
Recombinant Immunomodulating Lentogenic or Mesogenic Oncolytic Newcastle Disease Virus for Treatment of Pancreatic Adenocarcinoma

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
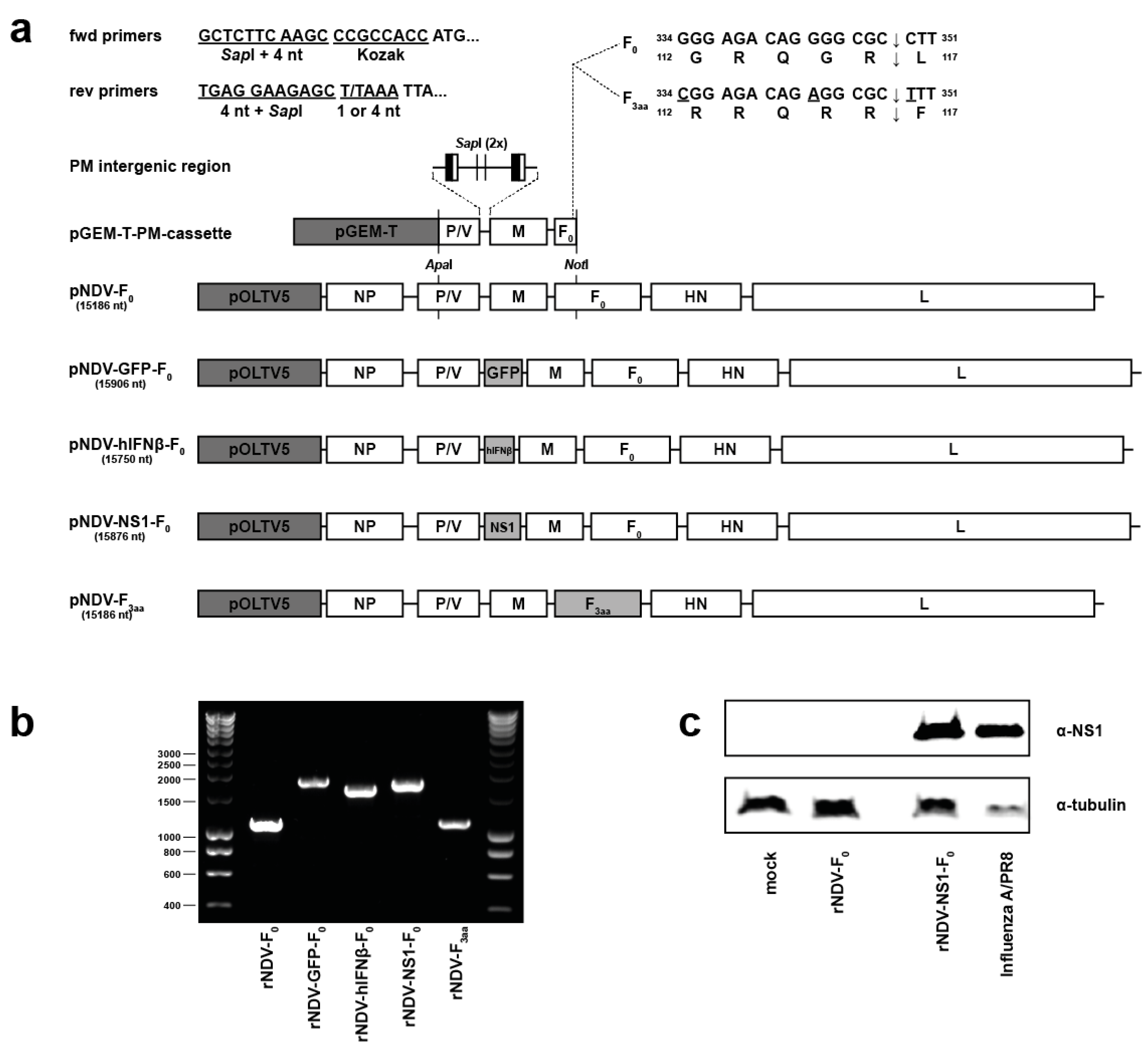
2.1. pNDV Cloning
2.2. Recombinant Virus Rescue
2.3. Titration of rNDV
2.4. Characterization of rNDV
2.5. Cell Lines and Culture Conditions
2.6. Replication Curves
2.7. RNA Isolation and Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR) for hIFNβ mRNA
2.8. IFN Measurement with Luciferase Bioassay
2.9. Cytotoxicity Assay
2.10. Ethics Statement
2.11. Animals and Experimental Design
3. Results
3.1. Cloning, Rescue and Characterization of Recombinant NDVs
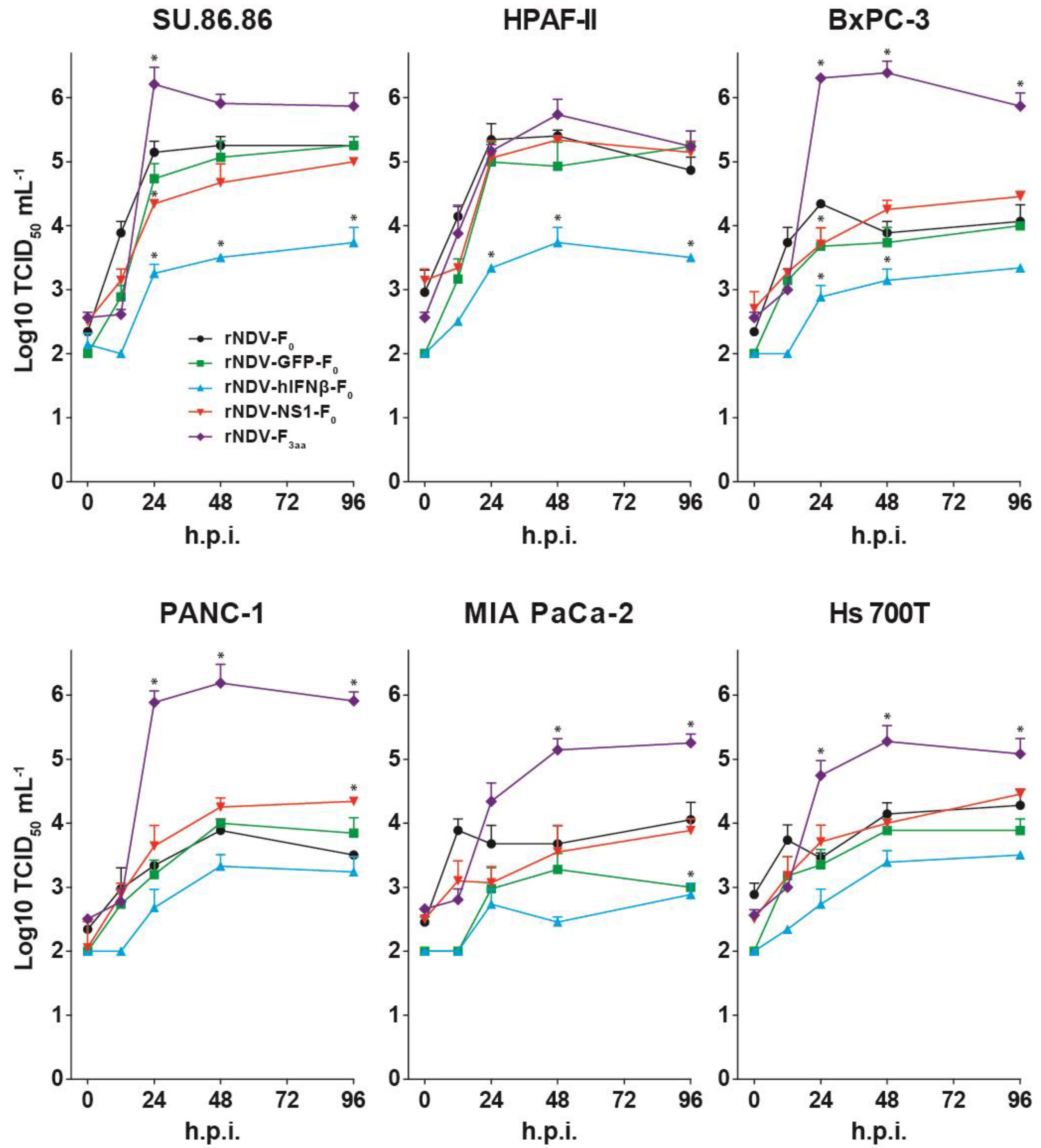
3.2. Replication Kinetics of rNDVs

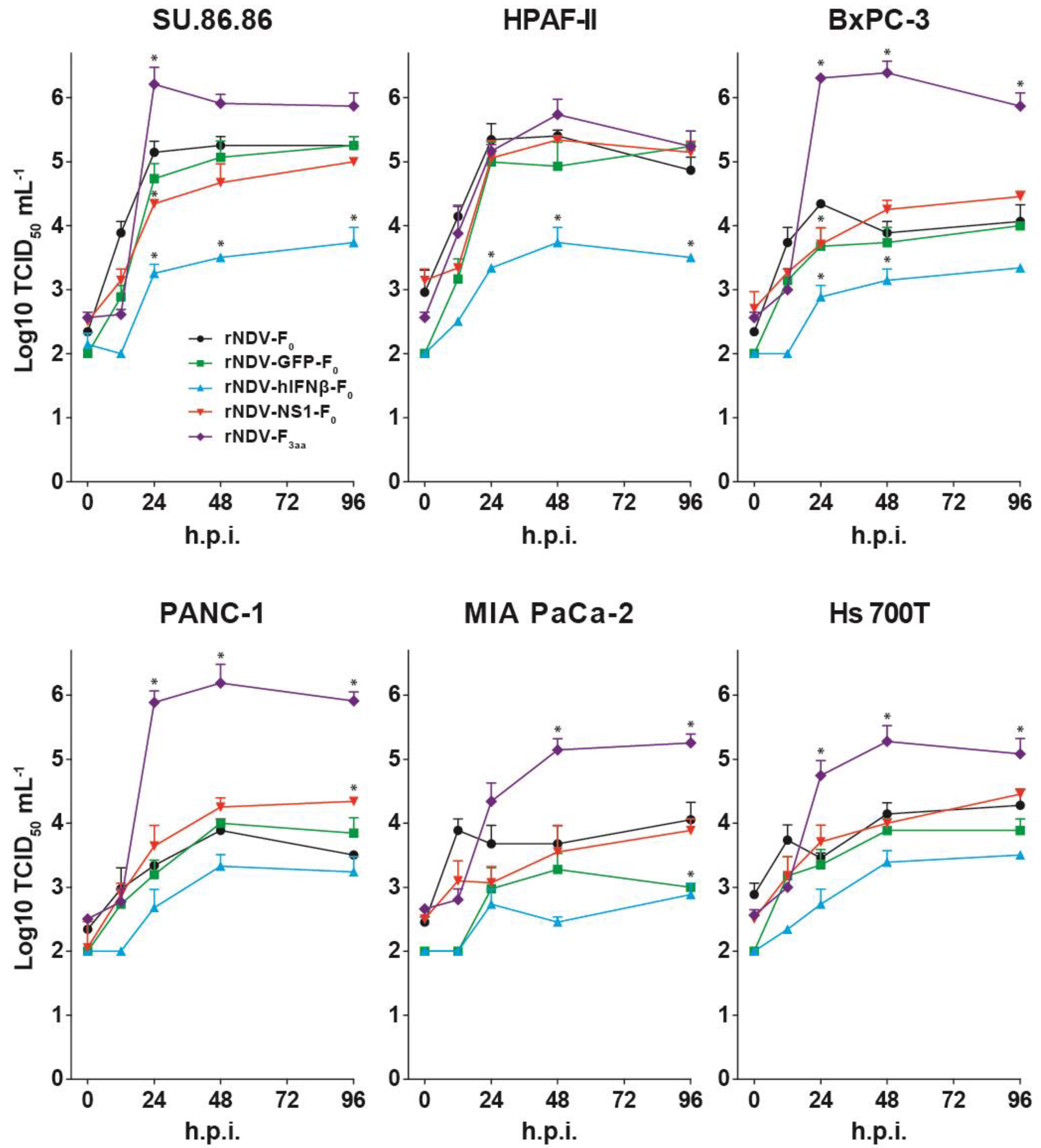
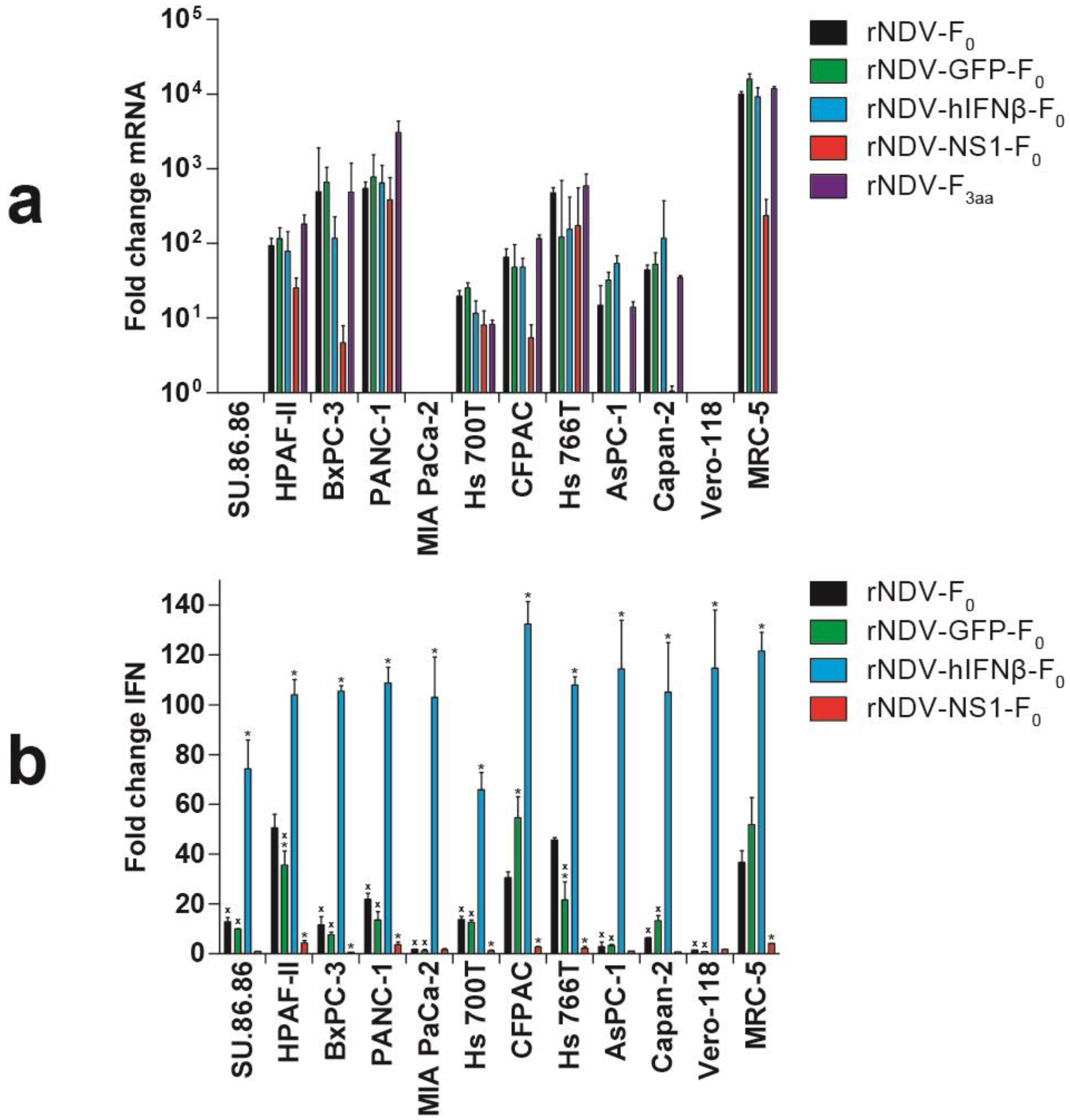
3.3. Modulation of IFN Response by rNDVs
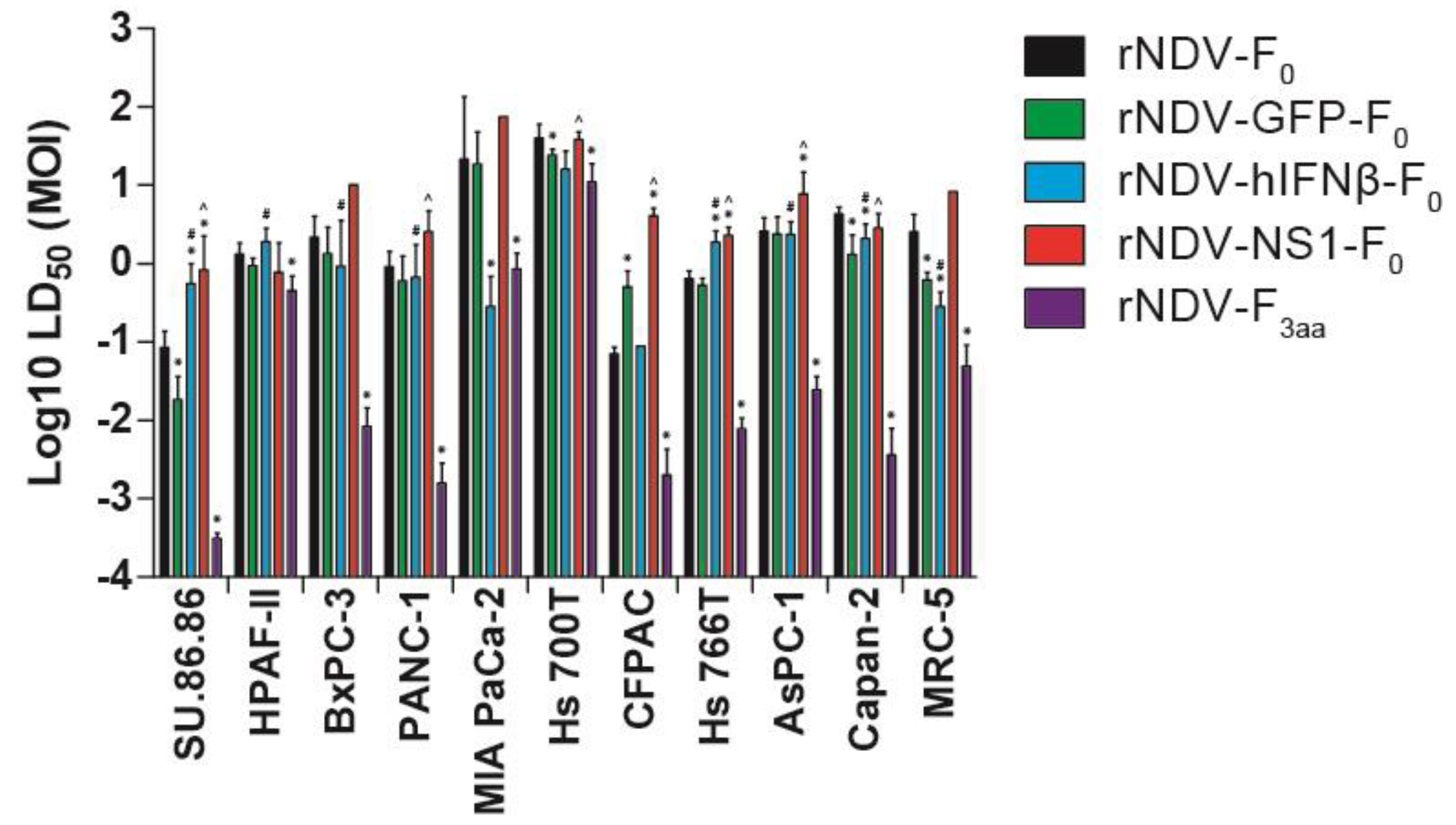
3.4. In Vitro Cytotoxicity


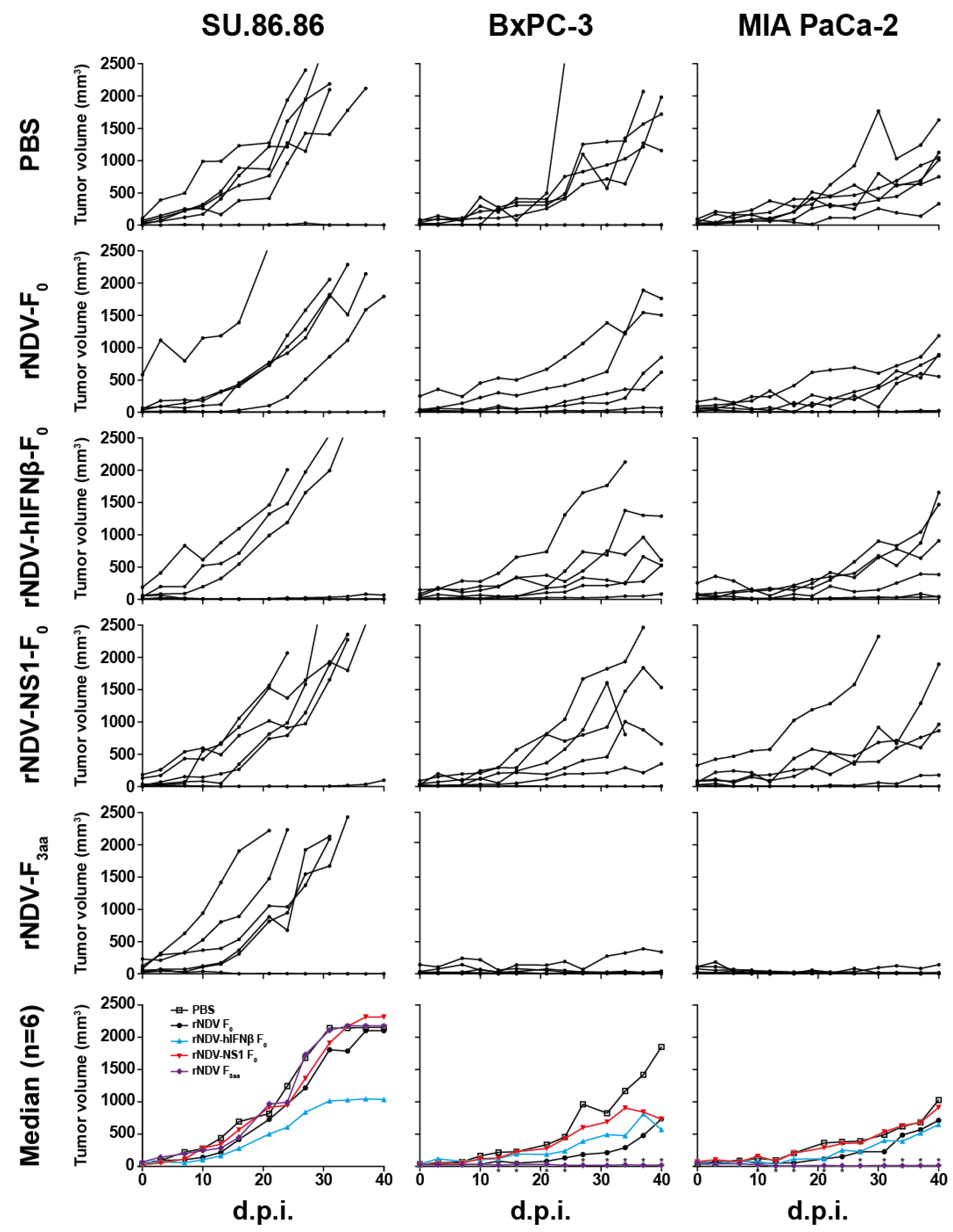
3.5. In Vivo Efficacy: rNDV-F3aa Effective in Multiple Models
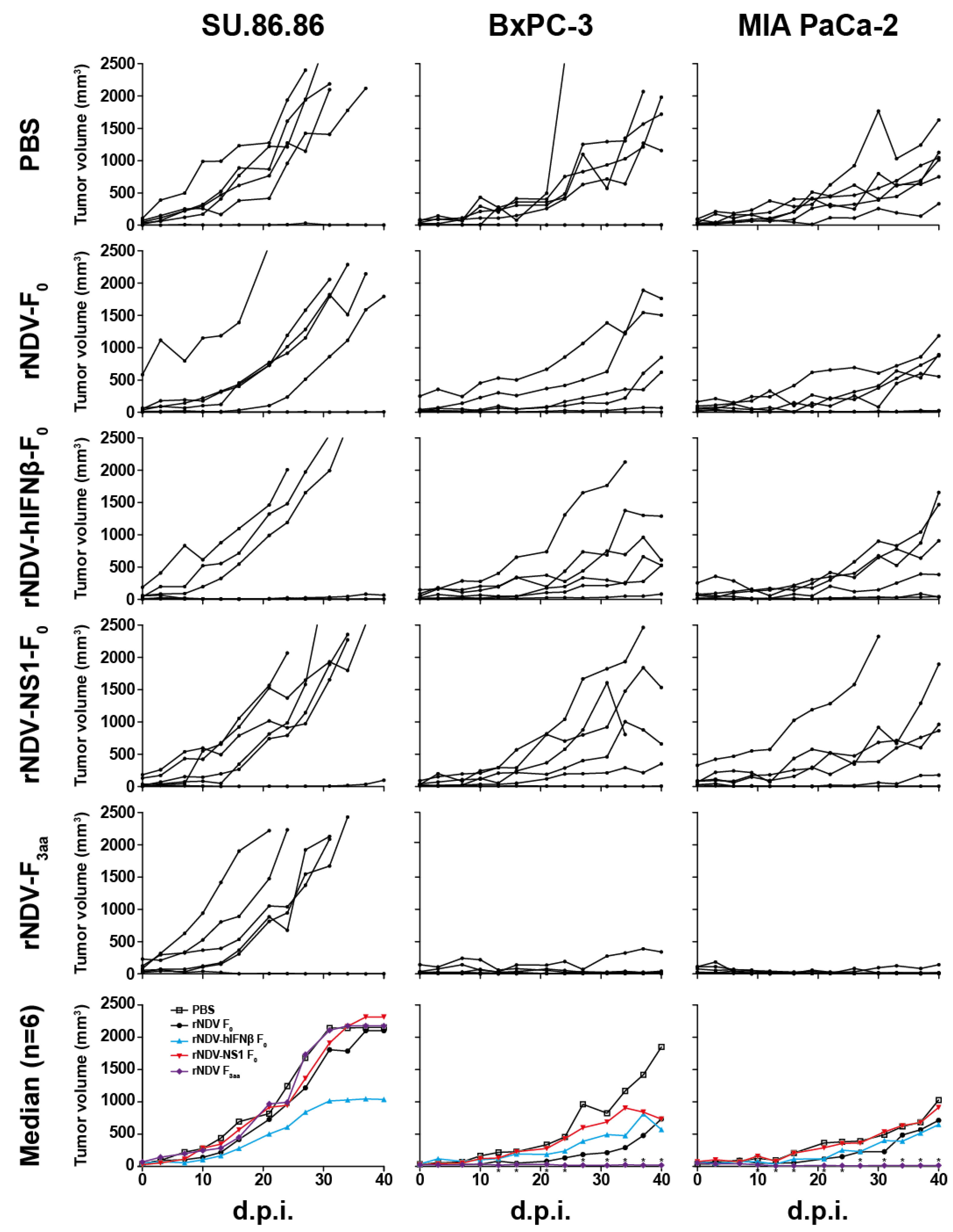
