
Dynamic Viral Glycoprotein Machines: Approaches for Probing Transient States That Drive Membrane Fusion

{kind=link}
{kind=link}
{kind=link}
Abstract
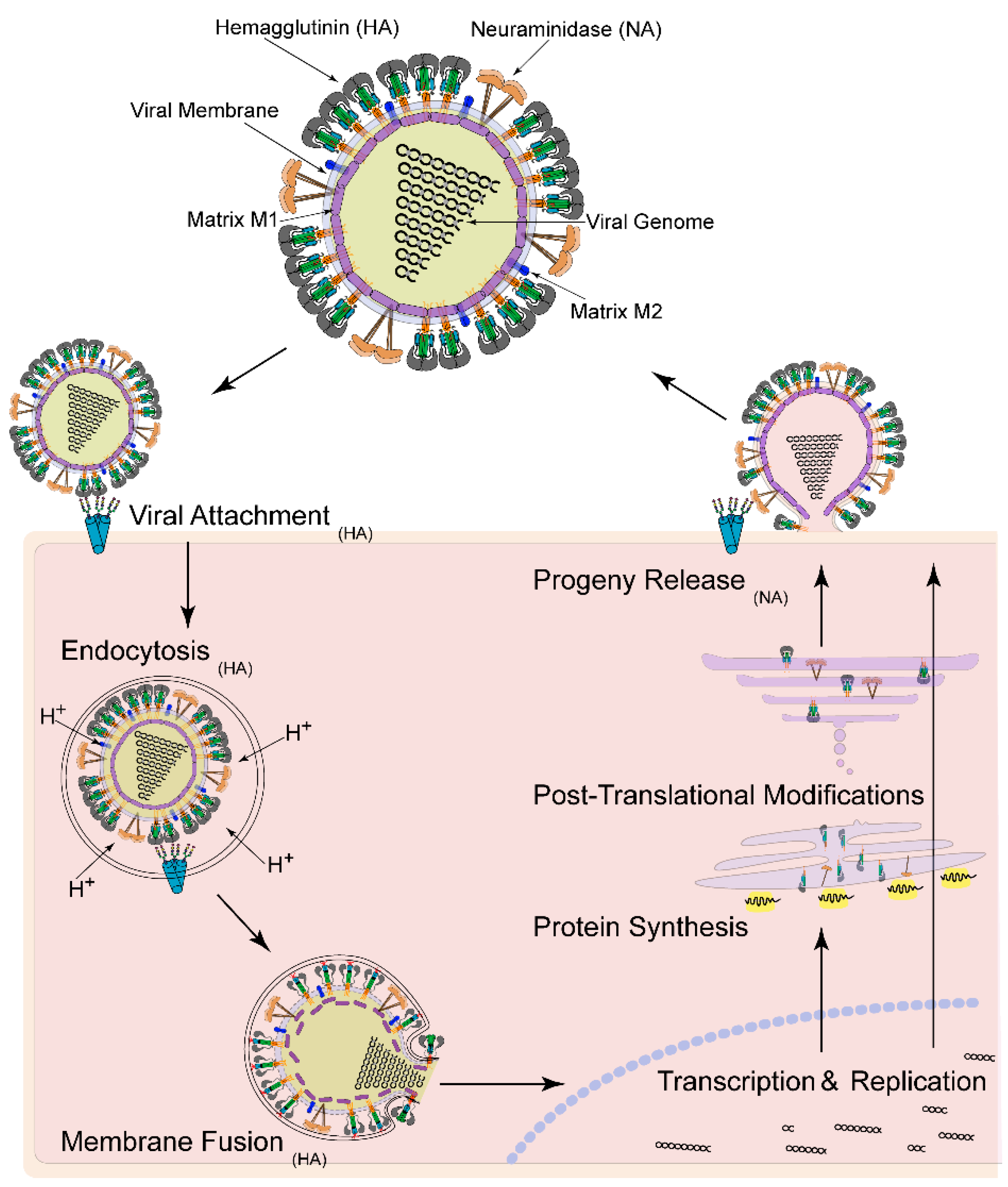
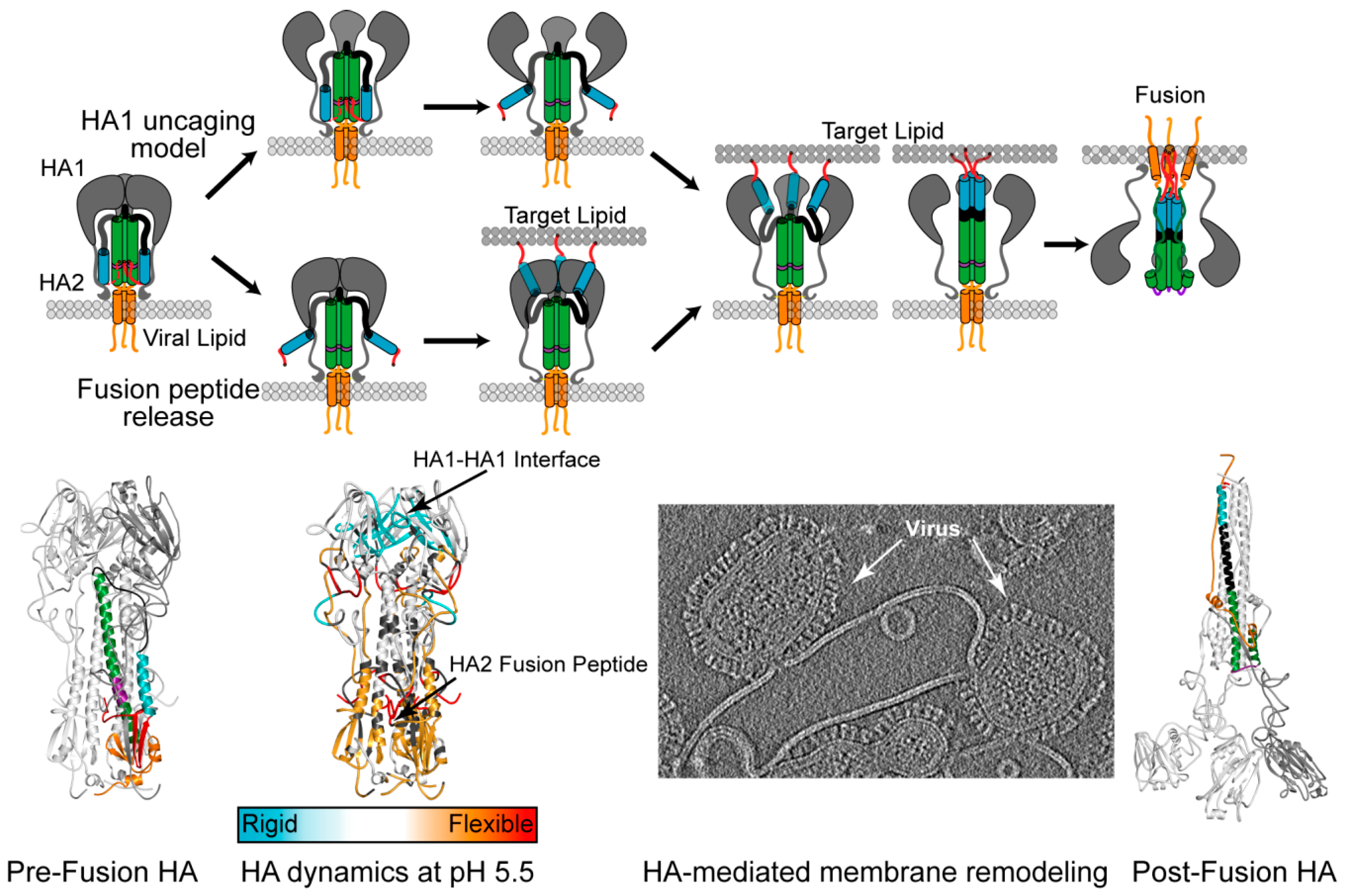
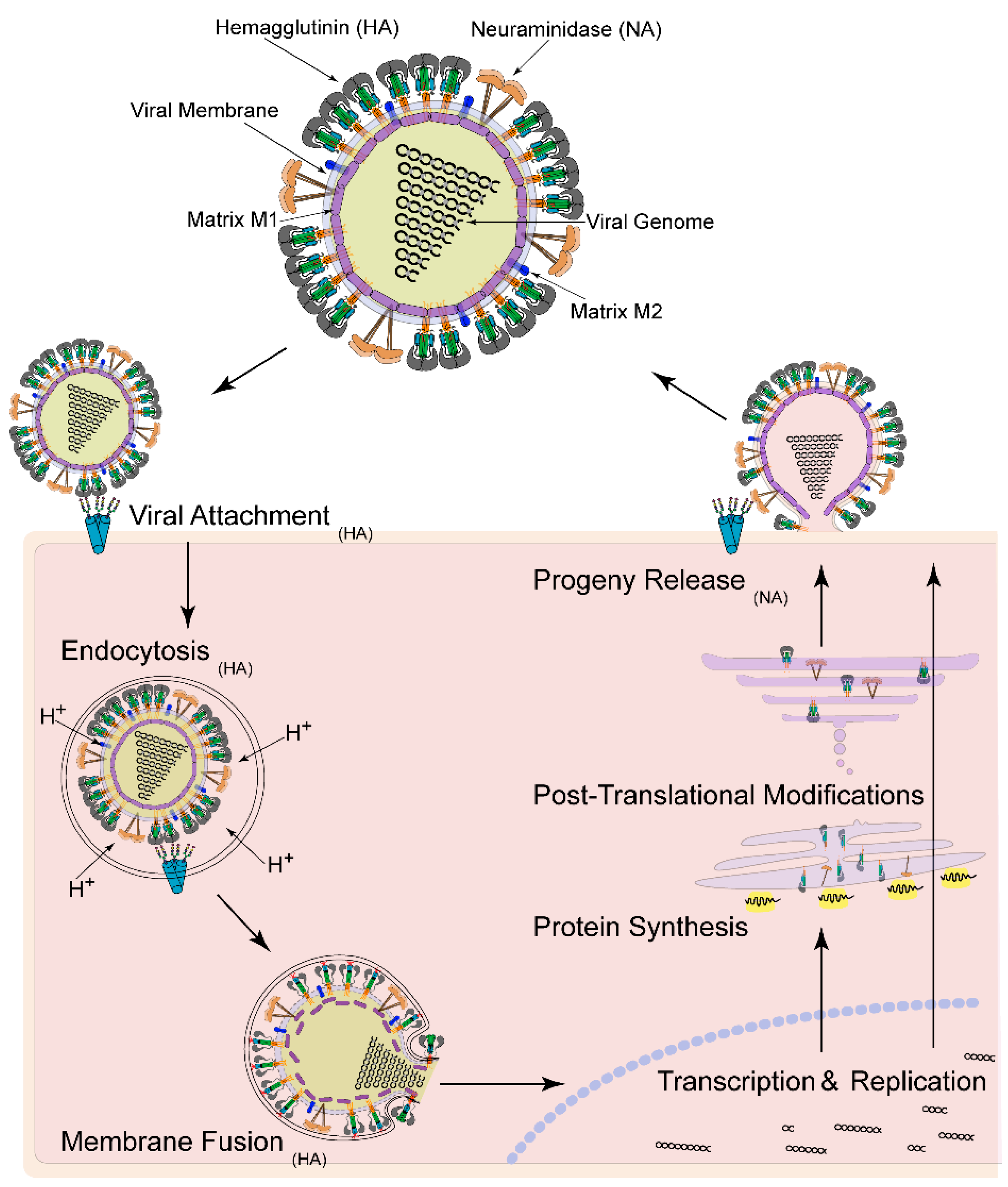
:1. Introduction
2. Solution-Based Biophysical Approaches

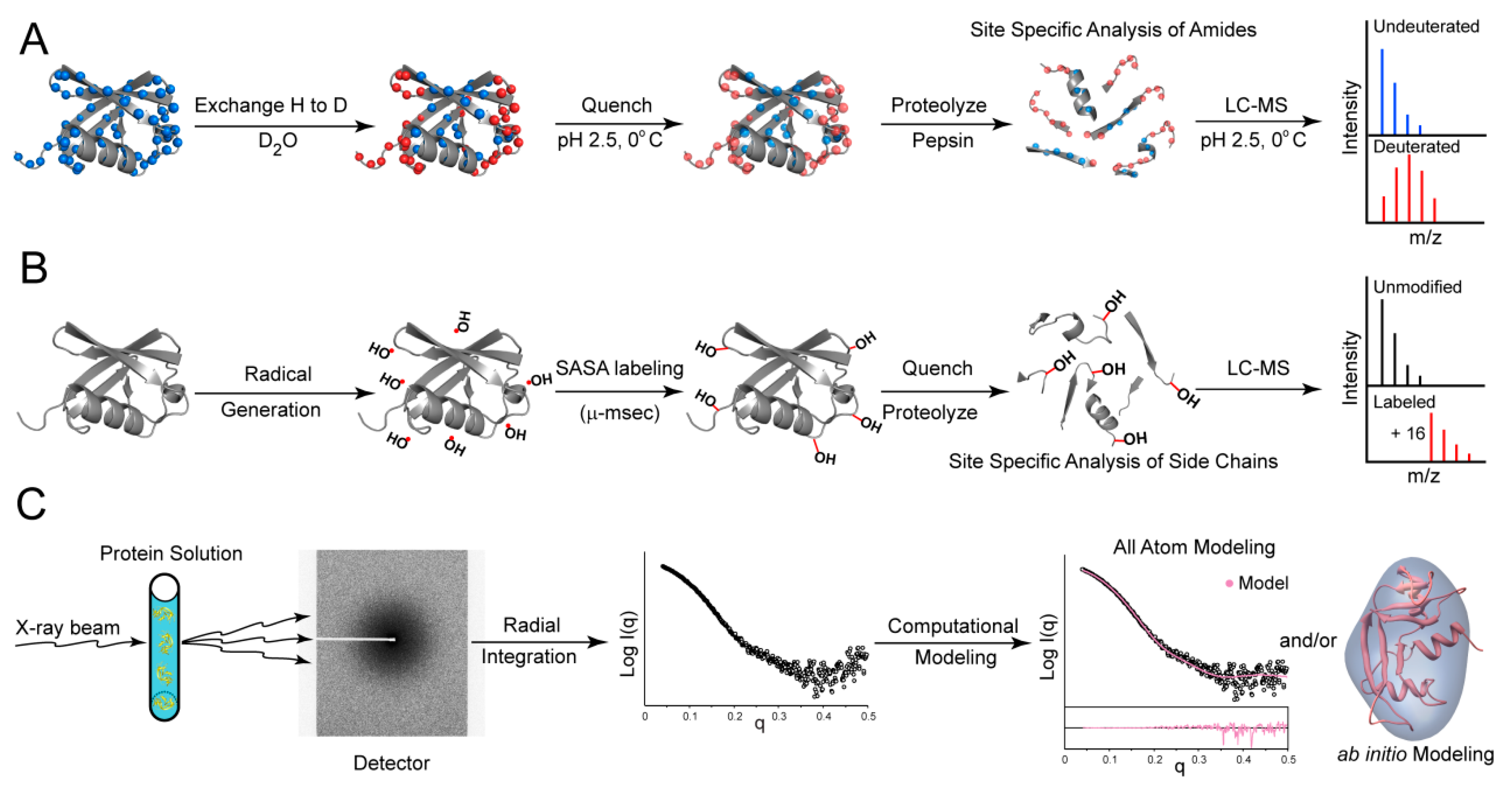
3. Hydrogen-Deuterium Exchange Mass Spectrometry (HDX-MS)

4. Oxidative Labeling With Mass Spectrometry
5. Small Angle X-Ray Scattering (SAXS)
6. Electron Microscopy (EM)
7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Garcia, N.K.; Guttman, M.; Ebner, J.L.; Lee, K.K. Dynamic Changes during Acid-Induced Activation of Influenza Hemagglutinin. Structure 2015, 23, 665–676. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Garcia, N.K.; Cupo, A.; Matsui, T.; Julien, J.P.; Sanders, R.W.; Wilson, I.A.; Moore, J.P.; Lee, K.K. CD4-induced activation in a soluble HIV-1 Env trimer. Structure 2014, 22, 974–984. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Cupo, A.; Julien, J.P.; Sanders, R.W.; Wilson, I.A.; Moore, J.P.; Lee, K.K. Antibody potency relates to the ability to recognize the closed, pre-fusion form of HIV Env. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Bartesaghi, A.; Borgnia, M.J.; Sapiro, G.; Subramaniam, S. Molecular architecture of native HIV-1 gp120 trimers. Nature 2008, 455, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.E.; Borgnia, M.J.; Kuybeda, O.; Schauder, D.M.; Bartesaghi, A.; Frank, G.A.; Sapiro, G.; Milne, J.L.; Subramaniam, S. Structural mechanism of trimeric HIV-1 envelope glycoprotein activation. PLoS Pathog. 2012, 8, e1002797. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.; Borgnia, M.J.; Shi, D.; Bartesaghi, A.; He, H.; Pejchal, R.; Kang, Y.K.; Depetris, R.; Marozsan, A.J.; Sanders, R.W.; et al. Trimeric HIV-1 glycoprotein gp140 immunogens and native HIV-1 envelope glycoproteins display the same closed and open quaternary molecular architectures. Proc. Natl. Acad. Sci. USA 2011, 108, 11440–11445. [Google Scholar] [CrossRef] [PubMed]
- Lyumkis, D.; Julien, J.P.; de Val, N.; Cupo, A.; Potter, C.S.; Klasse, P.J.; Burton, D.R.; Sanders, R.W.; Moore, J.P.; Carragher, B.; et al. Cryo-EM structure of a fully glycosylated soluble cleaved HIV-1 envelope trimer. Science 2013, 342, 1484–1490. [Google Scholar] [CrossRef] [PubMed]
- Julien, J.P.; Cupo, A.; Sok, D.; Stanfield, R.L.; Lyumkis, D.; Deller, M.C.; Klasse, P.J.; Burton, D.R.; Sanders, R.W.; Moore, J.P.; et al. Crystal structure of a soluble cleaved HIV-1 envelope trimer. Science 2013, 342, 1477–1483. [Google Scholar] [CrossRef] [PubMed]
- Depetris, R.S.; Julien, J.P.; Khayat, R.; Lee, J.H.; Pejchal, R.; Katpally, U.; Cocco, N.; Kachare, M.; Massi, E.; David, K.B.; et al. Partial enzymatic deglycosylation preserves the structure of cleaved recombinant HIV-1 envelope glycoprotein trimers. J. Biol. Chem. 2012, 287, 24239–24254. [Google Scholar] [CrossRef] [PubMed]
- Sanders, R.W.; Derking, R.; Cupo, A.; Julien, J.P.; Yasmeen, A.; de Val, N.; Kim, H.J.; Blattner, C.; de la Pena, A.T.; Korzun, J.; et al. A next-generation cleaved, soluble HIV-1 Env trimer, BG505 SOSIP.664 gp140, expresses multiple epitopes for broadly neutralizing but not non-neutralizing antibodies. PLoS Pathog. 2013, 9, e1003618. [Google Scholar] [CrossRef] [PubMed]
- Ivanovic, T.; Choi, J.L.; Whelan, S.P.; van Oijen, A.M.; Harrison, S.C. Influenza-virus membrane fusion by cooperative fold-back of stochastically induced hemagglutinin intermediates. eLife 2013, 2, e00333. [Google Scholar] [CrossRef] [PubMed]
- Munro, J.B.; Gorman, J.; Ma, X.; Zhou, Z.; Arthos, J.; Burton, D.R.; Koff, W.C.; Courter, J.R.; Smith, A.B., 3rd; Kwong, P.D.; et al. Conformational dynamics of single HIV-1 envelope trimers on the surface of native virions. Science 2014, 346, 759–763. [Google Scholar] [CrossRef] [PubMed]
- Costello, D.A.; Whittaker, G.R.; Daniel, S. Variations in pH sensitivity, acid stability, and fusogenicity of three influenza virus H3 subtypes. J. Virol. 2015, 89, 350–360. [Google Scholar] [CrossRef] [PubMed]
- Pancera, M.; Zhou, T.; Druz, A.; Georgiev, I.S.; Soto, C.; Gorman, J.; Huang, J.; Acharya, P.; Chuang, G.Y.; Ofek, G.; et al. Structure and immune recognition of trimeric pre-fusion HIV-1 Env. Nature 2014, 514, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Davenport, T.M.; Guttman, M.; Guo, W.; Cleveland, B.; Kahn, M.; Hu, S.L.; Lee, K.K. Isolate-specific differences in the conformational dynamics and antigenicity of HIV-1 gp120. J. Virol. 2013, 87, 10855–10873. [Google Scholar] [CrossRef] [PubMed]
- Bale, S.; Dias, J.M.; Fusco, M.L.; Hashiguchi, T.; Wong, A.C.; Liu, T.; Keuhne, A.I.; Li, S.; Woods, V.L., Jr.; Chandran, K.; et al. Structural basis for differential neutralization of ebolaviruses. Viruses 2012, 4, 447–470. [Google Scholar] [CrossRef] [PubMed]
- Bale, S.; Liu, T.; Li, S.; Wang, Y.; Abelson, D.; Fusco, M.; Woods, V.L., Jr.; Saphire, E.O. Ebola virus glycoprotein needs an additional trigger, beyond proteolytic priming for membrane fusion. PLoS Negl. Trop. Dis. 2011, 5, e1395. [Google Scholar] [CrossRef] [PubMed]
- Wilson, I.A.; Skehel, J.J.; Wiley, D.C. Structure of the haemagglutinin membrane glycoprotein of influenza virus at 3 Å resolution. Nature 1981, 289, 366–373. [Google Scholar] [CrossRef]
