
Modeling Viral Infectious Diseases and Development of Antiviral Therapies Using Human Induced Pluripotent Stem Cell-Derived Systems



Abstract
:1. Introduction
2. Induced Pluripotent Stem Cell-Derived Models of Diseases
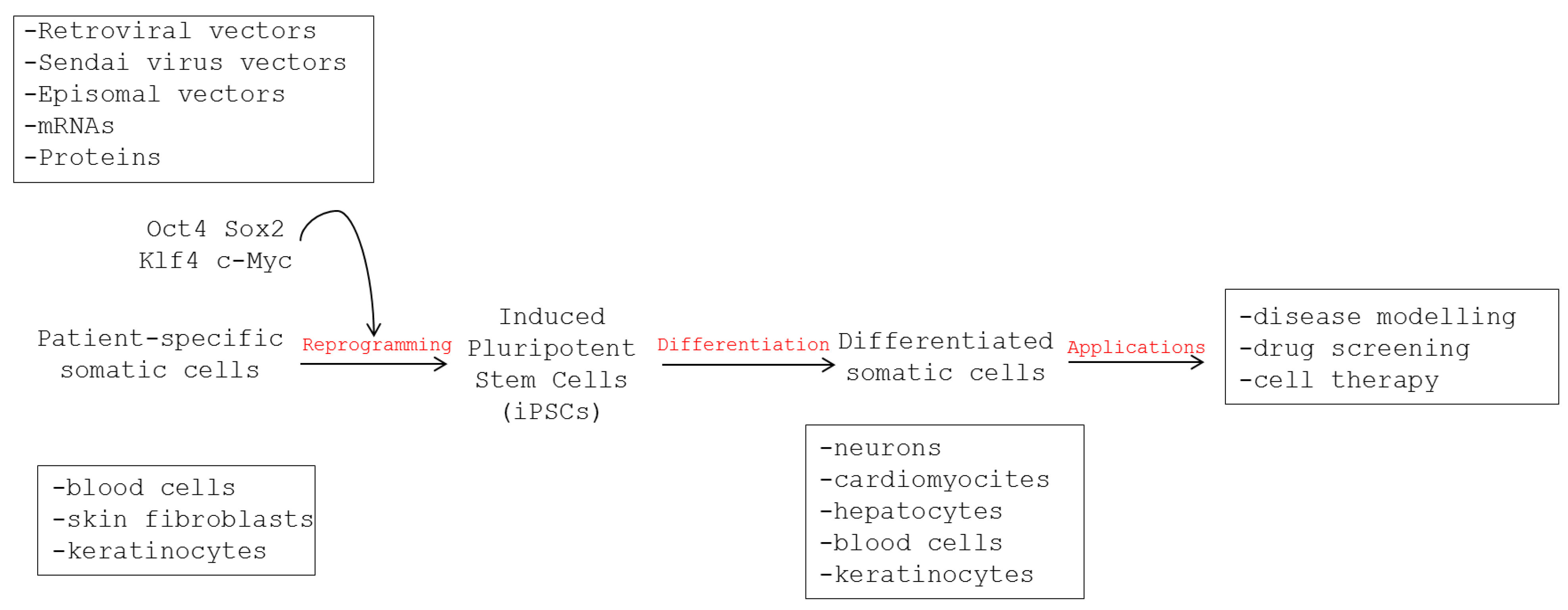
2.1. Generation of iPSCs
2.2. Features of iPSCs
2.3. Disease Modeling Using Human iPSCs
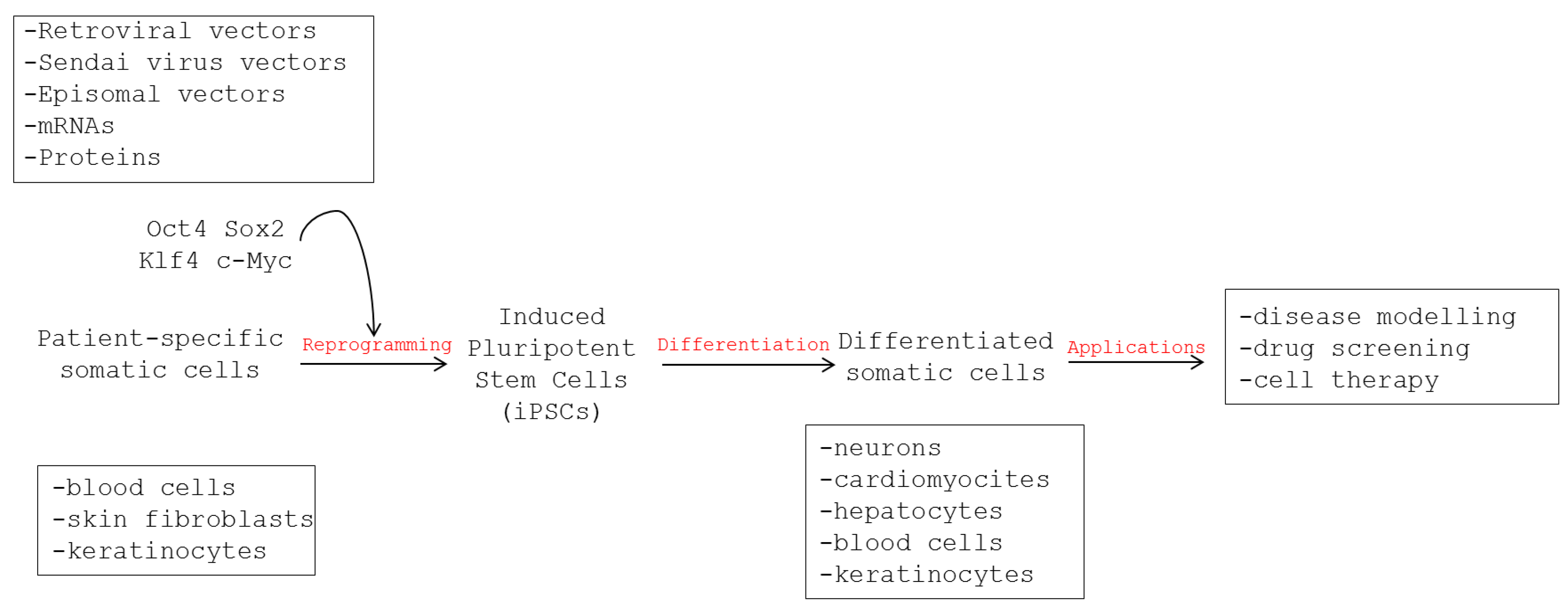
3. Use of Human iPSCs to Model Viral Infections
3.1. Human Cytomegalovirus Infection
3.2. Herpes Simplex Virus and Varicella Zoster Virus Infection of Neural Cells
3.3. Hepatitis B Virus Infection
3.4. Hepatitis C Virus Infection
3.5. Enterovirus Myocarditis

{kind=link}
| Disease Model | Virus | iPSC-Derived Cells | Findings | Refs |
|---|---|---|---|---|
| Encephalitis | HCMV | Neural stem cells, neural progenitor cells, neurons | Neural progenitor cells, but not neurons, are permissive for lytic HCMV replication | [56] |
| Encephalitis | HCMV | Neural stem cells, neural progenitor cells, neurons | Neural stem cells allow persistent HCMV infection; neurons are permissive for lytic replication | [55] |
| Encephalitis | HSV, VZV | Neural progenitor cells, sensory neurons | Neural progenitor cells and sensory neurons are permissive to productive HSV and VZV infection | [57] |
| Hepatitis | HBV | Hepatic progenitor cells, differentiated hepatocytes | Fully differentiated hepatocyte-like cells support productive HBV infection | [62] |
| Hepatitis | HCV | Hepatic progenitor cells, differentiated hepatocytes | Hepatic progenitor cells and differentiated hepatocytes are permissive for HCV infection | [77,78] |
| Hepatitis | HCV | Hepatic progenitor cells, differentiated hepatocytes | Liver-like cells can be engrafted in the liver of transgenic mice and persistently infected by HCV | [79,80,81] |
| Myocarditis | Coxsackievirus | Cardiomyocytes | Cardiomyocytes are susceptible to coxsackievirus infection | [85] |
4. Modeling Human Susceptibility to Viral Infectious Diseases Using iPSC-derived Systems
4.1. Herpes Simplex Virus Encephalitis
4.2. Severe Influenza
| Disease | Genetic Defect | Disease Traits in Patients | Phenotype in Human iPSC-Derived Cells | Rescue and Drug Testing | Refs |
|---|---|---|---|---|---|
| HSV encephalitis | Inactivating mutations of TLR3 and UNC93B1 | Predisposition to develop encephalitis during primary HSV-1 infection | Impaired IFN response to HSV infection and increased HSV replication in patient-specific neurons and oligodendrocytes | Gene addition; interferon | [91] |
| Severe influenza | Inactivating mutations of IRF7 | Development of acute respiratory distress syndrome during influenza virus infection | Impaired IFN response and increased influenza virus replication in patient-specific pulmonary epithelial cells | Interferon | [92] |
6. Conclusions
| Virus | Antiviral Strategy | iPSC-Derived Target Cells | Results | Refs |
|---|---|---|---|---|
| HIV | Disruption of integrated HIV genome | T cells, monocytes/ macrophages | HIV-targeted CRISPR/Cas9 disrupts reverse-transcribed and integrated HIV genome | [101] |
| HIV | Enhanced immune response | NK cells | Antiviral activity of iPSC-derived NKs; engineering a HIV chimeric CD4/CD3ζ receptor enhances NK activity against HIV | [102,103] |
| HIV | Viral receptor inactivation | Monocytes/ macrophages | Knockdown of CCR5 by shRNA; introduction of the CCR5Δ32 mutation by genome editing confers resistance to HIV-1 infection | [104,105,107] |
| HIV | Downregulation of viral cofactors by shRNAs | Monocytes/ macrophages | Inhibition of CDK2 and TRIM5α inhibits HIV-1 transcription | [105,108] |
| HCV | Downregulation of viral cofactors by siRNAs | Differentiated hepatocytes | Inhibition of cyclophilin A and PIaKIIIa inhibits HCV replication in hepatocytes | [109] |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Heymann, D.L.; Chen, L.; Takemi, K.; Fidler, D.P.; Tappero, J.W.; Thomas, M.J.; Kenyon, T.A.; Frieden, T.R.; Yach, D.; Nishtar, S.; et al. Global health security: The wider lessons from the west African Ebola virus disease epidemic. Lancet 2015, 385, 1884–1901. [Google Scholar] [CrossRef]