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Sultana, A.; Smith, C.T.; Cunningham, D.; Starling, N.; Neoptolemos, J.P.; Ghaneh, P. Meta-analyses of chemotherapy for locally advanced and metastatic pancreatic cancer. J. Clin. Oncol. 2007, 25, 2607–2615. [Google Scholar] [CrossRef] [PubMed]
- Neoptolemos, J.P.; Stocken, D.D.; Bassi, C.; Ghaneh, P.; Cunningham, D.; Goldstein, D.; Padbury, R.; Moore, M.J.; Gallinger, S.; Mariette, C.; et al. Adjuvant chemotherapy with fluorouracil plus folinic acid vs gemcitabine following pancreatic cancer resection: A randomized controlled trial. JAMA 2010, 304, 1073–1081. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.E.; Diamond, L.C.; Mackay, H.H.; Sabachewsky, L. Influence of hemagglutinating viruses on tumor cell suspensions. Ii. Newcastle disease virus and ehrlich carcinoma. Proc. Soc. Exp. Biol. Med. 1952, 81, 498–501. [Google Scholar] [CrossRef] [PubMed]
- Prince, A.M.; Ginsberg, H.S. Studies on the cytotoxic effect of newcastle disease virus (NDV) on ehrlich ascites tumor cells. Ii. The mechanism and significance of in vitro recovery from the effect of ndv. J. Immunol. 1957, 79, 107–112. [Google Scholar] [PubMed]
- Prince, A.M.; Ginsberg, H.S. Studies on the cytotoxic effect of newcastle disease virus (NDV) on ehrlich ascites tumor cells. I. Characteristics of the virus-cell interaction. J. Immunol. 1957, 79, 94–106. [Google Scholar] [PubMed]