- Bullough, P.A.; Hughson, F.M.; Skehel, J.J.; Wiley, D.C. Structure of influenza haemagglutinin at the pH of membrane fusion. Nature 1994, 371, 37–43. [Google Scholar] [CrossRef] [PubMed]
- Gigant, B.; Barbey-Martin, C.; Bizebard, T.; Fleury, D.; Daniels, R.; Skehel, J.J.; Knossow, M. A neutralizing antibody Fab-influenza haemagglutinin complex with an unprecedented 2:1 stoichiometry: characterization and crystallization. Acta Crystallogr. D Biol. Crystallogr. 2000, 56, 1067–1069. [Google Scholar] [CrossRef] [PubMed]
- Knossow, M.; Gaudier, M.; Douglas, A.; Barrere, B.; Bizebard, T.; Barbey, C.; Gigant, B.; Skehel, J.J. Mechanism of neutralization of influenza virus infectivity by antibodies. Virology 2002, 302, 294–298. [Google Scholar] [CrossRef] [PubMed]
- Barbey-Martin, C.; Gigant, B.; Bizebard, T.; Calder, L.J.; Wharton, S.A.; Skehel, J.J.; Knossow, M. An antibody that prevents the hemagglutinin low pH fusogenic transition. Virology 2002, 294, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Bizebard, T.; Gigant, B.; Rigolet, P.; Rasmussen, B.; Diat, O.; Bosecke, P.; Wharton, S.A.; Skehel, J.J.; Knossow, M. Structure of influenza virus haemagglutinin complexed with a neutralizing antibody. Nature 1995, 376, 92–94. [Google Scholar] [CrossRef] [PubMed]
- Corti, D.; Voss, J.; Gamblin, S.J.; Codoni, G.; Macagno, A.; Jarrossay, D.; Vachieri, S.G.; Pinna, D.; Minola, A.; Vanzetta, F.; et al. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science 2011, 333, 850–856. [Google Scholar] [CrossRef] [PubMed]
- Dreyfus, C.; Ekiert, D.C.; Wilson, I.A. Structure of a classical broadly neutralizing stem antibody in complex with a pandemic H2 influenza virus hemagglutinin. J. Virol. 2013, 87, 7149–7154. [Google Scholar] [CrossRef] [PubMed]
- Ekiert, D.C.; Friesen, R.H.; Bhabha, G.; Kwaks, T.; Jongeneelen, M.; Yu, W.; Ophorst, C.; Cox, F.; Korse, H.J.; Brandenburg, B.; et al. A highly conserved neutralizing epitope on group 2 influenza A viruses. Science 2011, 333, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Ekiert, D.C.; Kashyap, A.K.; Steel, J.; Rubrum, A.; Bhabha, G.; Khayat, R.; Lee, J.H.; Dillon, M.A.; O'Neil, R.E.; Faynboym, A.M.; et al. Cross-neutralization of influenza A viruses mediated by a single antibody loop. Nature 2012, 489, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Lee, P.S.; Ohshima, N.; Stanfield, R.L.; Yu, W.L.; Iba, Y.; Okuno, Y.; Kurosawa, Y.; Wilson, I.A. Receptor mimicry by antibody F045–092 facilitates universal binding to the H3 subtype of influenza virus. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Krause, J.C.; McBride, R.; Paulson, J.C.; Crowe, J.E., Jr.; Wilson, I.A. A recurring motif for antibody recognition of the receptor-binding site of influenza hemagglutinin. Nat. Struct. Mol. Biol. 2013, 20, 363–370. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.; Cardone, G.; Winkler, D.C.; Heymann, J.B.; Brecher, M.; White, J.M.; Steven, A.C. Influenza virus pleiomorphy characterized by cryoelectron tomography. Proc. Natl. Acad. Sci. USA 2006, 103, 19123–19127. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.K. Architecture of a nascent viral fusion pore. EMBO J. 2010, 29, 1299–1311. [Google Scholar] [CrossRef] [PubMed]
- Fontana, J.; Cardone, G.; Heymann, J.B.; Winkler, D.C.; Steven, A.C. Structural Changes in Influenza Virus at Low pH Characterized by Cryo-Electron Tomography. J. Virol. 2012, 86, 2919–2929. [Google Scholar] [CrossRef] [PubMed]
- Fontana, J.; Steven, A.C. At low pH, influenza virus matrix protein M1 undergoes a conformational change prior to dissociating from the membrane. J. Virol. 2013, 87, 5621–5628. [Google Scholar] [CrossRef] [PubMed]
- Calder, L.J.; Wasilewski, S.; Berriman, J.A.; Rosenthal, P.B. Structural organization of a filamentous influenza A virus. Proc. Natl. Acad. Sci. USA 2010, 107, 10685–10690. [Google Scholar] [CrossRef] [PubMed]
- Carr, C.M.; Kim, P.S. A spring-loaded mechanism for the conformational change of influenza hemagglutinin. Cell 1993, 73, 823–832. [Google Scholar] [CrossRef]
- Baker, D.; Agard, D.A. Influenza hemagglutinin: kinetic control of protein function. Structure 1994, 2, 907–910. [Google Scholar] [CrossRef]
- Carr, C.M.; Chaudhry, C.; Kim, P.S. Influenza hemagglutinin is spring-loaded by a metastable native conformation. Proc. Natl. Acad. Sci. USA 1997, 94, 14306–14313. [Google Scholar] [CrossRef] [PubMed]
- Remeta, D.P.; Krumbiegel, M.; Minetti, C.A.; Puri, A.; Ginsburg, A.; Blumenthal, R. Acid-induced changes in thermal stability and fusion activity of influenza hemagglutinin. Biochemistry 2002, 41, 2044–2054. [Google Scholar] [CrossRef] [PubMed]
- Ruigrok, R.W.; Martin, S.R.; Wharton, S.A.; Skehel, J.J.; Bayley, P.M.; Wiley, D.C. Conformational changes in the hemagglutinin of influenza virus which accompany heat-induced fusion of virus with liposomes. Virology 1986, 155, 484–497. [Google Scholar] [CrossRef]
- Korte, T.; Ludwig, K.; Krumbiegel, M.; Zirwer, D.; Damaschun, G.; Herrmann, A. Transient changes of the conformation of hemagglutinin of influenza virus at low pH detected by time-resolved circular dichroism spectroscopy. J. Biol. Chem. 1997, 272, 9764–9770. [Google Scholar] [PubMed]
