
Where in the Cell Are You? Probing HIV-1 Host Interactions through Advanced Imaging Techniques

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
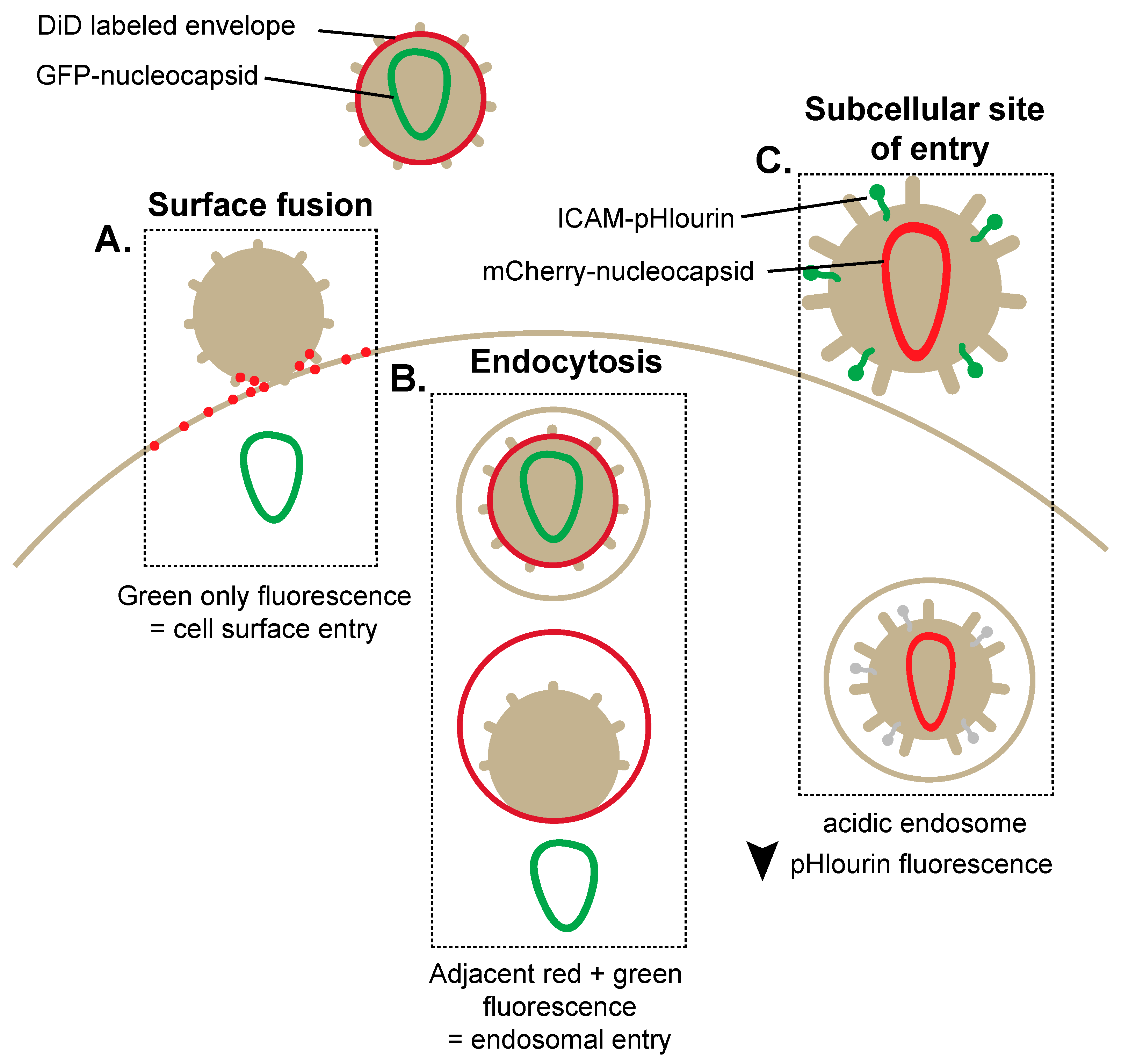
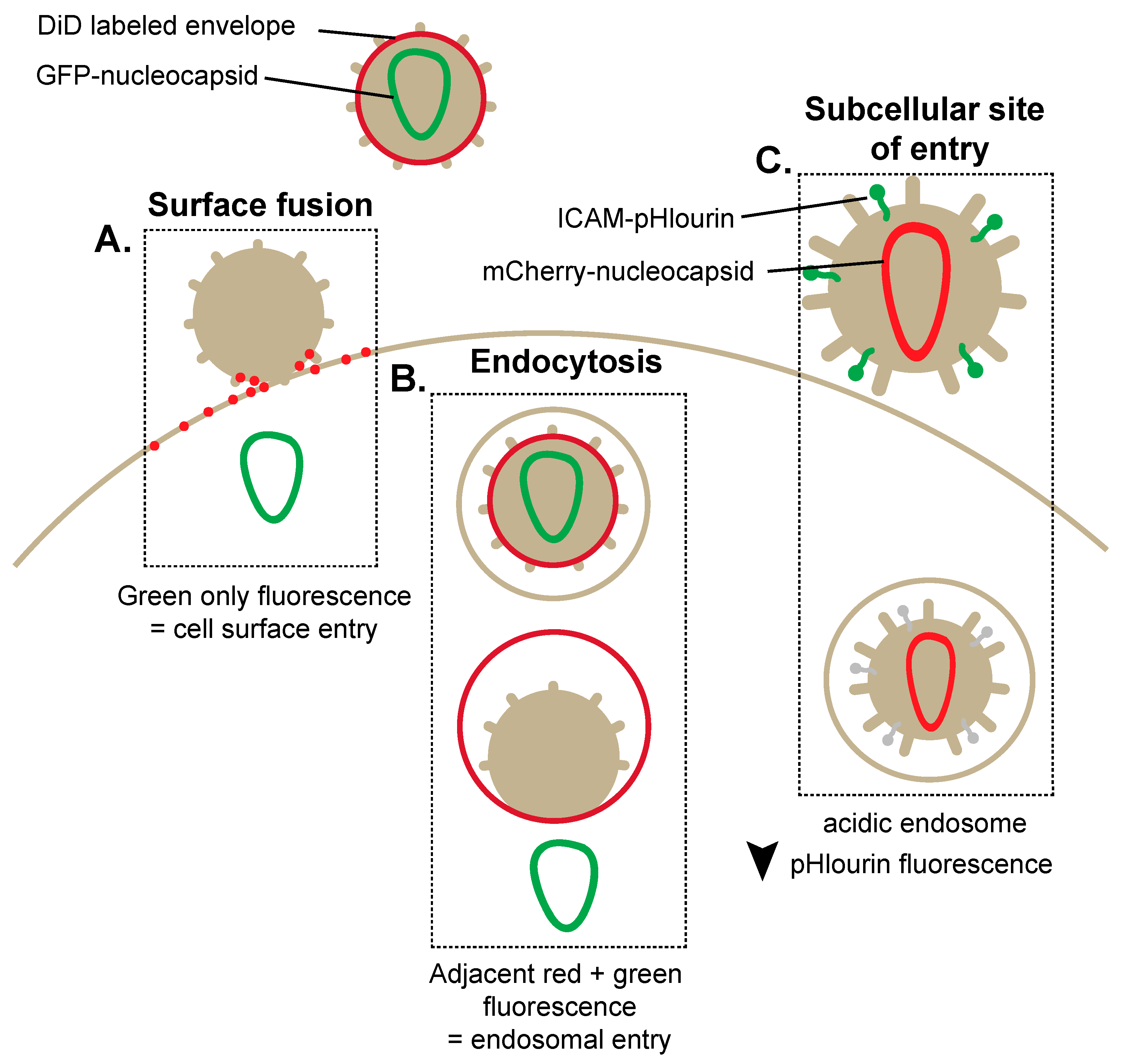
2. Breaking and Entering: Imaging Mechanisms of HIV-1 Entry
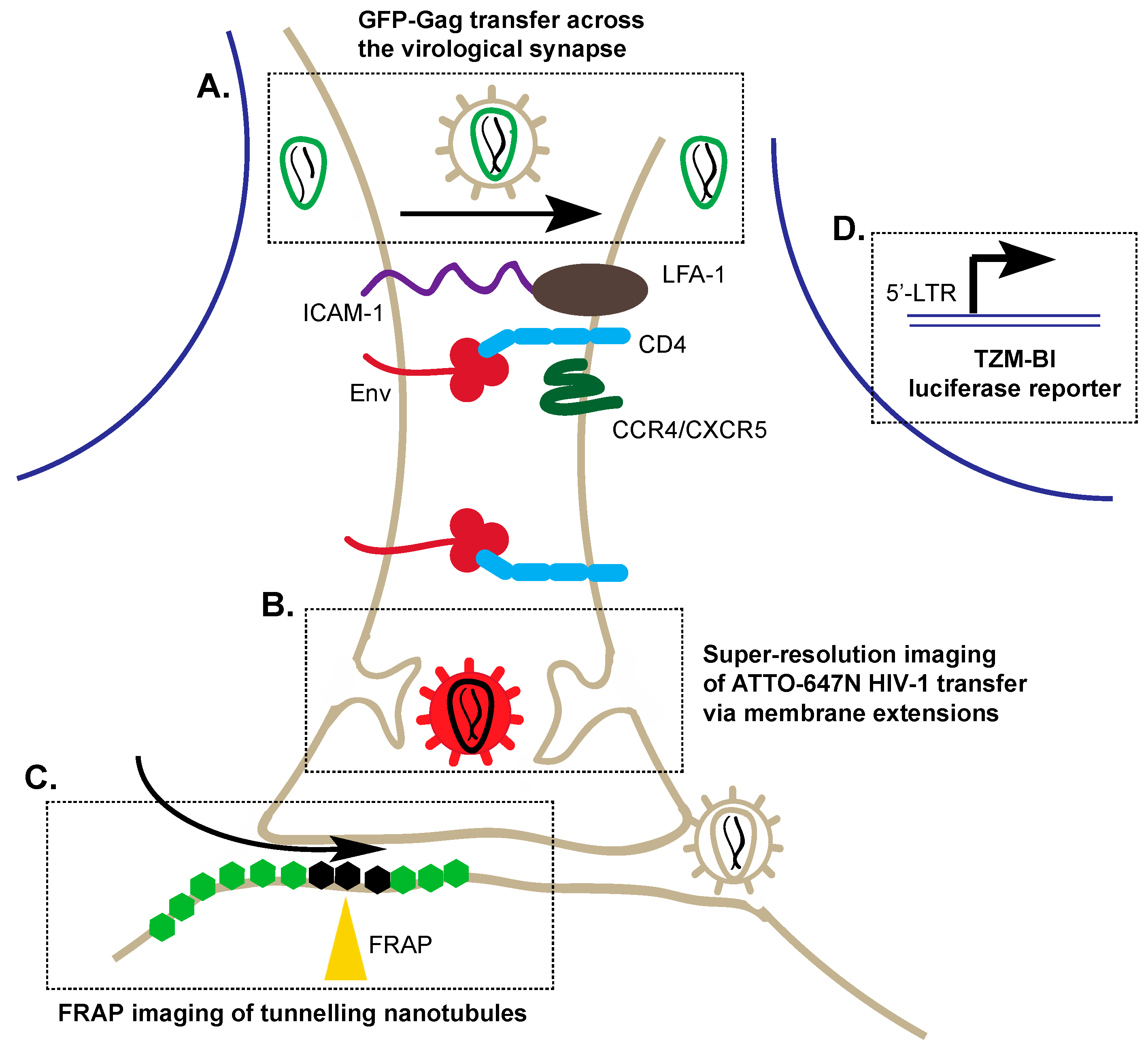
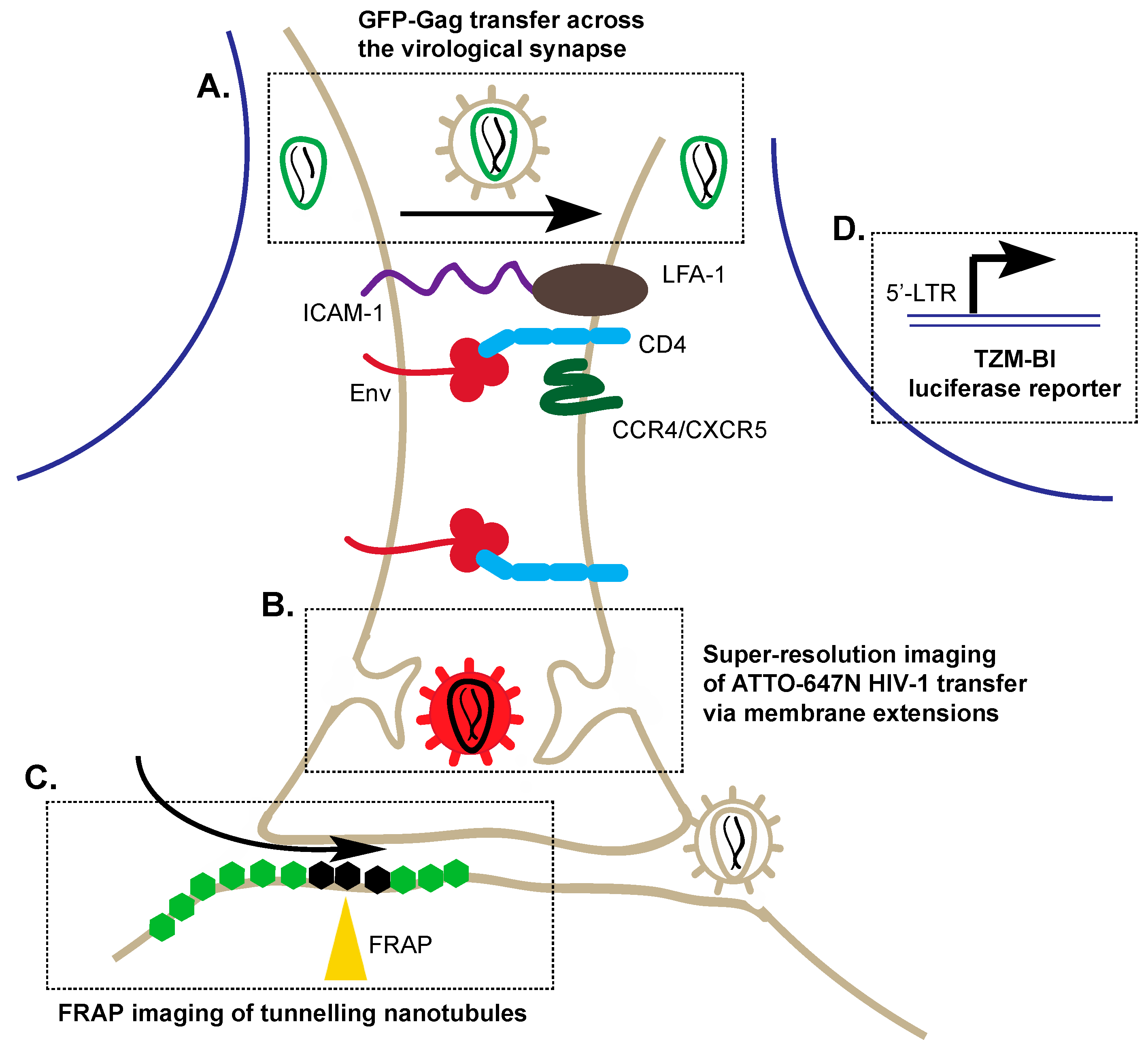
3. Alternative Transmission of HIV-1: Cell–Cell Viral Entry
3.1. Application of Co-Cultures to Study Cell–Cell Transmission
3.2. Super-Resolution Imaging of Cell–Cell Transmission
3.3. Fluorescence Recovery after Photobleaching (FRAP)
3.4. Reporters to Examine Cell–Cell Transmission
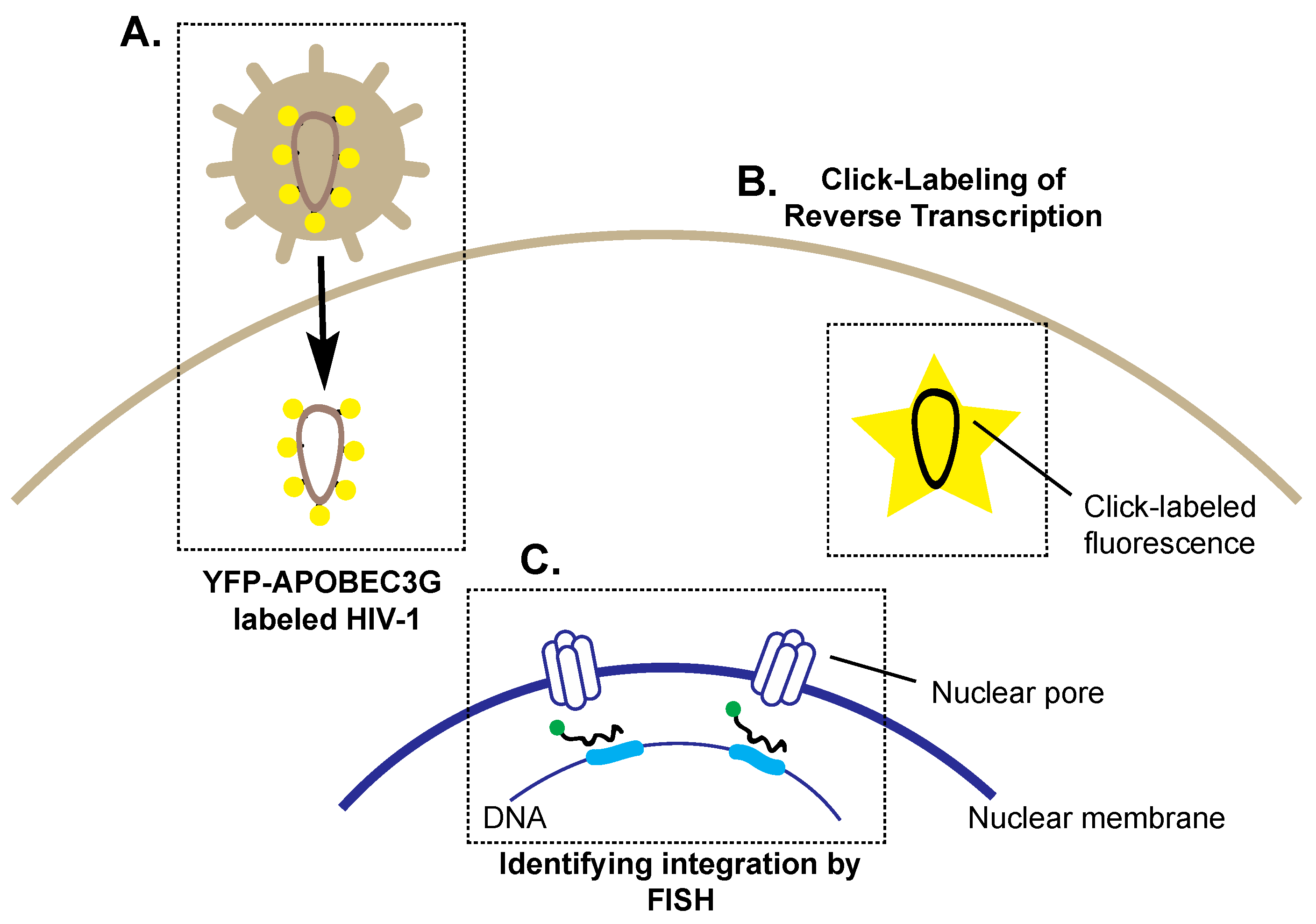
4. Preparing for Landing: Imaging HIV-1 Nuclear Entry and Integration
4.1. Imaging Reverse Transcription and Nuclear Entry
4.2. Imaging Integration and Latency
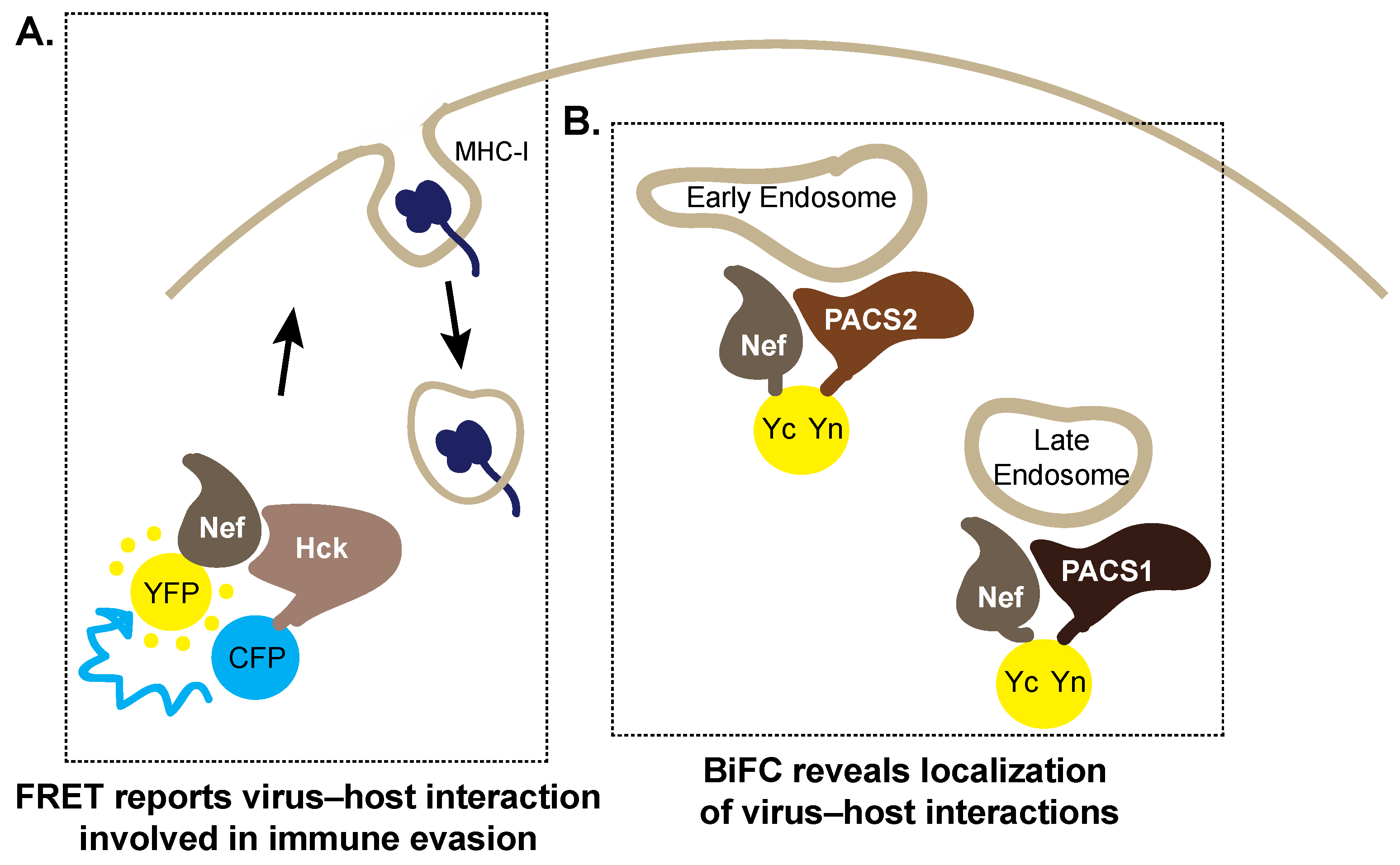
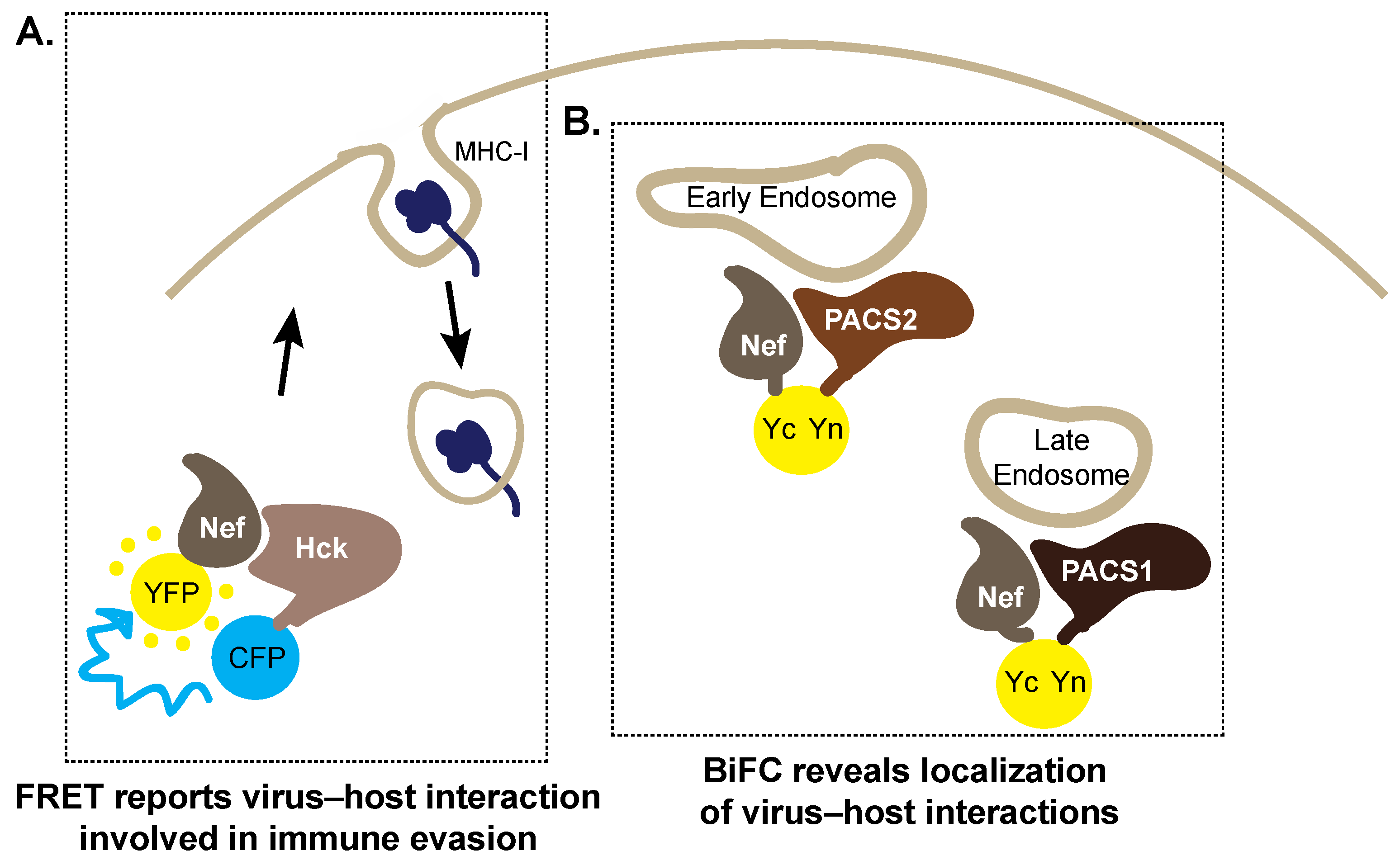
5. Trespassing into Enemy Territory: Imaging Host Cell Hijacking by HIV-1 Nef
5.1. Forster Resonance Energy Transfer
5.2. Bimolecular Fluorescence Complementation
5.3. Ground State Depletion Super-Resolution Microscopy
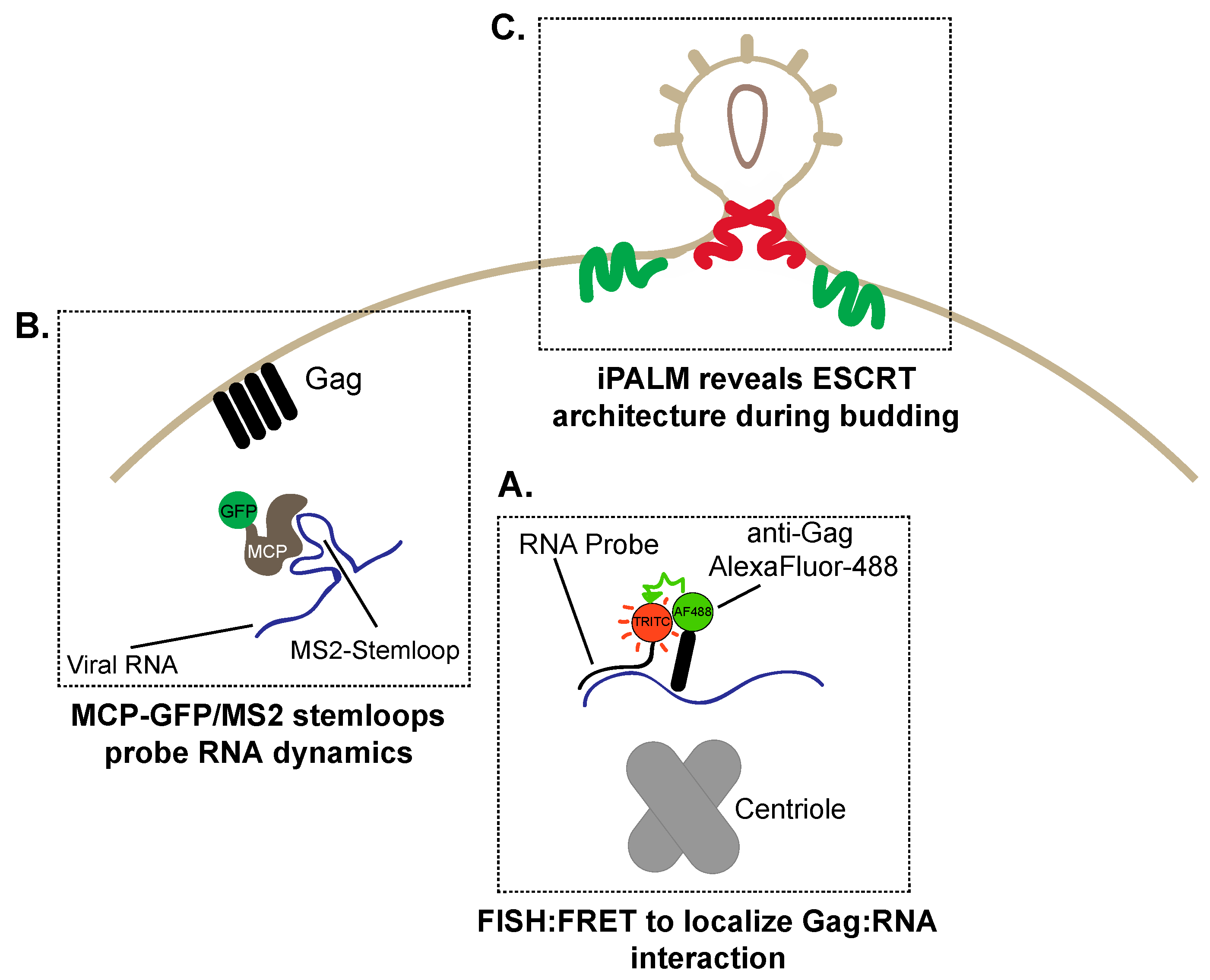
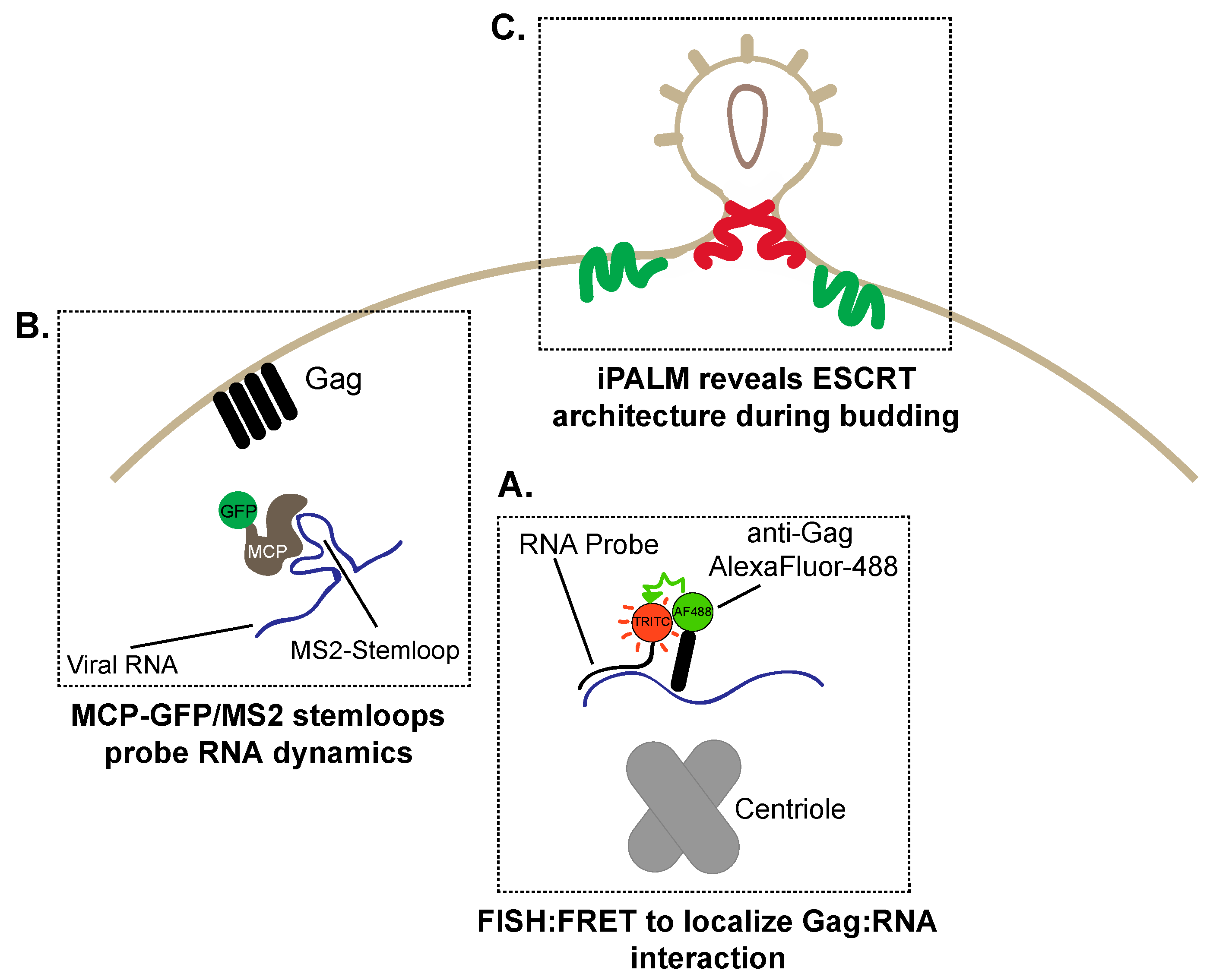
6. The Great Escape: Imaging HIV-1 Assembly and Budding
6.1. Visualizing RNA Trafficking to HIV-1 Assembly Sites
6.2. Super-Resolution Imaging of HIV-1 Budding
7. Applications of Cell-Culture Based Microscopy Assays for Drug Discovery
8. Future Directions
9. Conclusions
Acknowledgments
Conflicts of Interest
References
- Edwards, R.A.; Rohwer, F. Viral metagenomics. Nat. Rev. Microbiol. 2005, 3, 504–510. [Google Scholar] [CrossRef] [PubMed]
- Frankel, A.D.; Young, J.A. HIV-1: Fifteen proteins and an RNA. Annu. Rev. Biochem. 1998, 67, 1–25. [Google Scholar] [CrossRef] [PubMed]
- Stein, B.S.; Gowda, S.D.; Lifson, J.D.; Penhallow, R.C.; Bensch, K.G.; Engleman, E.G. pH-independent HIV entry into CD4-positive T cells via virus envelope fusion to the plasma membrane. Cell 1987, 49, 659–668. [Google Scholar] [CrossRef]
- Bleul, C.C.; Wu, L.; Hoxie, J.A.; Springer, T.A.; Mackay, C.R. The HIV coreceptors CXCR4 and CCR5 are differentially expressed and regulated on human T lymphocytes. Proc. Natl. Acad. Sci. USA 1997, 94, 1925–1930. [Google Scholar] [CrossRef] [PubMed]
- Salzwedel, K.; West, J.T.; Hunter, E. A conserved tryptophan-rich motif in the membrane-proximal region of the human immunodeficiency virus type 1 GP41 ectodomain is important for Env-mediated fusion and virus infectivity. J. Virol. 1999, 73, 2469–2480. [Google Scholar] [PubMed]
- McDonald, D.; Vodicka, M.A.; Lucero, G.; Svitkina, T.M.; Borisy, G.G.; Emerman, M.; Hope, T.J. Visualization of the intracellular behavior of HIV in living cells. J. Cell Biol. 2002, 159, 441–452. [Google Scholar] [CrossRef] [PubMed]
- Miyauchi, K.; Kim, Y.; Latinovic, O.; Morozov, V.; Melikyan, G.B. HIV enters cells via endocytosis and dynamin-dependent fusion with endosomes. Cell 2009, 137, 433–444. [Google Scholar] [CrossRef] [PubMed]
- Sood, C.; Marin, M.; Mason, C.S.; Melikyan, G.B. Visualization of Content Release from Cell Surface-Attached Single HIV-1 Particles Carrying an Extra-Viral Fluorescent pH-Sensor. PLoS ONE 2016, 11, e0148944. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; He, Z.; Tan, T.; Li, W.; Zhang, Z.-P.; Song, S.; Zhang, X.; Hu, Q.; Zhou, P.; Wu, Y. Real-Time Imaging of Single HIV-1 Disassembly with Multicolor Viral Particles. ACS Nano 2016, 10, 6273–6282. [Google Scholar] [CrossRef] [PubMed]
- Blumenthal, R.; Durell, S.; Viard, M. HIV entry and envelope glycoprotein-mediated fusion. J. Biol. Chem. 2012, 287, 40841–40849. [Google Scholar] [CrossRef] [PubMed]
