
Antiviral Potential of a Novel Compound CW-33 against Enterovirus A71 via Inhibition of Viral 2A Protease

 and
and 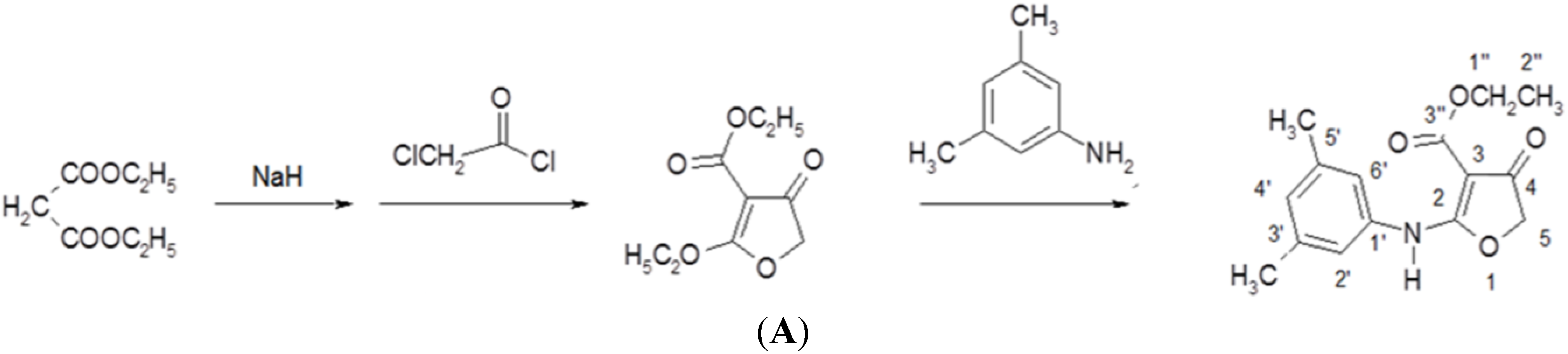{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Viruses and Cells
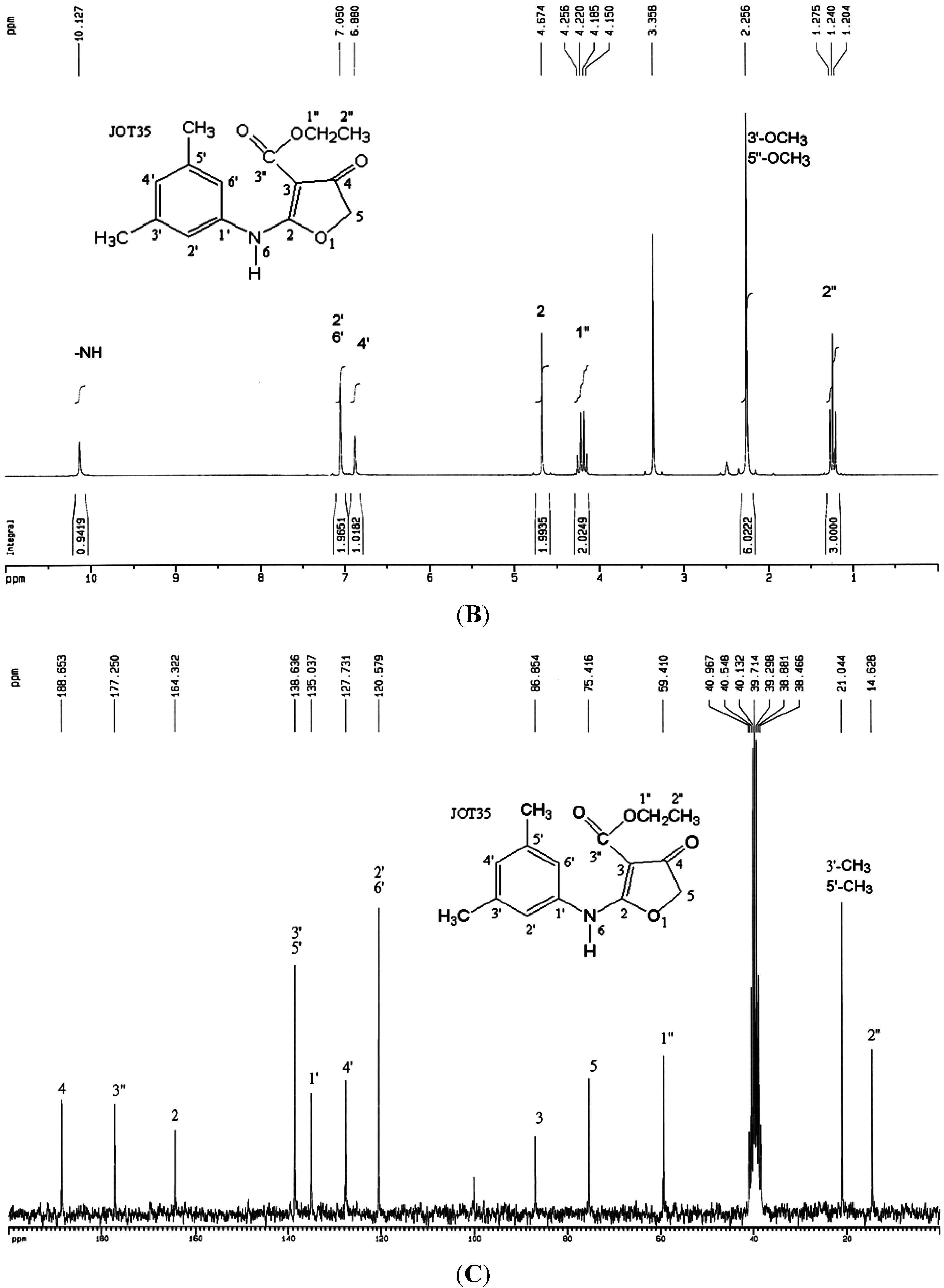
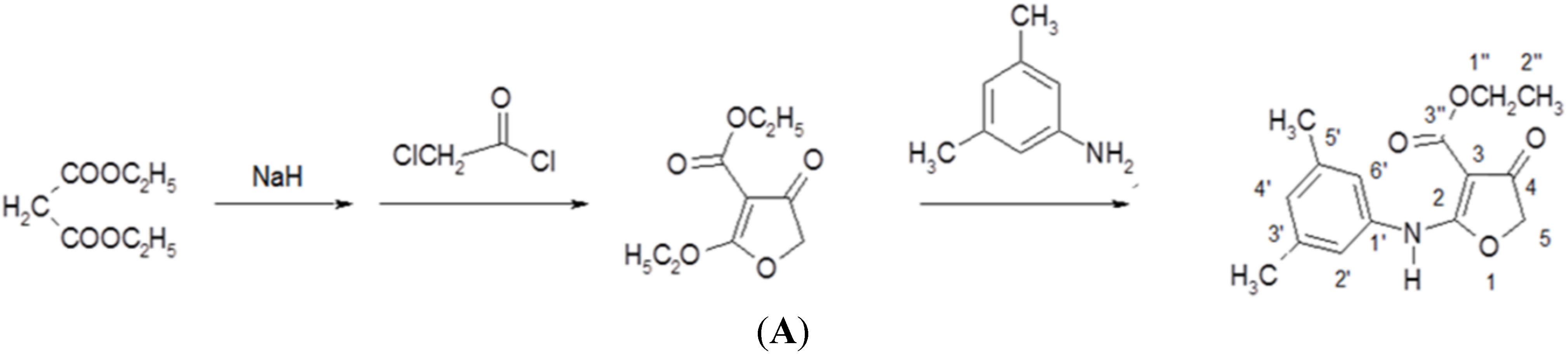
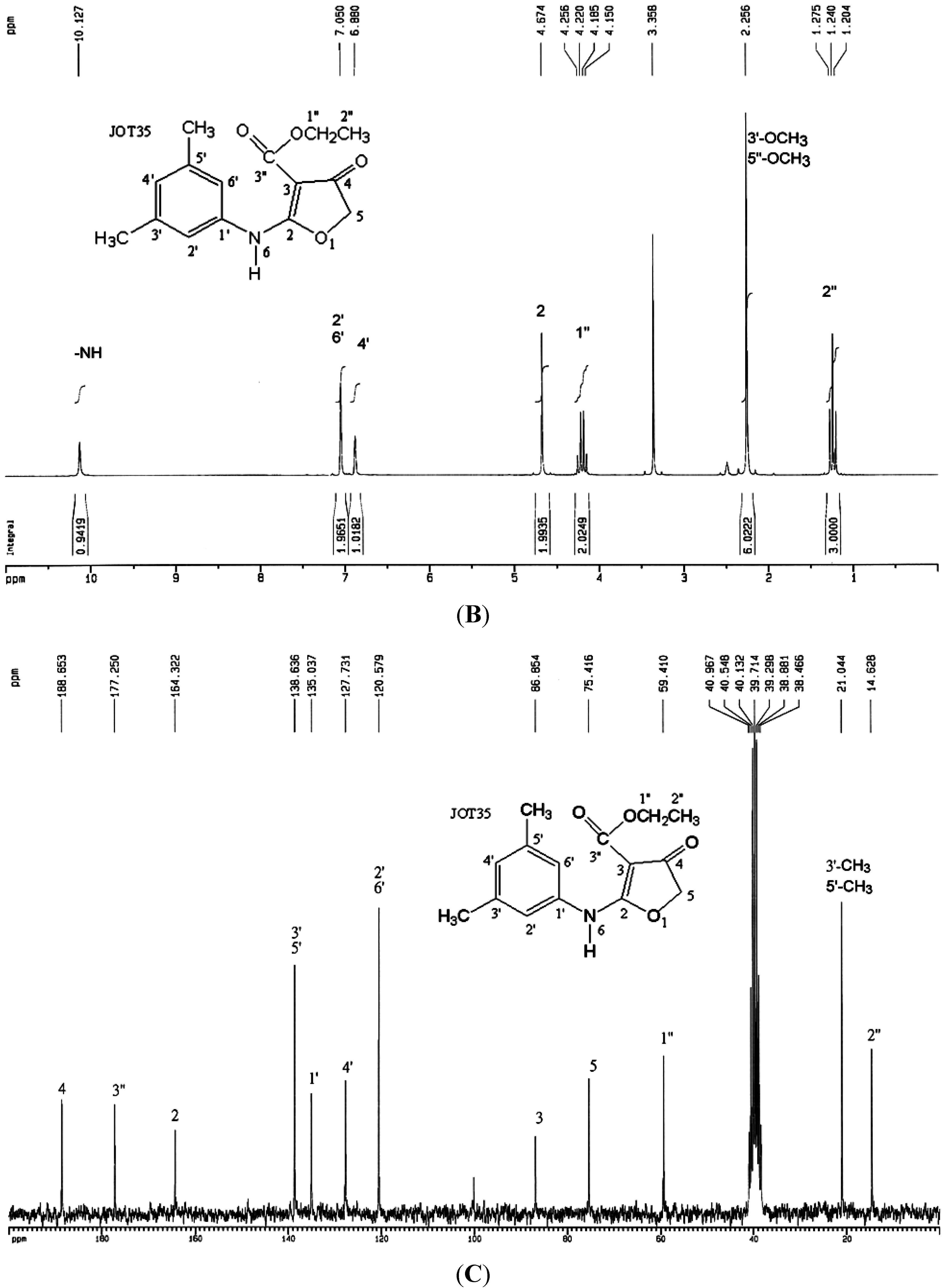
2.2. Synthesis of Compound CW-33
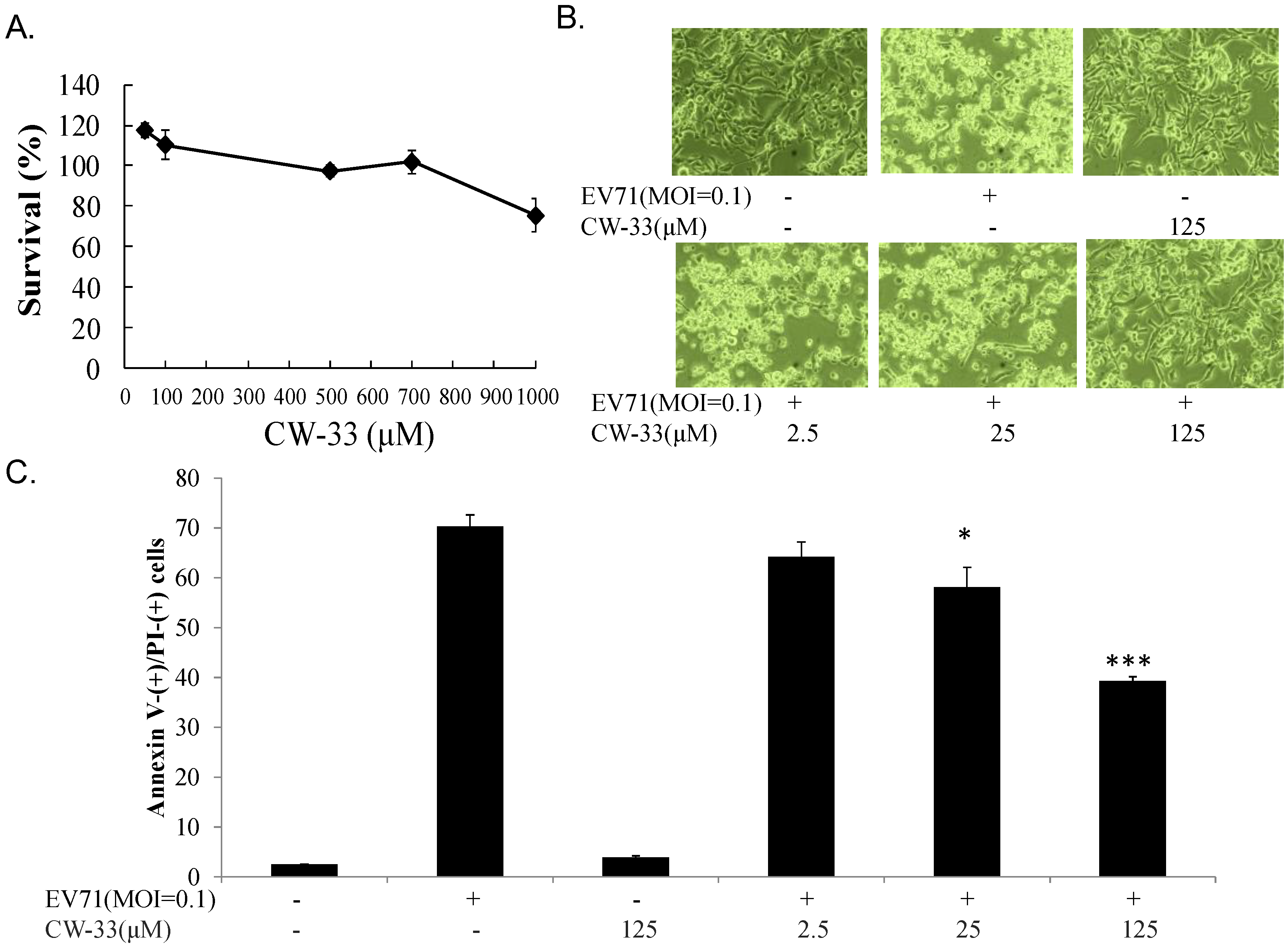
2.3. Cell Cytotoxicity Assay
2.4. Cytopathic Effect (CPE) Reduction and Virus Yield Assays
2.5. Plaque Reduction Assay for 50% Inhibitory Concentration (IC50)
2.6. Cell Cycle Analysis of Flow Cytometry
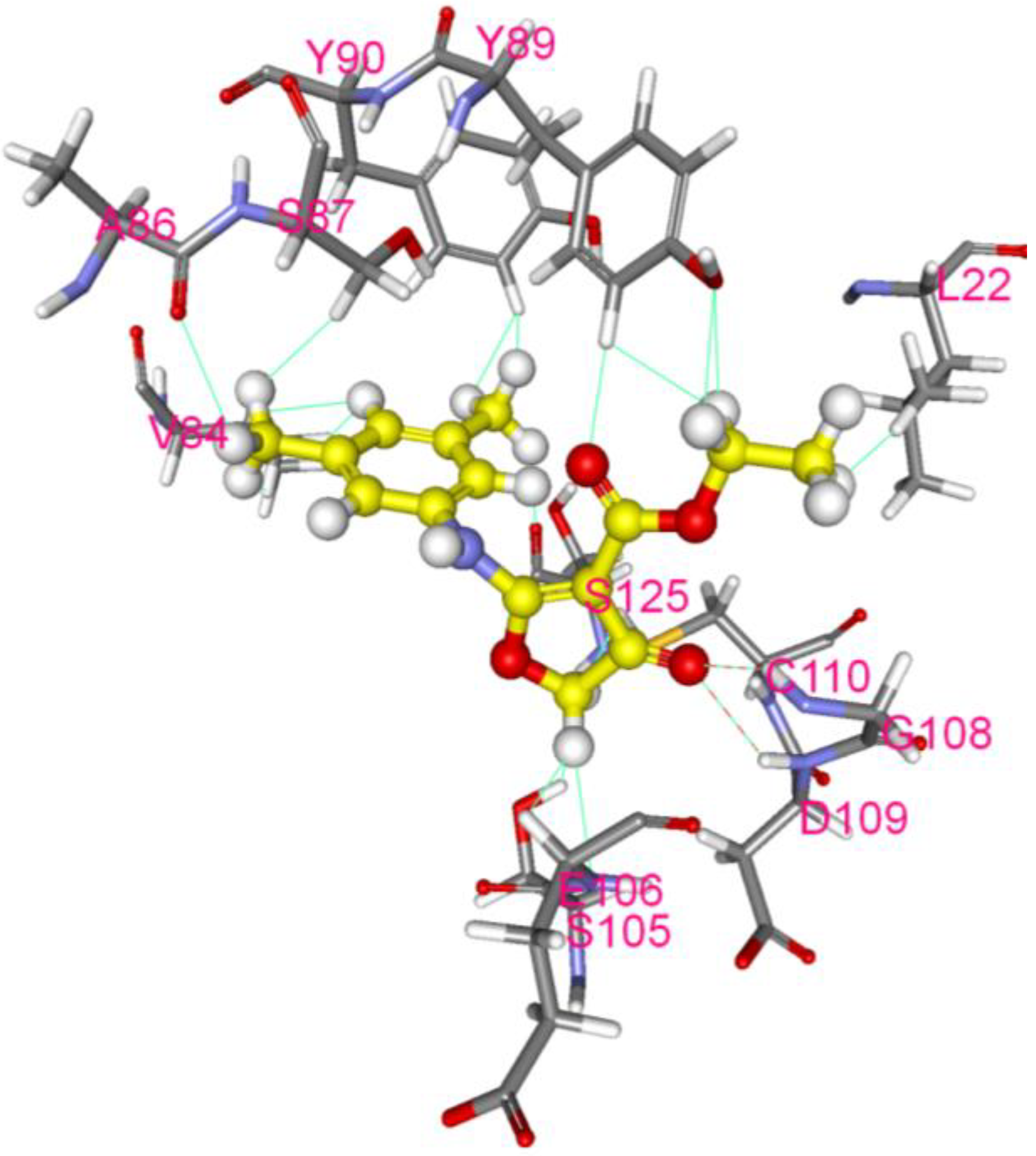
2.7. Molecular Docking
2.8. In Vitro Enzymatic Assay of Recombinant 2A Protease
2.9. Western Blot Analysis
2.10. Real-Time Reverse Transcription-Polymerase Chain Reaction (RT-PCR)
2.11. Statistical Analysis
3. Results
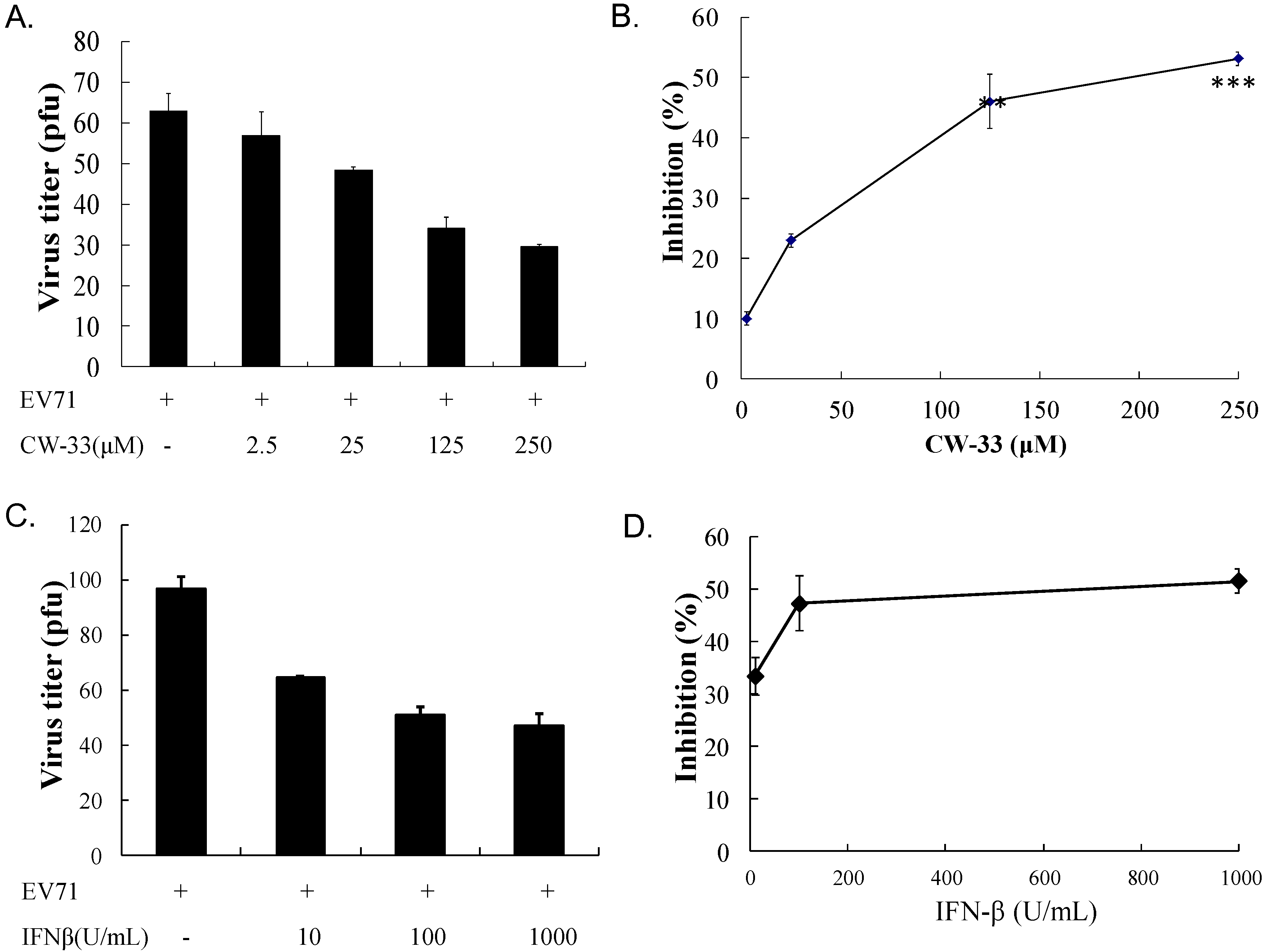
3.1. Antiviral Activity of CW-33 against EV-A71


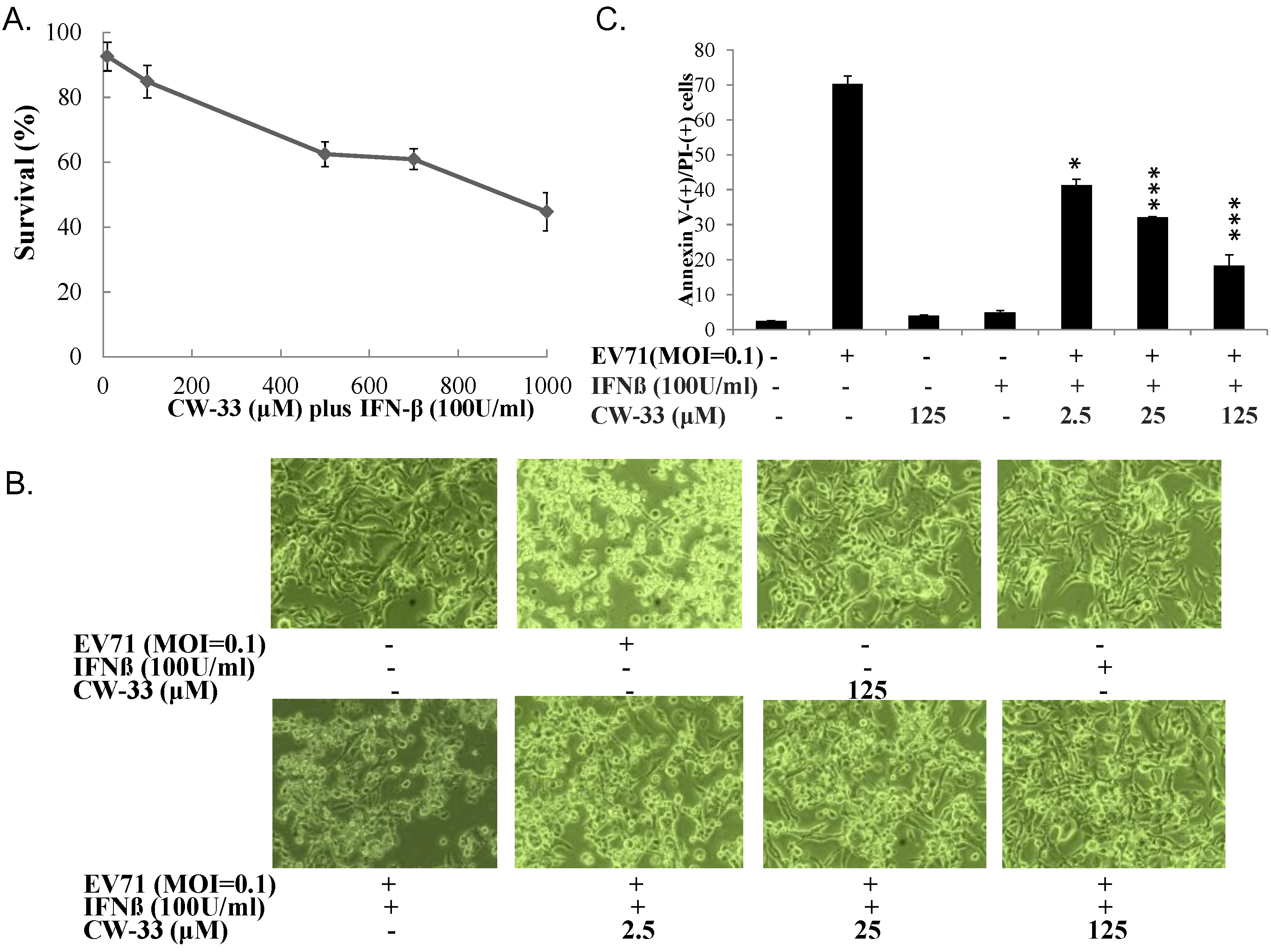
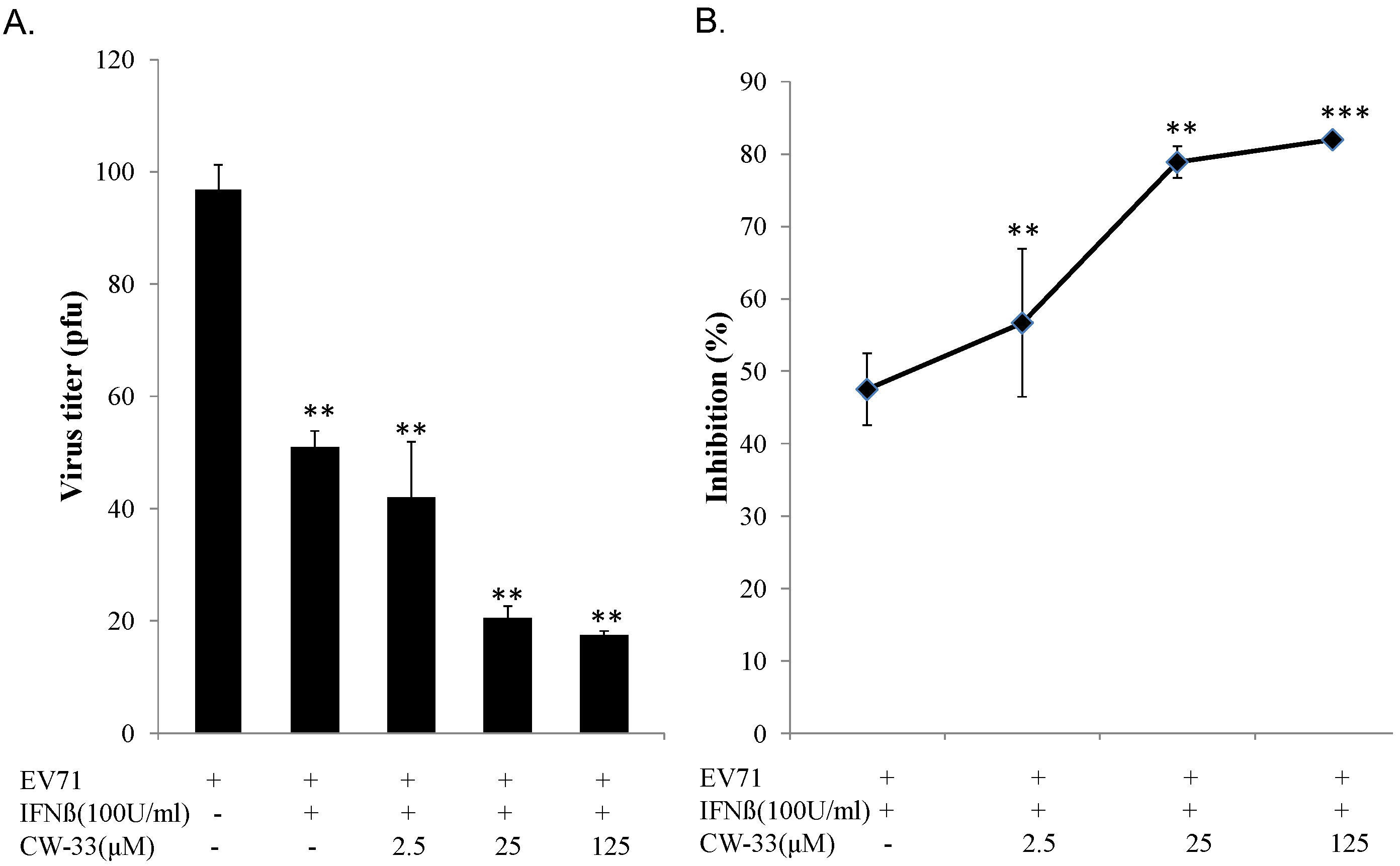
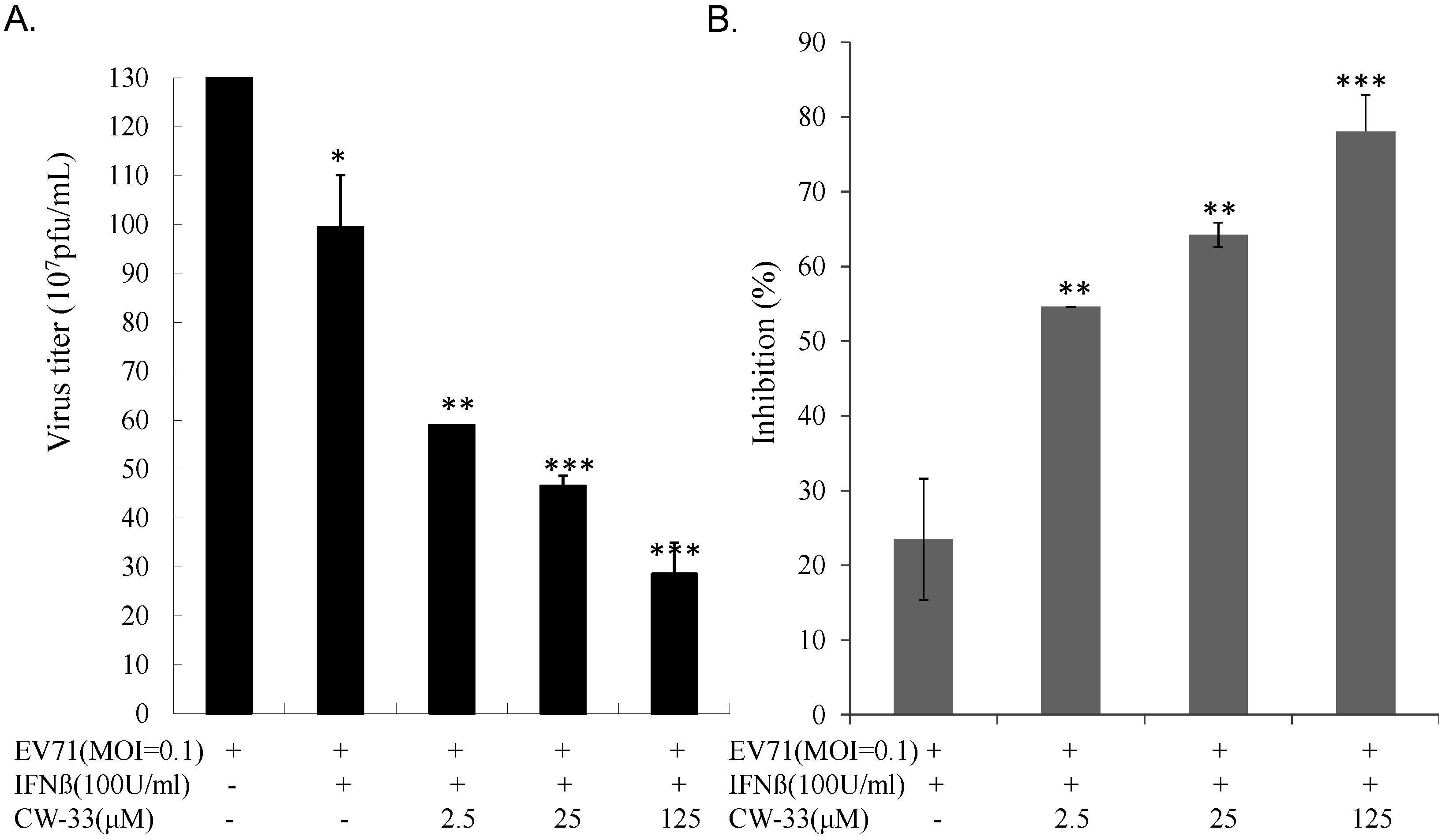
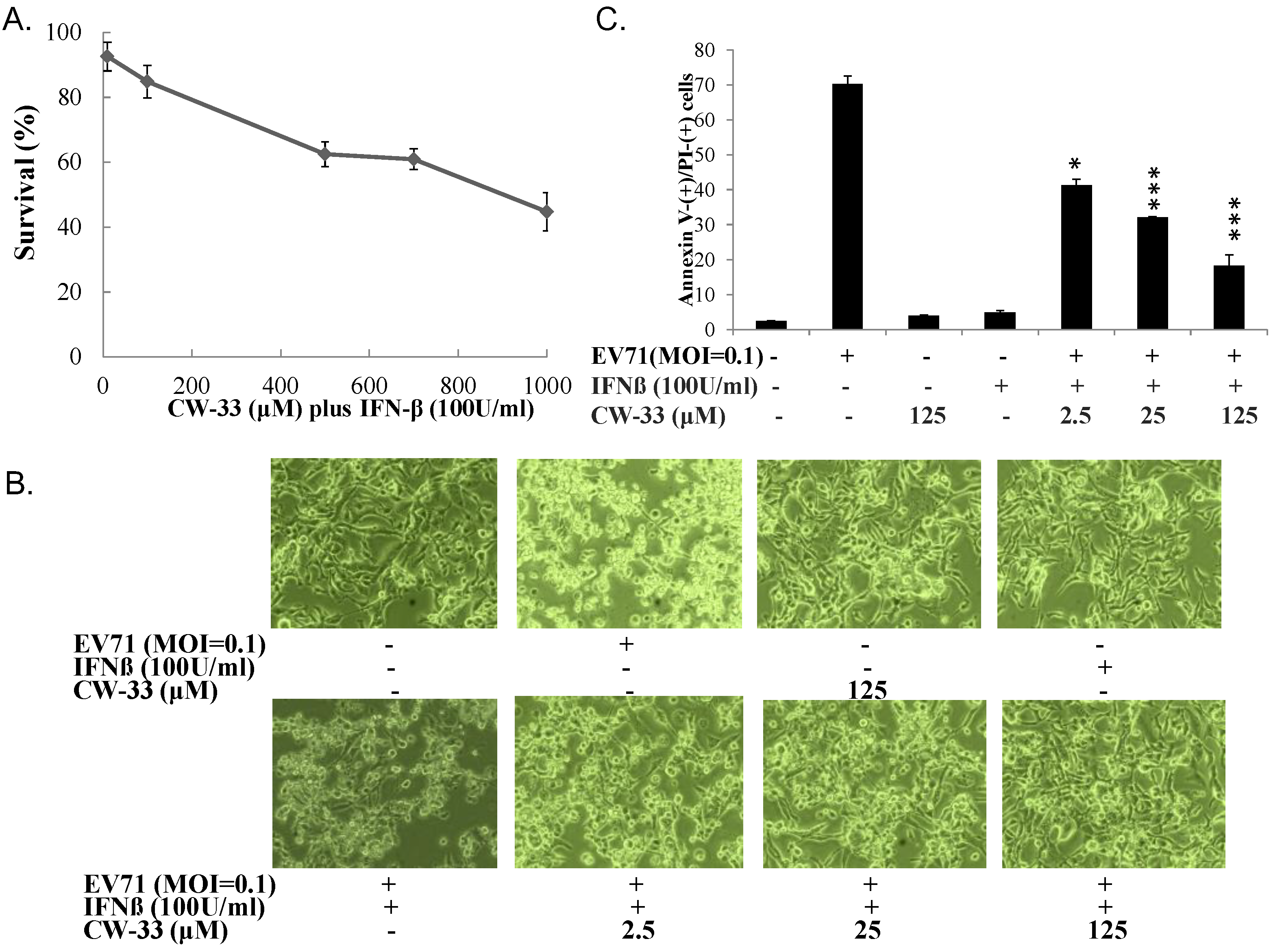
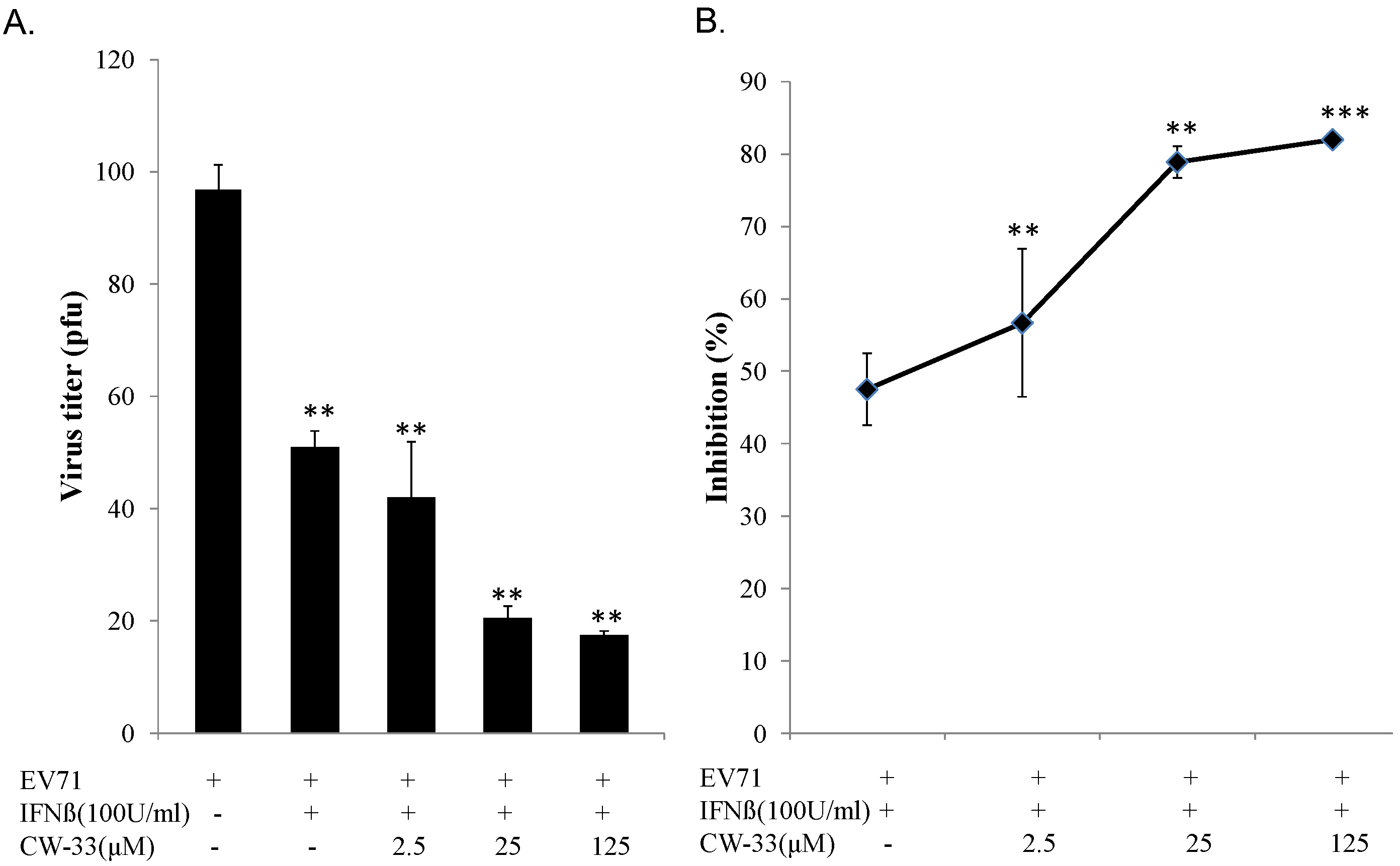
