs. J. Virol. 2003, 77, 9399–9411. [Google Scholar] [CrossRef] [PubMed]
- Nicola, A.V.; Hou, J.; Major, E.O.; Straus, S.E. Herpes simplex virus type 1 enters human epidermal keratinocytes, but not neurons, via a pH-dependent endocytic pathway. J. Virol. 2005, 79, 7609–7616. [Google Scholar] [CrossRef] [PubMed]
- Mercer, J.; Schelhaas, M.; Helenius, A. Virus entry by endocytosis. Annu. Rev. Biochem. 2010, 79, 803–833. [Google Scholar] [CrossRef] [PubMed]
- Valiya Veettil, M.; Sadagopan, S.; Kerur, N.; Chakraborty, S.; Chandran, B. Interaction of c-Cbl with myosin IIA regulates Bleb associated macropinocytosis of Kaposi’s sarcoma-associated herpesvirus. PLoS Pathog. 2010, 6, e1001238. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; ValiyaVeettil, M.; Sadagopan, S.; Paudel, N.; Chandran, B. c-Cbl-mediated selective virus-receptor translocations into lipid rafts regulate productive Kaposi’s sarcoma-associated herpesvirus infection in endothelial cells. J. Virol. 2011, 85, 12410–12430. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, C.; Valiya-Veettil, M.; Dutta, D.; Chakraborty, S.; Chandran, B. CIB1 synergizes with EphrinA2 to regulate Kaposi’s sarcoma-associated herpesvirus macropinocytic entry in human microvascular dermal endothelial cells. PLoS Pathog. 2014, 10, e1003941. [Google Scholar] [CrossRef] [PubMed]
- Veettil, M.V.; Kumar, B.; Ansari, M.A.; Dutta, D.; Iqbal, J.; Gjyshi, O.; Bottero, V.; Chandran, B. ESCRT-0 component Hrs promotes macropinocytosis of Kaposi’s sarcoma-associated herpesvirus in human dermal microvascular endothelial cells. J. Virol. 2016, 90, 3860–3872. [Google Scholar] [CrossRef] [PubMed]
- Kumar, B.; Dutta, D.; Iqbal, J.; Ansari, M.A.; Roy, A.; Chikoti, L.; Pisano, G.; Veettil, M.V.; Chandran, B. ESCRT-I protein Tsg101 plays a role in the post-macropinocytic trafficking and infection of endothelial cells by Kaposi’s sarcoma-associated herpesvirus. PLoS Pathog. 2016, 12, e1005960. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, E.; Belouzard, S.; Goueslain, L.; Wakita, T.; Dubuisson, J.; Wychowski, C.; Rouille, Y. Hepatitis C virus entry depends on clathrin-mediated endocytosis. J. Virol. 2006, 80, 6964–6972. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharyya, S.; Warfield, K.L.; Ruthel, G.; Bavari, S.; Aman, M.J.; Hope, T.J. Ebola virus uses clathrin-mediated endocytosis as an entry pathway. Virology 2010, 401, 18–28. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Chakraborty, S.; Bandyopadhyay, C.; Valiya Veettil, M.; Ansari, M.A.; Singh, V.V.; Chandran, B. EphrinA2 regulates clathrin mediated KSHV endocytosis in fibroblast cells by coordinating integrin-associated signaling and c-Cbl directed polyubiquitination. PLoS Pathog. 2013, 9, e1003510. [Google Scholar] [CrossRef] [PubMed]
- Neipel, F.; Albrecht, J.C.; Fleckenstein, B. Cell-homologous genes in the Kaposi’s sarcoma-associated rhadinovirus human herpesvirus 8: Determinants of its pathogenicity? J. Virol. 1997, 71, 4187–4192. [Google Scholar] [PubMed]
- Cesarman, E.; Nador, R.G.; Bai, F.; Bohenzky, R.A.; Russo, J.J.; Moore, P.S.; Chang, Y.; Knowles, D.M. Kaposi’s sarcoma-associated herpesvirus contains G protein-coupled receptor and cyclin D homologs which are expressed in Kaposi’s sarcoma and malignant lymphoma. J. Virol. 1996, 70, 8218–8223. [Google Scholar] [PubMed]
