
Structure and Biophysical Properties of a Triple-Stranded Beta-Helix Comprising the Central Spike of Bacteriophage T4

Abstract
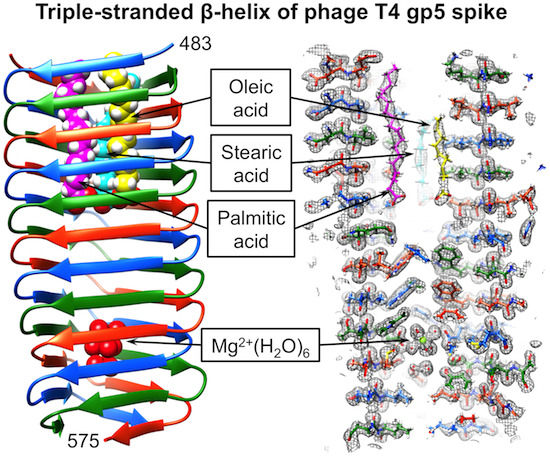
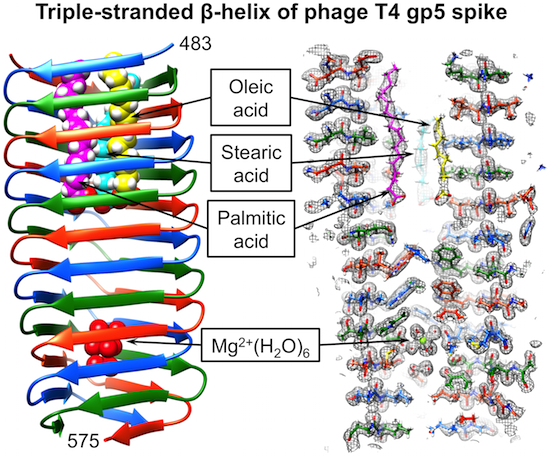
:
1. Introduction
2. Materials and Methods
2.1. Cloning, Expression and Purification
2.1.1. Gp5β-ABC, Gp5β-BC, Gp5β-C, and Gp5β-B Fragments
2.1.2. Gp5β-BC2 Fragment
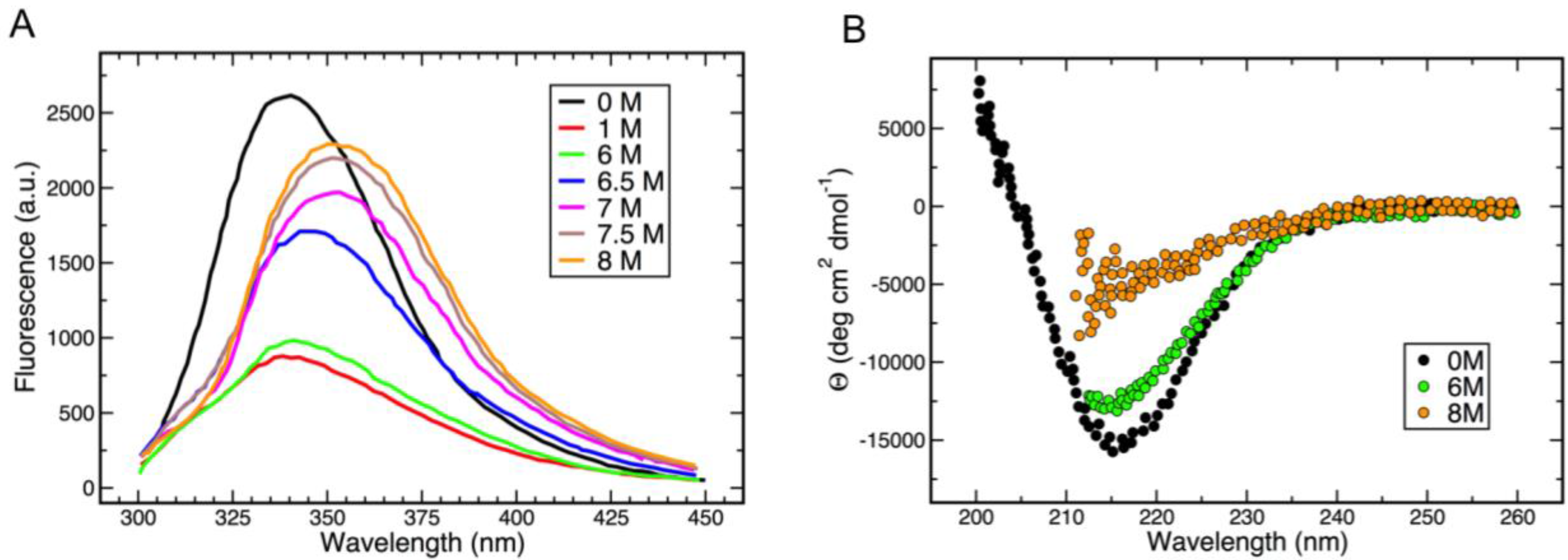
2.2. Circular Dichroism and Fluorescence Analysis
2.3. Analytical Ultracentrifugation
2.4. Crystallization and Structure Determination
2.4.1. Gp5β-BC

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gp5β-BC | Gp5β-BC2 | |
|---|---|---|
| Data Collection | ||
| Beamline | PXIII (SLS) | PXI (SLS) |
| Detector | MARMOSAIC 225mm CCD | DECTRIS PILATUS 6M |
| Wavelength (Å) | 0.900 | 1.000 |
| Oscillation angle (°) | 1.0 | 0.25 |
| Number of frames | 360 + 360 * | 990 |
| Space group | C2221 | P21 |
| Cell dimensions: a, b, c (Å), α, β, γ (°) | 57.61, 72.76, 130.49, 90.00, 90.00, 90.00 | 110.29, 73.79, 111.60, 90.00, 113.39, 90.00 |
| Number of polypeptide chains in the asymmetric unit | 3 | 9 |
| Resolution interval (Å) | 50.0–1.3 | 50.0–2.0 |
| Rsym (%) | 0.069 (0.299)# | 0.095 (0.363) |
| <I/σI> | 11.3 (3.6) | 6.7 (2.4) |
| Completeness (%) | 99.59 (87.0) | 96.6 (95.9) |
| Redundancy | 10.2 (4.3) | 2.4 (2.5) |
| Refinement | ||
| Number of reflections | ||
| Working | 69991 | 110537 |
| Test | 4689 | 5818 |
| Rwork/Rfree | 0.153/0.178 | 0.219/0.279 |
| B-factor (Å2) | 19.4 | 35.3 |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.0169 | 0.019 |
| Bond angles (°) | 0.0413 | 1.849 |
| Number of atoms | ||
| Protein | 2259 | 12217 |
| Solvent and ligands | 388 | 992 |
| Ramachandran plot (%) | ||
| Most favored | 97.4 | 98.9 |
| Additionally allowed | 2.6 | 1.1 |
| Outliers | 0.0 | 0.0 |
| PDB ID | 4JJ2 | 4OSD |
2.4.2. Gp5β-BC2
2.5. Extraction of Internal Compounds from gp5β-BC
2.6. Mass Spectrometry Analyses
2.6.1. ESI-QTOF-MS
2.6.2. GC-IE-MS
2.7. Sequence Analysis
3. Results
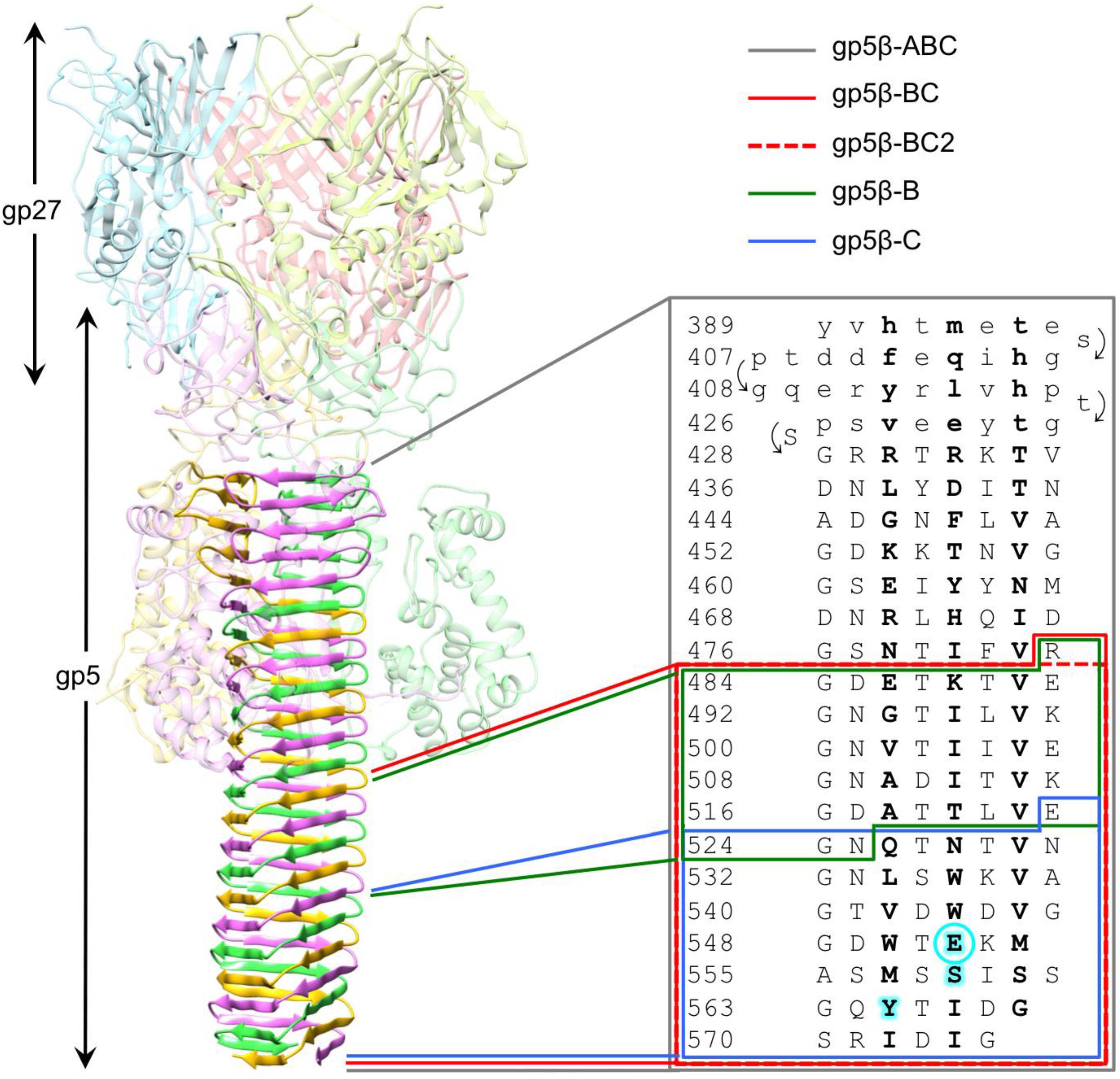
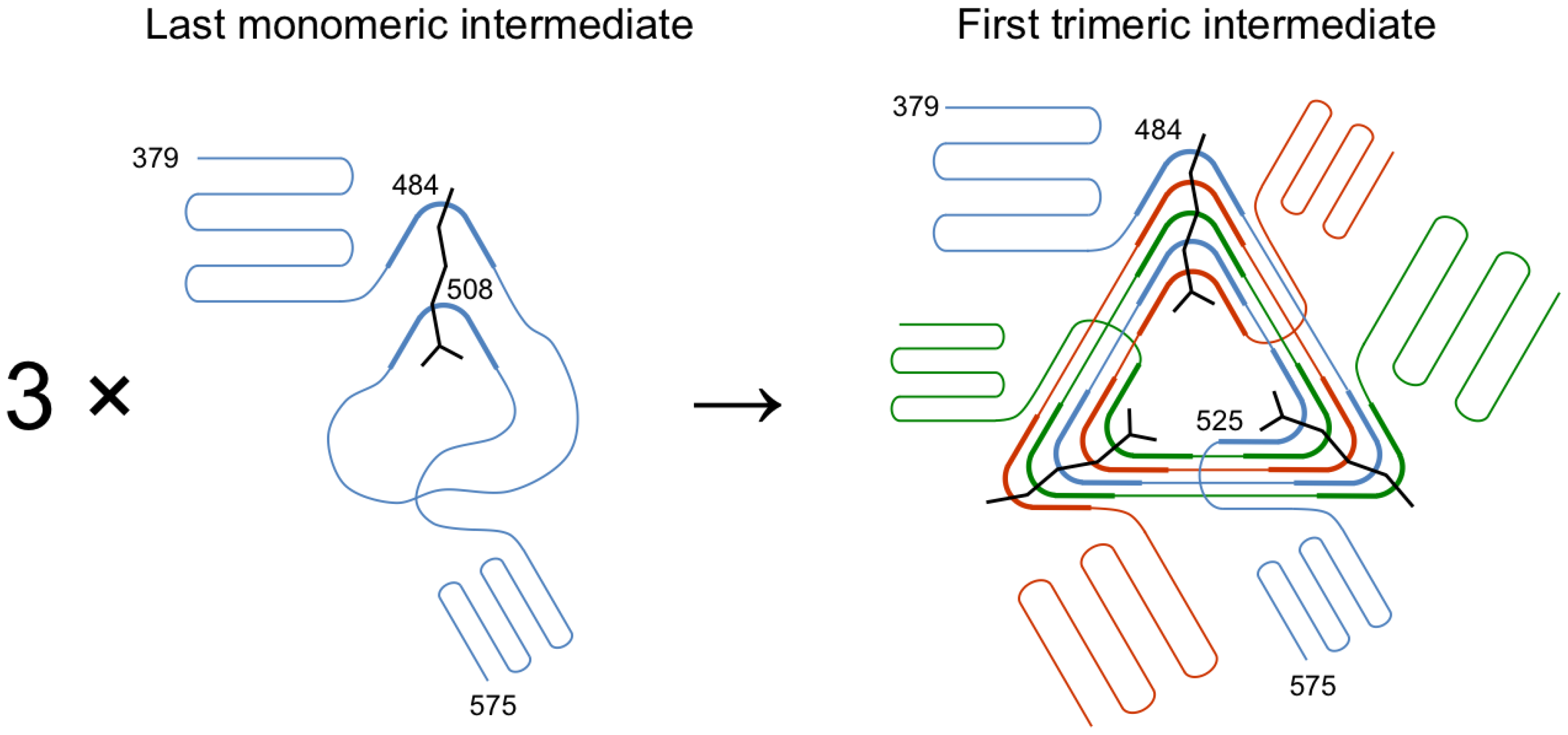
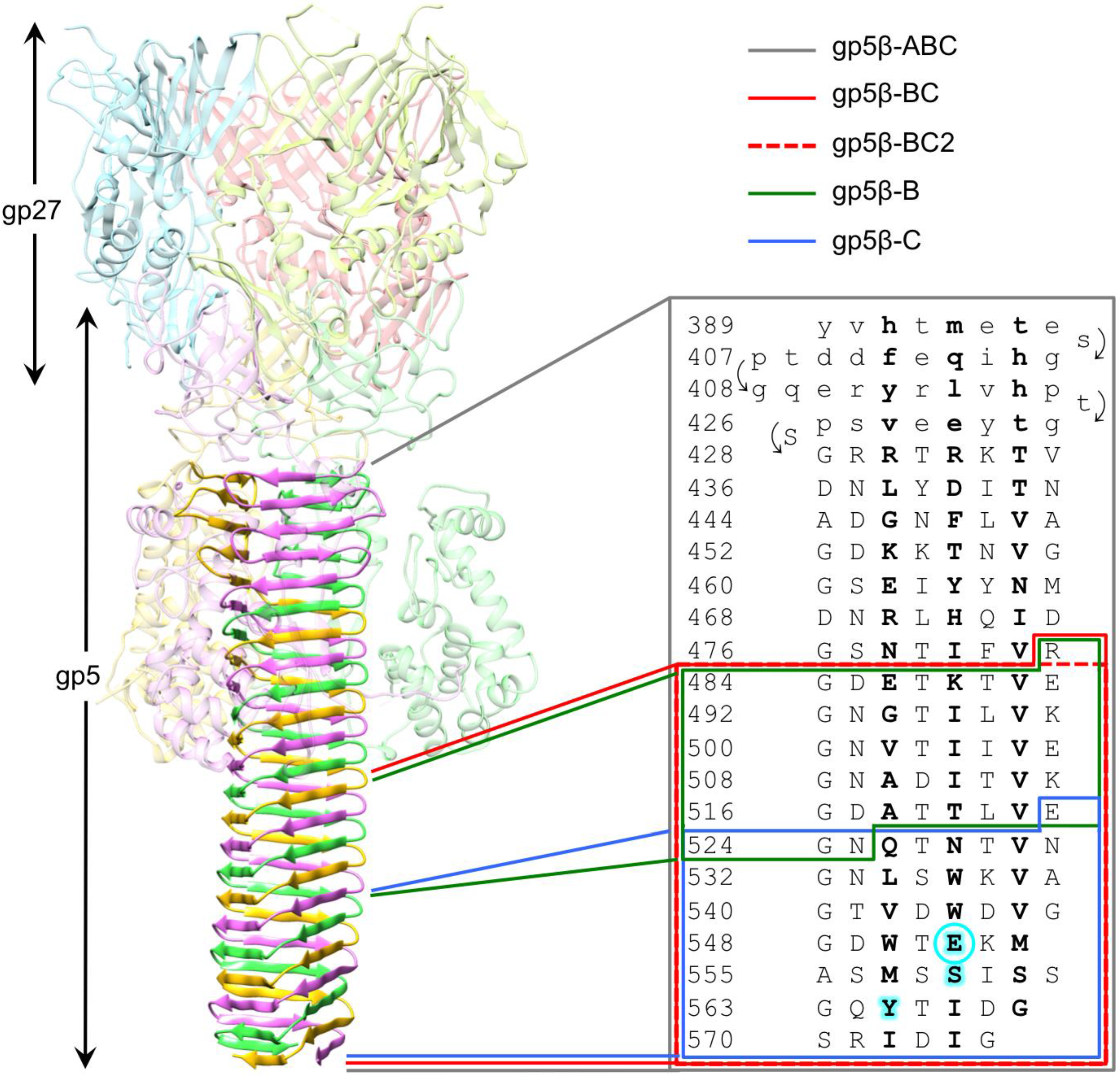
3.1. Design of β-Helix Fragments
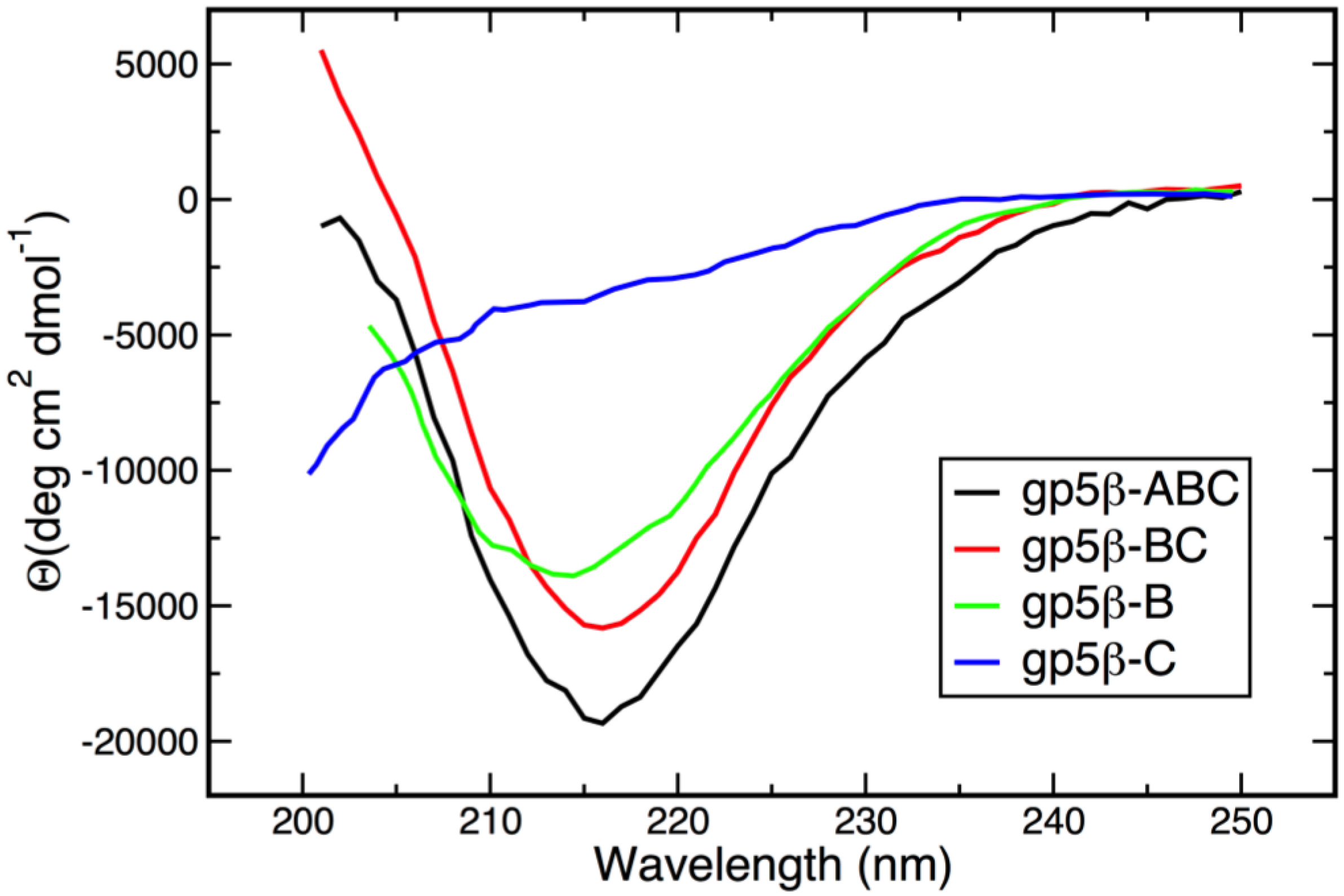
3.2. Secondary Structure and Oligomeric State of β-Helix Fragments