3.2. Synergistic Antiviral Activity of CW-33 in Combination with Interferon (IFN)-β

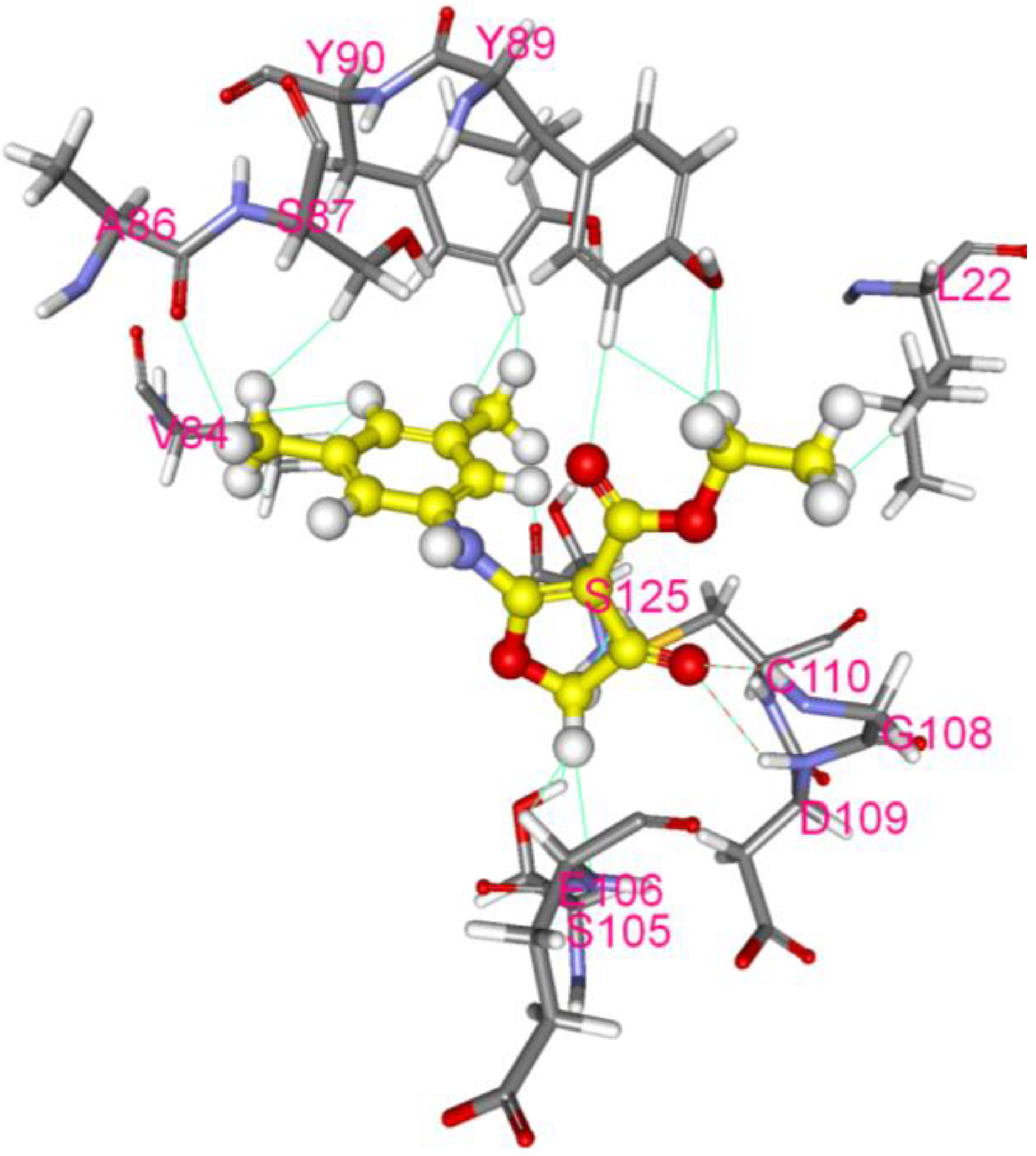
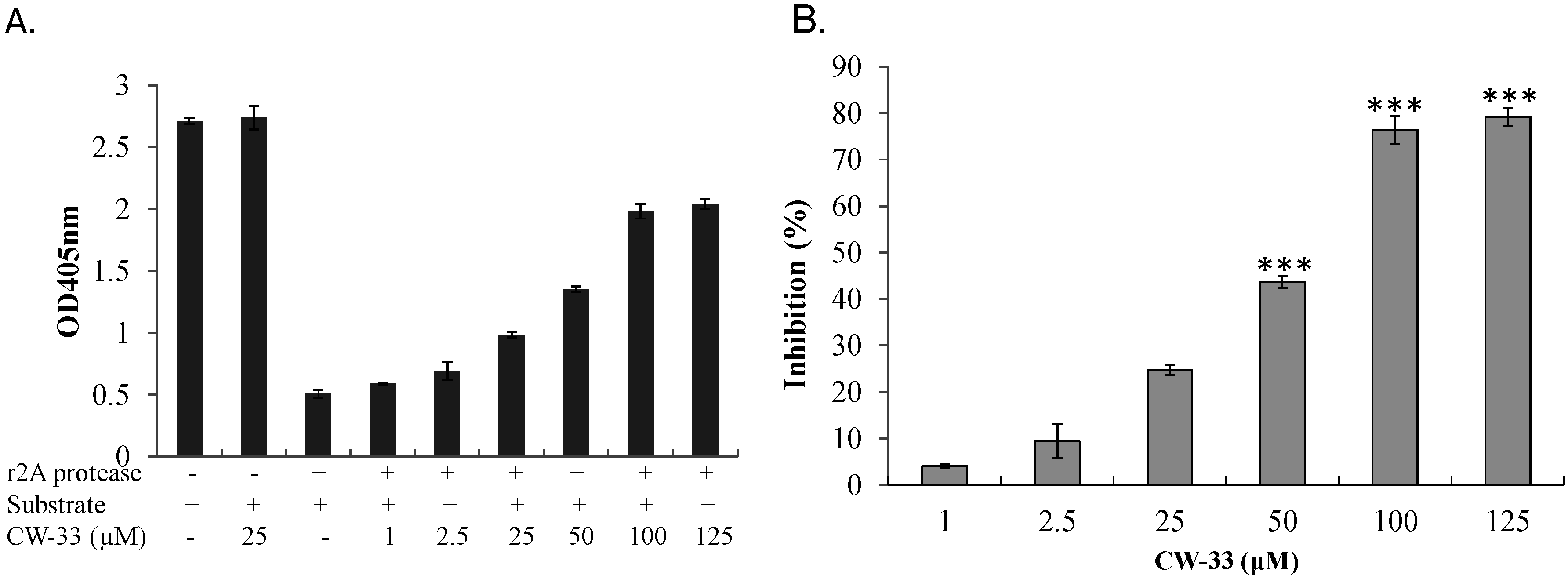
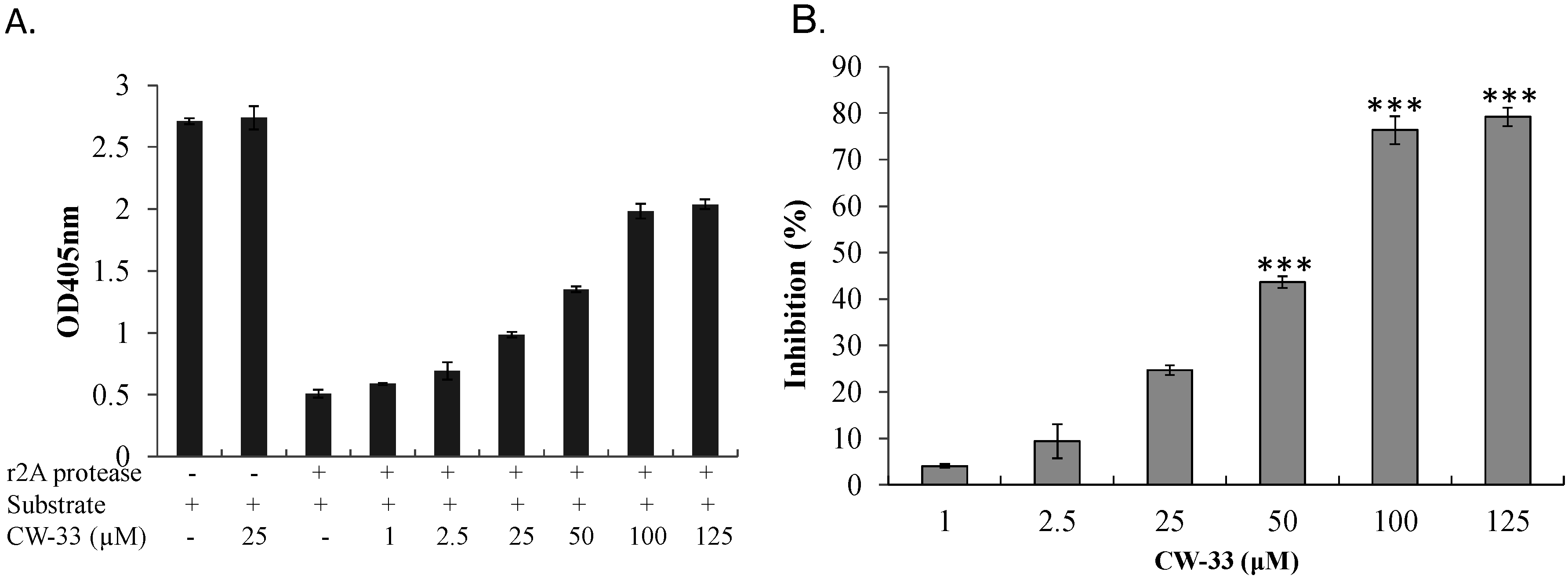
3.3. Inhibition of EV-A71 2A Protease by CW-33
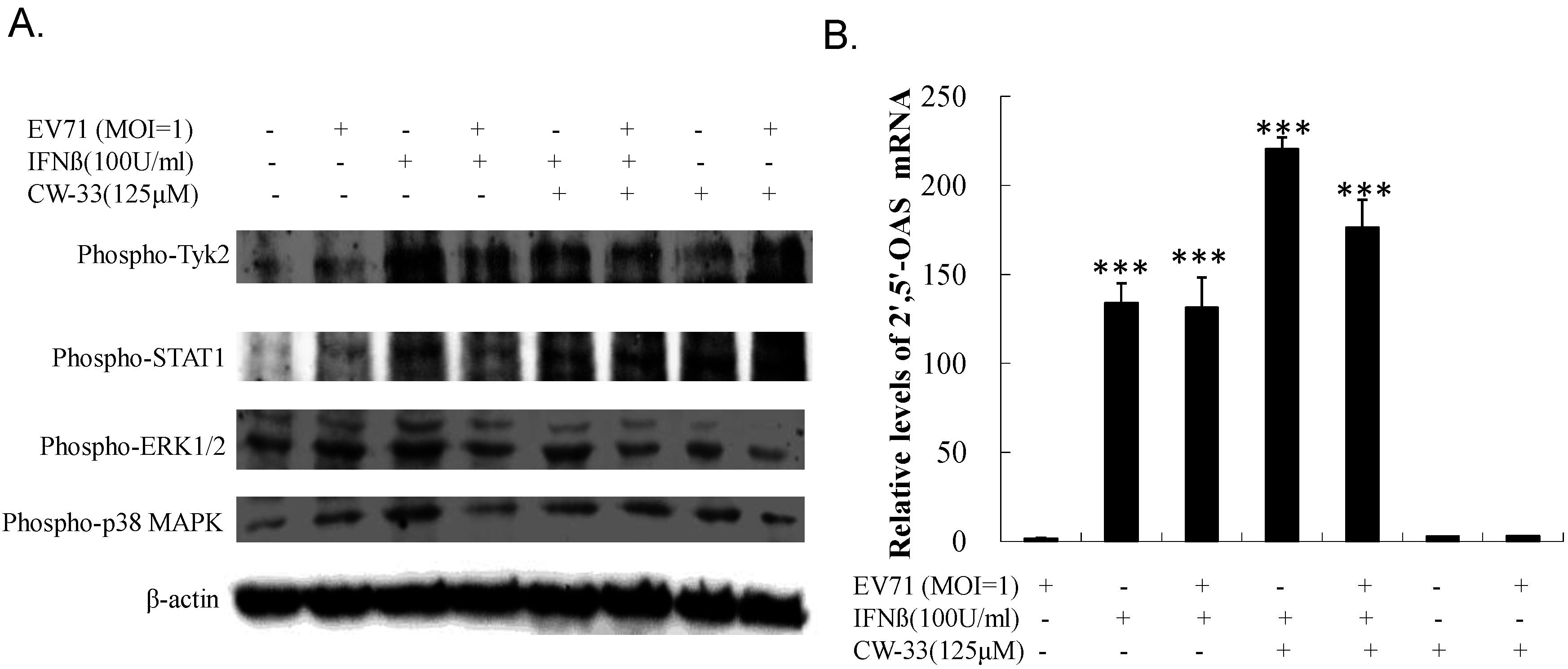
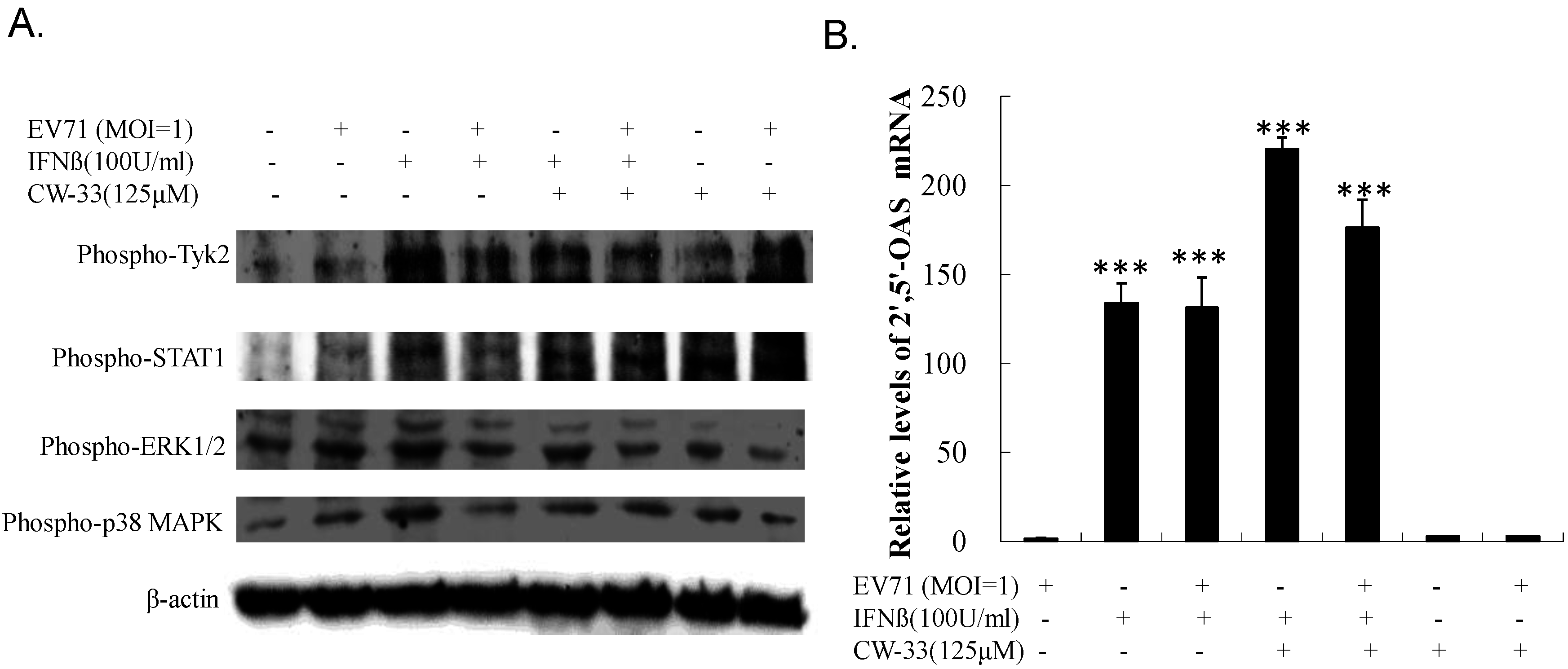
3.4. Recovery of IFN-Stimulated Tyk2/ STAT1 Signaling in Infected Cells by CW-33
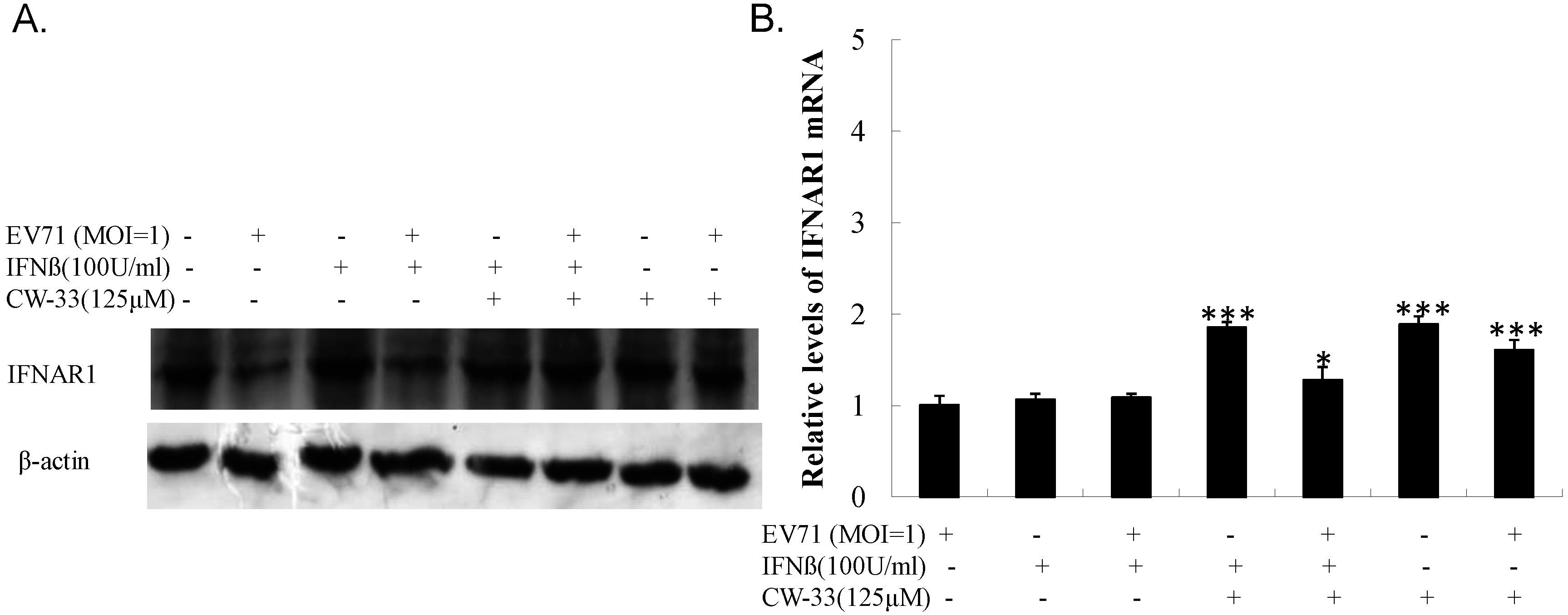
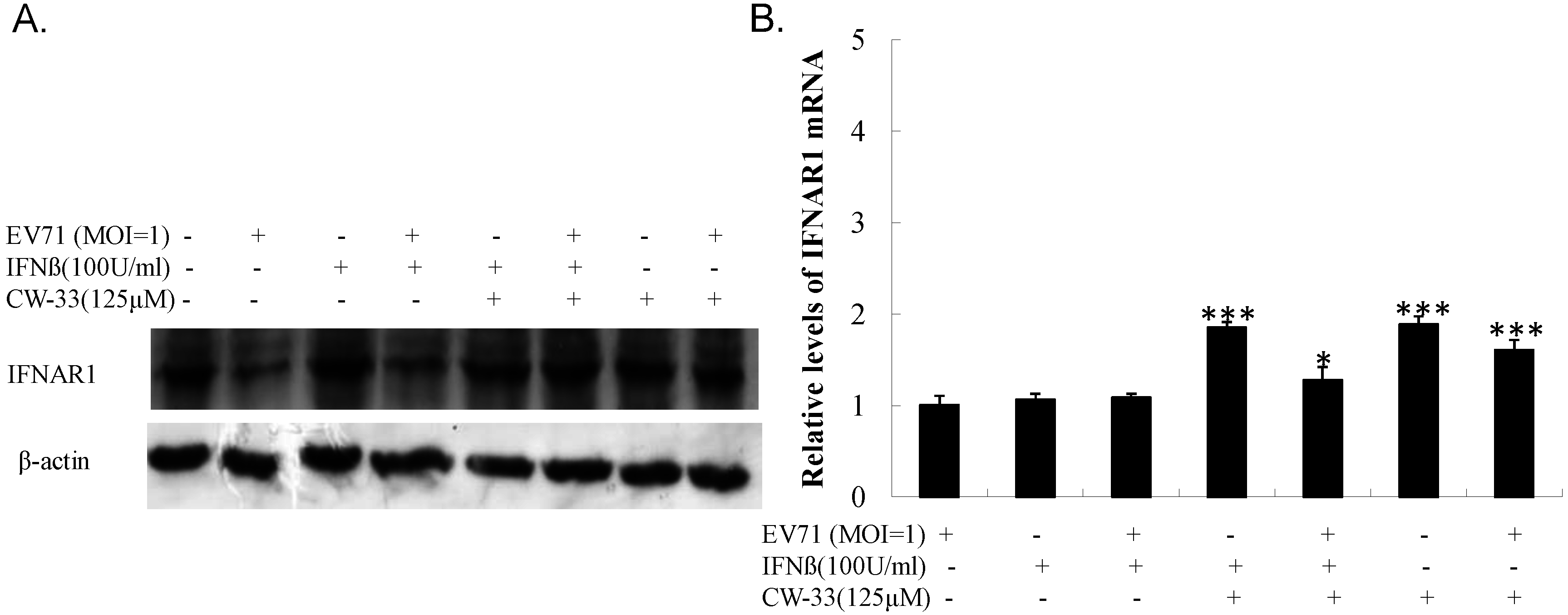
3.5. Inhibitory Effect on Viral 2A Protease-Mediated Cleavage of IFNAR1
4. Discussion
Acknowledgements
Transparency Declarations
Author Contributions
Abbreviations
Conflicts of Interest
References
- Lin, T.Y.; Twu, S.J.; Ho, M.S.; Chang, Y.L.; Lee, C.Y. Enterovirus 71 outbreaks, Taiwan: Occurrence and recognition. Emerg. Infect. Dis. 2003, 9, 291–293. [Google Scholar] [CrossRef] [PubMed]
- Melnick, J.L. The discovery of the enteroviruses and the classification of poliovirus among them. Biologicals 1993, 21, 305–309. [Google Scholar] [CrossRef] [PubMed]
- Mettenleiter, T.C.; Sobrino, F. Foot-and-Mouth Disease Virus. In Animal Viruses: Molecular Biology; Caister Academic Press: Norfolk, UK, 2008. [Google Scholar]