- Baghian, A.; Luftig, M.; Black, J.B.; Meng, Y.X.; Pau, C.P.; Voss, T.; Pellett, P.E.; Kousoulas, K.G. Glycoprotein B of human herpesvirus 8 is a component of the virion in a cleaved form composed of amino- and carboxyl-terminal fragments. Virology 2000, 269, 18–25. [Google Scholar] [CrossRef] [PubMed]
- Parravicini, C.; Chandran, B.; Corbellino, M.; Berti, E.; Paulli, M.; Moore, P.S.; Chang, Y. Differential viral protein expression in Kaposi’s sarcoma-associated herpesvirus-infected diseases: Kaposi’s sarcoma, primary effusion lymphoma, and multicentric Castleman’s disease. Am. J. Pathol. 2000, 156, 743–749. [Google Scholar] [CrossRef]
- Akula, S.M.; Pramod, N.P.; Wang, F.Z.; Chandran, B. Human herpesvirus 8 envelope-associated glycoprotein B interacts with heparan sulfate-like moieties. Virology 2001, 284, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Akula, S.M.; Wang, F.Z.; Vieira, J.; Chandran, B. Human herpesvirus 8 interaction with target cells involves heparan sulfate. Virology 2001, 282, 245–255. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.Z.; Akula, S.M.; Sharma-Walia, N.; Zeng, L.; Chandran, B. Human herpesvirus 8 envelope glycoprotein B mediates cell adhesion via its RGD sequence. J. Virol. 2003, 77, 3131–3147. [Google Scholar] [CrossRef] [PubMed]
- Akula, S.M.; Pramod, N.P.; Wang, F.Z.; Chandran, B. Integrin alpha3beta1 (CD 49c/29) is a cellular receptor for Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8) entry into the target cells. Cell 2002, 108, 407–419. [Google Scholar] [CrossRef]
- Veettil, M.V.; Sadagopan, S.; Sharma-Walia, N.; Wang, F.Z.; Raghu, H.; Varga, L.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus forms a multimolecular complex of integrins (alphaVbeta5, alphaVbeta3, and alpha3beta1) and CD98-xCT during infection of human dermal microvascular endothelial cells, and CD98-xCT is essential for the postentry stage of infection. J. Virol. 2008, 82, 12126–12144. [Google Scholar] [PubMed]
- Hensler, H.R.; Tomaszewski, M.J.; Rappocciolo, G.; Rinaldo, C.R.; Jenkins, F.J. Human herpesvirus 8 glycoprotein B binds the entry receptor DC-SIGN. Virus Res. 2014, 190, 97–103. [Google Scholar] [CrossRef] [PubMed]
- Sharma-Walia, N.; Naranatt, P.P.; Krishnan, H.H.; Zeng, L.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 envelope glycoprotein gB induces the integrin-dependent focal adhesion kinase-Src-phosphatidylinositol 3-kinase-rho GTPase signal pathways and cytoskeletal rearrangements. J. Virol. 2004, 78, 4207–4223. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, H.H.; Sharma-Walia, N.; Zeng, L.; Gao, S.J.; Chandran, B. Envelope glycoprotein gB of Kaposi’s sarcoma-associated herpesvirus is essential for egress from infected cells. J. Virol. 2005, 79, 10952–10967. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Puri, V.; Chandran, B. Characterization of human herpesvirus-8 K8.1A/B glycoproteins by monoclonal antibodies. Virology 1999, 262, 237–249. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Wang, R.; Sweat, A.; Goldstein, E.; Horvat, R.; Chandran, B. Comparison of human sera reactivities in immunoblots with recombinant human herpesvirus (HHV)-8 proteins associated with the latent (ORF73) and lytic (ORFs 65, K8.1A, and K8.1B) replicative cycles and in immunofluorescence assays with HHV-8-infected BCBL-1 cells. Virology 1999, 256, 381–392. [Google Scholar] [PubMed]
