
HCV Core Protein Uses Multiple Mechanisms to Induce Oxidative Stress in Human Hepatoma Huh7 Cells

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Materials
2.2. Plasmids
2.3. Cell Culture and Transfection
2.4. Immunoblot Analysis
2.5. Confocal Microscopy
2.6. Measurement of Reactive Oxygen Species
2.7. Quantitative RT-PCR (RT-qPCR)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Name | Primer/Probe | Sequence a |
|---|---|---|
| NOX1 | Forward primer | 5′-TTAACAGCACGCTGATCCTG-3′ |
| Reverse primer | 5′-CTGGAGAGAATGGAGGCAAG-3′ | |
| NOX4 | Forward primer | 5′-GCTGACGTTGCATGTTTCAG-3′ |
| Reverse primer | 5′-CGGGAGGGTGGGTATCTAA-3′ | |
| COX-2 | Forward primer | 5′-CCATGTCAAAACCGAGGTGTAT-3′ |
| Reverse primer | 5′-TCCGGTGTTGAGCAGTTTTCT-3′ | |
| TGFβ | Forward primer | 5′-GCAGCACGTGGAGCTGTA-3′ |
| Reverse primer | 5′-CAGCCGGTTGCTGAGGTA-3′ | |
| Ero1α | Forward primer | 5′-GCATTGAAGAAGGTGAGCAA-3′ |
| Reverse primer | 5′-ATCATGCTTGGTCCACTGAA-3′ | |
| CYP2E1 | Forward primer | 5′-GACTGTGGCCGACCTGTT-3′ |