- Oberste, M.S.; Maher, K.; Kilpatrick, D.R.; Flemister, M.R.; Brown, B.A.; Pallansch, M.A. Typing of human enteroviruses by partial sequencing of VP1. J. Clin. Microbiol. 1999, 37, 1288–1293. [Google Scholar] [PubMed]
- Wang, J.R.; Tuan, Y.C.; Tsai, H.P.; Yan, J.J.; Liu, C.C.; Su, I.J. Change of major genotype of enterovirus 71 in outbreaks of hand-foot-and-mouth disease in Taiwan between 1998 and 2000. J. Clin. Microbiol. 2002, 40, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Shang, L.; Xu, M.; Yin, Z. Antiviral drug discovery for the treatment of enterovirus 71 infections. Antivir. Res. 2013, 97, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Cooksley, W.G. The role of interferon therapy in hepatitis B. Med. Gen. Med. 2004, 6, 16. [Google Scholar]
- Shepherd, J.; Waugh, N.; Hewitson, P. Combination therapy (interferon alfa and ribavirin) in the treatment of chronic hepatitis C: A rapid and systematic review. Health Technol. Assess. 2000, 4, 1–67. [Google Scholar] [PubMed]
- Li, Z.H.; Li, C.M.; Ling, P.; Shen, F.H.; Chen, S.H.; Liu, C.C.; Yu, C.K.; Chen, S.H. Ribavirin reduces mortality in enterovirus 71-infected mice by decreasing viral replication. J. Infect. Dis. 2008, 197, 854–857. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.L.; Lee, Y.P.; Wang, Y.F.; Lei, H.Y.; Liu, C.C.; Wang, S.M.; Su, I.J.; Wang, J.R.; Yeh, T.M.; Chen, S.H.; et al. Type I interferons protect mice against enterovirus 71 infection. J. Gen. Virol. 2005, 86, 3263–3269. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Yi, L.; Zhao, J.; Yu, J.; Chen, Y.; Lin, M.C.; Kung, H.F.; He, M.L. Enterovirus 71 disrupts interferon signaling by reducing the level of interferon receptor 1. J. Virol. 2012, 86, 3767–3776. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Zhou, F.; Gu, B.; Dong, C.; Feng, D.; Xie, F.; Wang, J.; Zhang, C.; Cao, Q.; Deng, Y.; et al. In vitro and in vivo evaluation of ribavirin and pleconaril antiviral activity against enterovirus 71 infection. Arch. Virol. 2012, 157, 669–679. [Google Scholar] [CrossRef] [PubMed]
- Dragovich, P.S.; Prins, T.J.; Zhou, R.; Webber, S.E.; Marakovits, J.T.; Fuhrman, S.A.; Patick, A.K.; Matthews, D.A.; Lee, C.A.; Ford, C.E.; et al. Structure-based design, synthesis, and biological evaluation of irreversible human rhinovirus 3C protease inhibitors. 4. Incorporation of P1 lactam moieties as L-glutamine replacements. J. Med. Chem. 1999, 42, 1213–1224. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.N.; Song, Z.G.; Jiang, T.; Shi, B.S.; Hu, Y.W.; Yuan, Z.H. Rupintrivir is a promising candidate for treating severe cases of Enterovirus-71 infection. World J. Gastroenterol. 2010, 16, 201–209. [Google Scholar] [CrossRef] [PubMed]
- Shia, K.S.; Shih, S.R.; Chang, C.M. Imidazolidinone Compounds. US Patant US-6706739, 16 March 2004. [Google Scholar]
- Cui, S.; Wang, J.; Fan, T.; Qin, B.; Guo, L.; Lei, X.; Wang, M.; Jin, Q. Crystal structure of human enterovirus 71 3C protease. J. Mol. Biol. 2011, 408, 449–461. [Google Scholar] [CrossRef] [PubMed]
- Hung, H.C.; Wang, H.C.; Shih, S.R.; Teng, I.F.; Tseng, C.P.; Hsu, J.T. Synergistic inhibition of enterovirus 71 replication by interferon and rupintrivir. J. Infect. Dis. 2011, 203, 1784–1790. [Google Scholar] [CrossRef] [PubMed]
- Tarus, P.K.; Coombes, P.H.; Crouch, N.R.; Mulholland, D.A.; Moodley, B. Furoquinoline alkaloids from the southern African Rutaceae Teclea natalensis. Phytochemistry 2005, 66, 703–706. [Google Scholar] [CrossRef] [PubMed]
- Zhao, W.; Wolfender, J.L.; Hostettmann, K.; Xu, R.; Qin, G. Antifungal alkaloids and limonoid derivatives from Dictamnus dasycarpus. Phytochemistry 1998, 47, 7–11. [Google Scholar] [CrossRef]