- Chapman, S.J.; Hill, A.V. Human susceptibility to infectious disease. Nat. Rev. Genet. 2012, 13, 175–188. [Google Scholar] [PubMed]
- Lee, M.N.; Ye, C.; Villani, A.C.; Raj, T.; Li, W.; Eisenhaure, T.M.; Imboywa, S.H.; Chipendo, P.I.; Ran, F.A.; Slowikowski, K.; et al. Common genetic variants modulate pathogen-sensing responses in human dendritic cells. Science 2014, 343, 1246980. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Yamanaka, S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bellin, M.; Marchetto, M.C.; Gage, F.H.; Mummery, C.L. Induced pluripotent stem cells: The new patient? Nat. Rev. Mol. Cell Biol. 2012, 13, 713–726. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Tanabe, K.; Ohnuki, M.; Narita, M.; Ichisaka, T.; Tomoda, K.; Yamanaka, S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 2007, 131, 861–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, J.; Vodyanik, M.A.; Smuga-Otto, K.; Antosiewicz-Bourget, J.; Frane, J.L.; Tian, S.; Nie, J.; Jonsdottir, G.A.; Ruotti, V.; Stewart, R.; et al. Induced pluripotent stem cell lines derived from human somatic cells. Science 2007, 318, 1917–1920. [Google Scholar] [CrossRef] [PubMed]
- Schlaeger, T.M.; Daheron, L.; Brickler, T.R.; Entwisle, S.; Chan, K.; Cianci, A.; DeVine, A.; Ettenger, A.; Fitzgerald, K.; Godfrey, M.; et al. A comparison of non-integrating reprogramming methods. Nat. Biotechnol. 2015, 33, 58–63. [Google Scholar] [CrossRef] [PubMed]
- Martin, M.J.; Muotri, A.; Gage, F.; Varki, A. Human embryonic stem cells express an immunogenic nonhuman sialic acid. Nat. Med. 2005, 11, 228–232. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Narita, M.; Yokura, M.; Ichisaka, T.; Yamanaka, S. Huma induced pluripotent stem cells on autologous feeders. PLoS ONE 2009, 4, e8067. [Google Scholar] [CrossRef] [PubMed]
- Sugii, S.; Kida, Y.; Kawamura, T.; Suzuki, J.; Vassena, R.; Yin, Y.Q.; Lutz, M.K.; Berggren, W.T.; Izpisua Belmonte, J.C.; Evans, R.M. Human and mouseadipose-derived cells support feeder-independent induction of pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2010, 107, 3558–3563. [Google Scholar] [CrossRef] [PubMed]
- Du, S.H.; Tay, J.C.; Chen, C.; Tay, F.C.; Tan, W.K.; Li, Z.D.; Wang, S. Human iPS cell-derived fibroblast-like cells as feeder layers for iPS cell derivation and expansion. J. Biosci. Bioeng. 2015. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.T.; Lee, K.I.; Kim, D.W.; Hwang, D.Y. An ECM-based culture system for the generation and maintenance of xeno-free human iPS cells. Biomaterials 2013, 34, 1041–1050. [Google Scholar] [CrossRef] [PubMed]
- Groß, B.; Sgodda, M.; Rasche, M.; Schambach, A.; Göhring, G.; Schlegelberger, B.; Greber, B.; Linden, T.; Reinhardt, D.; Cantz, T.; et al. Improved generation of patient-specific induced pluripotent stem cells using a chemically-defined and matrigel-based approach. Curr. Mol. Med. 2013, 13, 765–776. [Google Scholar] [CrossRef] [PubMed]
- Sterneckert, J.L.; Reinhardt, P.; Schöler, H.R. Investigating human disease using stem cell models. Nat. Rev. Genet. 2014, 15, 625–639. [Google Scholar] [CrossRef] [PubMed]
- Doetschman, T.C.; Eistetter, H.; Katz, M.; Schmidt, W.; Kemler, R. The in vitro development of blastocyst-derived embryonic stem cell lines: Formation of visceral yolk sac, blood islands and myocardium. J. Embryol. Exp. Morphol. 1985, 87, 27–45. [Google Scholar] [PubMed]
- Imamura, T.; Cui, L.; Teng, R.; Johkura, K.; Okouchi, Y.; Asanuma, K.; Ogiwara, N.; Sasaki, K. Embryonic stem cell-derived embryoid bodies in three-dimensional culture system form hepatocyte-like cells in vitro and in vivo. Tissue Eng. 2004, 10, 1716–1724. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Kodama, H.; Honjo, T. Generation of lymphohematopoietic cells from embryonic stem cells in culture. Science 1994, 265, 1098–1101. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, S.; Nishikawa, S.; Hirashima, M.; Matsuyoshi, N.; Kodama, H. Progressive lineage analysis by cell sorting and culture identifies FLK+VE-cadherin cells at a diverging point of endothelial and hemopoietic lineages. Development 1998, 125, 1747–1757. [Google Scholar] [PubMed]