- Melikyan, G.B. HIV entry: A game of hide-and-fuse? Curr. Opin. Virol. 2014, 4, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Miyauchi, K.; Marin, M.; Melikyan, G.B. Visualization of retrovirus uptake and delivery into acidic endosomes. Biochem. J. 2011, 434, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Padilla-Parra, S.; Matos, P.M.; Kondo, N.; Marin, M.; Santos, N.C.; Melikyan, G.B. Quantitative imaging of endosome acidification and single retrovirus fusion with distinct pools of early endosomes. Proc. Natl. Acad. Sci. USA 2012, 109, 17627–17632. [Google Scholar] [CrossRef] [PubMed]
- Felts, R.L.; Narayan, K.; Estes, J.D.; Shi, D.; Trubey, C.M.; Fu, J.; Hartnell, L.M.; Ruthel, G.T.; Schneider, D.K.; Nagashima, K. 3D visualization of HIV transfer at the virological synapse between dendritic cells and T cells. Proc. Natl. Acad. Sci. USA 2010, 107, 13336–13341. [Google Scholar] [CrossRef] [PubMed]
- Groot, F.; Welsch, S.; Sattentau, Q.J. Efficient HIV-1 transmission from macrophages to T cells across transient virological synapses. Blood 2008, 111, 4660–4663. [Google Scholar] [CrossRef] [PubMed]
- Jolly, C.; Kashefi, K.; Hollinshead, M.; Sattentau, Q.J. HIV-1 cell to cell transfer across an Env-induced, actin-dependent synapse. J. Exp. Med. 2004, 199, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Jolly, C.; Mitar, I.; Sattentau, Q.J. Adhesion molecule interactions facilitate human immunodeficiency virus type 1-induced virological synapse formation between T cells. J. Virol. 2007, 81, 13916–13921. [Google Scholar] [CrossRef] [PubMed]
- Nejmeddine, M.; Barnard, A.L.; Tanaka, Y.; Taylor, G.P.; Bangham, C.R. Human T-lymphotropic virus, type 1, Tax protein triggers microtubule reorientation in the virological synapse. J. Biol. Chem. 2005, 280, 29653–29660. [Google Scholar] [CrossRef] [PubMed]
- Fackler, O.T.; Alcover, A.; Schwartz, O. Modulation of the immunological synapse: A key to HIV-1 pathogenesis? Nat. Rev. Immunol. 2007, 7, 310–317. [Google Scholar] [CrossRef] [PubMed]
- Vasiliver-Shamis, G.; Dustin, M.L.; Hioe, C.E. HIV-1 virological synapse is not simply a copycat of the immunological synapse. Viruses 2010, 2, 1239–1260. [Google Scholar] [CrossRef] [PubMed]
- McDonald, D.; Wu, L.; Bohks, S.M.; KewalRamani, V.N.; Unutmaz, D.; Hope, T.J. Recruitment of HIV and its receptors to dendritic cell-T cell junctions. Science 2003, 300, 1295–1297. [Google Scholar] [CrossRef] [PubMed]
- Garcia, E.; Pion, M.; Pelchen-Matthews, A.; Collinson, L.; Arrighi, J.F.; Blot, G.; Leuba, F.; Escola, J.M.; Demaurex, N.; Marsh, M. HIV-1 Trafficking to the Dendritic Cell-T-Cell Infectious Synapse Uses a Pathway of Tetraspanin Sorting to the Immunological Synapse. Traffic 2005, 6, 488–501. [Google Scholar] [CrossRef] [PubMed]
- Cameron, P.U.; Freudenthal, P.S.; Barker, J.M.; Gezelter, S.; Inaba, K.; Steinman, R.M. Dendritic cells exposed to human immunodeficiency virus type-1 transmit a vigorous cytopathic infection to CD4+ T cells. Science 1992, 257, 383–387. [Google Scholar] [CrossRef] [PubMed]
- Del Portillo, A.; Tripodi, J.; Najfeld, V.; Wodarz, D.; Levy, D.N.; Chen, B.K. Multiploid inheritance of HIV-1 during cell-to-cell infection. J. Virol. 2011, 85, 7169–7176. [Google Scholar] [CrossRef] [PubMed]
- Sattentau, Q. Avoiding the void: Cell-to-cell spread of human viruses. Nat. Rev. Microbiol. 2008, 6, 815–826. [Google Scholar] [CrossRef] [PubMed]
- Reh, L.; Magnus, C.; Schanz, M.; Weber, J.; Uhr, T.; Rusert, P.; Trkola, A. Capacity of broadly neutralizing antibodies to inhibit HIV-1 cell-cell transmission is strain-and epitope-dependent. PLoS Pathog. 2015, 11, e1004966. [Google Scholar] [CrossRef] [PubMed]
- Agosto, L.M.; Uchil, P.D.; Mothes, W. HIV cell-to-cell transmission: Effects on pathogenesis and antiretroviral therapy. Trends Microbiol. 2015, 23, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Hübner, W.; McNerney, G.P.; Chen, P.; Dale, B.M.; Gordon, R.E.; Chuang, F.Y.; Li, X.-D.; Asmuth, D.M.; Huser, T.; Chen, B.K. Quantitative 3D video microscopy of HIV transfer across T cell virological synapses. Science 2009, 323, 1743–1747. [Google Scholar] [CrossRef] [PubMed]