- Korte, T.; Ludwig, K.; Booy, F.P.; Blumenthal, R.; Herrmann, A. Conformational intermediates and fusion activity of influenza virus hemagglutinin. J. Virol. 1999, 73, 4567–4574. [Google Scholar] [PubMed]
- Puri, A.; Booy, F.P.; Doms, R.W.; White, J.M.; Blumenthal, R. Conformational changes and fusion activity of influenza virus hemagglutinin of the H2 and H3 subtypes: Effects of acid pretreatment. J. Virol. 1990, 64, 3824–3832. [Google Scholar] [PubMed]
- Floyd, D.L.; Ragains, J.R.; Skehel, J.J.; Harrison, S.C.; van Oijen, A.M. Single-particle kinetics of influenza virus membrane fusion. Proc. Natl. Acad. Sci. USA 2008, 105, 15382–15387. [Google Scholar] [CrossRef] [PubMed]
- Costello, D.A.; Lee, D.W.; Drewes, J.; Vasquez, K.A.; Kisler, K.; Wiesner, U.; Pollack, L.; Whittaker, G.R.; Daniel, S. Influenza virus-membrane fusion triggered by proton uncaging for single particle studies of fusion kinetics. Anal. Chem. 2012, 84, 8480–8489. [Google Scholar] [CrossRef] [PubMed]
- Hamilton, B.S.; Whittaker, G.R.; Daniel, S. Influenza virus-mediated membrane fusion: determinants of hemagglutinin fusogenic activity and experimental approaches for assessing virus fusion. Viruses 2012, 4, 1144–1168. [Google Scholar] [CrossRef] [PubMed]
- Lakadamyali, M.; Rust, M.J.; Babcock, H.P.; Zhuang, X. Visualizing infection of individual influenza viruses. Proc. Natl. Acad. Sci. USA 2003, 100, 9280–9285. [Google Scholar] [CrossRef] [PubMed]
- Rust, M.J.; Lakadamyali, M.; Zhang, F.; Zhuang, X. Assembly of endocytic machinery around individual influenza viruses during viral entry. Nat. Struct. Mol. Biol. 2004, 11, 567–573. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, N.; Sun, Y.; Sattentau, Q.; Thali, M.; Wu, D.; Denisova, G.; Gershoni, J.; Robinson, J.; Moore, J.; Sodroski, J. CD4-Induced conformational changes in the human immunodeficiency virus type 1 gp120 glycoprotein: consequences for virus entry and neutralization. J. Virol. 1998, 72, 4694–4703. [Google Scholar] [PubMed]
- Kwong, P.D.; Wyatt, R.; Robinson, J.; Sweet, R.W.; Sodroski, J.; Hendrickson, W.A. Structure of an HIV gp120 envelope glycoprotein in complex with the CD4 receptor and a neutralizing human antibody. Nature 1998, 393, 648–659. [Google Scholar] [PubMed]
- Wyatt, R.; Kwong, P.D.; Desjardins, E.; Sweet, R.W.; Robinson, J.; Hendrickson, W.A.; Sodroski, J.G. The antigenic structure of the HIV gp120 envelope glycoprotein. Nature 1998, 393, 705–711. [Google Scholar] [PubMed]
- Myszka, D.G.; Sweet, R.W.; Hensley, P.; Brigham-Burke, M.; Kwong, P.D.; Hendrickson, W.A.; Wyatt, R.; Sodroski, J.; Doyle, M.L. Energetics of the HIV gp120-CD4 binding reaction. Proc. Natl. Acad. Sci. USA 2000, 97, 9026–9031. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Kahn, M.; Garcia, N.K.; Hu, S.L.; Lee, K.K. Solution structure, conformational dynamics, and CD4-induced activation in full-length, glycosylated, monomeric HIV gp120. J. Virol. 2012, 86, 8750–8764. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Huang, C.C.; Coales, S.J.; Molnar, K.S.; Skinner, J.; Hamuro, Y.; Kwong, P.D. Local conformational stability of HIV-1 gp120 in unliganded and CD4-bound states as defined by amide hydrogen/deuterium exchange. J. Virol. 2010, 84, 10311–10321. [Google Scholar] [CrossRef] [PubMed]
- Paulson, J.C.; Sadler, J.E.; Hill, R.L. Restoration of specific myxovirus receptors to asialoerythrocytes by incorporation of sialic acid with pure sialyltransferases. J. Biol. Chem. 1979, 254, 2120–2124. [Google Scholar] [PubMed]
- Matlin, K.S.; Reggio, H.; Helenius, A.; Simons, K. Infectious entry pathway of influenza virus in a canine kidney cell line. J. Cell Biol. 1981, 91, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Skehel, J.J.; Wiley, D.C. Receptor binding and membrane fusion in virus entry: the influenza hemagglutinin. Annu. Rev. Biochem. 2000, 69, 531–569. [Google Scholar] [CrossRef] [PubMed]
- Wiley, D.C.; Skehel, J.J. The structure and function of the hemagglutinin membrane glycoprotein of influenza virus. Annu. Rev. Biochem. 1987, 56, 365–394. [Google Scholar] [CrossRef] [PubMed]
- Sauter, N.K.; Bednarski, M.D.; Wurzburg, B.A.; Hanson, J.E.; Whitesides, G.M.; Skehel, J.J.; Wiley, D.C. Hemagglutinins from two influenza virus variants bind to sialic acid derivatives with millimolar dissociation constants: a 500-MHz proton nuclear magnetic resonance study. Biochemistry 1989, 28, 8388–8396. [Google Scholar] [CrossRef] [PubMed]
- Sauter, N.K.; Hanson, J.E.; Glick, G.D.; Brown, J.H.; Crowther, R.L.; Park, S.J.; Skehel, J.J.; Wiley, D.C. Binding of influenza virus hemagglutinin to analogs of its cell-surface receptor, sialic acid: analysis by proton nuclear magnetic resonance spectroscopy and X-ray crystallography. Biochemistry 1992, 31, 9609–9621. [Google Scholar] [CrossRef] [PubMed]
- Hanson, J.E.; Sauter, N.K.; Skehel, J.J.; Wiley, D.C. Proton nuclear magnetic resonance studies of the binding of sialosides to intact influenza virus. Virology 1992, 189, 525–533. [Google Scholar] [CrossRef]
- Inglis, S.C.; Carroll, A.R.; Lamb, R.A.; Mahy, B.W. Polypeptides specified by the influenza virus genome I. Evidence for eight distinct gene products specified by fowl plague virus. Virology 1976, 74, 489–503. [Google Scholar] [CrossRef]