- Carpenter, L.; Carr, C.; Yang, C.T.; Stuckey, D.J.; Clarke, K.; Watt, S.M. Efficient differentiation of human induced pluripotent stem cells generates cardiac cells that provide protection following myocardial infarction in the rat. Stem Cells Dev. 2012, 21, 977–986. [Google Scholar] [CrossRef] [PubMed]
- Giobbe, G.G.; Zagallo, M.; Riello, M.; Serena, E.; Masi, G.; Barzon, L.; di Camillo, B.; Elvassore, N. Confined 3D microenvironment regulates early differentiation in human pluripotent stem cells. Biotechnol. Bioeng. 2012, 109, 3119–3132. [Google Scholar] [CrossRef] [PubMed]
- Takebe, T.; Sekine, K.; Enomura, M.; Koike, H.; Kimura, M.; Ogaeri, T.; Zhang, R.R.; Ueno, Y.; Zheng, Y.W.; Koike, N.; et al. Vascularized and functional human liver from an iPSC-derived organ bud transplant. Nature 2013, 499, 481–484. [Google Scholar] [CrossRef] [PubMed]
- Dvir, T.; Timko, B.P.; Brigham, M.D.; Naik, S.R.; Karajanagi, S.S.; Levy, O.; Jin, H.; Parker, K.K.; Langer, R.; Kohane, D.S. Nanowired three-dimensional cardiac patches. Nat. Nanotechnol. 2011, 6, 720–725. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.H.; Kim, Y.H.; Hebisch, M.; Sliwinski, C.; Lee, S.; D’Avanzo, C.; Chen, H.; Hooli, B.; Asselin, C.; Muffat, J.; et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature 2014, 515, 274–278. [Google Scholar] [CrossRef] [PubMed]
- Park, I.H.; Arora, N.; Huo, H.; Maherali, N.; Ahfeldt, T.; Shimamura, A.; Lensch, M.W.; Cowan, C.; Hochedlinger, K.; Daley, G.Q. Disease-specific induced pluripotent stem cells. Cell 2008, 134, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Tiscornia, G.; Vivas, E.L.; Izpisúa Belmonte, J.C. Diseases in a dish: Modeling human genetic disorders using induced pluripotent cells. Nat. Med. 2011, 17, 1570–1576. [Google Scholar] [CrossRef] [PubMed]
- Moretti, A.; Bellin, M.; Welling, A.; Jung, C.B.; Lam, J.T.; Bott-Flügel, L.; Dorn, T.; Goedel, A.; Höhnke, C.; Hofmann, F.; et al. Patient-specific induced pluripotent stem-cell models for long-QT syndrome. N. Engl. J. Med. 2010, 363, 1397–1409. [Google Scholar] [CrossRef] [PubMed]
- Itzhaki, I.; Maizels, L.; Huber, I.; Zwi-Dantsis, L.; Caspi, O.; Winterstern, A.; Feldman, O.; Gepstein, A.; Arbel, G.; Hammerman, H.; et al. Modelling the long QT syndrome with induced pluripotent stem cells. Nature 2011, 471, 225–229. [Google Scholar] [CrossRef] [PubMed]
- Ma, D.; Wei, H.; Lu, J.; Ho, S.; Zhang, G.; Sun, X.; Oh, Y.; Tan, S.H.; Ng, M.L.; Shim, W.; et al. Generation of patient-specific induced pluripotent stem cell-derived cardiomyocytes as a cellular model of arrhythmogenic right ventricular cardiomyopathy. Eur. Heart J. 2013, 34, 1122–1133. [Google Scholar] [CrossRef] [PubMed]
- Egashira, T.; Yuasa, S.; Suzuki, T.; Aizawa, Y.; Yamakawa, H.; Matsuhashi, T.; Ohno, Y.; Tohyama, S.; Okata, S.; Seki, T.; et al. Disease characterization using LQTS-specific induced pluripotent stem cells. Cardiovasc. Res. 2012, 95, 419–429. [Google Scholar] [CrossRef] [PubMed]
- Lee, G.; Papapetrou, E.P.; Kim, H.; Chambers, S.M.; Tomishima, M.J.; Fasano, C.A.; Ganat, Y.M.; Menon, J.; Shimizu, F.; Viale, A.; et al. Modelling pathogenesis and treatment of familial dysautonomia using patient-specific iPSCs. Nature 2009, 461, 402–406. [Google Scholar] [CrossRef] [PubMed]
- Marchetto, M.C.; Carromeu, C.; Acab, A.; Yu, D.; Yeo, G.W.; Mu, Y.; Chen, G.; Gage, F.H.; Muotri, A.R. A model for neural development and treatment of Rett syndrome using human induced pluripotent stem cells. Cell 2010, 143, 527–539. [Google Scholar] [CrossRef] [PubMed]
- Devine, M.J.; Ryten, M.; Vodicka, P.; Thomson, A.J.; Burdon, T.; Houlden, H.; Cavaleri, F.; Nagano, M.; Drummond, N.J.; Taanman, J.W.; et al. Parkinson’s disease induced pluripotent stem cells with triplication of the α-synuclein locus. Nat. Commun. 2011, 2, e440. [Google Scholar] [CrossRef] [PubMed]
- Brennand, K.J.; Simone, A.; Jou, J.; Gelboin-Burkhart, C.; Tran, N.; Sangar, S.; Li, Y.; Mu, Y.; Chen, G.; Yu, D.; et al. Modelling schizophrenia using human induced pluripotent stem cells. Nature 2011, 473, 221–225. [Google Scholar] [CrossRef] [PubMed]