- Gardiner, J.C.; Mauer, E.J.; Sherer, N.M. HIV-1 Gag, Envelope, and extracellular determinants cooperate to regulate the stability and turnover of virological synapses. J. Virol. 2016, 90, 6583–6597. [Google Scholar] [CrossRef] [PubMed]
- Jolly, C.; Sattentau, Q.J. Human immunodeficiency virus type 1 virological synapse formation in T cells requires lipid raft integrity. J. Virol. 2005, 79, 12088–12094. [Google Scholar] [CrossRef] [PubMed]
- Sol-Foulon, N.; Sourisseau, M.; Porrot, F.; Thoulouze, M.I.; Trouillet, C.; Nobile, C.; Blanchet, F.; di Bartolo, V.; Noraz, N.; Taylor, N. ZAP-70 kinase regulates HIV cell-to-cell spread and virological synapse formation. EMBO J. 2007, 26, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Klar, T.A.; Jakobs, S.; Dyba, M.; Egner, A.; Hell, S.W. Fluorescence microscopy with diffraction resolution barrier broken by stimulated emission. Proc. Natl. Acad. Sci. USA 2000, 97, 8206–8210. [Google Scholar] [CrossRef] [PubMed]
- Willig, K.I.; Harke, B.; Medda, R.; Hell, S.W. STED microscopy with continuous wave beams. Nat. Methods 2007, 4, 915–918. [Google Scholar] [CrossRef] [PubMed]
- Donnert, G.; Keller, J.; Medda, R.; Andrei, M.A.; Rizzoli, S.O.; Lührmann, R.; Jahn, R.; Eggeling, C.; Hell, S.W. Macromolecular-scale resolution in biological fluorescence microscopy. Proc. Natl. Acad. Sci. USA 2006, 103, 11440–11445. [Google Scholar] [CrossRef] [PubMed]
- Heymann, J.A.; Shi, D.; Kim, S.; Bliss, D.; Milne, J.L.; Subramaniam, S. 3D imaging of mammalian cells with ion-abrasion scanning electron microscopy. J. Struct. Biol. 2009, 166, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Hildebrand, M.; Kim, S.; Shi, D.; Scott, K.; Subramaniam, S. 3D imaging of diatoms with ion-abrasion scanning electron microscopy. J. Struct. Biol. 2009, 166, 316–328. [Google Scholar] [CrossRef] [PubMed]
- Bennett, A.E.; Narayan, K.; Shi, D.; Hartnell, L.M.; Gousset, K.; He, H.; Lowekamp, B.C.; Yoo, T.S.; Bliss, D.; Freed, E.O. Ion-abrasion scanning electron microscopy reveals surface-connected tubular conduits in HIV-infected macrophages. PLoS Pathog. 2009, 5, e1000591. [Google Scholar] [CrossRef] [PubMed]
- Reits, E.A.; Neefjes, J.J. From fixed to FRAP: Measuring protein mobility and activity in living cells. Nat. Cell Biol. 2001, 3, E145–E147. [Google Scholar] [CrossRef] [PubMed]
- Roy, N.H.; Chan, J.; Lambelé, M.; Thali, M. Clustering and mobility of HIV-1 Env at viral assembly sites predict its propensity to induce cell-cell fusion. J. Virol. 2013, 87, 7516–7525. [Google Scholar] [CrossRef] [PubMed]
- Steffens, C.M.; Hope, T.J. Mobility of the human immunodeficiency virus (HIV) receptor CD4 and coreceptor CCR5 in living cells: Implications for HIV fusion and entry events. J. Virol. 2004, 78, 9573–9578. [Google Scholar] [CrossRef] [PubMed]
- Eugenin, E.; Gaskill, P.; Berman, J. Tunneling nanotubes (TNT) are induced by HIV-infection of macrophages: A potential mechanism for intercellular HIV trafficking. Cell. Immunol. 2009, 254, 142–148. [Google Scholar] [CrossRef] [PubMed]
- Lachambre, S.; Chopard, C.; Beaumelle, B. Preliminary characterisation of nanotubes connecting T-cells and their use by HIV-1. Biol. Cell 2014, 106, 394–404. [Google Scholar] [CrossRef] [PubMed]
- Sowinski, S.; Jolly, C.; Berninghausen, O.; Purbhoo, M.A.; Chauveau, A.; Köhler, K.; Oddos, S.; Eissmann, P.; Brodsky, F.M.; Hopkins, C. Membrane nanotubes physically connect T cells over long distances presenting a novel route for HIV-1 transmission. Nat. Cell Biol. 2008, 10, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Gerdes, H.-H.; Carvalho, R.N. Intercellular transfer mediated by tunneling nanotubes. Curr. Opin. Cell Biol. 2008, 20, 470–475. [Google Scholar] [CrossRef] [PubMed]