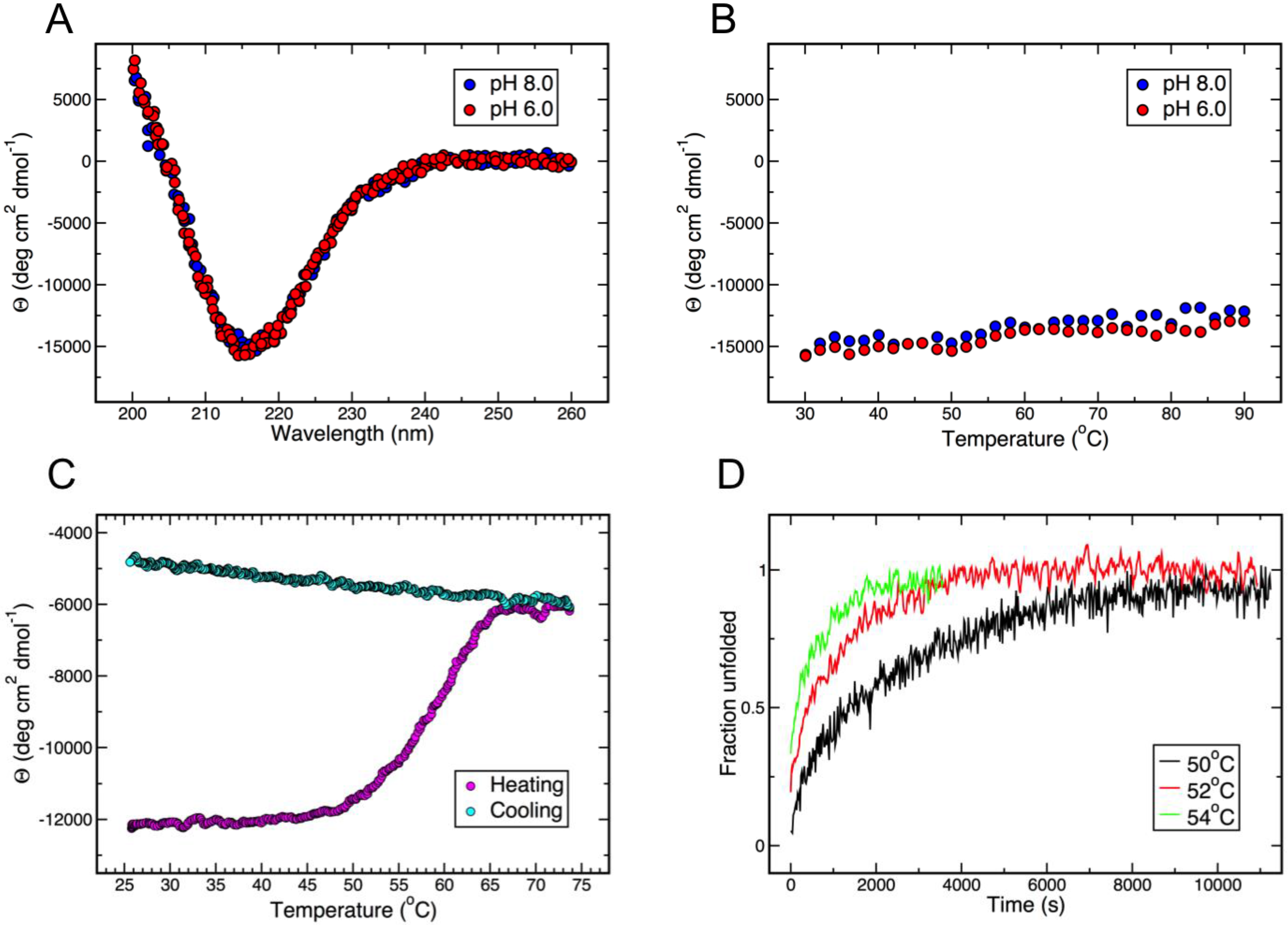
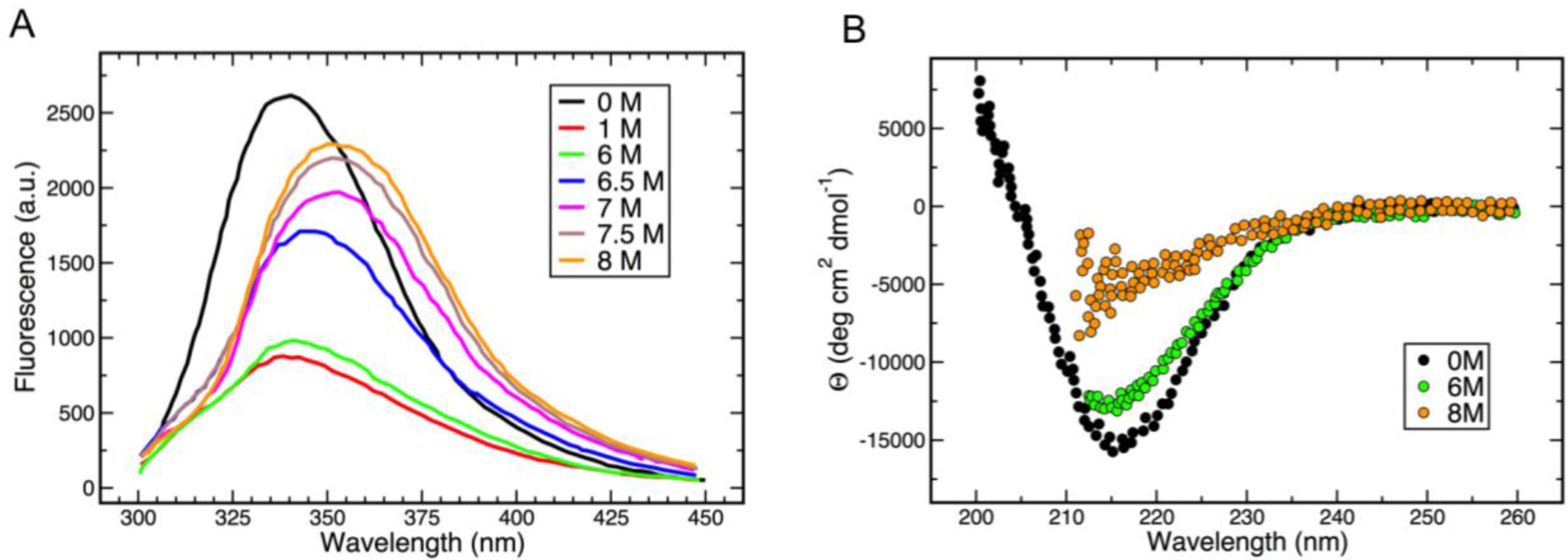
3.3. Thermal and Chemical Stability of gp5β-BC
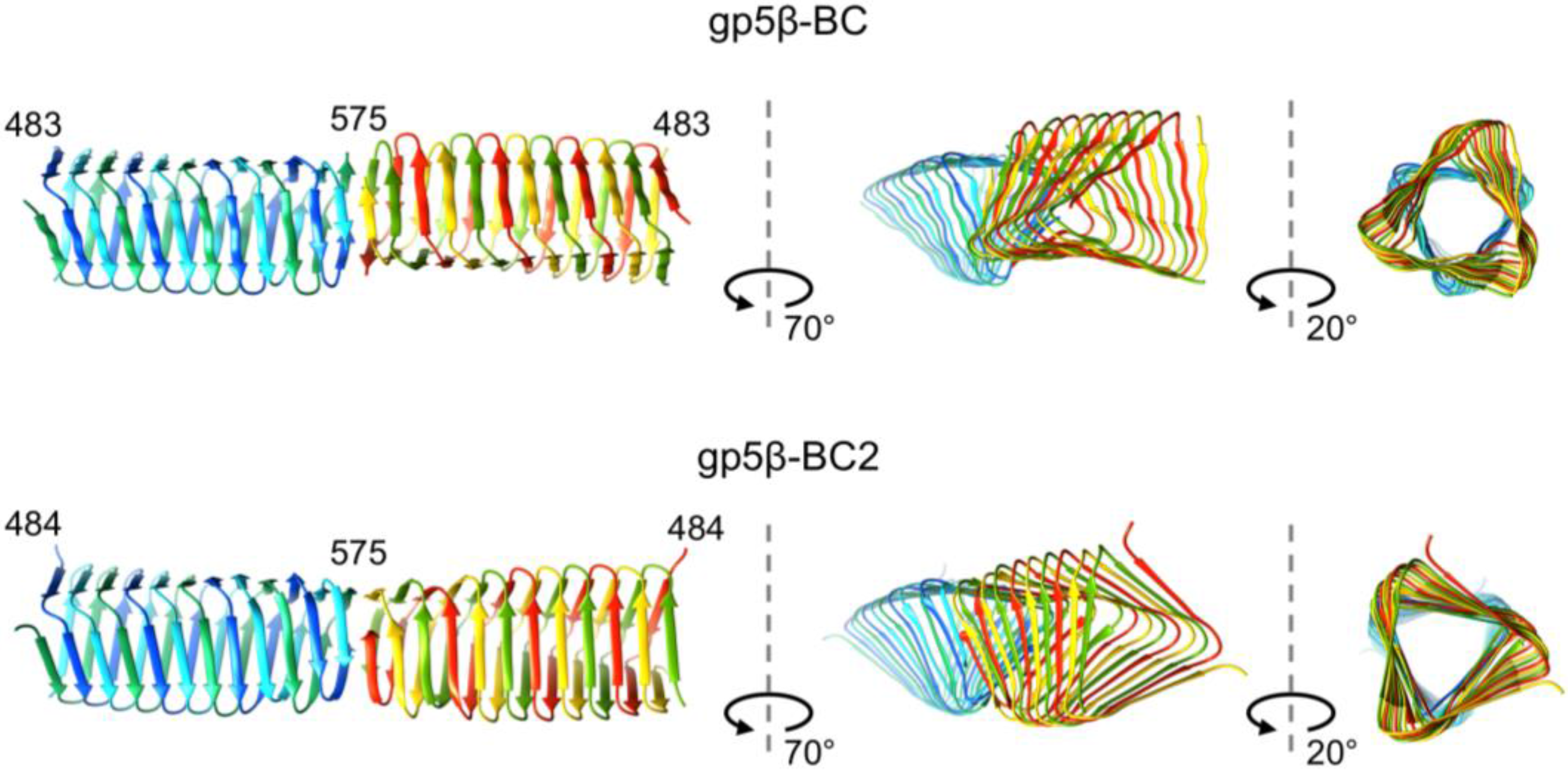
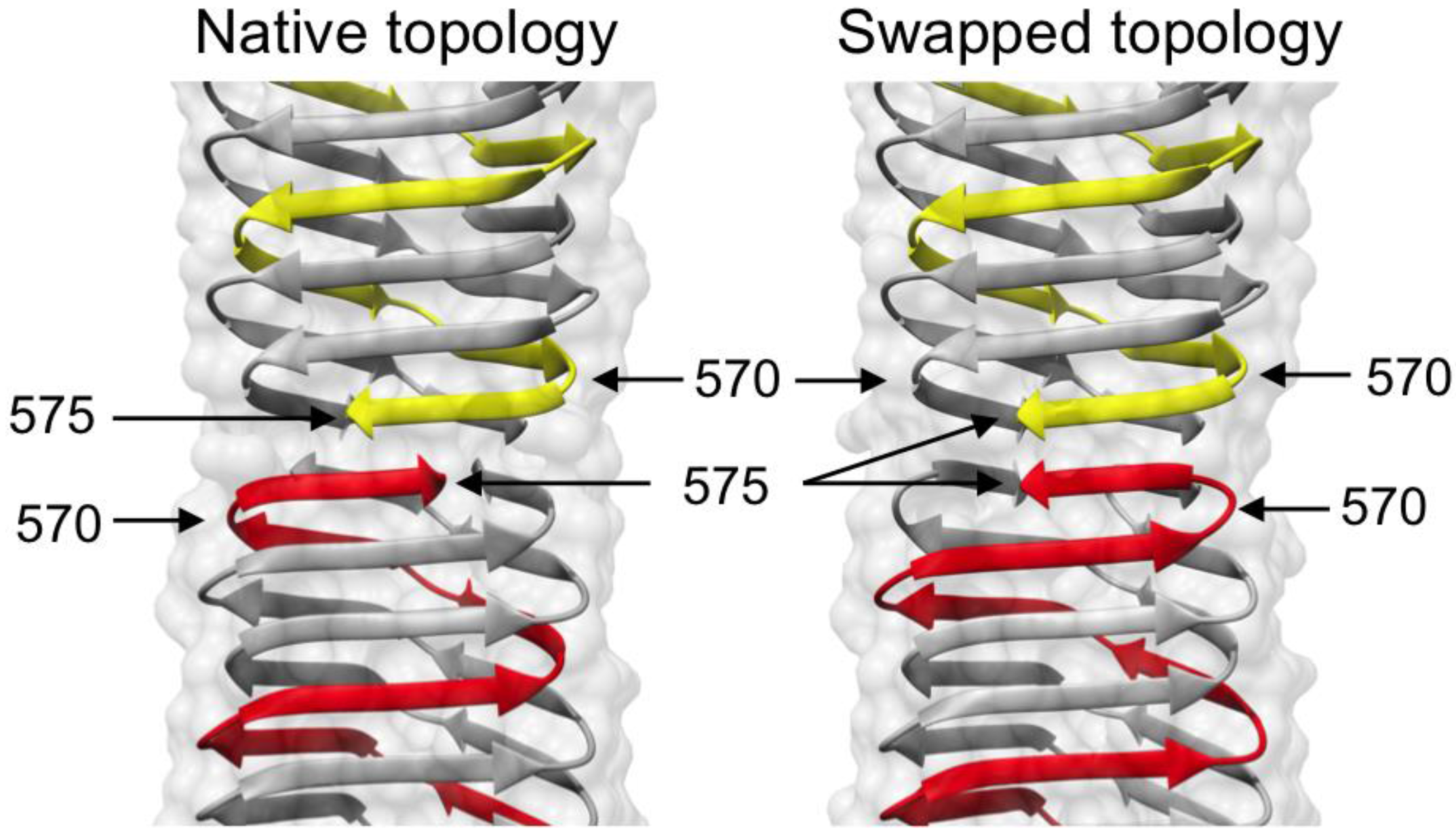
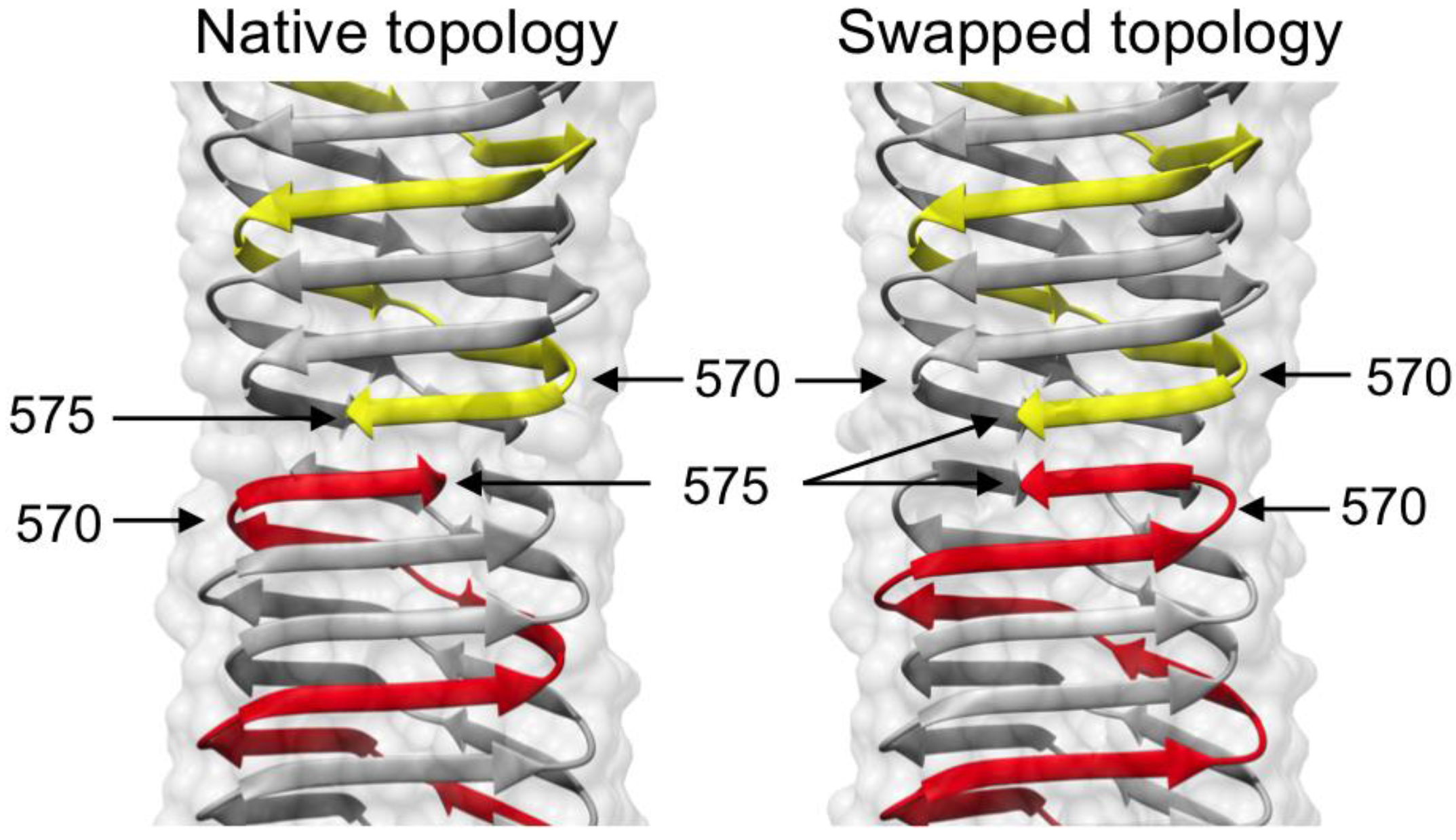
3.4. Crystal Structure of gp5β-BC and gp5β-BC2


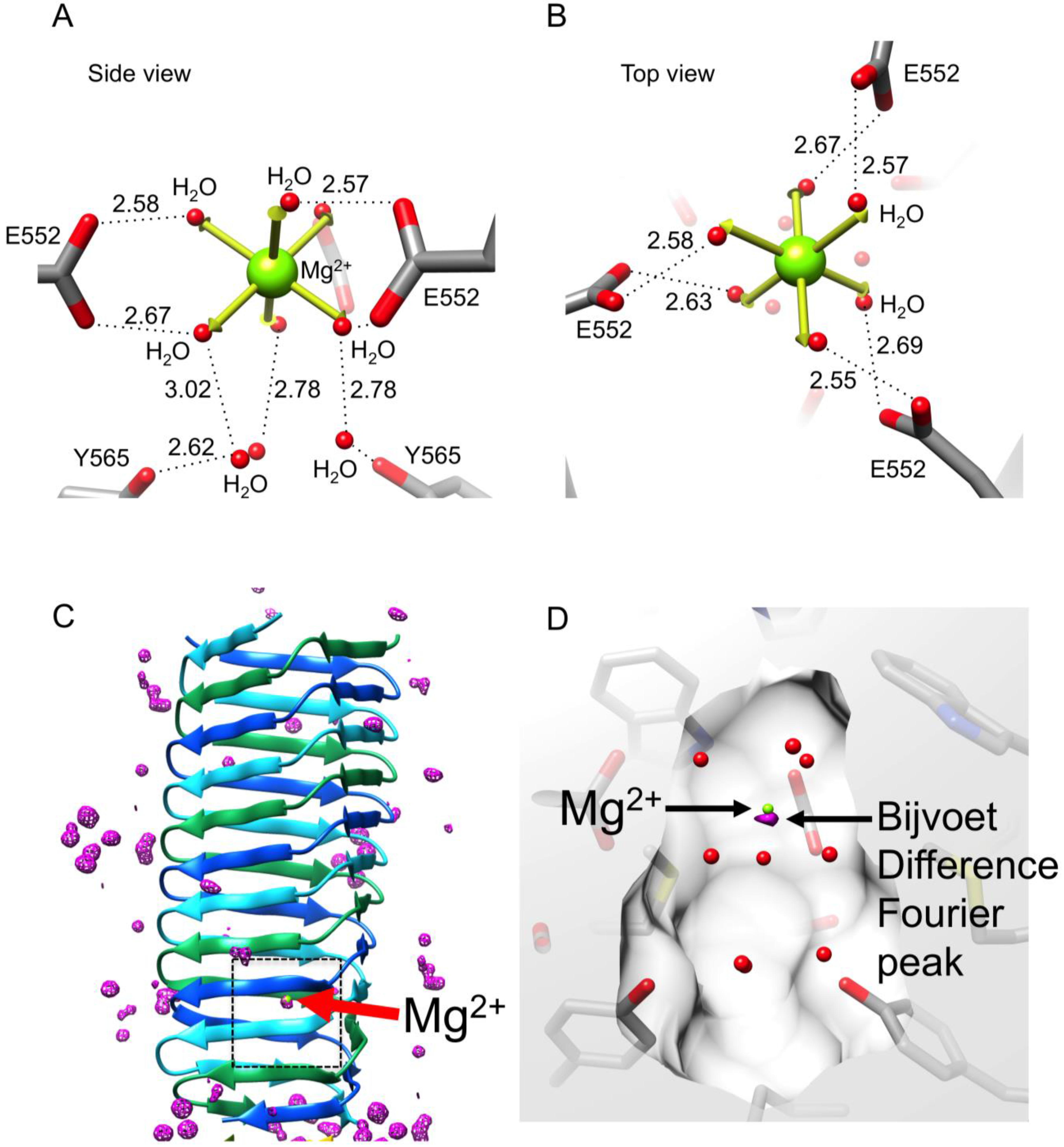
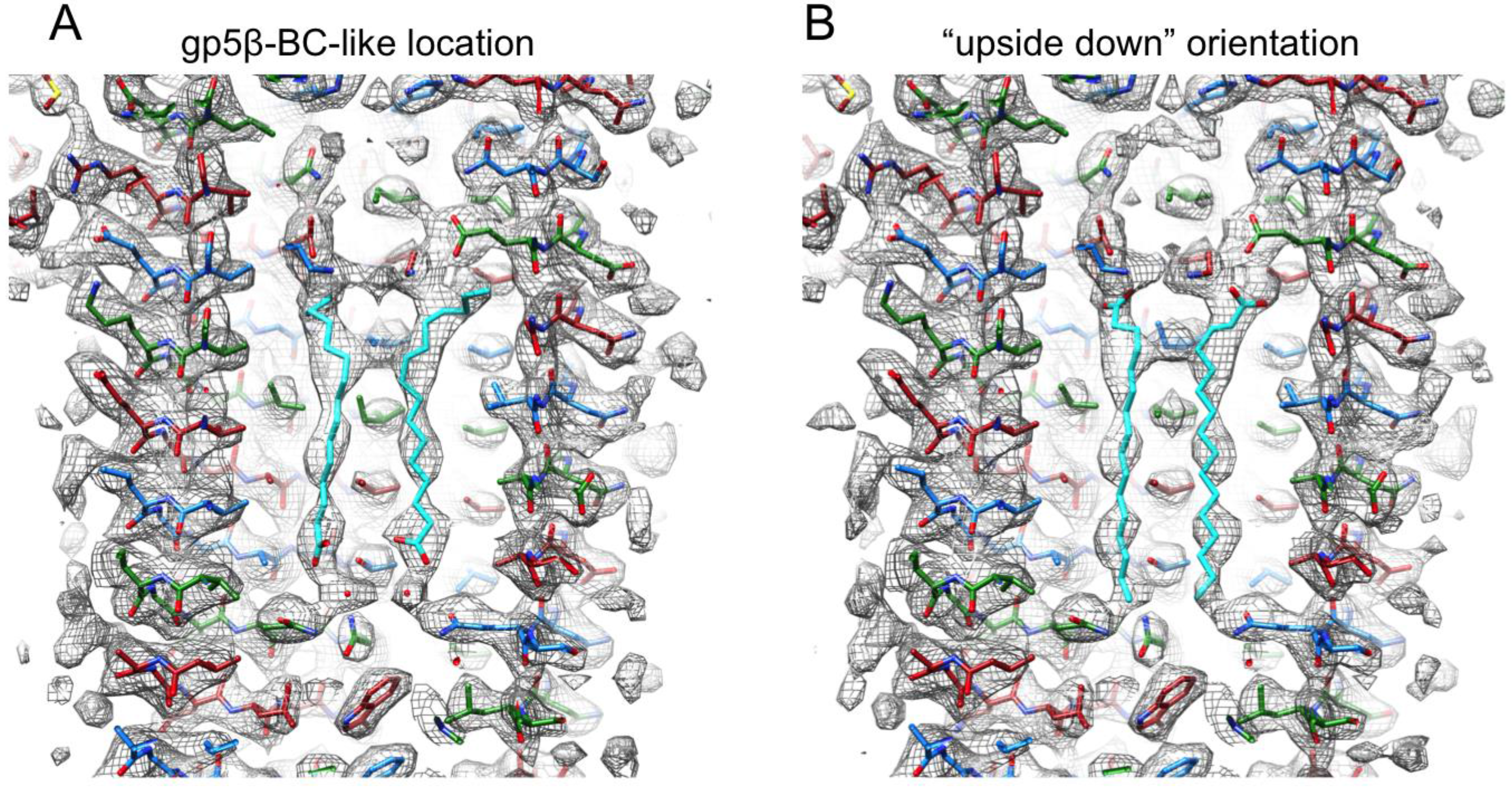
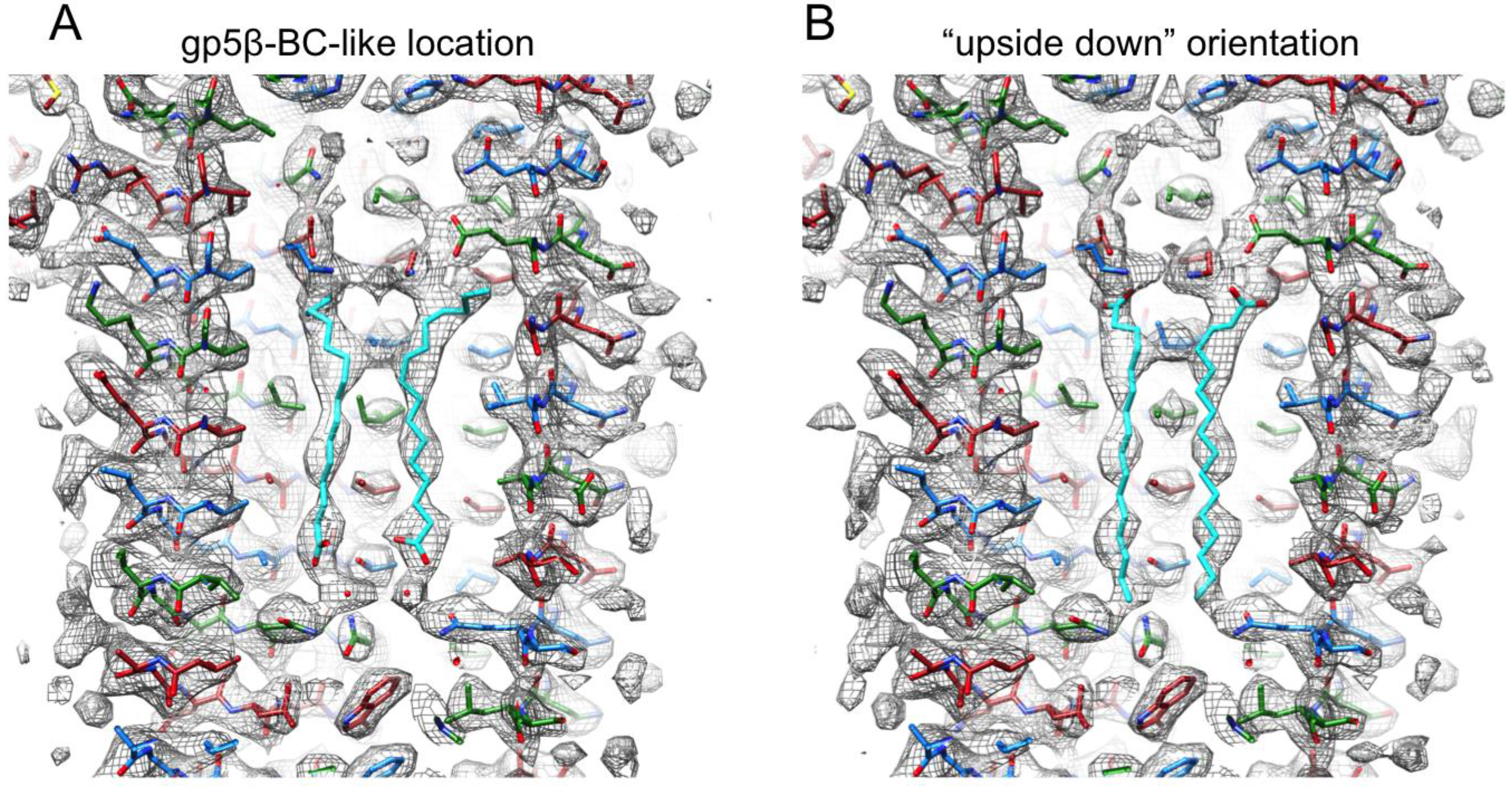
3.5. Identification of the Buried Metal Ion

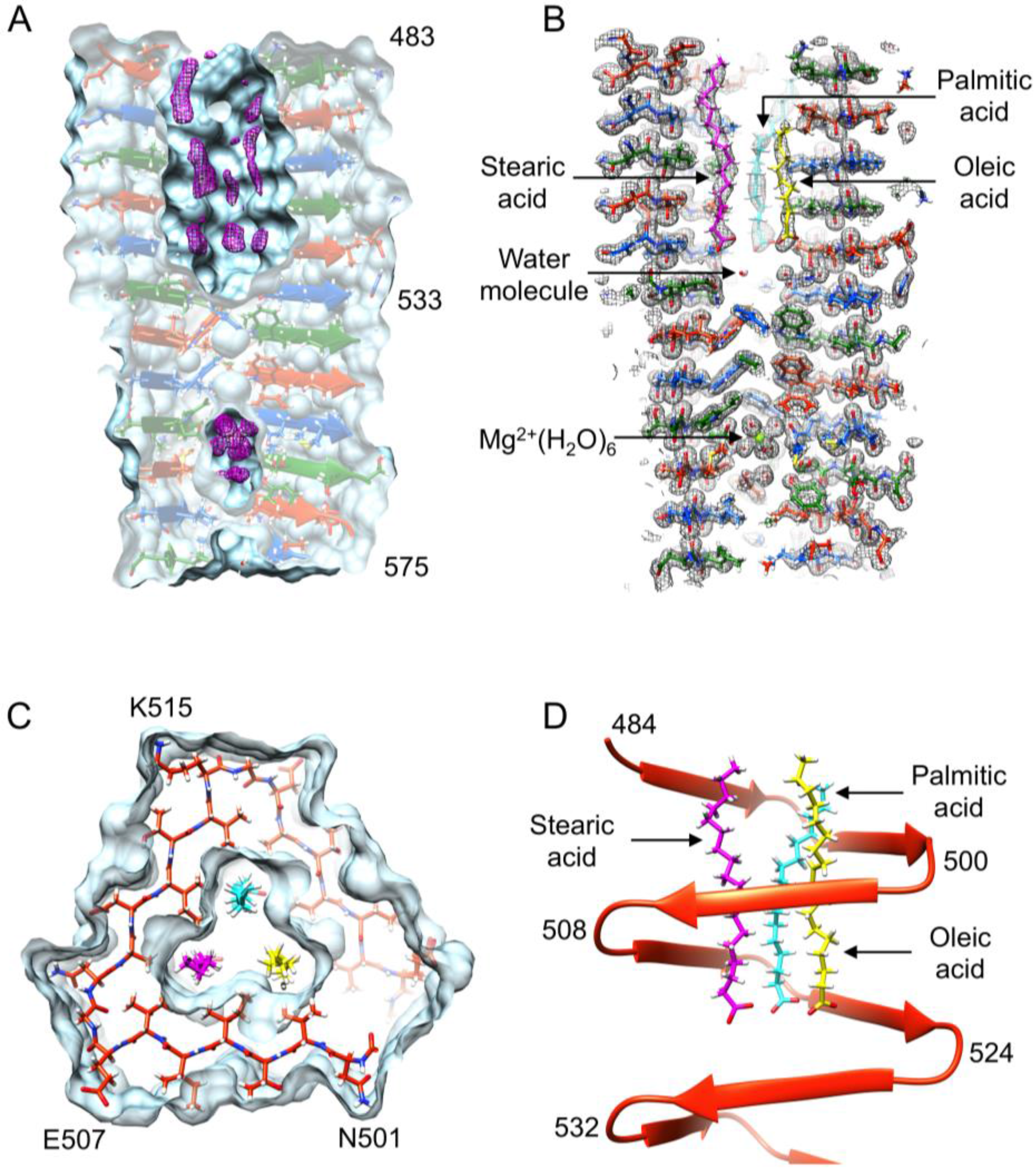
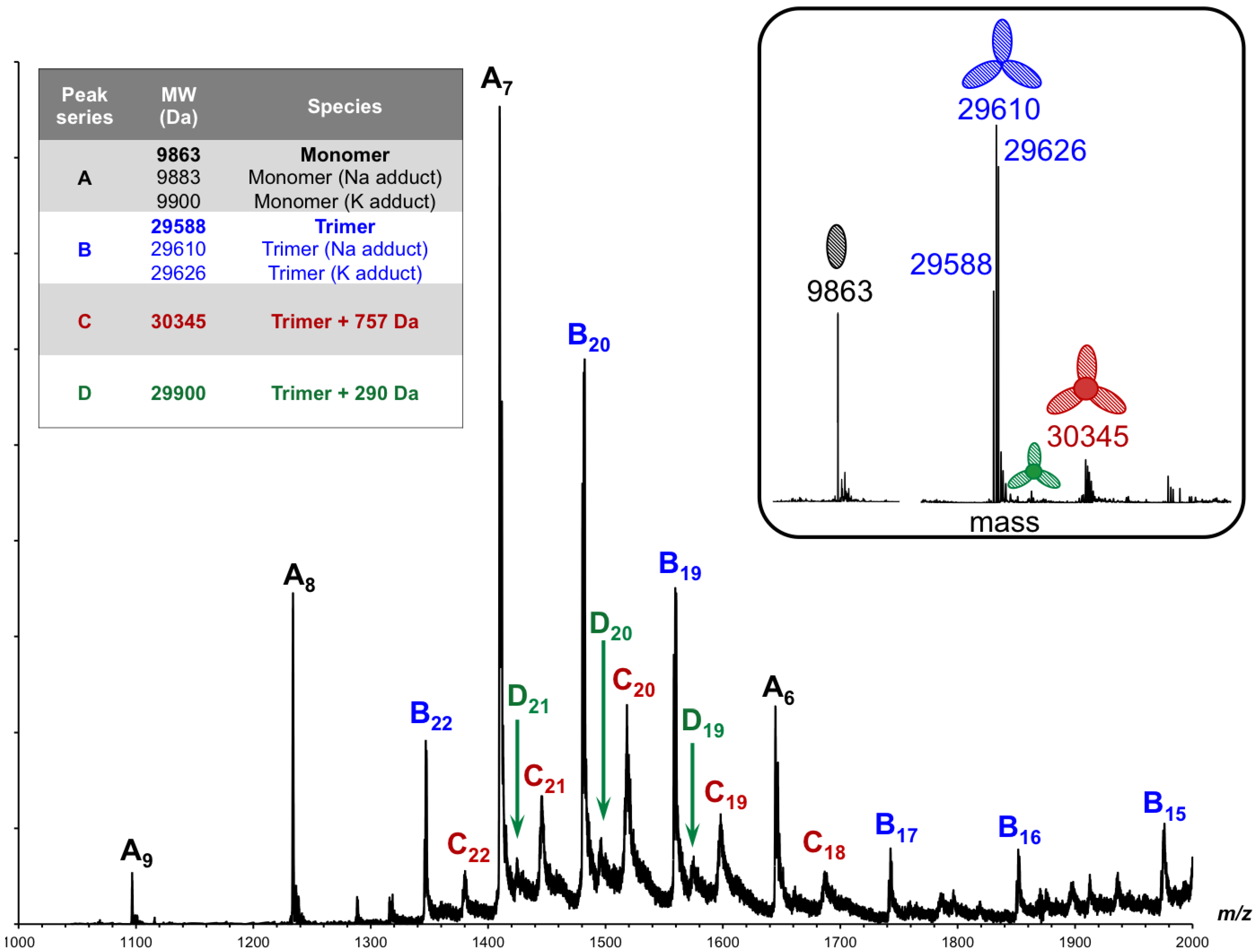
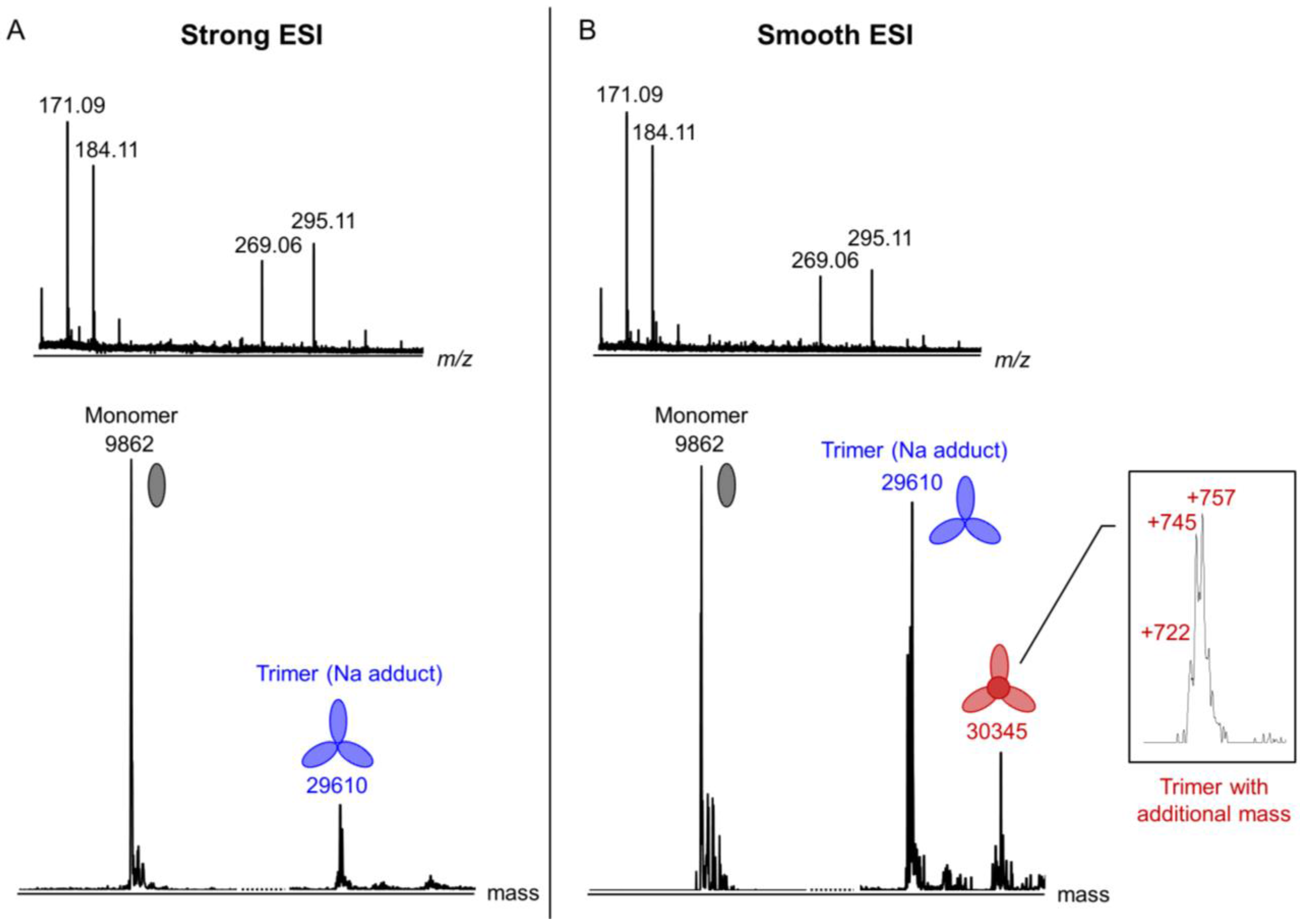
3.6. Identification of the Internal Compounds


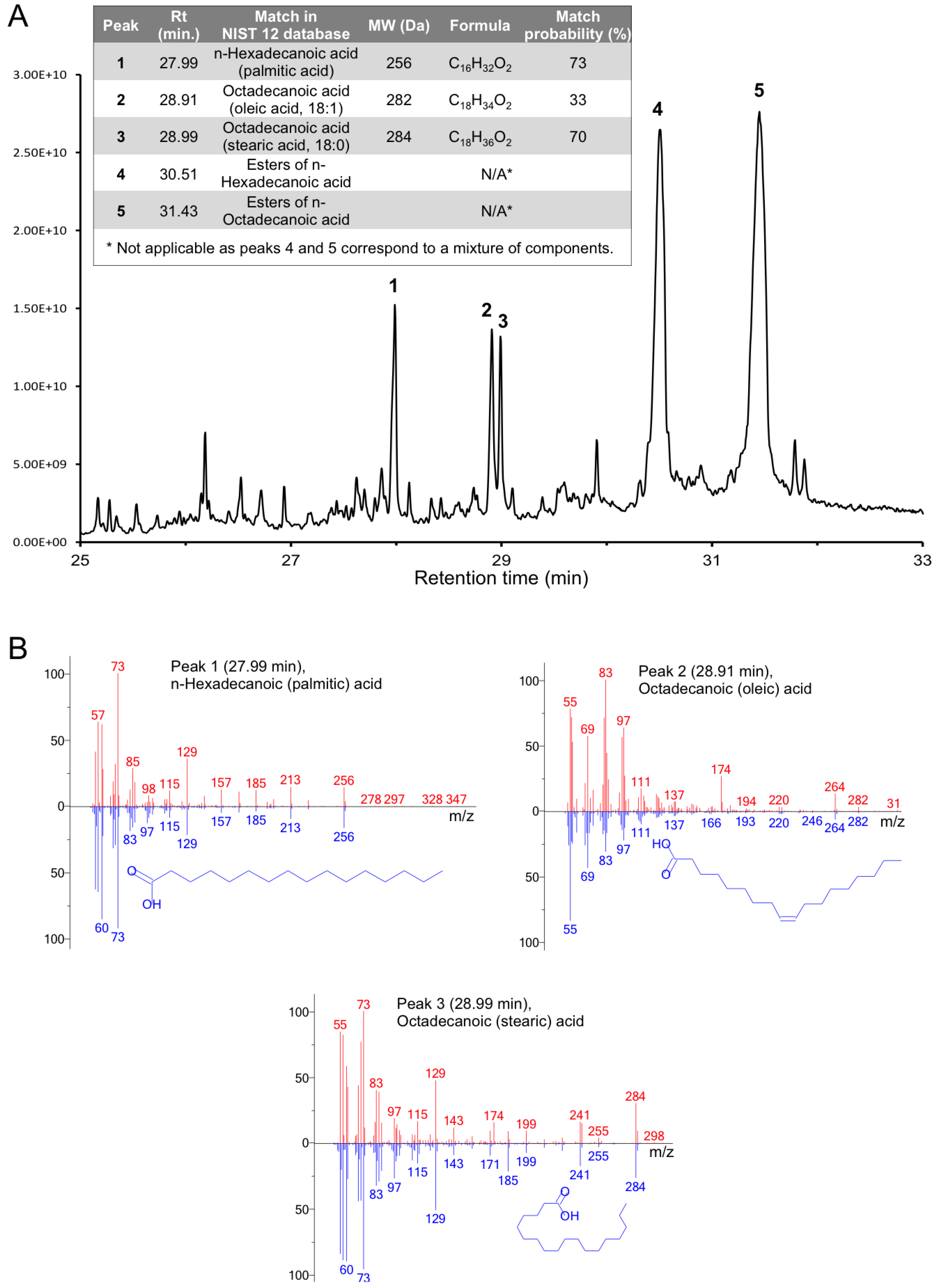
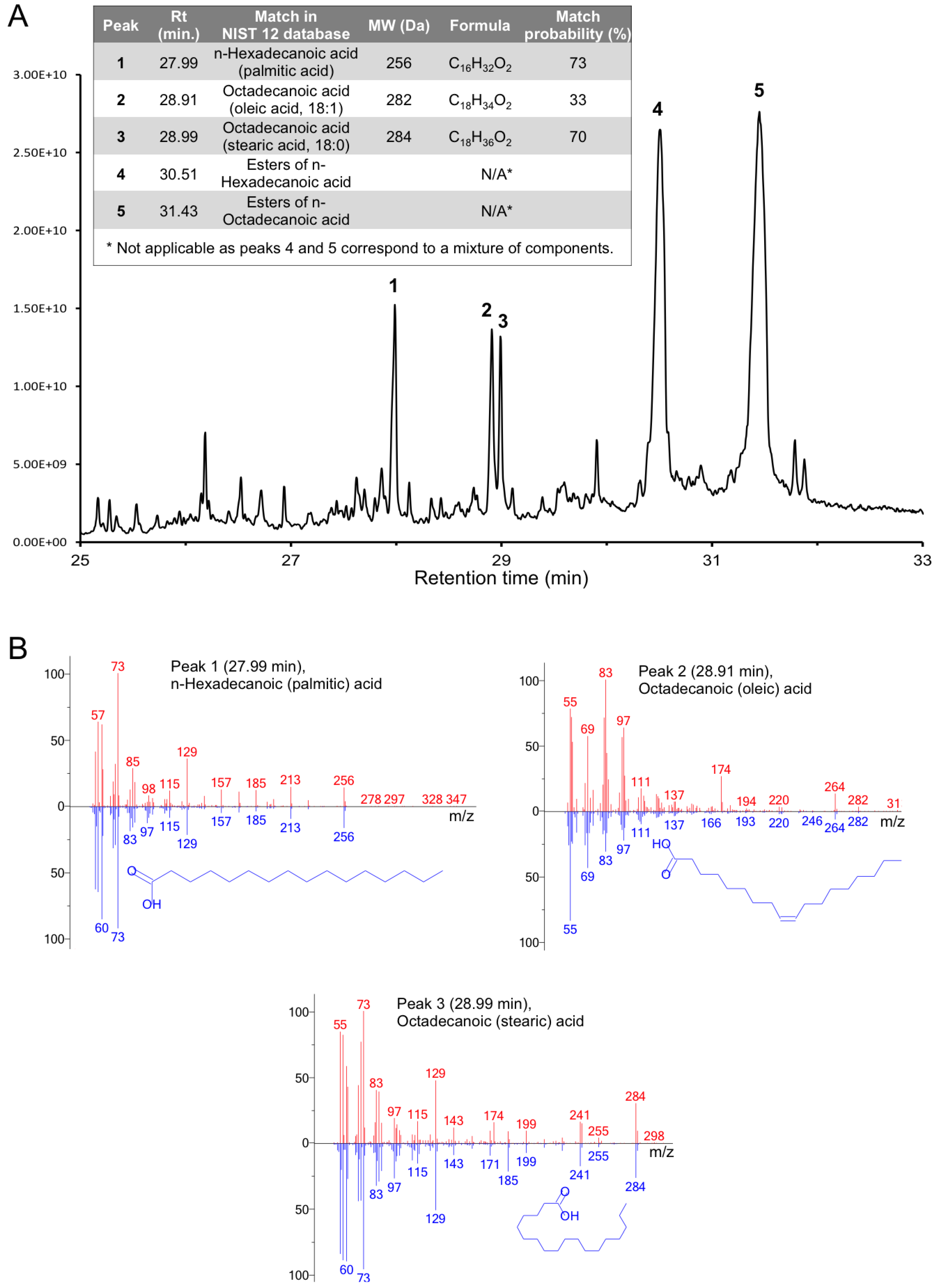
3.7. Full-Length Gp5 Contains Fatty Acids
4. Discussion
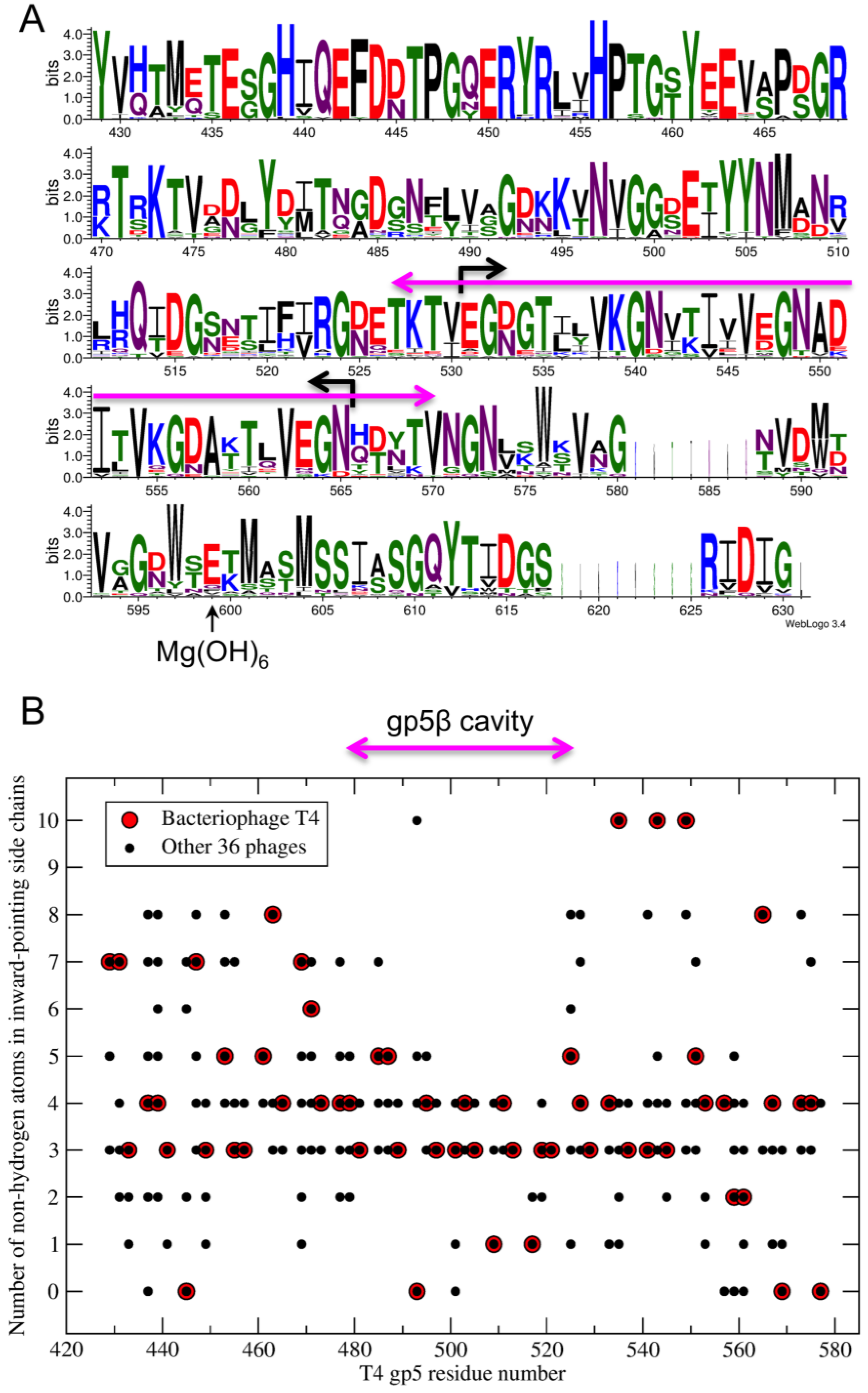

5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Leiman, P.G.; Arisaka, F.; van Raaij, M.J.; Kostyuchenko, V.A.; Aksyuk, A.A.; Kanamaru, S.; Rossmann, M.G. Morphogenesis of the t4 tail and tail fibers. Virol. J. 2010, 7, e355. [Google Scholar] [CrossRef] [PubMed]
- Leiman, P.G.; Shneider, M.M. Contractile tail machines of bacteriophages. Adv. Exp. Med. Biol. 2012, 726, 93–114. [Google Scholar] [PubMed]
- Kikuchi, Y.; King, J. Genetic control of bacteriophage T4 baseplate morphogenesis. II. Mutants unable to form the central part of the baseplate. J. Mol. Biol. 1975, 99, 673–694. [Google Scholar] [CrossRef]
- Kikuchi, Y.; King, J. Genetic control of bacteriophage T4 baseplate morphogenesis. III. Formation of the central plug and overall assembly pathway. J. Mol. Biol. 1975, 99, 695–716. [Google Scholar] [CrossRef]
- Kanamaru, S.; Gassner, N.C.; Ye, N.; Takeda, S.; Arisaka, F. The C-terminal fragment of the precursor tail lysozyme of bacteriophage T4 stays as a structural component of the baseplate after cleavage. J. Bacteriol. 1999, 181, 2739–2744. [Google Scholar] [PubMed]
- Leiman, P.G.; Kanamaru, S.; Mesyanzhinov, V.V.; Arisaka, F.; Rossmann, M.G. Structure and morphogenesis of bacteriophage T4. Cell. Mol. Life Sci. 2003, 60, 2356–2370. [Google Scholar] [CrossRef] [PubMed]