- Ruigrok, R.W.; Andree, P.J.; Hooft van Huysduynen, R.A.; Mellema, J.E. Characterization of three highly purified influenza virus strains by electron microscopy. J. Gen. Virol. 1984, 65, 799–802. [Google Scholar] [CrossRef] [PubMed]
- Skehel, J.J.; Bayley, P.M.; Brown, E.B.; Martin, S.R.; Waterfield, M.D.; White, J.M.; Wilson, I.A.; Wiley, D.C. Changes in the conformation of influenza virus hemagglutinin at the pH optimum of virus-mediated membrane fusion. Proc. Natl. Acad. Sci. USA 1982, 79, 968–972. [Google Scholar] [CrossRef] [PubMed]
- Stegmann, T.; White, J.M.; Helenius, A. Intermediates in influenza induced membrane fusion. EMBO J. 1990, 9, 4231–4241. [Google Scholar] [PubMed]
- Ruigrok, R.W.; Wrigley, N.G.; Calder, L.J.; Cusack, S.; Wharton, S.A.; Brown, E.B.; Skehel, J.J. Electron microscopy of the low pH structure of influenza virus haemagglutinin. EMBO J. 1986, 5, 41–49. [Google Scholar] [PubMed]
- Daniels, P.S.; Jeffries, S.; Yates, P.; Schild, G.C.; Rogers, G.N.; Paulson, J.C.; Wharton, S.A.; Douglas, A.R.; Skehel, J.J.; Wiley, D.C. The receptor-binding and membrane-fusion properties of influenza virus variants selected using anti-haemagglutinin monoclonal antibodies. EMBO J. 1987, 6, 1459–1465. [Google Scholar] [PubMed]
- Weissenhorn, W.; Dessen, A.; Harrison, S.C.; Skehel, J.J.; Wiley, D.C. Atomic structure of the ectodomain from HIV-1 gp41. Nature 1997, 387, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Skehel, J.J.; Wiley, D.C. N- and C-terminal residues combine in the fusion-pH influenza hemagglutinin HA(2) subunit to form an N cap that terminates the triple-stranded coiled coil. Proc. Natl. Acad. Sci. USA 1999, 96, 8967–8972. [Google Scholar] [CrossRef] [PubMed]
- Xu, R.; Wilson, I.A. Structural characterization of an early fusion intermediate of influenza virus hemagglutinin. J. Virol. 2011, 85, 5172–5182. [Google Scholar] [CrossRef] [PubMed]
- Leikina, E.; Ramos, C.; Markovic, I.; Zimmerberg, J.; Chernomordik, L.V. Reversible stages of the low-pH-triggered conformational change in influenza virus hemagglutinin. EMBO J. 2002, 21, 5701–5710. [Google Scholar] [CrossRef] [PubMed]
- Krumbiegel, M.; Herrmann, A.; Blumenthal, R. Kinetics of the low pH-induced conformational changes and fusogenic activity of influenza hemagglutinin. Biophys. J. 1994, 67(6), 2355–2360. [Google Scholar] [CrossRef]
- McLellan, J.S.; Chen, M.; Joyce, M.G.; Sastry, M.; Stewart-Jones, G.B.; Yang, Y.; Zhang, B.; Chen, L.; Srivatsan, S.; Zheng, A.; et al. Structure-based design of a fusion glycoprotein vaccine for respiratory syncytial virus. Science 2013, 342, 592–598. [Google Scholar] [CrossRef] [PubMed]
- Welch, B.D.; Liu, Y.; Kors, C.A.; Leser, G.P.; Jardetzky, T.S.; Lamb, R.A. Structure of the cleavage-activated prefusion form of the parainfluenza virus 5 fusion protein. Proc. Natl. Acad. Sci. USA 2012, 109, 16672–16677. [Google Scholar] [CrossRef] [PubMed]
- Yin, H.S.; Wen, X.; Paterson, R.G.; Lamb, R.A.; Jardetzky, T.S. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature 2006, 439, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Dias, J.M.; Kuehne, A.I.; Abelson, D.M.; Bale, S.; Wong, A.C.; Halfmann, P.; Muhammad, M.A.; Fusco, M.L.; Zak, S.E.; Kang, E.; et al. A shared structural solution for neutralizing ebolaviruses. Nat. Struct. Mol. Biol. 2011, 18, 1424–1427. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.E.; Fusco, M.L.; Hessell, A.J.; Oswald, W.B.; Burton, D.R.; Saphire, E.O. Structure of the Ebola virus glycoprotein bound to an antibody from a human survivor. Nature 2008, 454, 177–182. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Leaman, D.P.; Kim, A.S.; Torrents de la Pena, A.; Sliepen, K.; Yasmeen, A.; Derking, R.; Ramos, A.; de Taeye, S.W.; Ozorowski, G.; et al. Antibodies to a conformational epitope on gp41 neutralize HIV-1 by destabilizing the Env spike. Nat. Commun. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Hughson, F.M. Enveloped viruses: A common mode of membrane fusion? Curr. Biol. 1997, 7, R565–R569. [Google Scholar] [CrossRef]
- Malashkevich, V.N.; Schneider, B.J.; McNally, M.L.; Milhollen, M.A.; Pang, J.X.; Kim, P.S. Core structure of the envelope glycoprotein GP2 from Ebola virus at 1.9-Å resolution. Proc. Natl. Acad. Sci. USA 1999, 96, 2662–2667. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.C.; Fass, D.; Berger, J.M.; Kim, P.S. Core structure of gp41 from the HIV envelope glycoprotein. Cell 1997, 89, 263–273. [Google Scholar] [CrossRef]
- Weissenhorn, W.; Carfi, A.; Lee, K.H.; Skehel, J.J.; Wiley, D.C. Crystal structure of the Ebola virus membrane fusion subunit, GP2, from the envelope glycoprotein ectodomain. Mol. Cell 1998, 2, 605–616. [Google Scholar] [CrossRef]
- Konermann, L.; Vahidi, S.; Sowole, M.A. Mass spectrometry methods for studying structure and dynamics of biological macromolecules. Anal. Chem. 2014, 86, 213–232. [Google Scholar] [CrossRef] [PubMed]
- Orban, T.; Gupta, S.; Palczewski, K.; Chance, M.R. Visualizing water molecules in transmembrane proteins using radiolytic labeling methods. Biochemistry 2010, 49, 827–834. [Google Scholar] [CrossRef] [PubMed]
- Lim, X.X.; Chandramohan, A.; Anand, G.S. Temperature-dependent Conformational Dynamics in Whole Dengue Viral Particles by Hydrogen/Deuterium Exchange Mass Spectrometry. In Proceedings of the 63rd American Society for Mass spectrometry, St. Louis, MO, USA, 31 May-4 June 2015.