- Birkmann, A.; Mahr, K.; Ensser, A.; Yaguboglu, S.; Titgemeyer, F.; Fleckenstein, B.; Neipel, F. Cell surface heparan sulfate is a receptor for human herpesvirus 8 and interacts with envelope glycoprotein K8.1. J. Virol. 2001, 75, 11583–11593. [Google Scholar] [CrossRef] [PubMed]
- Wang, F.Z.; Akula, S.M.; Pramod, N.P.; Zeng, L.; Chandran, B. Human herpesvirus 8 envelope glycoprotein K8.1A interaction with the target cells involves heparan sulfate. J. Virol. 2001, 75, 7517–7527. [Google Scholar] [CrossRef] [PubMed]
- Mark, L.; Lee, W.H.; Spiller, O.B.; Villoutreix, B.O.; Blom, A.M. The Kaposi’s sarcoma-associated herpesvirus complement control protein (KCP) binds to heparin and cell surfaces via positively charged amino acids in CCP1–2. Mol. Immunol. 2006, 43, 1665–1675. [Google Scholar] [CrossRef] [PubMed]
- Hahn, A.; Birkmann, A.; Wies, E.; Dorer, D.; Mahr, K.; Sturzl, M.; Titgemeyer, F.; Neipel, F. Kaposi’s sarcoma-associated herpesvirus gH/gL: Glycoprotein export and interaction with cellular receptors. J. Virol. 2009, 83, 396–407. [Google Scholar] [CrossRef] [PubMed]
- Naranatt, P.P.; Akula, S.M.; Chandran, B. Characterization of gamma2-human herpesvirus-8 glycoproteins gH and gL. Arch. Virol. 2002, 147, 1349–1370. [Google Scholar] [CrossRef] [PubMed]
- Koyano, S.; Mar, E.C.; Stamey, F.R.; Inoue, N. Glycoproteins M and N of human herpesvirus 8 form a complex and inhibit cell fusion. J. Gen. Virol. 2003, 84, 1485–1491. [Google Scholar] [CrossRef] [PubMed]
- Kerur, N.; Veettil, M.V.; Sharma-Walia, N.; Sadagopan, S.; Bottero, V.; Paul, A.G.; Chandran, B. Characterization of entry and infection of monocytic THP-1 cells by Kaposi’s sarcoma associated herpesvirus (KSHV): Role of heparan sulfate, DC-SIGN, integrins and signaling. Virology 2010, 406, 103–116. [Google Scholar] [CrossRef] [PubMed]
- Kabir-Salmani, M.; Fukuda, M.N.; Kanai-Azuma, M.; Ahmed, N.; Shiokawa, S.; Akimoto, Y.; Sakai, K.; Nagamori, S.; Kanai, Y.; Sugihara, K.; et al. The membrane-spanning domain of CD98 heavy chain promotes alpha(v)beta3 integrin signals in human extravillous trophoblasts. Mol. Endocrinol. 2008, 22, 707–715. [Google Scholar] [CrossRef] [PubMed]
- Garrigues, H.J.; Rubinchikova, Y.E.; Dipersio, C.M.; Rose, T.M. Integrin αVβ3 Binds to the RGD motif of glycoprotein B of Kaposi’s sarcoma-associated herpesvirus and functions as an RGD-dependent entry receptor. J. Virol. 2008, 82, 1570–1580. [Google Scholar] [CrossRef] [PubMed]
- Rappocciolo, G.; Jenkins, F.J.; Hensler, H.R.; Piazza, P.; Jais, M.; Borowski, L.; Watkins, S.C.; Rinaldo, C.R., Jr. DC-SIGN is a receptor for human herpesvirus 8 on dendritic cells and macrophages. J. Immunol. 2006, 176, 1741–1749. [Google Scholar] [CrossRef] [PubMed]
- Hahn, A.S.; Kaufmann, J.K.; Wies, E.; Naschberger, E.; Panteleev-Ivlev, J.; Schmidt, K.; Holzer, A.; Schmidt, M.; Chen, J.; Konig, S.; et al. The ephrin receptor tyrosine kinase A2 is a cellular receptor for Kaposi’s sarcoma-associated herpesvirus. Nat. Med. 2012, 18, 961–966. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, H.H.; Sharma-Walia, N.; Streblow, D.N.; Naranatt, P.P.; Chandran, B. Focal adhesion kinase is critical for entry of Kaposi’s sarcoma-associated herpesvirus into target cells. J. Virol. 2006, 80, 1167–1180. [Google Scholar] [CrossRef] [PubMed]