- Kanamaru, S.; Leiman, P.G.; Kostyuchenko, V.A.; Chipman, P.R.; Mesyanzhinov, V.V.; Arisaka, F.; Rossmann, M.G. Structure of the cell-puncturing device of bacteriophage T4. Nature 2002, 415, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Mishra, P.; Prem Kumar, R.; Ethayathulla, A.S.; Singh, N.; Sharma, S.; Perbandt, M.; Betzel, C.; Kaur, P.; Srinivasan, A.; Bhakuni, V.; et al. Polysaccharide binding sites in hyaluronate lyase—Crystal structures of native phage-encoded hyaluronate lyase and its complexes with ascorbic acid and lactose. FEBS J. 2009, 276, 3392–3402. [Google Scholar] [CrossRef] [PubMed]
- Mitraki, A.; Papanikolopoulou, K.; van Raaij, M.J. Natural triple beta-stranded fibrous folds. Adv. Protein Chem. 2006, 73, 97–124. [Google Scholar] [PubMed]
- Bartual, S.G.; Otero, J.M.; Garcia-Doval, C.; Llamas-Saiz, A.L.; Kahn, R.; Fox, G.C.; van Raaij, M.J. Structure of the bacteriophage T4 long tail fiber receptor-binding tip. Proc. Natl. Acad. Sci. USA 2010, 107, 20287–20292. [Google Scholar] [CrossRef] [PubMed]
- Thomassen, E.; Gielen, G.; Schutz, M.; Schoehn, G.; Abrahams, J.P.; Miller, S.; van Raaij, M.J. The structure of the receptor-binding domain of the bacteriophage T4 short tail fibre reveals a knitted trimeric metal-binding fold. J. Mol. Biol. 2003, 331, 361–373. [Google Scholar] [CrossRef]
- Van Raaij, M.J.; Schoehn, G.; Burda, M.R.; Miller, S. Crystal structure of a heat and protease-stable part of the bacteriophage T4 short tail fibre. J. Mol. Biol. 2001, 314, 1137–1146. [Google Scholar] [CrossRef] [PubMed]
- Walter, M.; Fiedler, C.; Grassl, R.; Biebl, M.; Rachel, R.; Hermo-Parrado, X.L.; Llamas-Saiz, A.L.; Seckler, R.; Miller, S.; van Raaij, M.J. Structure of the receptor-binding protein of bacteriophage det7: A podoviral tail spike in a myovirus. J. Virol. 2008, 82, 2265–2273. [Google Scholar] [CrossRef] [PubMed]
- Steinbacher, S.; Seckler, R.; Miller, S.; Steipe, B.; Huber, R.; Reinemer, P. Crystal structure of p22 tailspike protein: Interdigitated subunits in a thermostable trimer. Science 1994, 265, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, S.; Campanacci, V.; Blangy, S.; Moineau, S.; Tegoni, M.; Cambillau, C. Modular structure of the receptor binding proteins of lactococcus lactis phages. The rbp structure of the temperate phage tp901-1. J. Biol. Chem. 2006, 281, 14256–14262. [Google Scholar] [CrossRef] [PubMed]