- Severino, V.G.; da Silva, M.F.; Lucarini, R.; Montanari, L.B.; Cunha, W.R.; Vinholis, A.H.; Martins, C.H. Determination of the antibacterial activity of crude extracts and compounds isolated from Hortia oreadica (Rutaceae) against oral pathogens. Braz. J. Microbiol. 2009, 40, 535–540. [Google Scholar] [CrossRef] [PubMed]
- Kiplimo, J.J.; Islam, M.S.; Koorbanally, N.A. A novel flavonoid and furoquinoline alkaloids from Vepris glomerata and their antioxidant activity. Nat. Prod. Commun. 2011, 6, 1847–1850. [Google Scholar] [PubMed]
- Wansi, J.D.; Mesaik, M.A.; Chiozem, D.D.; Devkota, K.P.; Gaboriaud-Kolar, N.; Lallemand, M.C.; Wandji, J.; Choudhary, M.I.; Sewald, N. Oxidative burst inhibitory and cytotoxic indoloquinazoline and furoquinoline alkaloids from Oricia suaveolens. J. Nat. Prod. 2008, 71, 1942–1945. [Google Scholar] [CrossRef] [PubMed]
- Kuo, S.C.; Huang, S.C.; Hung, L.J.; Cheng, H.E.; Lin, T.P.; Wu, C.H.; Ishii, K.; Nakamura, H. Studies of heterocyclic compounds. VIII. Synthesis, anti-inflammatory and antiallergic activities of N-alkyl-2,3,4,9-tetrahydrofuro[2,3-b]quinoline-3,4-diones and related compounds. J. Heterocycl. Chem. 1991, 28, 955–963. [Google Scholar] [CrossRef]