| Reverse primer | 5′-ACTACGACTGTGCCCTTGG-3′ | |
| β-actin | Forward primer | 5′-GATCATTGCTCCTCCTGAGC-3′ |
| Reverse primer | 5′-ACTCCTGCTTGCTGATCCAC-3′ | |
| Probe | 5′-[R6G]-CTCGCTGTCCACCTTCCAGCAGAT-[BHQ-1]-3′ |
2.8. RNA Interference
2.9. Statistical Analysis
3. Results
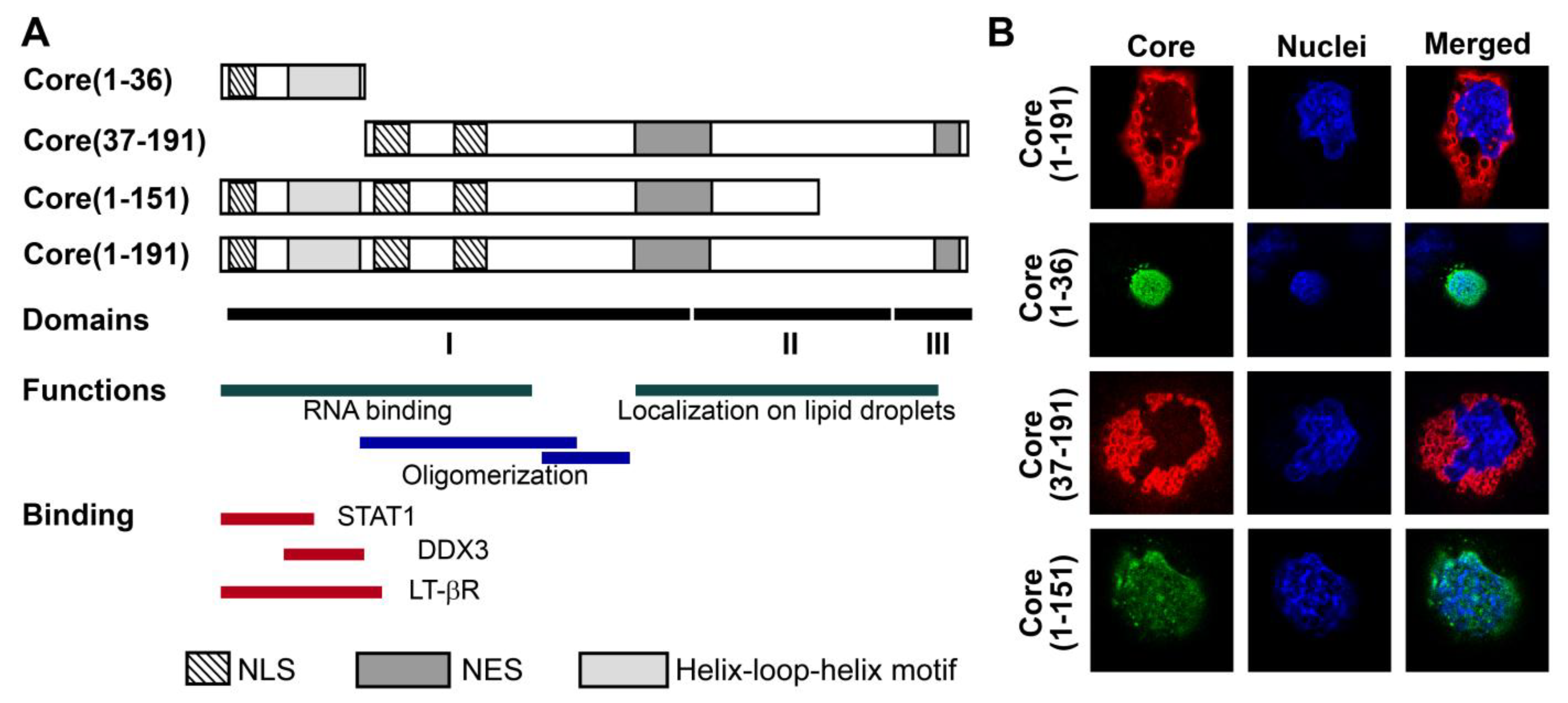
3.1. Panel of Truncated Forms of HCV Core
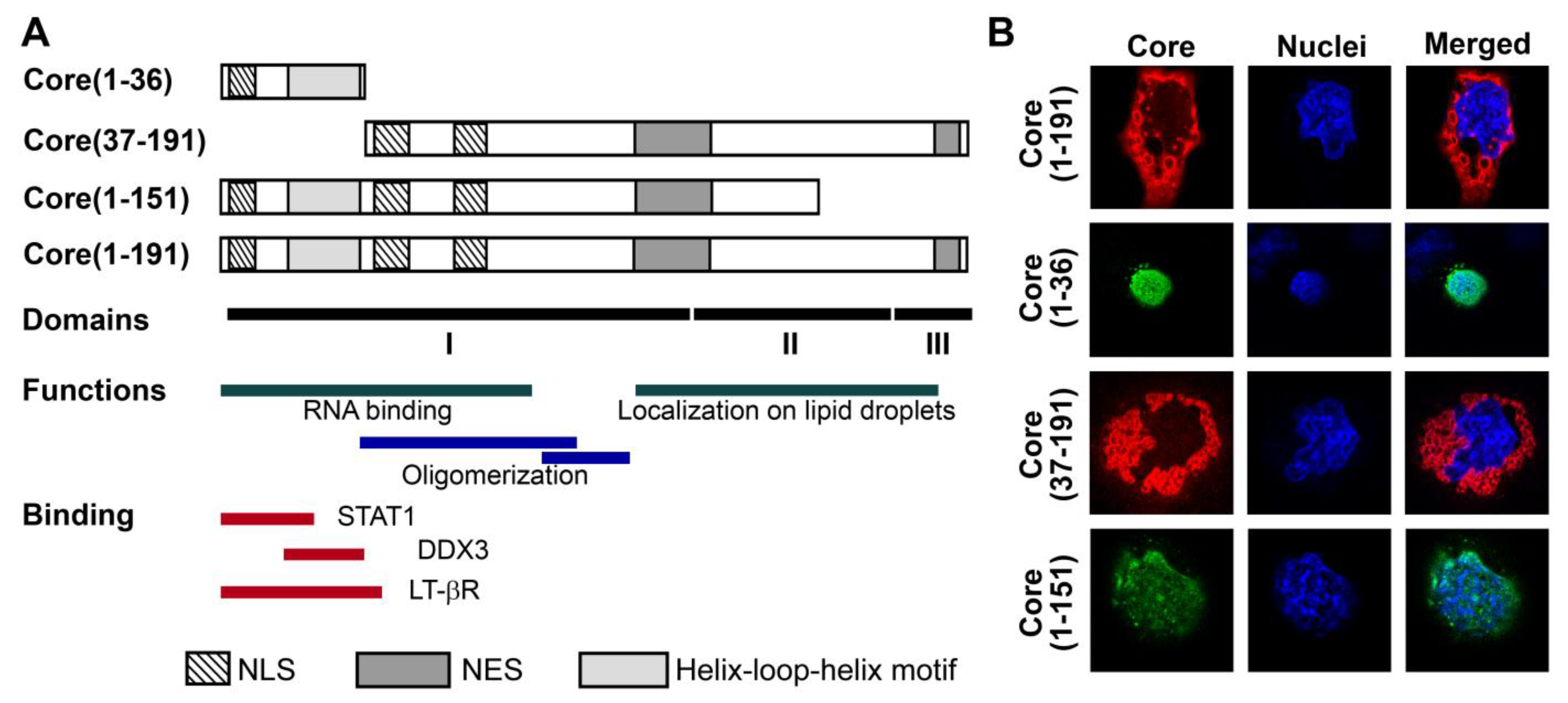
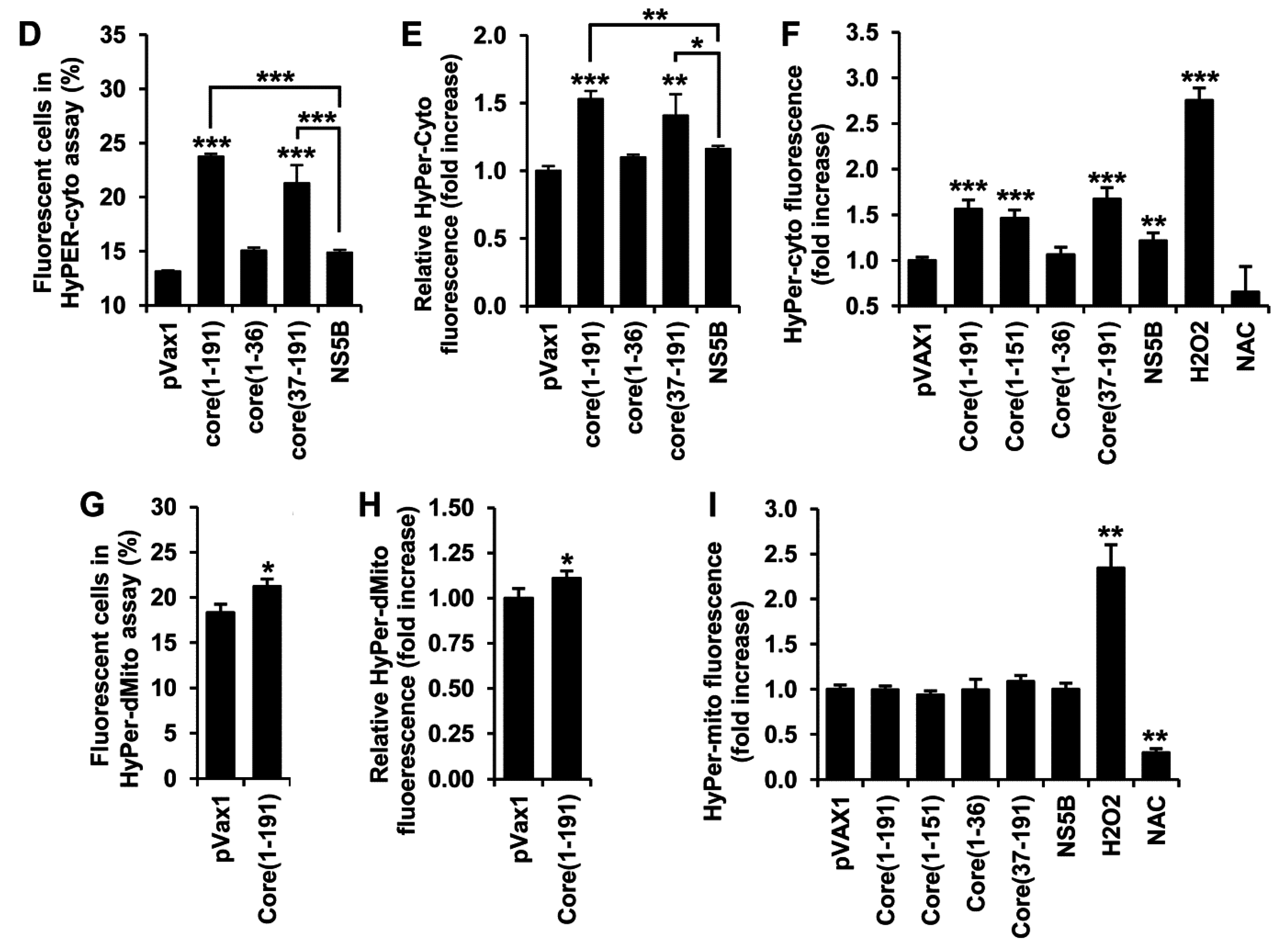
3.2. HCV Core Has Two Superoxide-Anion Inducing Domains