- Spinelli, S.; Desmyter, A.; Verrips, C.T.; de Haard, H.J.; Moineau, S.; Cambillau, C. Lactococcal bacteriophage p2 receptor-binding protein structure suggests a common ancestor gene with bacterial and mammalian viruses. Nat. Struct. Mol. Biol. 2006, 13, 85–89. [Google Scholar] [CrossRef] [PubMed]
- Ricagno, S.; Campanacci, V.; Blangy, S.; Spinelli, S.; Tremblay, D.; Moineau, S.; Tegoni, M.; Cambillau, C. Crystal structure of the receptor-binding protein head domain from Lactococcus lactis phage bil170. J. Virol. 2006, 80, 9331–9335. [Google Scholar] [CrossRef] [PubMed]
- Kajava, A.V.; Steven, A.C. Beta-rolls, beta-helices, and other beta-solenoid proteins. Adv. Protein Chem. 2006, 73, 55–96. [Google Scholar] [PubMed]
- Smith, N.L.; Taylor, E.J.; Lindsay, A.M.; Charnock, S.J.; Turkenburg, J.P.; Dodson, E.J.; Davies, G.J.; Black, G.W. Structure of a group a streptococcal phage-encoded virulence factor reveals a catalytically active triple-stranded beta-helix. Proc. Natl. Acad. Sci. USA 2005, 102, 17652–17657. [Google Scholar] [CrossRef] [PubMed]
- Stummeyer, K.; Dickmanns, A.; Muhlenhoff, M.; Gerardy-Schahn, R.; Ficner, R. Crystal structure of the polysialic acid-degrading endosialidase of bacteriophage k1f. Nat. Struct. Mol. Biol. 2005, 12, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Mosig, G.; Lin, G.W.; Franklin, J.; Fan, W.H. Functional relationships and structural determinants of two bacteriophage T4 lysozymes: A soluble (gene e) and a baseplate-associated (gene 5) protein. New Biol. 1989, 1, 171–179. [Google Scholar] [PubMed]
- Shneider, M.M.; Buth, S.A.; Ho, B.T.; Basler, M.; Mekalanos, J.J.; Leiman, P.G. Paar-repeat proteins sharpen and diversify the type vi secretion system spike. Nature 2013, 500, 350–353. [Google Scholar] [CrossRef] [PubMed]
- Sugimoto, K.; Kanamaru, S.; Iwasaki, K.; Arisaka, F.; Yamashita, I. Construction of a ball-and-spike protein supramolecule. Angew. Chem. Int. Ed. Engl. 2006, 45, 2725–2728. [Google Scholar] [CrossRef] [PubMed]
- Ueno, T.; Koshiyama, T.; Tsuruga, T.; Goto, T.; Kanamaru, S.; Arisaka, F.; Watanabe, Y. Bionanotube tetrapod assembly by in situ synthesis of a gold nanocluster with (gp5-his6)3 from bacteriophage T4. Angew. Chem. Int. Ed. Engl. 2006, 45, 4508–4512. [Google Scholar] [CrossRef] [PubMed]