- Israel, M.A.; Yuan, S.H.; Bardy, C.; Reyna, S.M.; Mu, Y.; Herrera, C.; Hefferan, M.P.; van Gorp, S.; Nazor, K.L.; Boscolo, F.S.; et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 2012, 482, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Rashid, S.T.; Corbineau, S.; Hannan, N.; Marciniak, S.J.; Miranda, E.; Alexander, G.; Huang-Doran, I.; Griffin, J.; Ahrlund-Richter, L.; Skepper, J.; et al. Modeling inherited metabolic disorders of the liver using human induced pluripotent stem cells. J. Clin. Investig. 2010, 120, 3127–3136. [Google Scholar] [CrossRef] [PubMed]
- Ghodsizadeh, A.; Taei, A.; Totonchi, M.; Seifinejad, A.; Gourabi, H.; Pournasr, B.; Aghdami, N.; Malekzadeh, R.; Almadani, N.; Salekdeh, G.H.; et al. Generation of liver disease-specific induced pluripotent stem cells along with efficient differentiation to functional hepatocyte-like cells. Stem Cell Rev. 2010, 6, 622–632. [Google Scholar] [CrossRef] [PubMed]
- Serena, E.; Cimetta, E.; Zatti, S.; Zaglia, T.; Zagallo, M.; Keller, G.; Elvassore, N. Micro-arrayed human embryonic stem cells-derived cardiomyocytes for in vitro functional assay. PLoS ONE 2012, 7, e48483. [Google Scholar] [CrossRef] [PubMed]
- Giobbe, G.G.; Michielin, F.; Luni, C.; Giulitti, S.; Martewicz, S.; Dupont, S.; Floreani, A.; Elvassore, N. Functional differentiation of human pluripotent stem cells on a chip. Nat. Methods 2015, 12, 637–640. [Google Scholar] [CrossRef] [PubMed]
- Luni, C.; Michielin, F.; Barzon, L.; Calabrò, V.; Elvassore, N. Stochastic model-assisted development of efficient low-dose viral transduction in microfluidics. Biophys. J. 2013, 104, 934–942. [Google Scholar] [CrossRef] [PubMed]
- Cimetta, E.; Franzoso, M.; Trevisan, M.; Serena, E.; Zambon, A.; Giulitti, S.; Barzon, L.; Elvassore, N. Microfluidic-driven viral infection on cell cultures: Theoretical and experimental study. Biomicrofluidics 2012, 6, 24127–2412712. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Lee, Y.K.; Schaefer, E.A.; Peters, D.T.; Veres, A.; Kim, K.; Kuperwasser, N.; Motola, D.L.; Meissner, T.B.; Hendriks, W.T.; et al. A TALEN genome-editing system for generating human stem cell-based disease models. Cell Stem Cell 2013, 12, 238–251. [Google Scholar] [CrossRef] [PubMed]
- Gerna, G.; Baldanti, F.; Revello, M.G. Pathogenesis of human cytomegalovirus infection and cellular targets. Hum. Immunol. 2004, 65, 381–386. [Google Scholar] [CrossRef] [PubMed]
- Goodrum, F.D.; Jordan, C.T.; High, K.; Shenk, T. Human cytomegalovirus gene expression during infection of primary hematopoietic progenitor cells: A model for latency. Proc. Natl. Acad. Sci. USA 2002, 99, 16255–16260. [Google Scholar] [CrossRef] [PubMed]
- Hargett, D.; Shenk, T.E. Experimental human cytomegalovirus latency in CD14+ monocytes. Proc. Natl. Acad. Sci. USA 2011, 107, 20039–20044. [Google Scholar] [CrossRef] [PubMed]
- Ibanez, C.E.; Schrier, R.; Ghazal, P.; Wiley, C.; Nelson, J.A. Human cytomegalovirus productively infects primary differentiated macrophages. J. Virol. 1991, 65, 6581–6588. [Google Scholar] [PubMed]
- Fish, K.N.; Soderberg-Naucler, C.; Mills, L.K.; Stenglein, S.; Nelson, J.A. Human cytomegalovirus persistently infects aortic endothelial cells. J. Virol. 1998, 72, 5661–5668. [Google Scholar] [PubMed]
- Hahn, G.; Revello, M.G.; Patrone, M.; Percivalle, E.; Campanini, G.; Sarasini, A.; Wagner, M.; Gallina, A.; Milanesi, G.; Koszinowski, U.; et al. Human cytomegalovirus UL131-128 genes are indispensable for virus growth in endothelial cells and virus transfer to leukocytes. J. Virol. 2004, 78, 10023–10033. [Google Scholar] [CrossRef] [PubMed]
- Bentz, G.L.; Jarquin-Pardo, M.; Chan, G.; Smith, M.S.; Sinzger, C.; Yurochko, A.D. Human cytomegalovirus (HCMV) infection of endothelial cells promotes naive monocyte extravasation and transfer of productive virus to enhance hematogenous dissemination of HCMV. J. Virol. 2006, 80, 11539–11555. [Google Scholar] [CrossRef] [PubMed]
- Weisblum, Y.; Panet, A.; Haimov-Kochman, R.; Wolf, D.G. Models of vertical cytomegalovirus (CMV) transmission and pathogenesis. Semin. Immunopathol. 2014, 36, 615–625. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, P.; Baraniak, I.; Reeves, M. The pathogenesis of human cytomegalovirus. J. Pathol. 2015, 235, 288–297. [Google Scholar] [CrossRef] [PubMed]