- Lorence, R.M.; Roberts, M.S.; O'Neil, J.D.; Groene, W.S.; Miller, J.A.; Mueller, S.N.; Bamat, M.K. Phase 1 clinical experience using intravenous administration of pv701, an oncolytic newcastle disease virus. Curr. Cancer Drug Targets 2007, 7, 157–167. [Google Scholar] [CrossRef] [PubMed]
- Freeman, A.I.; Zakay-Rones, Z.; Gomori, J.M.; Linetsky, E.; Rasooly, L.; Greenbaum, E.; Rozenman-Yair, S.; Panet, A.; Libson, E.; Irving, C.S.; et al. Phase I/II trial of intravenous NDV-HUJ oncolytic virus in recurrent glioblastoma multiforme. Mol. Ther. 2006, 13, 221–228. [Google Scholar] [CrossRef] [PubMed]
- Pecora, A.L.; Rizvi, N.; Cohen, G.I.; Meropol, N.J.; Sterman, D.; Marshall, J.L.; Goldberg, S.; Gross, P.; O'Neil, J.D.; Groene, W.S.; et al. Phase i trial of intravenous administration of pv701, an oncolytic virus, in patients with advanced solid cancers. J. Clin. Oncol. 2002, 20, 2251–2266. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V. Clinical trials of antitumor vaccination with an autologous tumor cell vaccine modified by virus infection: Improvement of patient survival based on improved antitumor immune memory. Cancer Immunol. Immunother. 2005, 54, 587–598. [Google Scholar] [CrossRef] [PubMed]