- Su, M.J.; Chang, G.J.; Wu, M.H.; Kuo, S.C. Electrophysiological basis for the antiarrhythmic action and positive inotropy of HA-7, a furoquinoline alkaloid derivative, in rat heart. Br. J. Pharmacol. 1997, 122, 1285–1298. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.C.; Lin, T.H.; Tsai, J.D.; Yang, Y.C.; Peng, C.T.; Shih, M.C.; Lin, Y.J.; Lin, C.W. Molecular epidemiology of the 2005 enterovirus 71 outbreak in central Taiwan. Scand. J. Infect. Dis. 2011, 43, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Huang, S.C.; Zhang, Y.; Lai, Z.R.; Kung, S.H.; Chang, Y.S.; Lin, C.W. Antiviral ability of Kalanchoe gracilis leaf extract against Enterovirus 71 and Coxsackievirus A16. Evid. Based Complement. Alternat. Med. 2012, 2012, e503165. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.Y.; Huang, S.C.; Lai, Z.R.; Ho, Y.L.; Jou, Y.J.; Kung, S.H.; Zhang, Y.; Chang, Y.S.; Lin, C.W. Eupafolin and Ethyl acetate fraction of Kalanchoe gracilis stem extract show potent antiviral activities against Enterovirus 71 and Coxsackievirus A16. Evid. Based Complement. Alternat. Med. 2013, 2013. [Google Scholar] [CrossRef]
- Mu, Z.; Wang, B.; Zhang, X.; Gao, X.; Qin, B.; Zhao, Z.; Cui, S. Crystal structure of 2A proteinase from hand, foot and mouth disease virus. J. Mol. Biol. 2013, 425, 4530–4543. [Google Scholar] [CrossRef] [PubMed]