- Xu, G.; Chance, M.R. Radiolytic modification and reactivity of amino acid residues serving as structural probes for protein footprinting. Anal. Chem. 2005, 77, 4549–4555. [Google Scholar] [CrossRef] [PubMed]
- Konermann, L.; Pan, Y. Exploring membrane protein structural features by oxidative labeling and mass spectrometry. Exp. Rev. Proteom. 2012, 9, 497–504. [Google Scholar] [CrossRef] [PubMed]
- Gau, B.C.; Sharp, J.S.; Rempel, D.L.; Gross, M.L. Fast photochemical oxidation of protein footprints faster than protein unfolding. Anal. Chem. 2009, 81, 6563–6571. [Google Scholar] [CrossRef] [PubMed]
- Iacob, R.E.; Murphy, J.P., 3rd; Engen, J.R. Ion mobility adds an additional dimension to mass spectrometric analysis of solution-phase hydrogen/deuterium exchange. Rapid Commun. Mass Spectrom. 2008, 22, 2898–2904. [Google Scholar] [CrossRef] [PubMed]
- Englander, S.W. Hydrogen exchange and mass spectrometry: A historical perspective. J Am Soc Mass Spectrom 2006, 17, 1481–1489. [Google Scholar] [CrossRef] [PubMed]
- Kiselar, J.G.; Chance, M.R. Future directions of structural mass spectrometry using hydroxyl radical footprinting. J. Mass Spectrom. 2010, 45, 1373–1382. [Google Scholar] [CrossRef] [PubMed]
- Coales, S.J.; Tuske, S.J.; Tomasso, J.C.; Hamuro, Y. Epitope mapping by amide hydrogen/deuterium exchange coupled with immobilization of antibody, on-line proteolysis, liquid chromatography and mass spectrometry. Rapid Commun. Mass Spectrom. 2009, 23, 639–647. [Google Scholar] [CrossRef] [PubMed]
- Jones, L.M.; J, B.S.; J, A.C.; Gross, M.L. Fast photochemical oxidation of proteins for epitope mapping. Anal. Chem. 2011, 83, 7657–7661. [Google Scholar] [CrossRef] [PubMed]
- Marcsisin, S.R.; Engen, J.R. Hydrogen exchange mass spectrometry: What is it and what can it tell us? Anal. Bioanal. Chem. 2010, 397, 967–972. [Google Scholar] [CrossRef] [PubMed]
- Hamuro, Y.; Coales, S.J.; Molnar, K.S.; Tuske, S.J.; Morrow, J.A. Specificity of immobilized porcine pepsin in H/D exchange compatible conditions. Rapid Commun. Mass Spectrom. 2008, 22, 1041–1046. [Google Scholar] [CrossRef] [PubMed]
- Syka, J.E.; Coon, J.J.; Schroeder, M.J.; Shabanowitz, J.; Hunt, D.F. Peptide and protein sequence analysis by electron transfer dissociation mass spectrometry. Proc. Natl. Acad. Sci. USA 2004, 101, 9528–9533. [Google Scholar] [CrossRef] [PubMed]
- Rand, K.D.; Pringle, S.D.; Morris, M.; Brown, J.M. Site-specific analysis of gas-phase hydrogen/deuterium exchange of peptides and proteins by electron transfer dissociation. Anal. Chem. 2012, 84, 1931–1940. [Google Scholar] [CrossRef] [PubMed]
- Rand, K.D.; Zehl, M.; Jensen, O.N.; Jorgensen, T.J. Protein hydrogen exchange measured at single-residue resolution by electron transfer dissociation mass spectrometry. Anal. Chem. 2009, 81, 5577–5584. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.M.; Kazazic, S.; Schaub, T.M.; Tipton, J.D.; Emmett, M.R.; Marshall, A.G. Enhanced digestion efficiency, peptide ionization efficiency, and sequence resolution for protein hydrogen/deuterium exchange monitored by Fourier transform ion cyclotron resonance mass spectrometry. Anal. Chem. 2008, 80, 9034–9041. [Google Scholar] [CrossRef] [PubMed]
- Kadek, A.; Mrazek, H.; Halada, P.; Rey, M.; Schriemer, D.C.; Man, P. Aspartic protease nepenthesin-1 as a tool for digestion in hydrogen/deuterium exchange mass spectrometry. Anal. Chem. 2014, 86, 4287–4294. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.Y.; Hudgens, J.W. Effects of desialylation on human alpha1-acid glycoprotein-ligand interactions. Biochemistry 2013, 52, 7127–7136. [Google Scholar] [CrossRef] [PubMed]
- Houde, D.; Arndt, J.; Domeier, W.; Berkowitz, S.; Engen, J.R. Characterization of IgG1 Conformation and Conformational Dynamics by Hydrogen/Deuterium Exchange Mass Spectrometry. Anal. Chem. 2009, 81, 2644–2651. [Google Scholar] [CrossRef] [PubMed]
- Bai, Y.; Milne, J.S.; Mayne, L.; Englander, S.W. Primary structure effects on peptide group hydrogen exchange. Proteins 1993, 17, 75–86. [Google Scholar] [CrossRef] [PubMed]
- DuBois, R.M.; Zaraket, H.; Reddivari, M.; Heath, R.J.; White, S.W.; Russell, C.J. Acid stability of the hemagglutinin protein regulates H5N1 influenza virus pathogenicity. PLoS Pathog. 2011, 7, e1002398. [Google Scholar] [CrossRef] [PubMed]
- White, J.M.; Wilson, I.A. Anti-peptide antibodies detect steps in a protein conformational change: Low-pH activation of the influenza virus hemagglutinin. J. Cell Biol. 1987, 105, 2887–2896. [Google Scholar] [CrossRef] [PubMed]
- Kemble, G.W.; Bodian, D.L.; Rose, J.; Wilson, I.A.; White, J.M. Intermonomer disulfide bonds impair the fusion activity of influenza virus hemagglutinin. J. Virol. 1992, 66, 4940–4950. [Google Scholar] [PubMed]
- Galloway, S.E.; Reed, M.L.; Russell, C.J.; Steinhauer, D.A. Influenza HA subtypes demonstrate divergent phenotypes for cleavage activation and pH of fusion: Implications for host range and adaptation. PLoS Pathog. 2013, 9, e1003151. [Google Scholar] [CrossRef] [PubMed]
- Byrd-Leotis, L.; Galloway, S.E.; Agbogu, E.; Steinhauer, D.A. Influenza hemagglutinin (HA) stem region mutations that stabilize or destabilize the structure of multiple HA subtypes. J. Virol. 2015, 89, 4504–4516. [Google Scholar] [CrossRef] [PubMed]
- Kwong, P.D.; Wyatt, R.; Majeed, S.; Robinson, J.; Sweet, R.W.; Sodroski, J.; Hendrickson, W.A. Structures of HIV-1 gp120 envelope glycoproteins from laboratory-adapted and primary isolates. Structure 2000, 8, 1329–1339. [Google Scholar] [CrossRef]
- Khayat, R.; Lee, J.H.; Julien, J.P.; Cupo, A.; Klasse, P.J.; Sanders, R.W.; Moore, J.P.; Wilson, I.A.; Ward, A.B. Structural Characterization of Cleaved, Soluble HIV-1 Envelope Glycoprotein Trimers. J. Virol. 2013, 87, 9865–9872. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.G.; Whidby, J.; Miller, M.T.; Scarborough, H.; Zatorski, A.V.; Cygan, A.; Price, A.A.; Yost, S.A.; Bohannon, C.D.; Jacob, J.; et al. Structure of the core ectodomain of the hepatitis C virus envelope glycoprotein 2. Nature 2014, 509, 381–384. [Google Scholar] [CrossRef] [PubMed]
- Ciferri, C.; Chandramouli, S.; Donnarumma, D.; Nikitin, P.A.; Cianfrocco, M.A.; Gerrein, R.; Feire, A.L.; Barnett, S.W.; Lilja, A.E.; Rappuoli, R.; et al. Structural and biochemical studies of HCMV gH/gL/gO and Pentamer reveal mutually exclusive cell entry complexes. Proc. Natl. Acad. Sci. USA 2015, 112, 1767–1772. [Google Scholar] [CrossRef] [PubMed]
- Weis, D.D.; Wales, T.E.; Engen, J.R.; Hotchko, M.; Ten Eyck, L.F. Identification and characterization of EX1 kinetics in H/D exchange mass spectrometry by peak width analysis. J. Am. Soc. Mass Spectrom 2006, 17, 1498–1509. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Lee, K.K. A functional interaction between gp41 and gp120 is observed for monomeric but not oligomeric, uncleaved HIV-1 Env gp140. J. Virol. 2013, 87, 11462–11475. [Google Scholar] [CrossRef] [PubMed]
- Wei, H.; Mo, J.; Tao, L.; Russell, R.J.; Tymiak, A.A.; Chen, G.; Iacob, R.E.; Engen, J.R. Hydrogen/deuterium exchange mass spectrometry for probing higher order structure of protein therapeutics: Methodology and applications. Drug Discov. Today 2014, 19, 95–102. [Google Scholar] [CrossRef] [PubMed]
- Visser, J.; Feuerstein, I.; Stangler, T.; Schmiederer, T.; Fritsch, C.; Schiestl, M. Physicochemical and functional comparability between the proposed biosimilar rituximab GP2013 and originator rituximab. BioDrugs 2013, 27, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Houde, D.; Berkowitz, S.A.; Engen, J.R. The utility of hydrogen/deuterium exchange mass spectrometry in biopharmaceutical comparability studies. J. Pharm. Sci. 2011, 100, 2071–2086. [Google Scholar] [CrossRef] [PubMed]
- Kiselar, J.G.; Maleknia, S.D.; Sullivan, M.; Downard, K.M.; Chance, M.R. Hydroxyl radical probe of protein surfaces using synchrotron X-ray radiolysis and mass spectrometry. Int. J. Radiat. Biol. 2002, 78, 101–114. [Google Scholar] [CrossRef] [PubMed]
- Maleknia, S.D.; Brenowitz, M.; Chance, M.R. Millisecond radiolytic modification of peptides by synchrotron X-rays identified by mass spectrometry. Anal. Chem. 1999, 71, 3965–3973. [Google Scholar] [CrossRef] [PubMed]
- Hambly, D.M.; Gross, M.L. Laser flash photolysis of hydrogen peroxide to oxidize protein solvent-accessible residues on the microsecond timescale. J. Am. Soc. Mass Spectrom. 2005, 16, 2057–2063. [Google Scholar] [CrossRef] [PubMed]
- Xu, G.; Chance, M.R. Hydroxyl radical-mediated modification of proteins as probes for structural proteomics. Chem. Rev. 2007, 107, 3514–3543. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Rempel, D.L.; Gau, B.C.; Gross, M.L. Fast photochemical oxidation of proteins and mass spectrometry follow submillisecond protein folding at the amino-acid level. J. Am. Chem. Soc. 2012, 134, 18724–18731. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Qin, Y.; Ilchenko, S.; Bohon, J.; Shi, W.; Cho, M.W.; Takamoto, K.; Chance, M.R. Structural analysis of a highly glycosylated and unliganded gp120-based antigen using mass spectrometry. Biochemistry 2010, 49, 9032–9045. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Sharp, J.S. Structural Analysis of HIV-1 gp120 and its Complex with Neutralizing Immunoglobulin G1 b12 using Hydroxyl Radical Protein Footprinting. In Proceedings of the 62nd American Society for Mass Spectrometry Conference, Baltimore, MD, USA, 15–19 June 2014.