- Cha, T.A.; Tom, E.; Kemble, G.W.; Duke, G.M.; Mocarski, E.S.; Spaete, R.R. Human cytomegalovirus clinical isolates carry at least 19 genes not found in laboratory strains. J. Virol. 1996, 70, 78–83. [Google Scholar] [PubMed]
- Wang, D.; Shenk, T. Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tropism. Proc. Natl. Acad. Sci. USA 2005, 102, 18153–18158. [Google Scholar] [CrossRef] [PubMed]
- Cosset, É.; Martinez, Y.; Preynat-Seauve, O.; Lobrinus, J.A.; Tapparel, C.; Cordey, S.; Peterson, H.; Petty, T.J.; Colaianna, M.; Tieng, V.; et al. Human three-dimensional engineered neural tissue reveals cellular and molecular events following cytomegalovirus infection. Biomaterials 2015, 53, 296–308. [Google Scholar]
- Belzile, J.P.; Stark, T.J.; Yeo, G.W.; Spector, D.H. Human cytomegalovirus infection of human embryonic stem cell-derived primitive neural stem cells is restricted at several steps but leads to the persistence of viral DNA. J. Virol. 2014, 88, 4021–4039. [Google Scholar] [CrossRef] [PubMed]
- D’Aiuto, L.; di Maio, R.; Heath, B.; Raimondi, G.; Milosevic, J.; Watson, A.M.; Bamne, M.; Parks, W.T.; Yang, L.; Lin, B.; et al. Human induced pluripotent stem cell-derived models to investigate human cytomegalovirus infection in neural cells. PLoS ONE 2012, 7, e49700. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.S.; Zhou, W.; Scott-McKean, J.J.; Emmerling, K.L.; Cai, G.Y.; Krah, D.L.; Costa, A.C.; Freed, C.R.; Levin, M.J. Human sensory neurons derived from induced pluripotent stem cells support varicella-zoster virus infection. PLoS ONE 2012, 7, e53010. [Google Scholar] [CrossRef] [PubMed]
- D’Aiuto, L.; Prasad, K.M.; Upton, C.H.; Viggiano, L.; Milosevic, J.; Raimondi, G.; McClain, L.; Chowdari, K.; Tischfield, J.; Sheldon, M.; et al. Persistent infection by HSV-1 is associated with changes in functional architecture of iPSC-derived neurons and brain activation patterns underlying working memory performance. Schizophr. Bull. 2015, 41, 123–132. [Google Scholar] [CrossRef] [PubMed]
- European Association for the Study of the Liver. EASL clinical practice guidelines: Management of chronic hepatitis B virus infection. J. Hepatol. 2012, 57, 167–185. [Google Scholar] [CrossRef] [PubMed]
- Gripon, P.; Diot, C.; Guguen-Guillouzo, C. Reproducible high level infection of cultured adult human hepatocytes by hepatitis B virus: Effect of polyethylene glycol on adsorption and penetration. Virology 1993, 192, 534–540. [Google Scholar] [CrossRef] [PubMed]
- Khetani, S.R.; Bhatiam, S.N. Microscale culture of human liver cells for drug development. Nat. Biotechnol. 2008, 26, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Shlomai, A.; Schwartz, R.E.; Ramanan, V.; Bhatta, A.; de Jong, Y.P.; Bhatia, S.N.; Rice, C.M. Modeling host interactions with hepatitis B virus using primary and induced pluripotent stem cell-derived hepatocellular systems. Proc. Natl. Acad. Sci. USA 2014, 111, 12193–12198. [Google Scholar] [CrossRef] [PubMed]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. Elife 2012, 1, e00049. [Google Scholar] [CrossRef] [PubMed]
- Ko, C.; Park, W.J.; Park, S.; Kim, S.; Windisch, M.P.; Ryu, W.S. The FDA approved drug irbesartan inhibits HBV-infection in HepG2 cells stably expressing sodium taurocholate co-transporting polypeptide. Antivir. Ther. 2015. [Google Scholar] [CrossRef] [PubMed]
- He, W.; Ren, B.; Mao, F.; Jing, Z.; Li, Y.; Liu, Y.; Peng, B.; Yan, H.; Qi, Y.; Sun, Y.; et al. Hepatitis D virus infection of mice expressing human sodium taurocholate co-transporting polypeptide. PLoS Pathog. 2015, 11, e1004840. [Google Scholar] [CrossRef] [PubMed]