- Cassel, W.A.; Murray, D.R. A ten-year follow-up on stage ii malignant melanoma patients treated postsurgically with newcastle disease virus oncolysate. Med. Oncol. Tumor Pharmacother. 1992, 9, 169–171. [Google Scholar] [PubMed]
- Sinkovics, J.G.; Horvath, J.C. Newcastle disease virus (ndv): Brief history of its oncolytic strains. J. Clin. Virol. 2000, 16, 1–15. [Google Scholar] [CrossRef]
- Peeters, B.P.; de Leeuw, O.S.; Koch, G.; Gielkens, A.L. Rescue of newcastle disease virus from cloned cdna: Evidence that cleavability of the fusion protein is a major determinant for virulence. J. Virol. 1999, 73, 5001–5009. [Google Scholar] [PubMed]
- Zamarin, D.; Palese, P. Oncolytic newcastle disease virus for cancer therapy: Old challenges and new directions. Future Microbiol. 2012, 7, 347–367. [Google Scholar] [CrossRef] [PubMed]
- Buijs, P.R.; van Eijck, C.H.; Hofland, L.J.; Fouchier, R.A.; van den Hoogen, B.G. Different responses of human pancreatic adenocarcinoma cell lines to oncolytic newcastle disease virus infection. Cancer Gene Ther. 2014, 21, 24–30. [Google Scholar] [CrossRef] [PubMed]