- Poor, T.A.; Jones, L.M.; Sood, A.; Leser, G.P.; Plasencia, M.D.; Rempel, D.L.; Jardetzky, T.S.; Woods, R.J.; Gross, M.L.; Lamb, R.A. Probing the paramyxovirus fusion (F) protein-refolding event from pre- to postfusion by oxidative footprinting. Proc. Natl. Acad. Sci. USA 2014, 111, E2596–E2605. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, G.; Kiselar, J.; He, Q.; Chance, M.R. Secondary reactions and strategies to improve quantitative protein footprinting. Anal. Chem. 2005, 77, 3029–3037. [Google Scholar] [CrossRef] [PubMed]
- Xie, B.; Sharp, J.S. Hydroxyl radical dosimetry for high flux hydroxyl radical protein footprinting applications using a simple optical detection method. Anal. Chem. 2015, 87, 10719–10723. [Google Scholar] [CrossRef] [PubMed]
- Putnam, C.D.; Hammel, M.; Hura, G.L.; Tainer, J.A. X-ray solution scattering (SAXS) combined with crystallography and computation: Defining accurate macromolecular structures, conformations and assemblies in solution. Q. Rev. Biophys. 2007, 40, 191–285. [Google Scholar] [CrossRef] [PubMed]
- Jacques, D.A.; Trewhella, J. Small-angle scattering for structural biology-Expanding the frontier while avoiding the pitfalls. Protein Sci. 2010, 19, 642–657. [Google Scholar] [CrossRef] [PubMed]
- Mylonas, E.; Svergun, D.I. Accuracy of molecular mass determination of proteins in solution by small-angle X-ray scattering. J. Appl. Crystallogr. 2007, 40, S245–S249. [Google Scholar] [CrossRef]
- Fischer, H.; de Oliveira Neto, M.; Napolitano, H.B.; Craievich, A.F.; Polikarpov, I. The molecular weight of proteins in solution can be determined from a single SAXS measurement on a relative scale. J. Appl. Cryst. 2010, 43, 101–109. [Google Scholar] [CrossRef]
- Petoukhov, M.V.; Svergun, D.I. Analysis of X-ray and neutron scattering from biomolecular solutions. Curr. Opin. Struc. Biol. 2007, 562–571. [Google Scholar] [CrossRef] [PubMed]
- Guttman, M.; Weinkam, P.; Sali, A.; Lee, K.K. All-atom ensemble modeling to analyze small-angle X-ray scattering of glycosylated proteins. Structure 2013, 21, 321–331. [Google Scholar] [CrossRef] [PubMed]
- Hammel, M.; Kriechbaum, M.; Gries, A.; Kostner, G.M.; Laggner, P.; Prassl, R. Solution structure of human and bovine β(2)-glycoprotein I revealed by small-angle X-ray scattering. J. Mol. Biol. 2002, 321, 85–97. [Google Scholar] [CrossRef]
- Svergun, D.I. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys. J. 1999, 76, 2879–2886. [Google Scholar] [CrossRef]
- Svergun, D.I.; Koch, M.H. Advances in structure analysis using small-angle scattering in solution. Curr. Opin. Struct. Biol. 2002, 12, 654–660. [Google Scholar] [CrossRef]
- Svergun, D.I.; Petoukhov, M.V.; Koch, M.H. Determination of domain structure of proteins from X-ray solution scattering. Biophys. J. 2001, 80, 2946–2953. [Google Scholar] [CrossRef]
- Forster, F.; Webb, B.; Krukenberg, K.A.; Tsuruta, H.; Agard, D.A.; Sali, A. Integration of small-angle X-ray scattering data into structural modeling of proteins and their assemblies. J. Mol. Biol. 2008, 382, 1089–1106. [Google Scholar] [CrossRef] [PubMed]
- Petoukhov, M.V.; Eady, N.A.J.; Brown, K.A.; Svergun, D.I. Addition of missing loops and domains to protein models by X-ray solution scattering. Biophys. J. 2002, 83, 3113–3125. [Google Scholar] [CrossRef]
- Hammel, M. Validation of macromolecular flexibility in solution by small-angle X-ray scattering (SAXS). Eur. Biophys. J. 2012, 41, 789–799. [Google Scholar] [CrossRef] [PubMed]
- Weinkam, P.; Pons, J.; Sali, A. Structure-based model of allostery predicts coupling between distant sites. Proc. Natl. Acad. Sci. USA 2012, 109, 4875–4880. [Google Scholar] [CrossRef] [PubMed]
- Schneidman-Duhovny, D.; Hammel, M.; Sali, A. FoXS: A web server for rapid computation and fitting of SAXS profiles. Nucleic Acids Res. 2010, 38, W540–W544. [Google Scholar] [CrossRef] [PubMed]
- Garg, R.; Anguita, J.; Krueger, J.K. Binding of full-length HIV-1 gp120 to CD4 induces structural reorientation around the gp120 core. Biophys. J. 2006, 91, L69–L71. [Google Scholar]
- Zheng, Y.; Doerschuk, P.C.; Johnson, J.E. Determination of three-dimensional low-resolution viral structure from solution X-ray scattering data. Biophys. J. 1995, 69, 619–639. [Google Scholar] [CrossRef]
- Perez, J.; Defrenne, S.; Witz, J.; Vachette, P. Detection and characterization of an intermediate conformation during the divalent ion-dependent swelling of tomato bushy stunt virus. Cell Mol. Biol. 2000, 46, 937–948. [Google Scholar] [PubMed]
- Canady, M.A.; Tsuruta, H.; Johnson, J.E. Analysis of rapid, large-scale protein quaternary structural changes: time-resolved X-ray solution scattering of Nudaurelia capensis omega virus (NomegaV) maturation. J. Mol. Biol. 2001, 311, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Aramayo, R.; Merigoux, C.; Larquet, E.; Bron, P.; Perez, J.; Dumas, C.; Vachette, P.; Boisset, N. Divalent ion-dependent swelling of tomato bushy stunt virus: A multi-approach study. Biochim. Biophys. Acta. 2005, 1724, 345–354. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.K.; Tsuruta, H.; Hendrix, R.W.; Duda, R.L.; Johnson, J.E. Cooperative reorganization of a 420 subunit virus capsid. J. Mol. Biol. 2005, 352, 723–735. [Google Scholar] [CrossRef] [PubMed]
- Kler, S.; Asor, R.; Li, C.; Ginsburg, A.; Harries, D.; Oppenheim, A.; Zlotnick, A.; Raviv, U. RNA encapsidation by SV40-derived nanoparticles follows a rapid two-state mechanism. J. Am. Chem. Soc. 2012, 134, 8823–8830. [Google Scholar] [CrossRef] [PubMed]
- Hashiguchi, T.; Fusco, M.L.; Bornholdt, Z.A.; Lee, J.E.; Flyak, A.I.; Matsuoka, R.; Kohda, D.; Yanagi, Y.; Hammel, M.; Crowe, J.E., Jr.; Saphire, E.O. Structural basis for Marburg virus neutralization by a cross-reactive human antibody. Cell 2015, 160, 904–912. [Google Scholar] [CrossRef] [PubMed]
- Julien, J.P.; Lee, J.H.; Cupo, A.; Murin, C.D.; Derking, R.; Hoffenberg, S.; Caulfield, M.J.; King, C.R.; Marozsan, A.J.; Klasse, P.J.; et al. Asymmetric recognition of the HIV-1 trimer by broadly neutralizing antibody PG9. Proc. Natl. Acad. Sci. USA 2013, 110, 4351–4356. [Google Scholar] [CrossRef] [PubMed]
- Leschziner, A.E.; Nogales, E. Visualizing flexibility at molecular resolution: analysis of heterogeneity in single-particle electron microscopy reconstructions. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 43–62. [Google Scholar] [CrossRef] [PubMed]
- Elmlund, D.; Elmlund, H. Cryogenic electron microscopy and single-particle analysis. Annu. Rev. Biochem. 2015, 84, 499–517. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.B.; Wilson, I.A. Insights into the trimeric HIV-1 envelope glycoprotein structure. Trends Biochem. Sci. 2015, 40, 101–107. [Google Scholar] [CrossRef] [PubMed]
- Lucic, V.; Forster, F.; Baumeister, W. Structural studies by electron tomography: From cells to molecules. Annu. Rev. Biochem. 2005, 74, 833–865. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S.; Bartesaghi, A.; Liu, J.; Bennett, A.E.; Sougrat, R. Electron tomography of viruses. Curr. Opin. Struct. Biol. 2007, 17, 596–602. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, K.; Cyrklaff, M. Structure of complex viruses and virus-infected cells by electron cryo tomography. Curr. Opin. Microbiol. 2006, 9, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, K.; Desai, P.; Winkler, D.C.; Heymann, J.B.; Belnap, D.M.; Baumeister, W.; Steven, A.C. Three-dimensional structure of herpes simplex virus from cryo-electron tomography. Science 2003, 302, 1396–1398. [Google Scholar] [CrossRef] [PubMed]