- Yokoi, N.; Inaba, H.; Terauchi, M.; Stieg, A.Z.; Sanghamitra, N.J.; Koshiyama, T.; Yutani, K.; Kanamaru, S.; Arisaka, F.; Hikage, T.; et al. Construction of robust bio-nanotubes using the controlled self-assembly of component proteins of bacteriophage T4. Small 2010, 6, 1873–1879. [Google Scholar] [CrossRef]
- Yokoi, N.; Miura, Y.; Huang, C.Y.; Takatani, N.; Inaba, H.; Koshiyama, T.; Kanamaru, S.; Arisaka, F.; Watanabe, Y.; Kitagawa, S.; et al. Dual modification of a triple-stranded beta-helix nanotube with ru and re metal complexes to promote photocatalytic reduction of co2. Chem. Commun. 2011, 47, 2074–2076. [Google Scholar] [CrossRef]
- Kammerer, R.A.; Schulthess, T.; Landwehr, R.; Lustig, A.; Fischer, D.; Engel, J. Tenascin-c hexabrachion assembly is a sequential two-step process initiated by coiled-coil alpha-helices. J. Biol. Chem. 1998, 273, 10602–10608. [Google Scholar] [CrossRef] [PubMed]
- Van Holde, K.E. Physical Biochemistry, 2nd ed.; Prentice Hall: Englewood Cliff, NJ, USA, 1985; pp. 93–136. [Google Scholar]
- Otwinowski, Z.; Minor, W. Processing of X-ray diffraction data collected in oscillation mode. Meth. Enzymol. 1997, 276, 307–326. [Google Scholar]
- Vagin, A.; Teplyakov, A. Molrep: An automated program for molecular replacement. J. Appl. Crystallogr. 1997, 30, 1022–1025. [Google Scholar] [CrossRef]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the ccp4 suite and current developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Sheldrick, G.M. A short history of shelx. Acta Crystallogr. Sect. A Found. Crystallogr. 2008, 64, 112–122. [Google Scholar] [CrossRef] [PubMed]
- Emsley, P.; Lohkamp, B.; Scott, W.G.; Cowtan, K. Features and development of coot. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 486–501. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. Integration, scaling, space-group assignment and post-refinement. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 133–144. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, W. Xds. Acta Crystallogr. Sect. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [PubMed]
- McCoy, A.J.; Grosse-Kunstleve, R.W.; Adams, P.D.; Winn, M.D.; Storoni, L.C.; Read, R.J. Phaser crystallographic software. J. Appl. Crystallogr. 2007, 40, 658–674. [Google Scholar] [CrossRef] [PubMed]