- Veettil, M.V.; Sharma-Walia, N.; Sadagopan, S.; Raghu, H.; Sivakumar, R.; Naranatt, P.P.; Chandran, B. RhoA-GTPase facilitates entry of Kaposi’s sarcoma-associated herpesvirus into adherent target cells in a Src-dependent manner. J. Virol. 2006, 80, 11432–11446. [Google Scholar] [CrossRef] [PubMed]
- Naranatt, P.P.; Akula, S.M.; Zien, C.A.; Krishnan, H.H.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus induces the phosphatidylinositol 3-kinase-PKC-zeta-MEK-ERK signaling pathway in target cells early during infection: Implications for infectivity. J. Virol. 2003, 77, 1524–1539. [Google Scholar] [CrossRef] [PubMed]
- Bandyopadhyay, C.; Veettil, M.V.; Dutta, S.; Chandran, B. p130Cas scaffolds the signalosome to direct adaptor-effector cross talk during Kaposi’s sarcoma-associated herpesvirus trafficking in human microvascular dermal endothelial cells. J. Virol. 2014, 88, 13858–13878. [Google Scholar] [CrossRef] [PubMed]
- Garrigues, H.J.; DeMaster, L.K.; Rubinchikova, Y.E.; Rose, T.M. KSHV attachment and entry are dependent on αVβ3 integrin localized to specific cell surface microdomains and do not correlate with the presence of heparan sulfate. Virology 2014, 464–465, 118–133. [Google Scholar] [CrossRef] [PubMed]
- Walker, L.R.; Hussein, H.A.; Akula, S.M. Disintegrin-like domain of glycoprotein B regulates Kaposi’s sarcoma-associated herpesvirus infection of cells. J. Gen. Virol. 2014, 95, 1770–1782. [Google Scholar] [CrossRef] [PubMed]
- Kaleeba, J.A.; Berger, E.A. Kaposi’s sarcoma-associated herpesvirus fusion-entry receptor: Cystine transporter xCT. Science 2006, 311, 1921–1924. [Google Scholar] [CrossRef] [PubMed]
- Fenczik, C.A.; Zent, R.; Dellos, M.; Calderwood, D.A.; Satriano, J.; Kelly, C.; Ginsberg, M.H. Distinct domains of CD98hc regulate integrins and amino acid transport. J. Biol. Chem. 2001, 276, 8746–8752. [Google Scholar] [CrossRef] [PubMed]
- Feral, C.C.; Zijlstra, A.; Tkachenko, E.; Prager, G.; Gardel, M.L.; Slepak, M.; Ginsberg, M.H. CD98hc (SLC3A2) participates in fibronectin matrix assembly by mediating integrin signaling. J. Cell Biol. 2007, 178, 701–711. [Google Scholar] [CrossRef] [PubMed]
- Geijtenbeek, T.B.; van Kooyk, Y. DC-SIGN: A novel HIV receptor on DCs that mediates HIV-1 transmission. Curr. Top. Microbiol. Immunol. 2003, 276, 31–54. [Google Scholar] [PubMed]
- Samreen, B.; Khaliq, S.; Ashfaq, U.A.; Khan, M.; Afzal, N.; Shahzad, M.A.; Riaz, S.; Jahan, S. Hepatitis C virus entry: Role of host and viral factors. Infect. Genet. Evol. 2012, 12, 1699–1709. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Sanchez, E.; Altmeyer, R.; Amara, A.; Schwartz, O.; Fieschi, F.; Virelizier, J.L.; Arenzana-Seisdedos, F.; Despres, P. Dendritic-cell-specific ICAM3-grabbing non-integrin is essential for the productive infection of human dendritic cells by mosquito-cell-derived dengue viruses. EMBO Rep. 2003, 4, 723–728. [Google Scholar] [CrossRef] [PubMed]
- Lozach, P.Y.; Kuhbacher, A.; Meier, R.; Mancini, R.; Bitto, D.; Bouloy, M.; Helenius, A. DC-SIGN as a receptor for phleboviruses. Cell Host Microbe 2011, 10, 75–88. [Google Scholar] [CrossRef] [PubMed]
- Soilleux, E.J.; Morris, L.S.; Trowsdale, J.; Coleman, N.; Boyle, J.J. Human atherosclerotic plaques express DC-SIGN, a novel protein found on dendritic cells and macrophages. J. Pathol. 2002, 198, 511–516. [Google Scholar] [CrossRef] [PubMed]