- Cyrklaff, M.; Risco, C.; Fernandez, J.J.; Jimenez, M.V.; Esteban, M.; Baumeister, W.; Carrascosa, J.L. Cryo-electron tomography of vaccinia virus. Proc. Natl. Acad. Sci. USA 2005, 102, 2772–2777. [Google Scholar] [CrossRef] [PubMed]
- Barcena, M.; Oostergetel, G.T.; Bartelink, W.; Faas, F.G.; Verkleij, A.; Rottier, P.J.; Koster, A.J.; Bosch, B.J. Cryo-electron tomography of mouse hepatitis virus: Insights into the structure of the coronavirion. Proc. Natl. Acad. Sci. USA 2009, 106, 582–587. [Google Scholar] [CrossRef] [PubMed]
- Loney, C.; Mottet-Osman, G.; Roux, L.; Bhella, D. Paramyxovirus ultrastructure and genome packaging: cryo-electron tomography of sendai virus. J. Virol. 2009, 83, 8191–8197. [Google Scholar] [CrossRef] [PubMed]
- Guichard, P.; Krell, T.; Chevalier, M.; Vaysse, C.; Adam, O.; Ronzon, F.; Marco, S. Three dimensional morphology of rabies virus studied by cryo-electron tomography. J. Struct. Biol. 2011, 176, 32–40. [Google Scholar] [CrossRef] [PubMed]
- Beniac, D.R.; Melito, P.L.; Devarennes, S.L.; Hiebert, S.L.; Rabb, M.J.; Lamboo, L.L.; Jones, S.M.; Booth, T.F. The organisation of Ebola virus reveals a capacity for extensive, modular polyploidy. PloS ONE 2012, 7, e29608. [Google Scholar] [CrossRef] [PubMed]
- Catanese, M.T.; Uryu, K.; Kopp, M.; Edwards, T.J.; Andrus, L.; Rice, W.J.; Silvestry, M.; Kuhn, R.J.; Rice, C.M. Ultrastructural analysis of hepatitis C virus particles. Proc. Natl. Acad. Sci. USA 2013, 110, 9505–9510. [Google Scholar] [CrossRef] [PubMed]
- Liljeroos, L.; Krzyzaniak, M.A.; Helenius, A.; Butcher, S.J. Architecture of respiratory syncytial virus revealed by electron cryotomography. Proc. Natl. Acad. Sci. USA 2013, 110, 11133–11138. [Google Scholar] [CrossRef] [PubMed]
- Kiss, G.; Holl, J.M.; Williams, G.M.; Alonas, E.; Vanover, D.; Lifland, A.W.; Gudheti, M.; Guerrero-Ferreira, R.C.; Nair, V.; Yi, H.; et al. Structural analysis of respiratory syncytial virus reveals the position of M2–1 between the matrix protein and the ribonucleoprotein complex. J. Virol. 2014, 88, 7602–7617. [Google Scholar] [CrossRef] [PubMed]
- Liljeroos, L.; Huiskonen, J.T.; Ora, A.; Susi, P.; Butcher, S.J. Electron cryotomography of measles virus reveals how matrix protein coats the ribonucleocapsid within intact virions. Proc. Natl. Acad. Sci. USA 2011, 108, 18085–18090. [Google Scholar] [CrossRef] [PubMed]
- Battisti, A.J.; Yoder, J.D.; Plevka, P.; Winkler, D.C.; Prasad, V.M.; Kuhn, R.J.; Frey, T.K.; Steven, A.C.; Rossmann, M.G. Cryo-electron tomography of rubella virus. J. Virol. 2012, 86, 11078–11085. [Google Scholar] [CrossRef] [PubMed]
- Libersou, S.; Albertini, A.A.; Ouldali, M.; Maury, V.; Maheu, C.; Raux, H.; de Haas, F.; Roche, S.; Gaudin, Y.; Lepault, J. Distinct structural rearrangements of the VSV glycoprotein drive membrane fusion. J. Cell Biol. 191, 199–210. [CrossRef] [PubMed]
- Cao, S.; Zhang, W. Characterization of an early-stage fusion intermediate of Sindbis virus using cryoelectron microscopy. Proc. Natl. Acad. Sci. USA 2013, 110, 13362–13367. [Google Scholar] [CrossRef] [PubMed]
- Maurer, U.E.; Sodeik, B.; Grunewald, K. Native 3D intermediates of membrane fusion in herpes simplex virus 1 entry. Proc. Natl. Acad. Sci. USA 2008, 105, 10559–10564. [Google Scholar] [CrossRef] [PubMed]
- Bharat, T.A.; Riches, J.D.; Kolesnikova, L.; Welsch, S.; Krahling, V.; Davey, N.; Parsy, M.L.; Becker, S.; Briggs, J.A. Cryo-electron tomography of Marburg virus particles and their morphogenesis within infected cells. PLoS Biol. 2011, 9, e1001196. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vijayakrishnan, S.; Loney, C.; Jackson, D.; Suphamungmee, W.; Rixon, F.J.; Bhella, D. Cryotomography of budding influenza A virus reveals filaments with diverse morphologies that mostly do not bear a genome at their distal end. PLoS Pathog. 2013, 9, e1003413. [Google Scholar] [CrossRef] [PubMed]
- Carlson, L.A.; Briggs, J.A.; Glass, B.; Riches, J.D.; Simon, M.N.; Johnson, M.C.; Muller, B.; Grunewald, K.; Krausslich, H.G. Three-dimensional analysis of budding sites and released virus suggests a revised model for HIV-1 morphogenesis. Cell Host Microbe 2008, 4, 592–599. [Google Scholar] [CrossRef] [PubMed]
- De Marco, A.; Muller, B.; Glass, B.; Riches, J.D.; Krausslich, H.G.; Briggs, J.A. Structural analysis of HIV-1 maturation using cryo-electron tomography. PLoS Pathog. 2010, 6, e1001215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plevka, P.; Battisti, A.J.; Junjhon, J.; Winkler, D.C.; Holdaway, H.A.; Keelapang, P.; Sittisombut, N.; Kuhn, R.J.; Steven, A.C.; Rossmann, M.G. Maturation of flaviviruses starts from one or more icosahedrally independent nucleation centres. EMBO Rep. 2011, 12, 602–606. [Google Scholar] [CrossRef] [PubMed]
- Plevka, P.; Battisti, A.J.; Sheng, J.; Rossmann, M.G. Mechanism for maturation-related reorganization of flavivirus glycoproteins. J. Struct. Biol. 2014, 185, 27–31. [Google Scholar] [CrossRef] [PubMed]
- Chlanda, P.; Schraidt, O.; Kummer, S.; Riches, J.; Oberwinkler, H.; Prinz, S.; Krausslich, H.G.; Briggs, J.A. Structural Analysis of the Roles of Influenza A Virus Membrane-Associated Proteins in Assembly and Morphology. J. Virol. 2015, 89, 8957–8966. [Google Scholar] [CrossRef] [PubMed]
- Gui, L.; Jurgens, E.M.; Ebner, J.L.; Porotto, M.; Moscona, A.; Lee, K.K. Electron tomography imaging of surface glycoproteins on human parainfluenza virus 3: Association of receptor binding and fusion proteins before receptor engagement. mBio 2015, 6, e02393–14. [Google Scholar] [CrossRef] [PubMed]
- Gruenke, J.A.; Armstrong, R.T.; Newcomb, W.W.; Brown, J.C.; White, J.M. New insights into the spring-loaded conformational change of influenza virus hemagglutinin. J. Virol. 2002, 76, 4456–4466. [Google Scholar] [CrossRef] [PubMed]
- Briggs, J.A. Structural biology in situ--the potential of subtomogram averaging. Curr. Opin. Struct. Biol. 2013, 23, 261–267. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Chertova, E.; Bess, J., Jr.; Lifson, J.D.; Arthur, L.O.; Liu, J.; Taylor, K.A.; Roux, K.H. Electron tomography analysis of envelope glycoprotein trimers on HIV and simian immunodeficiency virus virions. Proc. Natl. Acad. Sci. USA 2003, 100, 15812–15817. [Google Scholar] [CrossRef] [PubMed]
- Forster, F.; Medalia, O.; Zauberman, N.; Baumeister, W.; Fass, D. Retrovirus envelope protein complex structure in situ studied by cryo-electron tomography. Proc. Natl. Acad. Sci. USA 2005, 102, 4729–4734. [Google Scholar] [CrossRef] [PubMed]
- Zanetti, G.; Briggs, J.A.; Grunewald, K.; Sattentau, Q.J.; Fuller, S.D. Cryo-electron tomographic structure of an immunodeficiency virus envelope complex in situ. PLoS Pathog. 2006, 2, e83. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Liu, J.; Bess, J., Jr.; Chertova, E.; Lifson, J.D.; Grise, H.; Ofek, G.A.; Taylor, K.A.; Roux, K.H. Distribution and three-dimensional structure of AIDS virus envelope spikes. Nature 2006, 441, 847–852. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S. The SIV surface spike imaged by electron tomography: One leg or three? PLoS Pathog. 2006, 2, e91. [Google Scholar] [CrossRef] [PubMed]