- Murshudov, G.N.; Skubak, P.; Lebedev, A.A.; Pannu, N.S.; Steiner, R.A.; Nicholls, R.A.; Winn, M.D.; Long, F.; Vagin, A.A. Refmac5 for the refinement of macromolecular crystal structures. Acta Crystallogr. Sect. D Biol. Crystallogr. 2011, 67, 355–367. [Google Scholar] [CrossRef] [PubMed]
- Folch, J.; Lees, M.; Sloane Stanley, G.H. A simple method for the isolation and purification of total lipides from animal tissues. J. Biol. Chem. 1957, 226, 497–509. [Google Scholar] [PubMed]
- Sreerama, N.; Woody, R.W. Circular dichroism of peptides and proteins. In Circular Dichroism: Principles and Applications, 2nd ed.; Berova, N., Nakanishi, K., Woody, R.W., Eds.; John Wiley & Sons, Inc.: New York, NY, USA, 2000. [Google Scholar]
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 2007, 372, 774–797. [Google Scholar] [CrossRef] [PubMed]
- Adams, P.D.; Afonine, P.V.; Bunkoczi, G.; Chen, V.B.; Echols, N.; Headd, J.J.; Hung, L.W.; Jain, S.; Kapral, G.J.; Grosse Kunstleve, R.W.; et al. The phenix software for automated determination of macromolecular structures. Methods 2011, 55, 94–106. [Google Scholar] [CrossRef] [PubMed]
- Hsin, K.; Sheng, Y.; Harding, M.M.; Taylor, P.; Walkinshaw, M.D. Mespeus: A database of the geometry of metal sites in proteins. J. Appl. Crystallogr. 2008, 41, 963–968. [Google Scholar] [CrossRef]
- Potier, N.; Billas, I.M.L.; Steinmetz, A.; Schaeffer, C.; van Dorsselaer, A.; Moras, D.; Renaud, J.P. Using nondenaturing mass spectrometry to detect fortuitous ligands in orphan nuclear receptors. Protein Sci. 2003, 12, 725–733. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Su, B.; Wang, X. Study on the noncovalent interactions of saikosaponins and cytochrome C by electrospray ionization mass spectrometry. Rapid Commun. Mass Spectrom. 2012, 26, 719–727. [Google Scholar] [CrossRef] [PubMed]
- Last, A.M.; Robinson, C.V. Protein folding and interactions revealed by mass spectrometry. Curr. Opin. Chem. Biol. 1999, 3, 564–570. [Google Scholar] [CrossRef]
- Tito, M.A.; Miller, J.; Walker, N.; Griffin, K.F.; Williamson, E.D.; Despeyroux-Hill, D.; Titball, R.W.; Robinson, C.V. Probing molecular interactions in intact antibody: Antigen complexes, an electrospray time-of-flight mass spectrometry approach. Biophys. J. 2001, 81, 3503–3509. [Google Scholar] [CrossRef]