- Santarelli, R.; Gonnella, R.; di Giovenale, G.; Cuomo, L.; Capobianchi, A.; Granato, M.; Gentile, G.; Faggioni, A.; Cirone, M. STAT3 activation by KSHV correlates with IL-10, IL-6 and IL-23 release and an autophagic block in dendritic cells. Sci. Rep. 2014. [Google Scholar] [CrossRef] [PubMed]
- Pasquale, E.B. Eph receptor signalling casts a wide net on cell behaviour. Nat. Rev. Mol. Cell Biol. 2005, 6, 462–475. [Google Scholar] [CrossRef] [PubMed]
- Himanen, J.P.; Saha, N.; Nikolov, D.B. Cell-cell signaling via Eph receptors and ephrins. Curr. Opin. Cell Biol. 2007, 19, 534–542. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Veettil, M.V.; Bottero, V.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus interacts with EphrinA2 receptor to amplify signaling essential for productive infection. Proc. Natl. Acad. Sci. USA 2012, 109, E1163–E1172. [Google Scholar] [CrossRef] [PubMed]
- Giancotti, F.G. Complexity and specificity of integrin signalling. Nat. Cell Biol. 2000, 2, E13–E14. [Google Scholar] [CrossRef] [PubMed]
- Giancotti, F.G. A structural view of integrin activation and signaling. Dev. Cell 2003, 4, 149–151. [Google Scholar] [CrossRef]
- Calderwood, D.A.; Shattil, S.J.; Ginsberg, M.H. Integrins and actin filaments: Reciprocal regulation of cell adhesion and signaling. J. Biol. Chem. 2000, 275, 22607–22610. [Google Scholar] [CrossRef] [PubMed]
- Raghu, H.; Sharma-Walia, N.; Veettil, M.V.; Sadagopan, S.; Caballero, A.; Sivakumar, R.; Varga, L.; Bottero, V.; Chandran, B. Lipid rafts of primary endothelial cells are essential for Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8-induced phosphatidylinositol 3-kinase and RhoA-GTPases critical for microtubule dynamics and nuclear delivery of viral DNA but dispensable for binding and entry. J. Virol. 2007, 81, 7941–7959. [Google Scholar] [PubMed]
- Hall, A.; Nobes, C.D. Rho GTPases: Molecular switches that control the organization and dynamics of the actin cytoskeleton. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2000, 355, 965–970. [Google Scholar] [CrossRef] [PubMed]
- Ishizaki, T.; Morishima, Y.; Okamoto, M.; Furuyashiki, T.; Kato, T.; Narumiya, S. Coordination of microtubules and the actin cytoskeleton by the Rho effector mDia1. Nat. Cell Biol 2001, 3, 8–14. [Google Scholar] [PubMed]
- Greene, W.; Gao, S.J. Actin dynamics regulate multiple endosomal steps during Kaposi’s sarcoma-associated herpesvirus entry and trafficking in endothelial cells. PLoS Pathog. 2009, 5, e1000512. [Google Scholar] [CrossRef] [PubMed]
- Thien, C.B.; Langdon, W.Y. Cbl: Many adaptations to regulate protein tyrosine kinases. Nat. Rev. Mol. Cell Biol. 2001, 2, 294–307. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, M.H.; Dikic, I. The Cbl interactome and its functions. Nat. Rev. Mol. Cell Biol 2005, 6, 907–918. [Google Scholar] [CrossRef] [PubMed]
- Greene, W.; Zhang, W.; He, M.; Witt, C.; Ye, F.; Gao, S.J. The ubiquitin/proteasome system mediates entry and endosomal trafficking of Kaposi’s sarcoma-associated herpesvirus in endothelial cells. PLoS Pathog. 2012, 8, e1002703. [Google Scholar] [CrossRef] [PubMed]
- Naik, M.U.; Naik, U.P. Calcium-and integrin-binding protein regulates focal adhesion kinase activity during platelet spreading on immobilized fibrinogen. Blood 2003, 102, 3629–3636. [Google Scholar] [CrossRef] [PubMed]