- Morak, M.J.; van Koetsveld, P.M.; Kanaar, R.; Hofland, L.J.; van Eijck, C.H. Type I interferons as radiosensitisers for pancreatic cancer. Eur. J. Cancer 2011, 47, 1938–1945. [Google Scholar] [CrossRef] [PubMed]
- Vitale, G.; van Eijck, C.H.; van Koetsveld Ing, P.M.; Erdmann, J.I.; Speel, E.J.; van der Wansem Ing, K.; Mooij, D.M.; Colao, A.; Lombardi, G.; Croze, E.; et al. Type i interferons in the treatment of pancreatic cancer: Mechanisms of action and role of related receptors. Ann. Surg. 2007, 246, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Levy, D.E.; Garcia-Sastre, A. The virus battles: Ifn induction of the antiviral state and mechanisms of viral evasion. Cytokine Growth Factor Rev. 2001, 12, 143–156. [Google Scholar] [CrossRef]
- Booy, S.; van Eijck, C.H.; Dogan, F.; van Koetsveld, P.M.; Hofland, L.J. Influence of type-i interferon receptor expression level on the response to type-I interferons in human pancreatic cancer cells. J. Cell. Mol. Med. 2014, 18, 492–502. [Google Scholar] [CrossRef] [PubMed]
- Zamarin, D.; Holmgaard, R.B.; Subudhi, S.K.; Park, J.S.; Mansour, M.; Palese, P.; Merghoub, T.; Wolchok, J.D.; Allison, J.P. Localized oncolytic virotherapy overcomes systemic tumor resistance to immune checkpoint blockade immunotherapy. Sci. Transl. Med. 2014, 6, 226ra232. [Google Scholar] [CrossRef] [PubMed]