- Kitova, E.N.; El-Hawiet, A.; Schnier, P.D.; Klassen, J.S. Reliable determinations of protein-ligand interactions by direct esi-ms measurements. Are we there yet? J. Am. Soc. Mass Spectrom. 2012, 23, 431–441. [Google Scholar] [CrossRef] [PubMed]
- Stein, S.E. (Director) Mass Spectra by NIST Mass Spec Data Center. In NIST Chemistry WebBook; NIST Standard Reference Database Number 69; Linstrom, P.J.; Mallard, W.G. (Eds.) National Institute of Standards and Technology: Gaithersburg, MD, USA. Available online: http://webbook.nist.gov (accessed on 12 February 2014).
- Letarov, A.V.; Londer, Y.Y.; Boudko, S.P.; Mesyanzhinov, V.V. The carboxy-terminal domain initiates trimerization of bacteriophage T4 fibritin. Biochemistry 1999, 64, 817–823. [Google Scholar] [PubMed]
- Bartual, S.G.; Otero, J.M.; Garcia-Doval, C.; Llamas-Saiz, A.L.; Kahn, R.; Fox, G.C.; van Raaij, M.J. Structure of the bacteriophage T4 long tail fiber receptor-binding tip. Proc. Natl. Acad. Sci. USA 2010, 107, 20287–20292. [Google Scholar] [CrossRef] [PubMed]
- Van Raaij, M.J.; Mitraki, A.; Lavigne, G.; Cusack, S. A triple beta-spiral in the adenovirus fibre shaft reveals a new structural motif for a fibrous protein. Nature 1999, 401, 935–938. [Google Scholar] [PubMed]
- Boudko, S.P.; Engel, J.; Bachinger, H.P. The crucial role of trimerization domains in collagen folding. Int. J. Biochem. Cell Biol. 2012, 44, 21–32. [Google Scholar] [CrossRef] [PubMed]
- Browning, C.; Shneider, M.M.; Bowman, V.D.; Schwarzer, D.; Leiman, P.G. Phage pierces the host cell membrane with the iron-loaded spike. Structure 2012, 20, 326–339. [Google Scholar] [CrossRef] [PubMed]
- Squeglia, F.; Bachert, B.; De Simone, A.; Lukomski, S.; Berisio, R. The crystal structure of the streptococcal collagen-like protein 2 globular domain from invasive m3-type group a streptococcus shows significant similarity to immunomodulatory hiv protein gp41. J. Biol. Chem. 2014, 289, 5122–5133. [Google Scholar] [CrossRef] [PubMed]
- Williams, S.R.; Gebhart, D.; Martin, D.W.; Scholl, D. Retargeting r-type pyocins to generate novel bactericidal protein complexes. Appl. Environ. Microbiol. 2008, 74, 3868–3876. [Google Scholar] [CrossRef] [PubMed]