- Sherer, N.M.; Lehmann, M.J.; Jimenez-Soto, L.F.; Horensavitz, C.; Pypaert, M.; Mothes, W. Retroviruses can establish filopodial bridges for efficient cell-to-cell transmission. Nat. Cell Biol. 2007, 9, 310–315. [Google Scholar] [CrossRef] [PubMed]
- Wei, X.; Decker, J.M.; Liu, H.; Zhang, Z.; Arani, R.B.; Kilby, J.M.; Saag, M.S.; Wu, X.; Shaw, G.M.; Kappes, J.C. Emergence of resistant human immunodeficiency virus type 1 in patients receiving fusion inhibitor (T-20) monotherapy. Antimicrob. Agents Chemother. 2002, 46, 1896–1905. [Google Scholar] [CrossRef] [PubMed]
- Abela, I.A.; Berlinger, L.; Schanz, M.; Reynell, L.; Günthard, H.F.; Rusert, P.; Trkola, A. Cell-cell transmission enables HIV-1 to evade inhibition by potent CD4bs directed antibodies. PLoS Pathog. 2012, 8, e1002634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sloan, R.D.; Kuhl, B.D.; Mesplède, T.; Münch, J.; Donahue, D.A.; Wainberg, M.A. Productive entry of HIV-1 during cell-to-cell transmission via dynamin-dependent endocytosis. J. Virol. 2013, 87, 8110–8123. [Google Scholar] [CrossRef] [PubMed]
- Engelman, A.; Mizuuchi, K.; Craigie, R. HIV-1 DNA integration: Mechanism of viral DNA cleavage and DNA strand transfer. Cell 1991, 67, 1211–1221. [Google Scholar] [CrossRef]
- Miller, M.D.; Farnet, C.M.; Bushman, F.D. Human immunodeficiency virus type 1 preintegration complexes: Studies of organization and composition. J. Virol. 1997, 71, 5382–5390. [Google Scholar] [PubMed]
- Alexaki, A.; Liu, Y.; Wigdahl, B. Cellular reservoirs of HIV-1 and their role in viral persistence. Curr. HIV Res. 2008, 6, 388–400. [Google Scholar] [CrossRef] [PubMed]
- Ambrose, Z.; Aiken, C. HIV-1 uncoating: Connection to nuclear entry and regulation by host proteins. Virology 2014, 454, 371–379. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.A.; Terwilliger, E.F.; Jalinoos, Y.; Proulx, J.; Sodroski, J.G.; Haseltine, W.A. Identification of HIV-1 Vpr product and function. JAIDS J. Acquir. Immune Defic. Syndr. 1990, 3, 11–18. [Google Scholar]
- Desai, T.M.; Marin, M.; Melikyan, G.B. Real-Time Imaging Reveals that HIV-1 Vpr Dissociates from the Core and Accumulates in the Nucleus after Viral Fusion. Biophys. J. 2015, 108, 408a. [Google Scholar] [CrossRef]
- Popov, S.; Rexach, M.; Zybarth, G.; Reiling, N.; Lee, M.A.; Ratner, L.; Lane, C.M.; Moore, M.S.; Blobel, G.; Bukrinsky, M. Viral protein R regulates nuclear import of the HIV-1 pre-integration complex. EMBO J. 1998, 17, 909–917. [Google Scholar] [CrossRef] [PubMed]
- Price, A.J.; Fletcher, A.J.; Schaller, T.; Elliott, T.; Lee, K.; KewalRamani, V.N.; Chin, J.W.; Towers, G.J.; James, L.C. CPSF6 defines a conserved capsid interface that modulates HIV-1 replication. PLoS Pathog. 2012, 8, e1002896. [Google Scholar] [CrossRef] [PubMed]
- Bukrinsky, M.I.; Sharova, N.; Dempsey, M.P.; Stanwick, T.L.; Bukrinskaya, A.G.; Haggerty, S.; Stevenson, M. Active nuclear import of human immunodeficiency virus type 1 preintegration complexes. Proc. Natl. Acad. Sci. USA 1992, 89, 6580–6584. [Google Scholar] [CrossRef] [PubMed]
- Marini, B.; Kertesz-Farkas, A.; Ali, H.; Lucic, B.; Lisek, K.; Manganaro, L.; Pongor, S.; Luzzati, R.; Recchia, A.; Mavilio, F. Nuclear architecture dictates HIV-1 integration site selection. Nature 2015, 521, 227–231. [Google Scholar] [CrossRef] [PubMed]
- Burdick, R.C.; Hu, W.-S.; Pathak, V.K. Nuclear import of APOBEC3F-labeled HIV-1 preintegration complexes. Proc. Natl. Acad. Sci. USA 2013, 110, E4780–E4789. [Google Scholar] [CrossRef] [PubMed]
- Desimmie, B.A.; Delviks-Frankenberrry, K.A.; Burdick, R.C.; Qi, D.; Izumi, T.; Pathak, V.K. Multiple APOBEC3 restriction factors for HIV-1 and one Vif to rule them all. J. Mol. Biol. 2014, 426, 1220–1245. [Google Scholar] [CrossRef] [PubMed]