- Lavanchy, D. The global burden of hepatitis C. Liver Int. 2009, 29, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Date, T.; Miyamoto, M.; Furusaka, A.; Tokushige, K.; Mizokami, M.; Wakita, T. Efficient replication of the genotype 2a hepatitis C virus subgenomic replicon. Gastroenterology 2003, 125, 1808–1817. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Evans, M.J.; Syder, A.J.; Wölk, B.; Tellinghuisen, T.L.; Liu, C.C.; Maruyama, T.; Hynes, R.O.; Burton, D.R.; McKeating, J.A.; et al. Complete replication of hepatitis C virus in cell culture. Science 2005, 309, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Wakita, T.; Pietschmann, T.; Kato, T.; Date, T.; Miyamoto, M.; Zhao, Z.; Murthy, K.; Habermann, A.; Kräusslich, H.G.; Mizokami, M.; et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 2005, 11, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Heller, T.; Saito, S.; Auerbach, J.; Williams, T.; Moreen, T.R.; Jazwinski, A.; Cruz, B.; Jeurkar, N.; Sapp, R.; Luo, G.; et al. An in vitro model of hepatitis C virion production. Proc. Natl. Acad. Sci. USA 2005, 102, 2579–2583. [Google Scholar] [CrossRef] [PubMed]
- Kato, T.; Matsumura, T.; Heller, T.; Saito, S.; Sapp, R.K.; Murthy, K.; Wakita, T.; Liang, T.J. Production of infectious hepatitis C virus of various genotypes in cell cultures. J. Virol. 2007, 81, 4405–4411. [Google Scholar] [CrossRef] [PubMed]
- Ploss, A.; Khetani, S.R.; Jones, C.T.; Syder, A.J.; Trehan, K.; Gaysinskaya, V.A.; Mu, K.; Ritola, K.; Rice, C.M.; Bhatia, S.N. Persistent hepatitis C virus infection in microscale primary human hepatocyte cultures. Proc. Natl. Acad. Sci. USA 2010, 107, 3141–3145. [Google Scholar] [CrossRef] [PubMed]
- Podevin, P.; Carpentier, A.; Pène, V.; Aoudjehane, L.; Carrière, M.; Zaïdi, S.; Hernandez, C.; Calle, V.; Méritet, J.F.; Scatton, O.; et al. Production of infectious hepatitis C virus in primary cultures of human adult hepatocytes. Gastroenterology 2010, 139, 1355–1364. [Google Scholar] [CrossRef] [PubMed]
- Thomas, E.; Gonzalez, V.D.; Li, Q.; Modi, A.A.; Chen, W.; Noureddin, M.; Rotman, Y.; Liang, T.J. HCV infection induces a unique hepatic innate immune response associated with robust production of type III interferons. Gastroenterology 2012, 142, 978–988. [Google Scholar] [CrossRef] [PubMed]
- Mercer, D.F.; Schiller, D.E.; Elliott, J.F.; Douglas, D.N.; Hao, C.; Rinfret, A.; Addison, W.R.; Fischer, K.P.; Churchill, T.A.; Lakey, J.R.; et al. Hepatitis C virus replication in mice with chimeric human livers. Nat. Med. 2001, 7, 927–933. [Google Scholar] [CrossRef] [PubMed]
- Tesfaye, A.; Stift, J.; Maric, D.; Cui, Q.; Dienes, H.P.; Feinstone, S.M. Chimeric mouse model for the infection of hepatitis B and C viruses. PLoS ONE 2013, 8, e77298. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Robotham, J.M.; Lee, E.; Dalton, S.; Kneteman, N.M.; Gilbert, D.M.; Tang, H. Productive hepatitis C virus infection of stem cell-derived hepatocytes reveals a critical transition to viral permissiveness during differentiation. PLoS Pathog. 2012, 8, e1002617. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, R.E.; Trehan, K.; Andrus, L.; Sheahan, T.P.; Ploss, A.; Duncan, S.A.; Rice, C.M.; Bhatia, S.N. Modeling hepatitis C virus infection using human induced pluripotent stem cells. Proc. Natl. Acad. Sci. USA 2012, 109, 2544–2548. [Google Scholar] [CrossRef] [PubMed]
- Carpentier, A.; Tesfaye, A.; Chu, V.; Nimgaonkar, I.; Zhang, F.; Lee, S.B.; Thorgeirsson, S.S.; Feinstone, S.M.; Liang, T.J. Engrafted human stem cell-derived hepatocytes establish an infectious HCV murine model. J. Clin. Investig. 2014, 124, 4953–4964. [Google Scholar] [CrossRef] [PubMed]
- Weglarz, T.C.; Degen, J.L.; Sandgren, E.P. Hepatocyte transplantation into diseased mouse liver. Kinetics of parenchymal repopulation and identification of the proliferative capacity of tetraploid and octaploid hepatocytes. Am. J. Pathol. 2000, 157, 1963–1974. [Google Scholar] [CrossRef]
- Liu, H.; Kim, Y.; Sharkis, S.; Marchionni, L.; Jang, Y.Y. In vivo liver regeneration potential of human induced pluripotent stem cells from diverse origins. Sci. Transl. Med. 2011, 3, 82ra39. [Google Scholar] [CrossRef] [PubMed]
- Moriguchi, H.; Chung, R.T.; Sato, C. An identification of novel therapy for human hepatocellular carcinoma by using human induced pluripotent stem cells. Hepatology 2010, 51, 1090–1091. [Google Scholar] [CrossRef] [PubMed]