- Schwarzer, D.; Stummeyer, K.; Haselhorst, T.; Freiberger, F.; Rode, B.; Grove, M.; Scheper, T.; von Itzstein, M.; Muhlenhoff, M.; Gerardy-Schahn, R. Proteolytic release of the intramolecular chaperone domain confers processivity to endosialidase f. J. Biol. Chem. 2009, 284, 9465–9474. [Google Scholar] [CrossRef] [PubMed]
- Steinmetz, M.O.; Jelesarov, I.; Matousek, W.M.; Honnappa, S.; Jahnke, W.; Missimer, J.H.; Frank, S.; Alexandrescu, A.T.; Kammerer, R.A. Molecular basis of coiled-coil formation. Proc. Natl. Acad. Sci. USA 2007, 104, 7062–7067. [Google Scholar] [CrossRef] [PubMed]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. Weblogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef] [PubMed]
- Chenna, R.; Sugawara, H.; Koike, T.; Lopez, R.; Gibson, T.J.; Higgins, D.G.; Thompson, J.D. Multiple sequence alignment with the clustal series of programs. Nucleic Acids Res. 2003, 31, 3497–3500. [Google Scholar] [CrossRef] [PubMed]
- Traub, W.; Piez, K.A. The chemistry and structure of collagen. Adv. Protein Chem. 1971, 25, 243–352. [Google Scholar] [PubMed]
- Bella, J.; Eaton, M.; Brodsky, B.; Berman, H.M. Crystal and molecular structure of a collagen-like peptide at 1.9 a resolution. Science 1994, 266, 75–81. [Google Scholar] [CrossRef] [PubMed]
- Lupas, A. Coiled coils: New structures and new functions. Trends Biochem. Sci. 1996, 21, 375–382. [Google Scholar] [CrossRef]
- Inaba, H.; Kanamaru, S.; Arisaka, F.; Kitagawa, S.; Ueno, T. Semi-synthesis of an artificial scandium(iii) enzyme with a beta-helical bio-nanotube. Dalton Trans. 2012, 41, 11424–11427. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buth, S.A.; Menin, L.; Shneider, M.M.; Engel, J.; Boudko, S.P.; Leiman, P.G. Structure and Biophysical Properties of a Triple-Stranded Beta-Helix Comprising the Central Spike of Bacteriophage T4. Viruses 2015, 7, 4676-4706. https://doi.org/10.3390/v7082839
Buth SA, Menin L, Shneider MM, Engel J, Boudko SP, Leiman PG. Structure and Biophysical Properties of a Triple-Stranded Beta-Helix Comprising the Central Spike of Bacteriophage T4. Viruses. 2015; 7(8):4676-4706. https://doi.org/10.3390/v7082839
Chicago/Turabian StyleButh, Sergey A., Laure Menin, Mikhail M. Shneider, Jürgen Engel, Sergei P. Boudko, and Petr G. Leiman. 2015. "Structure and Biophysical Properties of a Triple-Stranded Beta-Helix Comprising the Central Spike of Bacteriophage T4" Viruses 7, no. 8: 4676-4706. https://doi.org/10.3390/v7082839