- Bishop, K.N.; Verma, M.; Kim, E.-Y.; Wolinsky, S.M.; Malim, M.H. APOBEC3G inhibits elongation of HIV-1 reverse transcripts. PLoS Pathog. 2008, 4, e1000231. [Google Scholar] [CrossRef] [PubMed]
- Peliska, J.A.; Benkovic, S.J. Mechanism of DNA strand transfer reactions catalyzed by HIV-1 reverse transcriptase. Science 1992, 258, 1112–1118. [Google Scholar] [CrossRef] [PubMed]
- Charneau, P.; Mirambeau, G.; Roux, P.; Paulous, S.; Buc, H.; Clavel, F. HIV-1 reverse transcription. A termination step at the center of the genome. J. Mol. Biol. 1994, 241, 651–662. [Google Scholar] [CrossRef] [PubMed]
- Salic, A.; Mitchison, T.J. A chemical method for fast and sensitive detection of DNA synthesis in vivo. Proc. Natl. Acad. Sci. USA 2008, 105, 2415–2420. [Google Scholar] [CrossRef] [PubMed]
- Peng, K.; Muranyi, W.; Glass, B.; Laketa, V.; Yant, S.R.; Tsai, L.; Cihlar, T.; Müller, B.; Kräusslich, H.-G. Quantitative microscopy of functional HIV post-entry complexes reveals association of replication with the viral capsid. Elife 2014, 3, e04114. [Google Scholar] [CrossRef] [PubMed]
- König, R.; Zhou, Y.; Elleder, D.; Diamond, T.L.; Bonamy, G.M.; Irelan, J.T.; Chiang, C.Y.; Tu, B.P.; de Jesus, P.D.; Lilley, C.E. Global analysis of host-pathogen interactions that regulate early-stage HIV-1 replication. Cell 2008, 135, 49–60. [Google Scholar] [CrossRef] [PubMed]
- Siliciano, R.F.; Greene, W.C. HIV latency. Cold Spring Harb. Perspect. Med. 2011, 1, a007096. [Google Scholar] [CrossRef] [PubMed]
- Ho, Y.-C.; Shan, L.; Hosmane, N.N.; Wang, J.; Laskey, S.B.; Rosenbloom, D.I.; Lai, J.; Blankson, J.N.; Siliciano, J.D.; Siliciano, R.F. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell 2013, 155, 540–551. [Google Scholar] [CrossRef] [PubMed]
- Price, C. Fluorescence in situ hybridization. Blood Rev. 1993, 7, 127–134. [Google Scholar] [CrossRef]
- Schröder, A.R.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.R.; Bushman, F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell 2002, 110, 521–529. [Google Scholar] [CrossRef]
- Barr, S.D.; Ciuffi, A.; Leipzig, J.; Shinn, P.; Ecker, J.R.; Bushman, F.D. HIV integration site selection: Targeting in macrophages and the effects of different routes of viral entry. Mol. Ther. 2006, 14, 218–225. [Google Scholar] [CrossRef] [PubMed]
- Lelek, M.; Casartelli, N.; Pellin, D.; Rizzi, E.; Souque, P.; Severgnini, M.; Di Serio, C.; Fricke, T.; Diaz-Griffero, F.; Zimmer, C. Chromatin organization at the nuclear pore favours HIV replication. Nat. Commun. 2015, 6, 6483. [Google Scholar] [CrossRef] [PubMed]
- Malim, M.H.; Emerman, M. HIV-1 accessory proteins—ensuring viral survival in a hostile environment. Cell Host Microbe 2008, 3, 388–398. [Google Scholar] [CrossRef] [PubMed]
- Pawlak, E.N.; Dikeakos, J.D. HIV-1 Nef: A master manipulator of the membrane trafficking machinery mediating immune evasion. Biochim. Biophys. Acta Gen. Subj. 2015, 1850, 733–741. [Google Scholar] [CrossRef] [PubMed]
- Collins, K.L.; Chen, B.K.; Kalams, S.A.; Walker, B.D.; Baltimore, D. HIV-1 Nef protein protects infected primary cells against killing by cytotoxic T lymphocytes. Nature 1998, 391, 397–401. [Google Scholar] [PubMed]
- Garcia, J.V.; Miller, A.D. Serine phosphorylation-independent downregulation of cell-surface CD4 by Nef. Nature 1991, 350, 508–511. [Google Scholar] [CrossRef] [PubMed]
- Sekar, R.B.; Periasamy, A. Fluorescence resonance energy transfer (FRET) microscopy imaging of live cell protein localizations. J. Cell Biol. 2003, 160, 629–633. [Google Scholar] [CrossRef] [PubMed]