- Bowles, N.E.; Richardson, P.J.; Olsen, E.G.; Archard, L.C. Detection of Coxsackie-B-virus-specific RNA sequences in myocardial biopsy samples from patients with myocarditis and dilated cardiomyopathy. Lancet 1986, 1, 1120–1123. [Google Scholar] [CrossRef]
- Muehlenbachs, A.; Bhatnagar, J.; Zaki, S.R. Tissue tropism, pathology and pathogenesis of enterovirus infection. J. Pathol. 2015, 235, 217–228. [Google Scholar] [CrossRef] [PubMed]
- Sharma, A.; Marceau, C.; Hamaguchi, R.; Burridge, P.W.; Rajarajan, K.; Churko, J.M.; Wu, H.; Sallam, K.I.; Matsa, E.; Sturzu, A.C.; et al. Human induced pluripotent stem cell-derived cardiomyocytes as an in vitro model for coxsackievirus B3-induced myocarditis and antiviral drug screening platform. Circ. Res. 2014, 115, 556–566. [Google Scholar] [CrossRef] [PubMed]
- Sancho-Shimizu, V.; Perez de Diego, R.; Jouanguy, E.; Zhang, S.Y.; Casanova, J.L. Inborn errors of anti-viral interferon immunity in humans. Curr. Opin. Virol. 2011, 1, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Casrouge, A.; Zhang, S.Y.; Eidenschenk, C.; Jouanguy, E.; Puel, A.; Yang, K.; Alcais, A.; Picard, C.; Mahfoufi, N.; Nicolas, N.; et al. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science 2006, 314, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Y.; Jouanguy, E.; Ugolini, S.; Smahi, A.; Elain, G.; Romero, P.; Segal, D.; Sancho-Shimizu, V.; Lorenzo, L.; Puel, A.; et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science 2007, 317, 1522–1527. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Audry, M.; Ciancanelli, M.; Alsina, L.; Azevedo, J.; Herman, M.; Anguiano, E.; Sancho-Shimizu, V.; Lorenzo, L.; Pauwels, E.; et al. Herpes simplex virus encephalitis in a patient with complete TLR3 deficiency: TLR3 is otherwise redundant in protective immunity. J. Exp. Med. 2011, 208, 2083–2098. [Google Scholar] [CrossRef] [PubMed]
- Herman, M.; Ciancanelli, M.; Ou, Y.H.; Lorenzo, L.; Klaudel-Dreszler, M.; Pauwels, E.; Sancho-Shimizu, V.; Pérez de Diego, R.; Abhyankar, A.; Israelsson, E.; et al. Heterozygous TBK1 mutations impair TLR3 immunity and underlie herpes simplex encephalitis of childhood. J. Exp. Med. 2012, 209, 1567–1582. [Google Scholar] [CrossRef] [PubMed]
- Lafaille, F.G.; Pessach, I.M.; Zhang, S.Y.; Ciancanelli, M.J.; Herman, M.; Abhyankar, A.; Ying, S.W.; Keros, S.; Goldstein, P.A.; Mostoslavsky, G.; et al. Impaired intrinsic immunity to HSV-1 in human iPSC-derived TLR3-deficient CNS cells. Nature 2012, 491, 769–773. [Google Scholar] [CrossRef] [PubMed]
- Ciancanelli, M.J.; Huang, S.X.; Luthra, P.; Garner, H.; Itan, Y.; Volpi, S.; Lafaille, F.G.; Trouillet, C.; Schmolke, M.; Albrecht, R.A.; et al. Infectious disease. Life-threatening influenza and impaired interferon amplification in human IRF7 deficiency. Science 2015, 348, 448–453. [Google Scholar] [CrossRef] [PubMed]
- Barrangou, R.; Fremaux, C.; Deveau, H.; Richards, M.; Boyaval, P.; Moineau, S.; Romero, D.A.; Horvath, P. CRISPR provides acquired resistance against viruses in prokaryotes. Science 2007, 315, 1709–1712. [Google Scholar] [CrossRef] [PubMed]
- Jinek, M.; Chylinski, K.; Fonfara, I.; Hauer, M.; Doudna, J.A.; Charpentier, E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 2012, 337, 816–821. [Google Scholar] [CrossRef] [PubMed]
- Cong, L.; Ran, F.A.; Cox, D.; Lin, S.; Barretto, R.; Habib, N.; Hsu, P.D.; Wu, X.; Jiang, W.; Marraffini, L.A.; Zhang, F. Multiplex genome engineering using CRISPR/Cas systems. Science 2013, 339, 819–823. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.W.; Kim, S.; Kim, J.M.; Kim, J.S. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat. Biotechnol. 2013, 31, 230–232. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, J.S. A guide to genome engineering with programmable nucleases. Nat. Rev. Genet. 2014, 15, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Qu, X.; Wang, P.; Ding, D.; Li, L.; Wang, H.; Ma, L.; Zhou, X.; Liu, S.; Lin, S.; Wang, X.; et al. Zinc-finger-nucleases mediate specific and efficient excision of HIV-1 proviral DNA from infected and latently infected human T cells. Nucleic Acids Res. 2013, 41, 7771–7782. [Google Scholar] [CrossRef] [PubMed]