- Naik, M.U.; Naik, U.P. Contra-regulation of calcium- and integrin-binding protein 1-induced cell migration on fibronectin by PAK1 and MAP kinase signaling. J. Cell. Biochem. 2011, 112, 3289–3299. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Lipp, P.; Bootman, M.D. The versatility and universality of calcium signalling. Nat. Rev. Mol. Cell Biol. 2000, 1, 11–21. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.C.; Zhou, F.C.; Nithianantham, S.; Chandran, B.; Yu, X.L.; Weinberg, A.; Gao, S.J. Kaposi’s sarcoma-associated herpesvirus induces rapid release of angiopoietin-2 from endothelial cells. J. Virol. 2013, 87, 6326–6335. [Google Scholar] [CrossRef] [PubMed]
- Bozym, R.A.; Morosky, S.A.; Kim, K.S.; Cherry, S.; Coyne, C.B. Release of intracellular calcium stores facilitates coxsackievirus entry into polarized endothelial cells. PLoS Pathog. 2010, 6, e1001135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheshenko, N.; del Rosario, B.; Woda, C.; Marcellino, D.; Satlin, L.M.; Herold, B.C. Herpes simplex virus triggers activation of calcium-signaling pathways. J. Cell Biol. 2003, 163, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Uemura, N.; Griffin, J.D. The adapter protein Crkl links Cbl to C3G after integrin ligation and enhances cell migration. J. Biol. Chem. 1999, 274, 37525–37532. [Google Scholar] [CrossRef] [PubMed]
- Li, E.; Stupack, D.G.; Brown, S.L.; Klemke, R.; Schlaepfer, D.D.; Nemerow, G.R. Association of p130CAS with phosphatidylinositol-3-OH kinase mediates adenovirus cell entry. J. Biol. Chem. 2000, 275, 14729–14735. [Google Scholar] [CrossRef] [PubMed]
- Simons, K.; Toomre, D. Lipid rafts and signal transduction. Nat. Rev. Mol. Cell Biol. 2000, 1, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Naranatt, P.P.; Krishnan, H.H.; Smith, M.S.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus modulates microtubule dynamics via RhoA-GTP-diaphanous 2 signaling and utilizes the dynein motors to deliver its DNA to the nucleus. J. Virol. 2005, 79, 1191–1206. [Google Scholar] [CrossRef] [PubMed]
- Naranatt, P.P.; Krishnan, H.H.; Svojanovsky, S.R.; Bloomer, C.; Mathur, S.; Chandran, B. Host gene induction and transcriptional reprogramming in Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8)-infected endothelial, fibroblast, and B cells: Insights into modulation events early during infection. Cancer Res. 2004, 64, 72–84. [Google Scholar] [CrossRef] [PubMed]
- Ma, Q.; Cavallin, L.E.; Leung, H.J.; Chiozzini, C.; Goldschmidt-Clermont, P.J.; Mesri, E.A. A role for virally induced reactive oxygen species in Kaposi’s sarcoma herpesvirus tumorigenesis. Antioxid. Redox Signal. 2013, 18, 80–90. [Google Scholar] [CrossRef] [PubMed]
- Bottero, V.; Chakraborty, S.; Chandran, B. Reactive oxygen species are induced by Kaposi’s sarcoma-associated herpesvirus early during primary infection of endothelial cells to promote virus entry. J. Virol. 2013, 87, 1733–1749. [Google Scholar] [CrossRef] [PubMed]
- Gjyshi, O.; Bottero, V.; Veettil, M.V.; Dutta, S.; Singh, V.V.; Chikoti, L.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus induces Nrf2 during de novo infection of endothelial cells to create a microenvironment conducive to infection. PLoS Pathog. 2014, 10, e1004460. [Google Scholar] [CrossRef] [PubMed]
- Hurley, J.H.; Hanson, P.I. Membrane budding and scission by the ESCRT machinery: It’s all in the neck. Nat. Rev. Mol. Cell Biol. 2010, 11, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Demirov, D.G.; Ono, A.; Orenstein, J.M.; Freed, E.O. Overexpression of the N-terminal domain of TSG101 inhibits HIV-1 budding by blocking late domain function. Proc. Natl. Acad. Sci. USA 2002, 99, 955–960. [Google Scholar] [CrossRef] [PubMed]