- Roy, R.; Hohng, S.; Ha, T. A practical guide to single-molecule FRET. Nat. Methods 2008, 5, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Hung, C.-H.; Thomas, L.; Ruby, C.E.; Atkins, K.M.; Morris, N.P.; Knight, Z.A.; Scholz, I.; Barklis, E.; Weinberg, A.D.; Shokat, K.M. HIV-1 Nef assembles a Src family kinase-ZAP-70/Syk-PI3K cascade to downregulate cell-surface MHC-I. Cell Host Microbe 2007, 1, 121–133. [Google Scholar] [CrossRef] [PubMed]
- Gerlach, H.; Laumann, V.; Martens, S.; Becker, C.F.; Goody, R.S.; Geyer, M. HIV-1 Nef membrane association depends on charge, curvature, composition and sequence. Nat. Chem. Biol. 2010, 6, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Atkins, K.M.; Thomas, L.; Youker, R.T.; Harriff, M.J.; Pissani, F.; You, H.; Thomas, G. HIV-1 Nef Binds PACS-2 to Assemble a Multikinase Cascade That Triggers Major Histocompatibility Complex Class I (MHC-I) Down-regulation analysis using short interfering RNA and knock-out mice. J. Biol. Chem. 2008, 283, 11772–11784. [Google Scholar] [CrossRef] [PubMed]
- Piguet, V.; Wan, L.; Borel, C.; Mangasarian, A.; Demaurex, N.; Thomas, G.; Trono, D. HIV-1 Nef protein binds to the cellular protein PACS-1 to downregulate class I major histocompatibility complexes. Nat. Cell Biol. 2000, 2, 163–167. [Google Scholar] [PubMed]
- Kerppola, T.K. Design and implementation of bimolecular fluorescence complementation (BiFC) assays for the visualization of protein interactions in living cells. Nat. Protoc. 2006, 1, 1278–1286. [Google Scholar] [CrossRef] [PubMed]
- Dikeakos, J.D.; Thomas, L.; Kwon, G.; Elferich, J.; Shinde, U.; Thomas, G. An interdomain binding site on HIV-1 Nef interacts with PACS-1 and PACS-2 on endosomes to down-regulate MHC-I. Mol. Biol. Cell 2012, 23, 2184–2197. [Google Scholar] [CrossRef] [PubMed]
- Dirk, B.S.; Jacob, R.A.; Johnson, A.L.; Pawlak, E.N.; Cavanagh, P.C.; Van Nynatten, L.; Haeryfar, S.M.; Dikeakos, J.D. Viral Bimolecular Fluorescence Complementation: A Novel Tool to Study Intracellular Vesicular Trafficking Pathways. PLoS ONE 2015, 10, e0125619. [Google Scholar] [CrossRef] [PubMed]
- Hell, S.W.; Kroug, M. Ground-state-depletion fluorscence microscopy: A concept for breaking the diffraction resolution limit. Appl. Phys. B 1995, 60, 495–497. [Google Scholar] [CrossRef]
- Fölling, J.; Bossi, M.; Bock, H.; Medda, R.; Wurm, C.A.; Hein, B.; Jakobs, S.; Eggeling, C.; Hell, S.W. Fluorescence nanoscopy by ground-state depletion and single-molecule return. Nat. Methods 2008, 5, 943–945. [Google Scholar] [CrossRef] [PubMed]
- Caetano, F.A.; Dirk, B.S.; Tam, J.H.; Cavanagh, P.C.; Goiko, M.; Ferguson, S.S.; Pasternak, S.H.; Dikeakos, J.D.; de Bruyn, J.R.; Heit, B. MIiSR: Molecular Interactions in Super-Resolution Imaging Enables the Analysis of Protein Interactions, Dynamics and Formation of Multi-protein Structures. PLoS Comput. Biol. 2015, 11, e1004634. [Google Scholar] [CrossRef] [PubMed]
- Roeth, J.F.; Williams, M.; Kasper, M.R.; Filzen, T.M.; Collins, K.L. HIV-1 Nef disrupts MHC-I trafficking by recruiting AP-1 to the MHC-I cytoplasmic tail. J. Cell Biol. 2004, 167, 903–913. [Google Scholar] [CrossRef] [PubMed]
- Dirk, B.S.; Heit, B.; Dikeakos, J.D. Visualizing Interactions Between HIV-1 Nef and Host Cellular Proteins Using Ground-State Depletion Microscopy. AIDS Res. Hum. Retrovir. 2015, 31, 671–672. [Google Scholar] [CrossRef] [PubMed]
- Sundquist, W.I.; Kräusslich, H.-G. HIV-1 assembly, budding, and maturation. Cold Spring Harb. Perspect. Med. 2012, 2, a006924. [Google Scholar] [CrossRef] [PubMed]
- Ono, A.; Demirov, D.; Freed, E.O. Relationship between human immunodeficiency virus type 1 Gag multimerization and membrane binding. J. Virol. 2000, 74, 5142–5150. [Google Scholar] [CrossRef] [PubMed]
- Morikawa, Y.; Goto, T.; Sano, K. In vitro assembly of human immunodeficiency virus type 1 Gag protein. J. Biol. Chem. 1999, 274, 27997–28002. [Google Scholar] [CrossRef] [PubMed]