- Ebina, H.; Misawa, N.; Kanemura, Y.; Koyanagi, Y. Harnessing the CRISPR/Cas9 system to disrupt latent HIV-1 provirus. Sci. Rep. 2013, 3, e2510. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, W.; Kaminski, R.; Yang, F.; Zhang, Y.; Cosentino, L.; Li, F.; Luo, B.; Alvarez-Carbonell, D.; Garcia-Mesa, Y.; Karn, J.; et al. RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc. Natl. Acad. Sci. USA 2014, 111, 11461–11466. [Google Scholar] [CrossRef] [PubMed]
- Liao, H.K.; Gu, Y.; Diaz, A.; Marlett, J.; Takahashi, Y.; Li, M.; Suzuki, K.; Xu, R.; Hishida, T.; Chang, C.J.; et al. Use of the CRISPR/Cas9 system as an intracellular defense against HIV-1 infection in human cells. Nat. Commun. 2015, 6, e6413. [Google Scholar] [CrossRef] [PubMed]
- Ni, Z.; Knorr, D.A.; Clouser, C.L.; Hexum, M.K.; Southern, P.; Mansky, L.M.; Park, I.H.; Kaufman, D.S. Human pluripotent stem cells produce natural killer cells that mediate anti-HIV-1 activity by utilizing diverse cellular mechanisms. J. Virol. 2011, 85, 43–50. [Google Scholar] [CrossRef] [PubMed]
- Ni, Z.; Knorr, D.A.; Bendzick, L.; Allred, J.; Kaufman, D.S. Expression of chimeric receptor CD4ζ by natural killer cells derived from human pluripotent stem cells improves in vitro activity but does not enhance suppression of HIV infection in vivo. Stem Cells 2014, 32, 1021–1031. [Google Scholar] [CrossRef] [PubMed]
- Kamata, M.; Liu, S.; Liang, M.; Nagaoka, Y.; Chen, I.S. Generation of human induced pluripotent stem cells bearing an anti-HIV transgene by a lentiviral vector carrying an internal murine leukemia virus promoter. Hum. Gene Ther. 2010, 21, 1555–1567. [Google Scholar] [CrossRef] [PubMed]
- Kambal, A.; Mitchell, G.; Cary, W.; Gruenloh, W.; Jung, Y.; Kalomoiris, S.; Nacey, C.; McGee, J.; Lindsey, M.; Fury, B.; et al. Generation of HIV-1 resistant and functional macrophages from hematopoietic stem cell-derived induced pluripotent stem cells. Mol. Ther. 2011, 19, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.S.; Javien, J.; Nolta, J.A.; Bauer, G. Preintegration HIV-1 inhibition by a combination lentiviral vector containing a chimeric TRIM5 α protein, a CCR5 shRNA, and a TAR decoy. Mol. Ther. 2009, 17, 2103–2114. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Wang, J.; Beyer, A.I.; Teque, F.; Cradick, T.J.; Qi, Z.; Chang, J.C.; Bao, G.; Muench, M.O.; Yu, J.; et al. Seamless modification of wild-type induced pluripotent stem cells to the natural CCR5Δ32 mutation confers resistance to HIV infection. Proc. Natl. Acad. Sci. USA 2014, 111, 9591–9596. [Google Scholar] [CrossRef] [PubMed]
- Jerebtsova, M.; Kumari, N.; Xu, M.; de Melo, G.B.; Niu, X.; Jeang, K.T.; Nekhai, S. HIV-1 resistant CDK2-knockdown macrophage-like cells generated from 293T cell-derived human induced pluripotent stem cells. Biology 2012, 1, 175–195. [Google Scholar] [CrossRef] [PubMed]
- Yang, F.; Robotham, J.M.; Nelson, H.B.; Irsigler, A.; Kenworthy, R.; Tang, H. Cyclophilin A is an essential cofactor for hepatitis C virus infection and the principal mediator of cyclosporine resistance in vitro. J. Virol. 2008, 82, 5269–5278. [Google Scholar] [CrossRef] [PubMed]
- Seeger, C.; Sohn, J.A. Targeting hepatitis B virus with CRISPR/Cas9. Mol. Ther. Nucleic Acids 2014, 3, e216. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Trevisan, M.; Sinigaglia, A.; Desole, G.; Berto, A.; Pacenti, M.; Palù, G.; Barzon, L. Modeling Viral Infectious Diseases and Development of Antiviral Therapies Using Human Induced Pluripotent Stem Cell-Derived Systems. Viruses 2015, 7, 3835-3856. https://doi.org/10.3390/v7072800
Trevisan M, Sinigaglia A, Desole G, Berto A, Pacenti M, Palù G, Barzon L. Modeling Viral Infectious Diseases and Development of Antiviral Therapies Using Human Induced Pluripotent Stem Cell-Derived Systems. Viruses. 2015; 7(7):3835-3856. https://doi.org/10.3390/v7072800
Chicago/Turabian StyleTrevisan, Marta, Alessandro Sinigaglia, Giovanna Desole, Alessandro Berto, Monia Pacenti, Giorgio Palù, and Luisa Barzon. 2015. "Modeling Viral Infectious Diseases and Development of Antiviral Therapies Using Human Induced Pluripotent Stem Cell-Derived Systems" Viruses 7, no. 7: 3835-3856. https://doi.org/10.3390/v7072800