- Yi, L.; He, Y.; Chen, Y.; Kung, H.F.; He, M.L. Potent inhibition of human enterovirus 71replication by type I interferon subtypes. Antivir. Ther. 2011, 16, 51–58. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.-Y.; Huang, A.-C.; Hour, M.-J.; Huang, S.-H.; Kung, S.-H.; Chen, C.-H.; Chen, I.-C.; Chang, Y.-S.; Lien, J.-C.; Lin, C.-W. Antiviral Potential of a Novel Compound CW-33 against Enterovirus A71 via Inhibition of Viral 2A Protease. Viruses 2015, 7, 3155-3171. https://doi.org/10.3390/v7062764
Wang C-Y, Huang A-C, Hour M-J, Huang S-H, Kung S-H, Chen C-H, Chen I-C, Chang Y-S, Lien J-C, Lin C-W. Antiviral Potential of a Novel Compound CW-33 against Enterovirus A71 via Inhibition of Viral 2A Protease. Viruses. 2015; 7(6):3155-3171. https://doi.org/10.3390/v7062764
Chicago/Turabian StyleWang, Ching-Ying, An-Cheng Huang, Mann-Jen Hour, Su-Hua Huang, Szu-Hao Kung, Chao-Hsien Chen, I-Chieh Chen, Yuan-Shiun Chang, Jin-Cherng Lien, and Cheng-Wen Lin. 2015. "Antiviral Potential of a Novel Compound CW-33 against Enterovirus A71 via Inhibition of Viral 2A Protease" Viruses 7, no. 6: 3155-3171. https://doi.org/10.3390/v7062764