- Van de Sandt, C.E.; Kreijtz, J.H.; Rimmelzwaan, G.F. Evasion of influenza a viruses from innate and adaptive immune responses. Viruses 2012, 4, 1438–1476. [Google Scholar] [CrossRef] [PubMed]
- Kochs, G.; Garcia-Sastre, A.; Martinez-Sobrido, L. Multiple anti-interferon actions of the influenza a virus ns1 protein. J. Virol. 2007, 81, 7011–7021. [Google Scholar] [CrossRef] [PubMed]
- Kortekaas, J.; Dekker, A.; de Boer, S.M.; Weerdmeester, K.; Vloet, R.P.; de Wit, A.A.; Peeters, B.P.; Moormann, R.J. Intramuscular inoculation of calves with an experimental newcastle disease virus-based vector vaccine elicits neutralizing antibodies against rift valley fever virus. Vaccine 2010, 28, 2271–2276. [Google Scholar] [CrossRef] [PubMed]
- Peeters, B.P.; Gruijthuijsen, Y.K.; de Leeuw, O.S.; Gielkens, A.L. Genome replication of newcastle disease virus: Involvement of the rule-of-six. Arch. Virol. 2000, 145, 1829–1845. [Google Scholar] [CrossRef] [PubMed]
- Vigil, A.; Park, M.S.; Martinez, O.; Chua, M.A.; Xiao, S.; Cros, J.F.; Martinez-Sobrido, L.; Woo, S.L.; Garcia-Sastre, A. Use of reverse genetics to enhance the oncolytic properties of newcastle disease virus. Cancer Res. 2007, 67, 8285–8292. [Google Scholar] [CrossRef] [PubMed]