- Garrus, J.E.; von Schwedler, U.K.; Pornillos, O.W.; Morham, S.G.; Zavitz, K.H.; Wang, H.E.; Wettstein, D.A.; Stray, K.M.; Cote, M.; Rich, R.L.; et al. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell 2001, 107, 55–65. [Google Scholar] [CrossRef]
- Shtanko, O.; Nikitina, R.A.; Altuntas, C.Z.; Chepurnov, A.A.; Davey, R.A. Crimean-Congo hemorrhagic fever virus entry into host cells occurs through the multivesicular body and requires ESCRT regulators. PLoS Pathog. 2014, 10, e1004390. [Google Scholar] [CrossRef] [PubMed]
- Silva-Ayala, D.; Lopez, T.; Gutierrez, M.; Perrimon, N.; Lopez, S.; Arias, C.F. Genome-wide RNAi screen reveals a role for the ESCRT complex in rotavirus cell entry. Proc. Natl. Acad. Sci. USA 2013, 110, 10270–10275. [Google Scholar] [CrossRef] [PubMed]
- Pasqual, G.; Rojek, J.M.; Masin, M.; Chatton, J.Y.; Kunz, S. Old world arenaviruses enter the host cell via the multivesicular body and depend on the endosomal sorting complex required for transport. PLoS Pathog. 2011, 7, e1002232. [Google Scholar] [CrossRef]
- Sharma-Walia, N.; Krishnan, H.H.; Naranatt, P.P.; Zeng, L.; Smith, M.S.; Chandran, B. ERK1/2 and MEK1/2 induced by Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) early during infection of target cells are essential for expression of viral genes and for establishment of infection. J. Virol. 2005, 79, 10308–10329. [Google Scholar] [CrossRef] [PubMed]
- Sadagopan, S.; Sharma-Walia, N.; Veettil, M.V.; Raghu, H.; Sivakumar, R.; Bottero, V.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus induces sustained NF-γB activation during de novo infection of primary human dermal microvascular endothelial cells that is essential for viral gene expression. J. Virol. 2007, 81, 3949–3968. [Google Scholar] [CrossRef] [PubMed]
- Gjyshi, O.; Roy, A.; Dutta, S.; Veettil, M.V.; Dutta, D.; Chandran, B. Activated Nrf2 interacts with Kaposi’s sarcoma-associated herpesvirus latency protein LANA-1 and host protein KAP1 to mediate global lytic gene repression. J. Virol. 2015, 89, 7874–7892. [Google Scholar] [CrossRef] [PubMed]
- Ciccia, A.; Elledge, S.J. The DNA damage response: Making it safe to play with knives. Mol. Cell 2010, 40, 179–204. [Google Scholar] [CrossRef] [PubMed]
- Turnell, A.S.; Grand, R.J. DNA viruses and the cellular DNA-damage response. J. Gen. Virol. 2012, 93, 2076–2097. [Google Scholar] [CrossRef] [PubMed]
- Singh, V.V.; Dutta, D.; Ansari, M.A.; Dutta, S.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus induces the ATM and H2AX DNA damage response early during de novo infection of primary endothelial cells, which play roles in latency establishment. J. Virol. 2014, 88, 2821–2834. [Google Scholar] [CrossRef] [PubMed]
- Kerur, N.; Veettil, M.V.; Sharma-Walia, N.; Bottero, V.; Sadagopan, S.; Otageri, P.; Chandran, B. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated herpesvirus infection. Cell Host Microbe 2011, 9, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Dutta, D.; Dutta, S.; Veettil, M.V.; Roy, A.; Ansari, M.A.; Iqbal, J.; Chikoti, L.; Kumar, B.; Johnson, K.E.; Chandran, B. BRCA1 regulates IFI16 mediated nuclear innate sensing of herpes viral DNA and subsequent induction of the innate inflammasome and interferon-beta responses. PLoS Pathog. 2015, 11, e1005030. [Google Scholar] [CrossRef] [PubMed]