- Harris, A.K.; Meyerson, J.R.; Matsuoka, Y.; Kuybeda, O.; Moran, A.; Bliss, D.; Das, S.R.; Yewdell, J.W.; Sapiro, G.; Subbarao, K.; Subramaniam, S. Structure and accessibility of HA trimers on intact 2009 H1N1 pandemic influenza virus to stem region-specific neutralizing antibodies. Proc. Natl. Acad. Sci. USA 2013, 110, 4592–4597. [Google Scholar] [CrossRef] [PubMed]
- Tran, E.E.; Simmons, J.A.; Bartesaghi, A.; Shoemaker, C.J.; Nelson, E.; White, J.M.; Subramaniam, S. Spatial localization of the Ebola virus glycoprotein mucin-like domain determined by cryo-electron tomography. J. Virol. 2014, 88, 10958–10962. [Google Scholar] [CrossRef] [PubMed]
- Bharat, T.A.; Davey, N.E.; Ulbrich, P.; Riches, J.D.; de Marco, A.; Rumlova, M.; Sachse, C.; Ruml, T.; Briggs, J.A. Structure of the immature retroviral capsid at 8 Å resolution by cryo-electron microscopy. Nature 2012, 487, 385–389. [Google Scholar] [CrossRef] [PubMed]
- Schur, F.K.; Hagen, W.J.; Rumlova, M.; Ruml, T.; Muller, B.; Krausslich, H.G.; Briggs, J.A. Structure of the immature HIV-1 capsid in intact virus particles at 8.8 Å resolution. Nature 2015, 517, 505–508. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Z.H. Atomic Resolution Cryo Electron Microscopy of Macromolecular Complexes. Adv. Protein Chem. Struct. 2011, 82, 1–35. [Google Scholar]
- Lee, J.H.; de Val, N.; Lyumkis, D.; Ward, A.B. Model Building and Refinement of a Natively Glycosylated HIV-1 Env Protein by High-Resolution Cryoelectron Microscopy. Structure 2015, 23, 1943–1951. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.; Avila-Sakar, A.; Kim, J.; Booth, D.S.; Greenberg, C.H.; Rossi, A.; Liao, M.; Li, X.; Alian, A.; Griner, S.L.; et al. Fabs enable single particle cryoEM studies of small proteins. Structure 2012, 20, 582–592. [Google Scholar] [CrossRef] [PubMed]
- Murin, C.D.; Fusco, M.L.; Bornholdt, Z.A.; Qiu, X.; Olinger, G.G.; Zeitlin, L.; Kobinger, G.P.; Ward, A.B.; Saphire, E.O. Structures of protective antibodies reveal sites of vulnerability on Ebola virus. Proc. Natl. Acad. Sci. USA 2014, 111, 17182–17187. [Google Scholar] [CrossRef] [PubMed]
- Hong, M.; Lee, P.S.; Hoffman, R.M.; Zhu, X.; Krause, J.C.; Laursen, N.S.; Yoon, S.I.; Song, L.; Tussey, L.; Crowe, J.E., Jr.; et al. Antibody recognition of the pandemic H1N1 Influenza virus hemagglutinin receptor binding site. J. Virol. 2013, 87, 12471–12480. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Giang, E.; Nieusma, T.; Kadam, R.U.; Cogburn, K.E.; Hua, Y.; Dai, X.; Stanfield, R.L.; Burton, D.R.; Ward, A.B.; Wilson, I.A.; Law, M. Hepatitis C virus E2 envelope glycoprotein core structure. Science 2013, 342, 1090–1094. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Lee, J.H.; Doores, K.J.; Murin, C.D.; Julien, J.P.; McBride, R.; Liu, Y.; Marozsan, A.; Cupo, A.; Klasse, P.J.; et al. Supersite of immune vulnerability on the glycosylated face of HIV-1 envelope glycoprotein gp120. Nat. Struct. Mol. Biol. 2013, 20, 796–803. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Kang, B.H.; Pancera, M.; Lee, J.H.; Tong, T.; Feng, Y.; Imamichi, H.; Georgiev, I.S.; Chuang, G.Y.; Druz, A.; et al. Broad and potent HIV-1 neutralization by a human antibody that binds the gp41-gp120 interface. Nature 2014, 515, 138–142. [Google Scholar] [CrossRef] [PubMed]
- Murin, C.D.; Julien, J.P.; Sok, D.; Stanfield, R.L.; Khayat, R.; Cupo, A.; Moore, J.P.; Burton, D.R.; Wilson, I.A.; Ward, A.B. Structure of 2G12 Fab2 in complex with soluble and fully glycosylated HIV-1 Env by negative-stain single-particle electron microscopy. J. Virol. 2014, 88, 10177–10188. [Google Scholar] [CrossRef] [PubMed]
- Yasmeen, A.; Ringe, R.; Derking, R.; Cupo, A.; Julien, J.P.; Burton, D.R.; Ward, A.B.; Wilson, I.A.; Sanders, R.W.; Moore, J.P.; Klasse, P.J. Differential binding of neutralizing and non-neutralizing antibodies to native-like soluble HIV-1 Env trimers, uncleaved Env proteins, and monomeric subunits. Retrovirology 2014, 11. [Google Scholar] [CrossRef] [PubMed]
- Flyak, A.I.; Ilinykh, P.A.; Murin, C.D.; Garron, T.; Shen, X.; Fusco, M.L.; Hashiguchi, T.; Bornholdt, Z.A.; Slaughter, J.C.; Sapparapu, G.; et al. Mechanism of human antibody-mediated neutralization of Marburg virus. Cell 2015, 160, 893–903. [Google Scholar] [CrossRef] [PubMed]
- Henderson, R. Avoiding the pitfalls of single particle cryo-electron microscopy: Einstein from noise. Proc. Natl. Acad. Sci. USA 2013, 110, 18037–18041. [Google Scholar] [CrossRef] [PubMed]
- Mao, Y.; Castillo-Menendez, L.R.; Sodroski, J.G. Reply to Subramaniam, van Heel, and Henderson: Validity of the cryo-electron microscopy structures of the HIV-1 envelope glycoprotein complex. Proc. Natl. Acad. Sci. USA 2013, 110, E4178–E4182. [Google Scholar] [CrossRef] [PubMed]
- Subramaniam, S. Structure of trimeric HIV-1 envelope glycoproteins. Proc. Natl. Acad. Sci. USA 2013, 110, E4172–E4174. [Google Scholar] [CrossRef] [PubMed]
- Wasilewski, S.; Calder, L.J.; Grant, T.; Rosenthal, P.B. Distribution of surface glycoproteins on influenza A virus determined by electron cryotomography. Vaccine 2012, 30, 7368–7373. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Sheng, J.; Austin, S.K.; Hoornweg, T.E.; Smit, J.M.; Kuhn, R.J.; Diamond, M.S.; Rossmann, M.G. Structure of acidic pH dengue virus showing the fusogenic glycoprotein trimers. J. Virol. 2015, 89, 743–750. [Google Scholar] [CrossRef] [PubMed]
- Kostyuchenko, V.A.; Zhang, Q.; Tan, J.L.; Ng, T.S.; Lok, S.M. Immature and mature dengue serotype 1 virus structures provide insight into the maturation process. J. Virol. 2013, 87, 7700–7707. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Ge, P.; Yu, X.; Brannan, J.M.; Bi, G.; Zhang, Q.; Schein, S.; Zhou, Z.H. Cryo-EM structure of the mature dengue virus at 3.5-Å resolution. Nat. Struct. Mol. Biol. 2013, 20, 105–110. [Google Scholar] [CrossRef] [PubMed]
- Kostyuchenko, V.A.; Chew, P.L.; Ng, T.S.; Lok, S.M. Near-atomic resolution cryo-electron microscopic structure of dengue serotype 4 virus. J. Virol. 2014, 88, 477–482. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Hryc, C.F.; Cong, Y.; Liu, X.; Jakana, J.; Gorchakov, R.; Baker, M.L.; Weaver, S.C.; Chiu, W. 4.4 Å cryo-EM structure of an enveloped alphavirus Venezuelan equine encephalitis virus. EMBO J. 2011, 30, 3854–3863. [Google Scholar] [CrossRef] [PubMed]
- Ward, A.B.; Sali, A.; Wilson, I.A. Biochemistry. Integrative structural biology. Science 2013, 339, 913–915. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garcia, N.K.; Lee, K.K. Dynamic Viral Glycoprotein Machines: Approaches for Probing Transient States That Drive Membrane Fusion. Viruses 2016, 8, 15. https://doi.org/10.3390/v8010015
Garcia NK, Lee KK. Dynamic Viral Glycoprotein Machines: Approaches for Probing Transient States That Drive Membrane Fusion. Viruses. 2016; 8(1):15. https://doi.org/10.3390/v8010015
Chicago/Turabian StyleGarcia, Natalie K., and Kelly K. Lee. 2016. "Dynamic Viral Glycoprotein Machines: Approaches for Probing Transient States That Drive Membrane Fusion" Viruses 8, no. 1: 15. https://doi.org/10.3390/v8010015