- Hirst, G.K. The quantitative determination of influenza virus and antibodies by means of red cell agglutination. J. Exp. Med. 1942, 75, 49–64. [Google Scholar] [CrossRef] [PubMed]
- De Wit, E.; Spronken, M.I.; Bestebroer, T.M.; Rimmelzwaan, G.F.; Osterhaus, A.D.; Fouchier, R.A. Efficient generation and growth of influenza virus a/pr/8/34 from eight cdna fragments. Virus Res. 2004, 103, 155–161. [Google Scholar] [CrossRef] [PubMed]
- Masters, J.R.; Thomson, J.A.; Daly-Burns, B.; Reid, Y.A.; Dirks, W.G.; Packer, P.; Toji, L.H.; Ohno, T.; Tanabe, H.; Arlett, C.F.; et al. Short tandem repeat profiling provides an international reference standard for human cell lines. Proc. Natl. Acad. Sci. USA 2001, 98, 8012–8017. [Google Scholar] [CrossRef] [PubMed]
- Herfst, S.; de Graaf, M.; Schickli, J.H.; Tang, R.S.; Kaur, J.; Yang, C.F.; Spaete, R.R.; Haller, A.A.; van den Hoogen, B.G.; Osterhaus, A.D.; et al. Recovery of human metapneumovirus genetic lineages a and b from cloned cDNA. J. Virol. 2004, 78, 8264–8270. [Google Scholar] [CrossRef] [PubMed]
- Hs00277188_s1. Available online: http://www.lifetechnologies.com/order/genome-database/details/gene-expression/Hs00277188_s1 (accessed on 9 June 2015).