- Ansari, M.A.; Dutta, S.; Veettil, M.V.; Dutta, D.; Iqbal, J.; Kumar, B.; Roy, A.; Chikoti, L.; Singh, V.V.; Chandran, B. Herpesvirus genome recognition induced acetylation of nuclear IFI16 is essential for its cytoplasmic translocation, inflammasome and IFN-beta responses. PLoS Pathog. 2015, 11, e1005019. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
| KSHV Receptors | Inhibitor/Treatment | Cell Type | Effect | References |
| Heparan sulfate | Pre-incubation of virus with soluble heparin | HMVEC-d, HFF, BJAB, HEK293, THP1 | Blocks virus binding to the cell | [21,41,43,44,45,51,54,55,57,58] |
| α3β1 integrin | Pre-treatment of cells with anti- α3β1 antibodies; Pre-incubation of virus with soluble α3β1 integrin | HMVEC-d, HFF, THP1 | Blocks virus entry and no effect on virus binding | [44,45,57] |
| αVβ3 integrin | Pre-treatment of cells with anti-αVβ3 antibodies; Pre-incubation of virus with soluble αVβ3 integrin | HMVEC-d, HFF, THP-1 | Blocks virus entry and no effect on virus binding | [45,57,59] |
| αVβ5 integrin | Pre-treatment of cells with anti-αVβ5 antibodies; Pre-incubation of virus with soluble αVβ5 integrin | HMVEC-d, HFF, THP-1 | Blocks virus entry and no effect on virus binding | [44,59] |
| xCT | Pre-treatment of cells with anti-xCT antibodies | HMVEC-d | Blocks virus gene expression and no effect on virus binding and entry | [44] |
| CD98 | Pre-treatment of cells with anti-CD98 antibodies | HMVEC-d | Blocks virus gene expression and no effect on virus binding and entry | [44] |
| DC-SIGN | Pre-treatment of cells with Mannan; Pre-treatment of cells with anti-DC-SIGN antibodies | B cells, THP-1 | Blocks virus binding and entry | [23,57,60] |
| EphA2R | Pre-treatment of cells with anti-EphA2R antibodies; Pre-incubation of virus with soluble EphA2 | HMVEC-d, HFF | Blocks virus entry and no effect on virus binding | [35,61] |
| Signal Molecules Induced by KSHV | Inhibitor/Treatment | Cell Type | Effect | References |
| FAK | Focal Adhesion Kinase (FAK)-related Non-kinase (FRANK) | DU17 mouse embryonic fibroblasts | Blocks virus entry | [62] |
| Src | SU6656 | HEK293 | Blocks virus entry | [63] |
| PI3K | LY294002 | HMVEC-d, HFF | Blocks virus entry | [64] |
| RhoA | Clostridium difficile toxin B | HMVEC-d, HFF | Blocks virus entry | [63] |
| PKCζ | myr-ζ | HFF | Blocks viral gene expression | [64] |
| CIB1 | shCIB1 | HMVEC-d | Blocks virus entry | [31] |
| p130Cas | shCas | HMVEC-d | Blocks nuclear viral genome delivery | [65] |
| Hrs | shHrs | HMVEC-d | Blocks virus entry | [32] |
| Tsg101 | siTsg101 | HMVEC-d | Blocks cytoplasmic KSHV trafficking and nuclear viral genome delivery | [33] |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kumar, B.; Chandran, B. KSHV Entry and Trafficking in Target Cells—Hijacking of Cell Signal Pathways, Actin and Membrane Dynamics. Viruses 2016, 8, 305. https://doi.org/10.3390/v8110305
Kumar B, Chandran B. KSHV Entry and Trafficking in Target Cells—Hijacking of Cell Signal Pathways, Actin and Membrane Dynamics. Viruses. 2016; 8(11):305. https://doi.org/10.3390/v8110305
Chicago/Turabian StyleKumar, Binod, and Bala Chandran. 2016. "KSHV Entry and Trafficking in Target Cells—Hijacking of Cell Signal Pathways, Actin and Membrane Dynamics" Viruses 8, no. 11: 305. https://doi.org/10.3390/v8110305