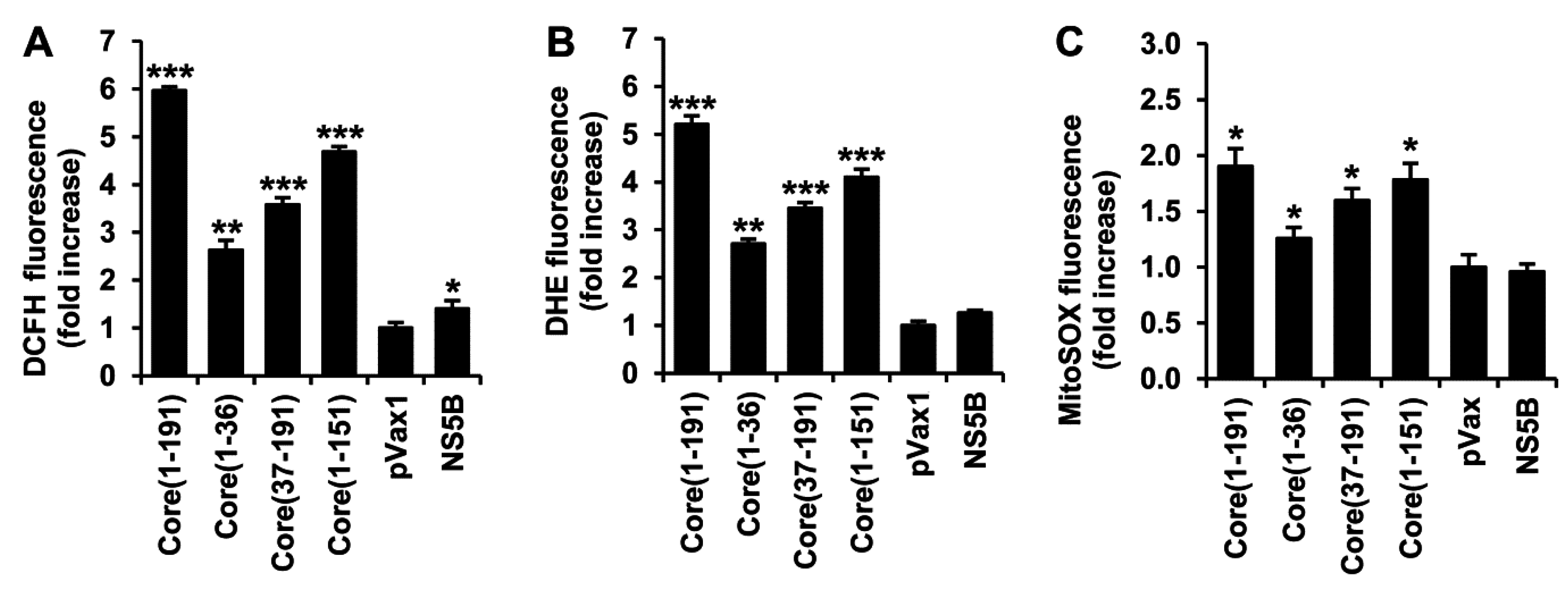
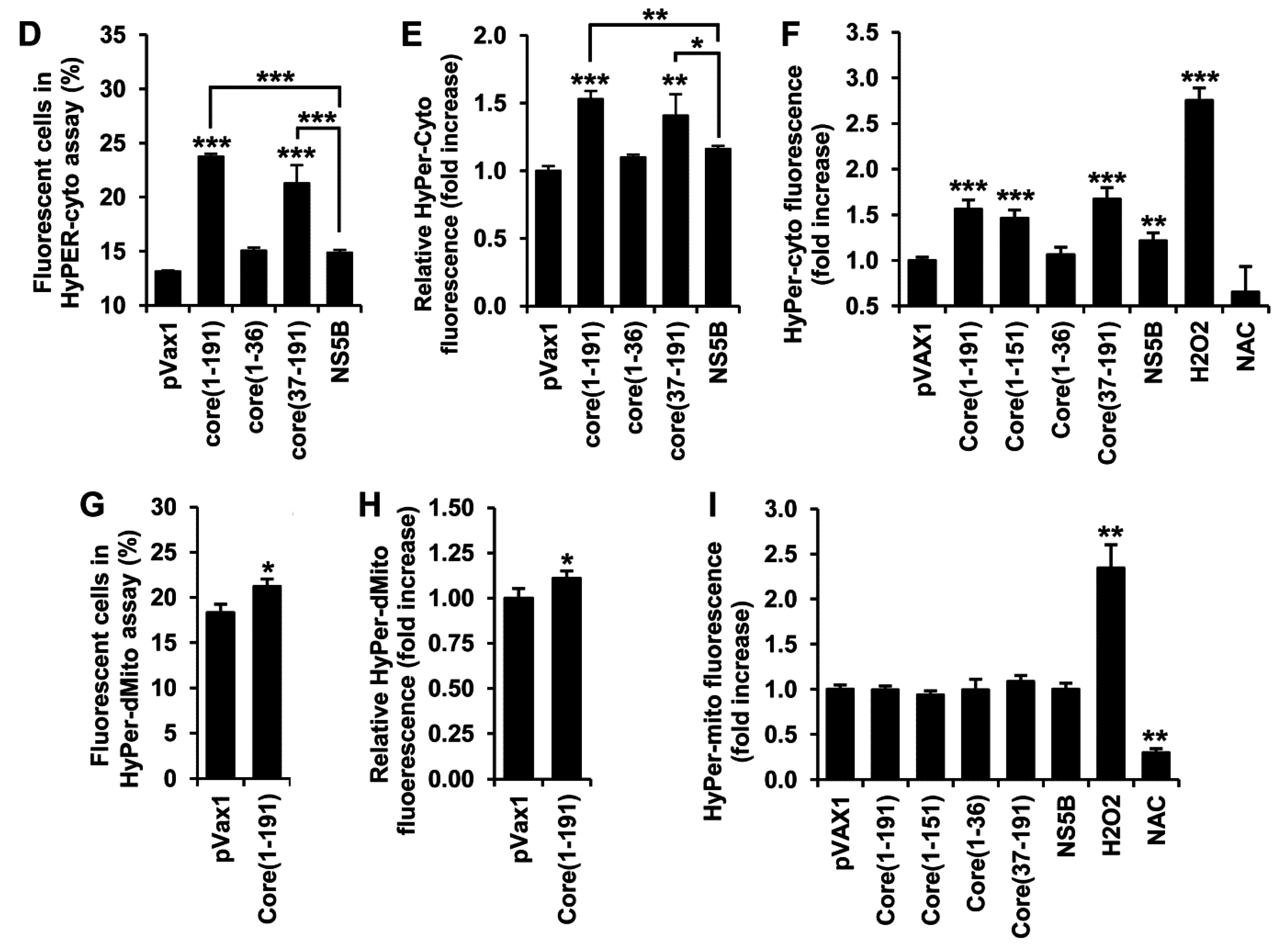
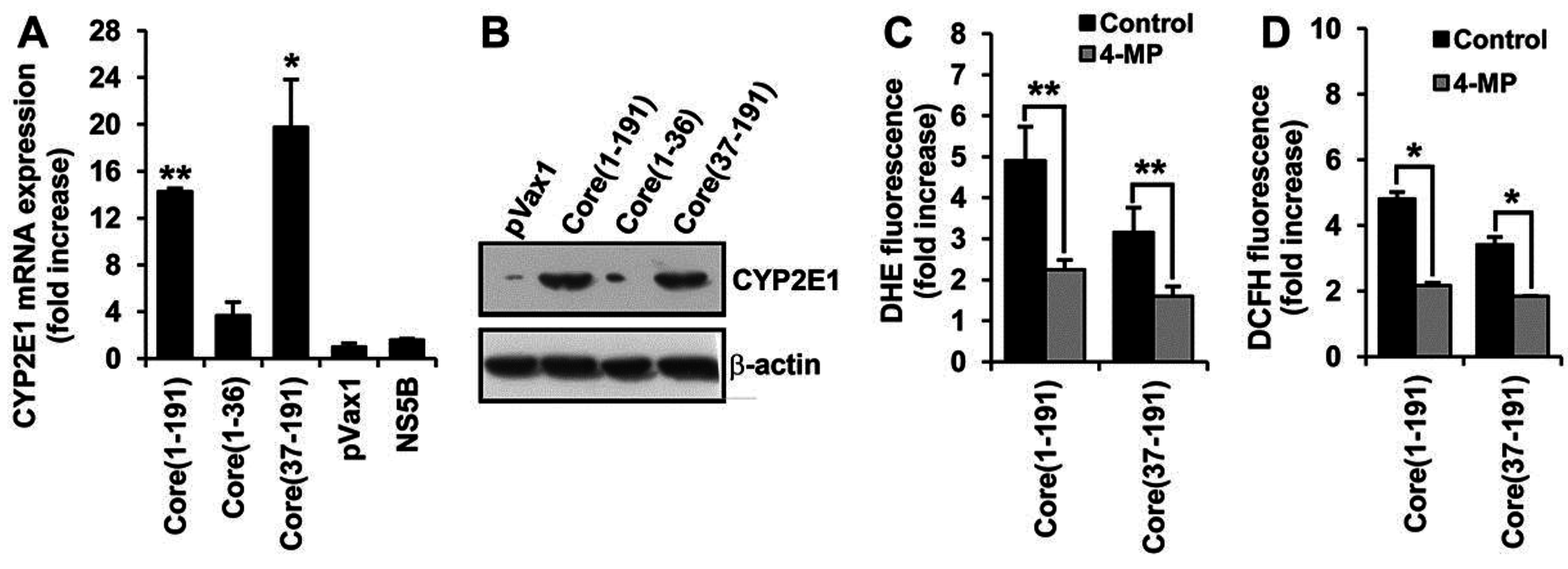
3.3. HCV Core Fragment aa 37-191 Induces a Strong Production of Hydrogen Peroxide
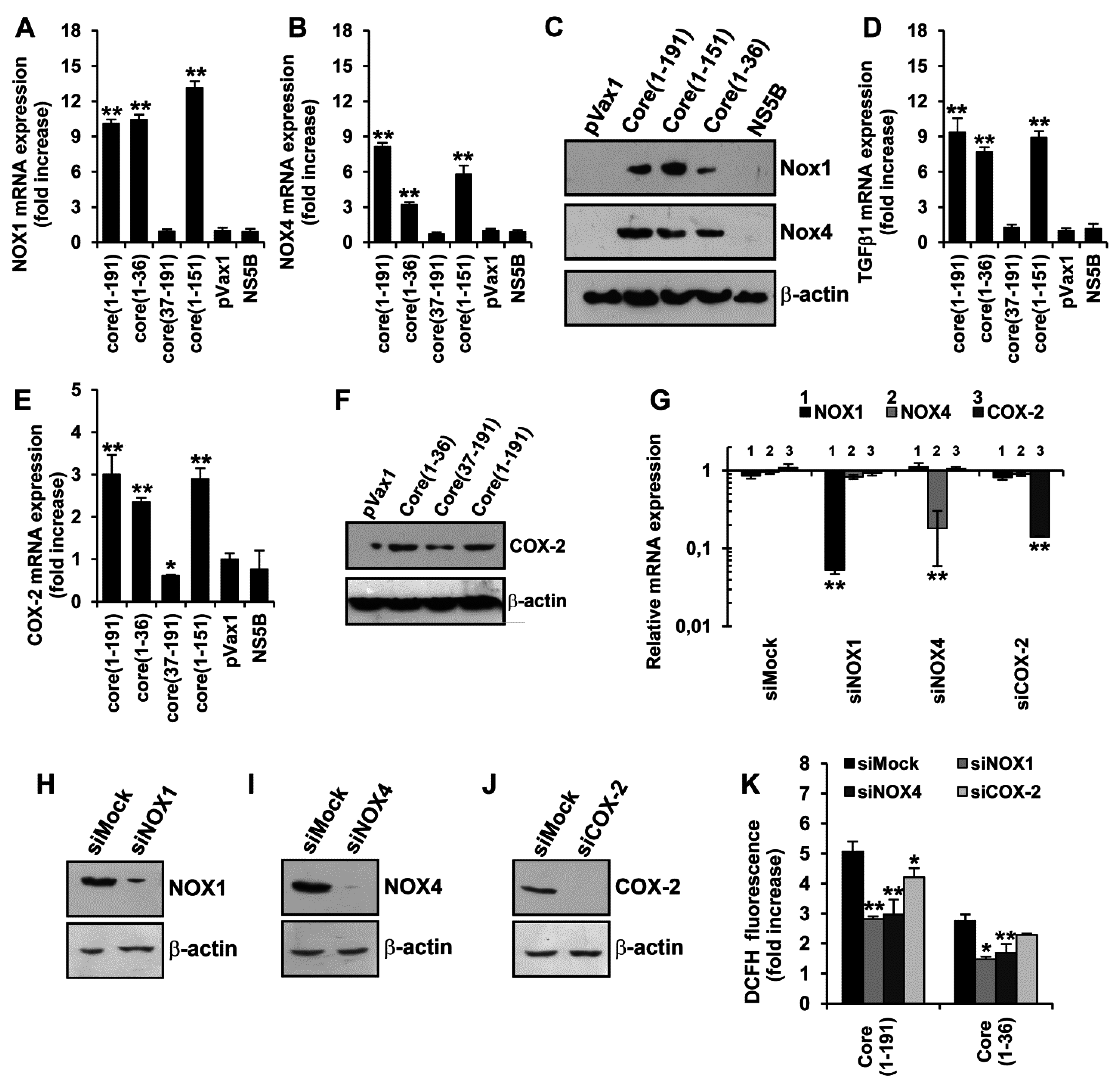