- Spann, K.M.; Tran, K.C.; Chi, B.; Rabin, R.L.; Collins, P.L. Suppression of the induction of alpha, beta, and lambda interferons by the ns1 and ns2 proteins of human respiratory syncytial virus in human epithelial cells and macrophages [corrected]. J. Virol. 2004, 78, 4363–4369. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-delta delta c(t)) method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef] [PubMed]
- Euhus, D.M.; Hudd, C.; LaRegina, M.C.; Johnson, F.E. Tumor measurement in the nude mouse. J. Surg. Oncol. 1986, 31, 229–234. [Google Scholar] [CrossRef] [PubMed]
- Tomayko, M.M.; Reynolds, C.P. Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother. Pharmacol. 1989, 24, 148–154. [Google Scholar] [CrossRef] [PubMed]
- Zamarin, D.; Martinez-Sobrido, L.; Kelly, K.; Mansour, M.; Sheng, G.; Vigil, A.; Garcia-Sastre, A.; Palese, P.; Fong, Y. Enhancement of oncolytic properties of recombinant newcastle disease virus through antagonism of cellular innate immune responses. Mol. Ther. 2009, 17, 697–706. [Google Scholar] [CrossRef] [PubMed]
- Zamarin, D.; Vigil, A.; Kelly, K.; Garcia-Sastre, A.; Fong, Y. Genetically engineered newcastle disease virus for malignant melanoma therapy. Gene Ther. 2009, 16, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Janke, M.; Fournier, P.; Schirrmacher, V. Recombinant newcastle disease virus expressing human interleukin-2 serves as a potential candidate for tumor therapy. Virus Res. 2008, 136, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Bai, F.; Niu, Z.; Tian, H.; Li, S.; Lv, Z.; Zhang, T.; Ren, G.; Li, D. Genetically-engineered newcastle disease virus expressing interleukin 2 is a potential drug candidate for cancer immunotherapy. Immunol. Lett. 2014, 159, 36–46. [Google Scholar] [CrossRef] [PubMed]
- Bian, H.; Fournier, P.; Moormann, R.; Peeters, B.; Schirrmacher, V. Selective gene transfer in vitro to tumor cells via recombinant newcastle disease virus. Cancer Gene Ther. 2005, 12, 295–303. [Google Scholar] [CrossRef] [PubMed]
- Shobana, R.; Samal, S.K.; Elankumaran, S. Prostate-Specific antigen-retargeted recombinant newcastle disease virus for prostate cancer virotherapy. J. Virol. 2013, 87, 3792–3800. [Google Scholar] [CrossRef] [PubMed]
- Altomonte, J.; Marozin, S.; Schmid, R.M.; Ebert, O. Engineered newcastle disease virus as an improved oncolytic agent against hepatocellular carcinoma. Mol. Ther. 2010, 18, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Silberhumer, G.R.; Brader, P.; Wong, J.; Serganova, I.S.; Gonen, M.; Gonzalez, S.J.; Blasberg, R.; Zamarin, D.; Fong, Y. Genetically engineered oncolytic newcastle disease virus effectively induces sustained remission of malignant pleural mesothelioma. Mol. Cancer Ther. 2010, 9, 2761–2769. [Google Scholar] [CrossRef] [PubMed]
- Song, K.Y.; Wong, J.; Gonzalez, L.; Sheng, G.; Zamarin, D.; Fong, Y. Antitumor efficacy of viral therapy using genetically engineered newcastle disease virus [ndv(f3aa)-gfp] for peritoneally disseminated gastric cancer. J. Mol. Med. 2010, 88, 589–596. [Google Scholar] [CrossRef] [PubMed]
- Li, P.; Chen, C.H.; Li, S.; Givi, B.; Yu, Z.; Zamarin, D.; Palese, P.; Fong, Y.; Wong, R.J. Therapeutic effects of a fusogenic newcastle disease virus in treating head and neck cancer. Head Neck 2011, 33, 1394–1399. [Google Scholar] [CrossRef] [PubMed]
- Obuchi, M.; Fernandez, M.; Barber, G.N. Development of recombinant vesicular stomatitis viruses that exploit defects in host defense to augment specific oncolytic activity. J. Virol. 2003, 77, 8843–8856. [Google Scholar] [CrossRef] [PubMed]
- Saloura, V.; Wang, L.C.; Fridlender, Z.G.; Sun, J.; Cheng, G.; Kapoor, V.; Sterman, D.H.; Harty, R.N.; Okumura, A.; Barber, G.N.; et al. Evaluation of an attenuated vesicular stomatitis virus vector expressing interferon-beta for use in malignant pleural mesothelioma: Heterogeneity in interferon responsiveness defines potential efficacy. Hum. Gene Ther. 2010, 21, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Naik, S.; Nace, R.; Barber, G.N.; Russell, S.J. Potent systemic therapy of multiple myeloma utilizing oncolytic vesicular stomatitis virus coding for interferon-beta. Cancer Gene Ther. 2012, 19, 443–450. [Google Scholar] [CrossRef] [PubMed]
- Kirn, D.H.; Wang, Y.; Le Boeuf, F.; Bell, J.; Thorne, S.H. Targeting of interferon-beta to produce a specific, multi-mechanistic oncolytic vaccinia virus. PLoS Med. 2007, 4, e353. [Google Scholar] [CrossRef] [PubMed]
- Moerdyk-Schauwecker, M.; Shah, N.R.; Murphy, A.M.; Hastie, E.; Mukherjee, P.; Grdzelishvili, V.Z. Resistance of pancreatic cancer cells to oncolytic vesicular stomatitis virus: Role of type I interferon signaling. Virology 2013, 436, 221–234. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Buijs, P.; Van Nieuwkoop, S.; Vaes, V.; Fouchier, R.; Van Eijck, C.; Hoogen, B.V.d. Recombinant Immunomodulating Lentogenic or Mesogenic Oncolytic Newcastle Disease Virus for Treatment of Pancreatic Adenocarcinoma. Viruses 2015, 7, 2980-2998. https://doi.org/10.3390/v7062756
Buijs P, Van Nieuwkoop S, Vaes V, Fouchier R, Van Eijck C, Hoogen BVd. Recombinant Immunomodulating Lentogenic or Mesogenic Oncolytic Newcastle Disease Virus for Treatment of Pancreatic Adenocarcinoma. Viruses. 2015; 7(6):2980-2998. https://doi.org/10.3390/v7062756
Chicago/Turabian StyleBuijs, Pascal, Stefan Van Nieuwkoop, Vincent Vaes, Ron Fouchier, Casper Van Eijck, and Bernadette Van den Hoogen. 2015. "Recombinant Immunomodulating Lentogenic or Mesogenic Oncolytic Newcastle Disease Virus for Treatment of Pancreatic Adenocarcinoma" Viruses 7, no. 6: 2980-2998. https://doi.org/10.3390/v7062756