- Poole, E.; Strappe, P.; Mok, H.P.; Hicks, R.; Lever, A.M.L. HIV-1 Gag–RNA Interaction Occurs at a Perinuclear/Centrosomal Site; Analysis by Confocal Microscopy and FRET. Traffic 2005, 6, 741–755. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Grunwald, D.; Sardo, L.; Galli, A.; Plisov, S.; Nikolaitchik, O.A.; Chen, D.; Lockett, S.; Larson, D.R.; Pathak, V.K. Cytoplasmic HIV-1 RNA is mainly transported by diffusion in the presence or absence of Gag protein. Proc. Natl. Acad. Sci. USA 2014, 111, E5205–E5213. [Google Scholar] [CrossRef] [PubMed]
- Houman, F.; Diaz-Torres, M.R.; Wright, A. Transcriptional antitermination in the BGL operon of E. coli is modulated by a specific RNA binding protein. Cell 1990, 62, 1153–1163. [Google Scholar] [CrossRef]
- Axelrod, D. Total internal reflection fluorescence microscopy in cell biology. Methods Enzymol. 2003, 361, 1–33. [Google Scholar] [PubMed]
- Jouvenet, N.; Neil, S.J.; Bess, C.; Johnson, M.C.; Virgen, C.A.; Simon, S.M.; Bieniasz, P.D. Plasma membrane is the site of productive HIV-1 particle assembly. PLoS Biol. 2006, 4, e435. [Google Scholar] [CrossRef] [PubMed]
- Jouvenet, N.; Bieniasz, P.D.; Simon, S.M. Imaging the biogenesis of individual HIV-1 virions in live cells. Nature 2008, 454, 236–240. [Google Scholar] [CrossRef] [PubMed]
- Jouvenet, N.; Simon, S.M.; Bieniasz, P.D. Imaging the interaction of HIV-1 genomes and Gag during assembly of individual viral particles. Proc. Natl. Acad. Sci. USA 2009, 106, 19114–19119. [Google Scholar] [CrossRef] [PubMed]
- Bao, G.; Rhee, W.J.; Tsourkas, A. Fluorescent probes for live-cell RNA detection. Annu. Rev. Biomed. Eng. 2009, 11, 25. [Google Scholar] [CrossRef] [PubMed]
- Johansson, H.E.; Dertinger, D.; LeCuyer, K.A.; Behlen, L.S.; Greef, C.H.; Uhlenbeck, O.C. A thermodynamic analysis of the sequence-specific binding of RNA by bacteriophage MS2 coat protein. Proc. Natl. Acad. Sci. USA 1998, 95, 9244–9249. [Google Scholar] [CrossRef] [PubMed]
- Jouvenet, N.; Zhadina, M.; Bieniasz, P.D.; Simon, S.M. Dynamics of ESCRT protein recruitment during retroviral assembly. Nat. Cell Biol. 2011, 13, 394–401. [Google Scholar] [CrossRef] [PubMed]
- Henne, W.M.; Buchkovich, N.J.; Emr, S.D. The ESCRT pathway. Dev. Cell 2011, 21, 77–91. [Google Scholar] [CrossRef] [PubMed]
- Carlson, L.-A.; Hurley, J.H. In vitro reconstitution of the ordered assembly of the endosomal sorting complex required for transport at membrane-bound HIV-1 Gag clusters. Proc. Natl. Acad. Sci. USA 2012, 109, 16928–16933. [Google Scholar] [CrossRef] [PubMed]
- Göttlinger, H.G.; Dorfman, T.; Sodroski, J.G.; Haseltine, W.A. Effect of mutations affecting the p6 Gag protein on human immunodeficiency virus particle release. Proc. Natl. Acad. Sci. USA 1991, 88, 3195–3199. [Google Scholar] [CrossRef] [PubMed]
- Hell, S.W. Toward fluorescence nanoscopy. Nat. Biotechnol. 2003, 21, 1347–1355. [Google Scholar] [CrossRef] [PubMed]
- Muranyi, W.; Malkusch, S.; Müller, B.; Heilemann, M.; Kräusslich, H.-G. Super-resolution microscopy reveals specific recruitment of HIV-1 envelope proteins to viral assembly sites dependent on the envelope C-terminal tail. PLoS Pathog. 2013, 9, e1003198. [Google Scholar] [CrossRef] [PubMed]
- Van Engelenburg, S.B.; Shtengel, G.; Sengupta, P.; Waki, K.; Jarnik, M.; Ablan, S.D.; Freed, E.O.; Hess, H.F.; Lippincott-Schwartz, J. Distribution of ESCRT machinery at HIV assembly sites reveals virus scaffolding of ESCRT subunits. Science 2014, 343, 653–656. [Google Scholar] [CrossRef] [PubMed]
- Shtengel, G.; Wang, Y.; Zhang, Z.; Goh, W.I.; Hess, H.F.; Kanchanawong, P. Imaging cellular ultrastructure by PALM, iPALM, and correlative iPALM-EM. Methods Cell Biol. 2013, 123, 273–294. [Google Scholar]
- Prescher, J.; Baumgärtel, V.; Ivanchenko, S.; Torrano, A.A.; Bräuchle, C.; Müller, B.; Lamb, D.C. Super-resolution imaging of ESCRT-proteins at HIV-1 assembly sites. PLoS Pathog. 2015, 11, e1004677. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.-G.; Zhang, X.; Ip, D.T.-M.; Yang, L.-M.; Zheng, Y.-T.; Wan, D.C.-C. Discovery of a novel HIV-1 integrase inhibitor from natural compounds through structure based virtual screening and cell imaging. FEBS Lett. 2014, 588, 3461–3468. [Google Scholar] [CrossRef] [PubMed]
- Sheik-Khalil, E.; Bray, M.-A.; Şahin, G.Ö.; Scarlatti, G.; Jansson, M.; Carpenter, A.E.; Fenyö, E.M. Automated image-based assay for evaluation of HIV neutralization and cell-to-cell fusion inhibition. BMC Infect. Dis. 2014, 14, 472. [Google Scholar] [CrossRef] [PubMed]
- Poe, J.A.; Vollmer, L.; Vogt, A.; Smithgall, T.E. Development and validation of a high-content bimolecular fluorescence complementation assay for small-molecule inhibitors of HIV-1 Nef dimerization. J. Biomol. Screen. 2013, 9, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Genovesio, A.; Kwon, Y.-J.; Windisch, M.P.; Kim, N.Y.; Choi, S.Y.; Kim, H.C.; Jung, S.; Mammano, F.; Perrin, V.; Boese, A.S. Automated genome-wide visual profiling of cellular proteins involved in HIV infection. J. Biomol. Screen. 2011, 16, 945–958. [Google Scholar] [CrossRef] [PubMed]
- Henriques, R.; Griffiths, C.; Hesper Rego, E.; Mhlanga, M.M. PALM and STORM: Unlocking live-cell super-resolution. Biopolymers 2011, 95, 322–331. [Google Scholar] [CrossRef] [PubMed]
- Gustafsson, N.; Culley, S.; Ashdown, G.; Owen, D.M.; Pereira, P.M.; Henriques, R. Fast live-cell conventional fluorophore nanoscopy with ImageJ through super-resolution radial fluctuations. Nat. Commun. 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Murooka, T.T.; Deruaz, M.; Marangoni, F.; Vrbanac, V.D.; Seung, E.; Von Andrian, U.H.; Tager, A.M.; Luster, A.D.; Mempel, T.R. HIV-infected T cells are migratory vehicles for viral dissemination. Nature 2012, 490, 283–287. [Google Scholar] [CrossRef] [PubMed]
- Symeonides, M.; Murooka, T.T.; Bellfy, L.N.; Roy, N.H.; Mempel, T.R.; Thali, M. HIV-1-induced small T cell syncytia can transfer virus particles to target cells through transient contacts. Viruses 2015, 7, 6590–6603. [Google Scholar] [CrossRef] [PubMed]
- Hübner, W.; Chen, P.; Del Portillo, A.; Liu, Y.; Gordon, R.E.; Chen, B.K. Sequence of human immunodeficiency virus type 1 (HIV-1) Gag localization and oligomerization monitored with live confocal imaging of a replication-competent, fluorescently tagged HIV-1. J. Virol. 2007, 81, 12596–12607. [Google Scholar] [CrossRef] [PubMed]
- Arhel, N.; Genovesio, A.; Kim, K.-A.; Miko, S.; Perret, E.; Olivo-Marin, J.-C.; Shorte, S.; Charneau, P. Quantitative four-dimensional tracking of cytoplasmic and nuclear HIV-1 complexes. Nat. Methods 2006, 3, 817–824. [Google Scholar] [CrossRef] [PubMed]
- Eckhardt, M.; Anders, M.; Muranyi, W.; Heilemann, M.; Krijnse-Locker, J.; Müller, B. A SNAP-tagged derivative of HIV-1—A versatile tool to study virus-cell interactions. PLoS ONE 2011, 6, e22007. [Google Scholar] [CrossRef] [PubMed]
- Masters, J.R. HeLa cells 50 years on: The good, the bad and the ugly. Nat. Rev. Cancer 2002, 2, 315–319. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dirk, B.S.; Van Nynatten, L.R.; Dikeakos, J.D. Where in the Cell Are You? Probing HIV-1 Host Interactions through Advanced Imaging Techniques. Viruses 2016, 8, 288. https://doi.org/10.3390/v8100288
Dirk BS, Van Nynatten LR, Dikeakos JD. Where in the Cell Are You? Probing HIV-1 Host Interactions through Advanced Imaging Techniques. Viruses. 2016; 8(10):288. https://doi.org/10.3390/v8100288
Chicago/Turabian StyleDirk, Brennan S., Logan R. Van Nynatten, and Jimmy D. Dikeakos. 2016. "Where in the Cell Are You? Probing HIV-1 Host Interactions through Advanced Imaging Techniques" Viruses 8, no. 10: 288. https://doi.org/10.3390/v8100288