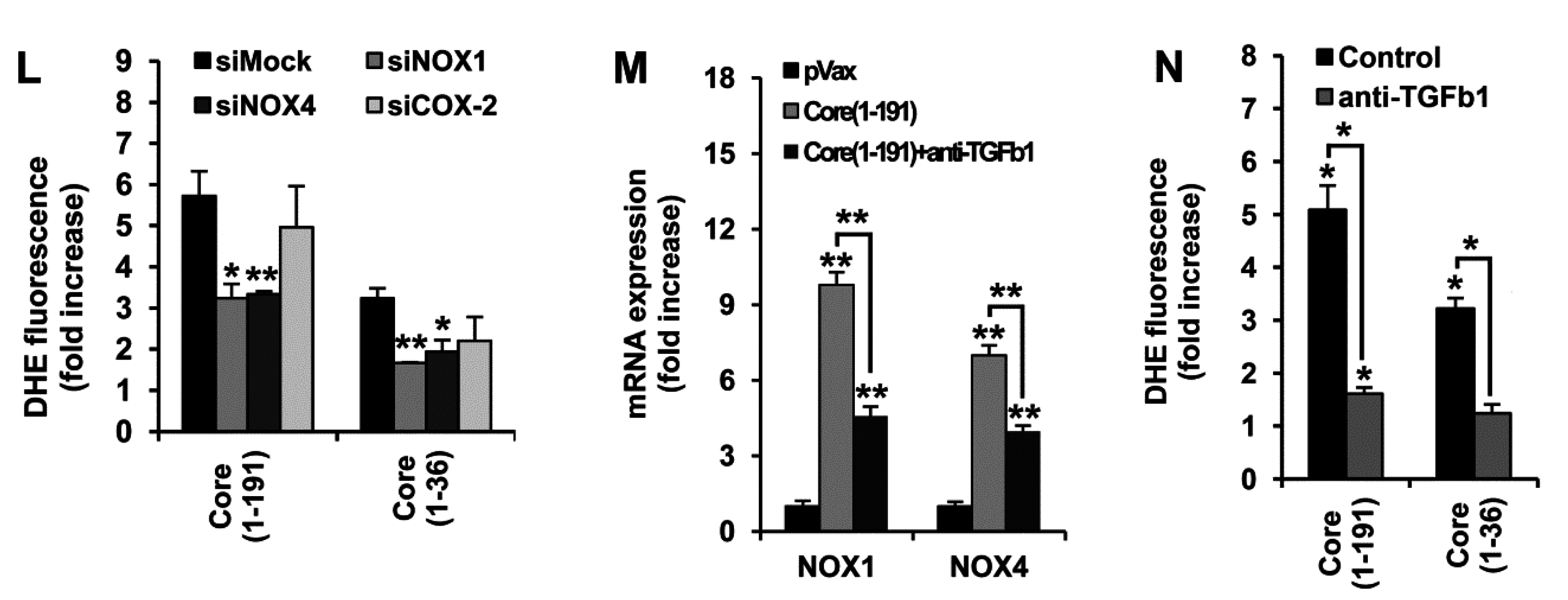
3.4. HCV Core Fragment aa 1–36 Mediates the Production of Superoxide Anion by Inducing the Expression of NOX1, NOX4, and TGFβ1
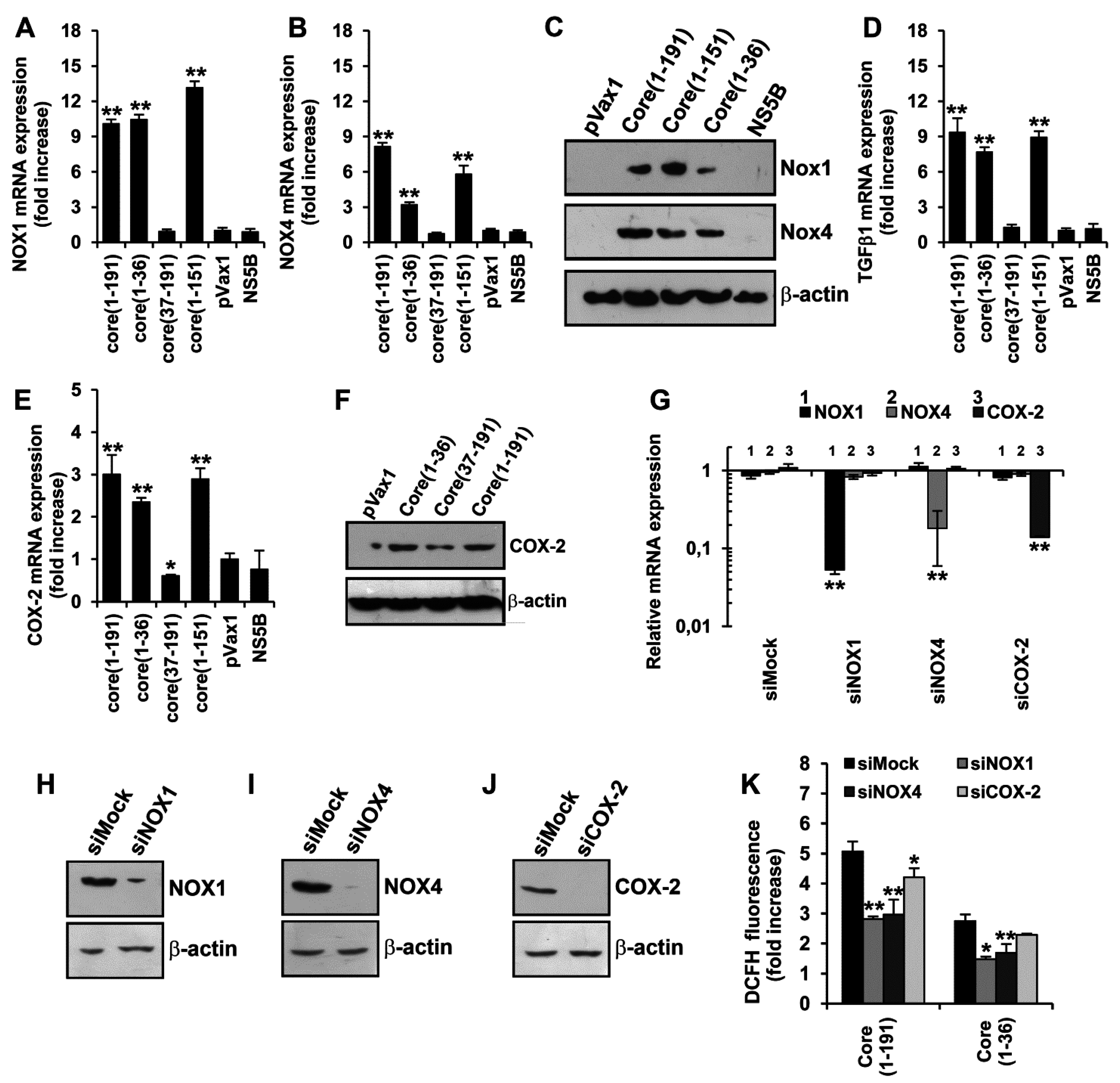

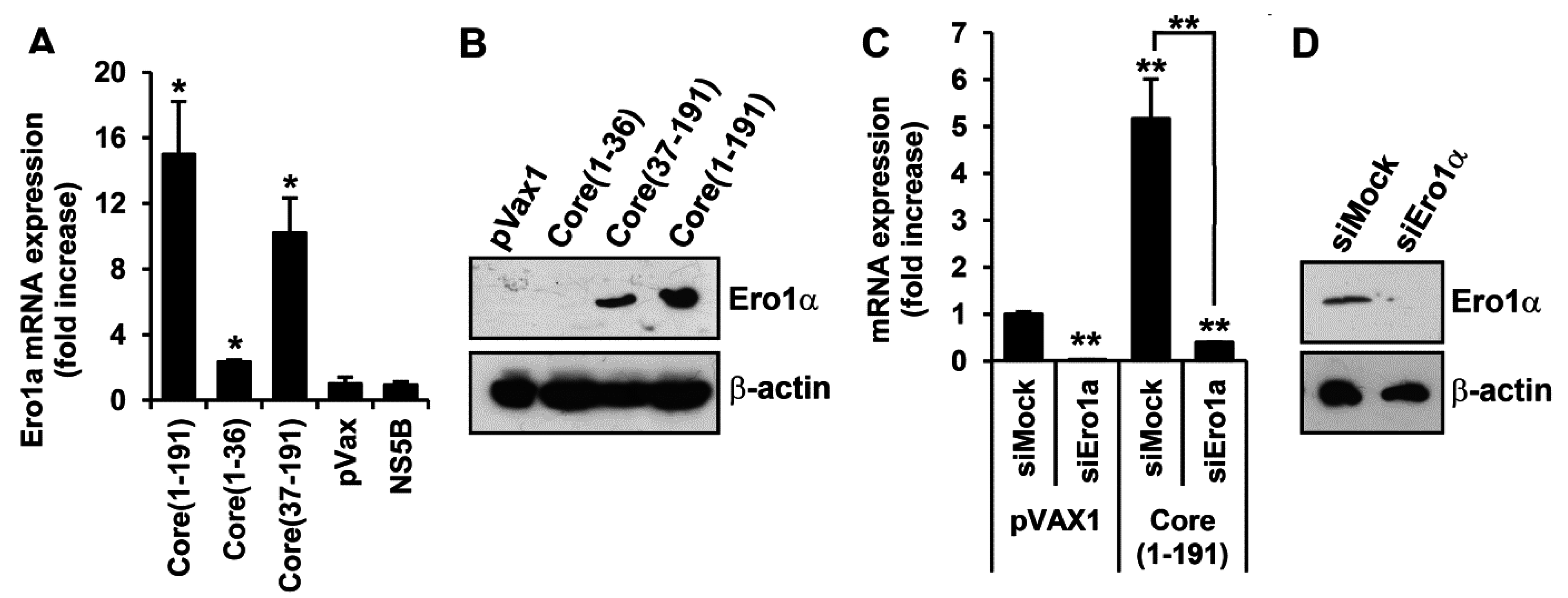
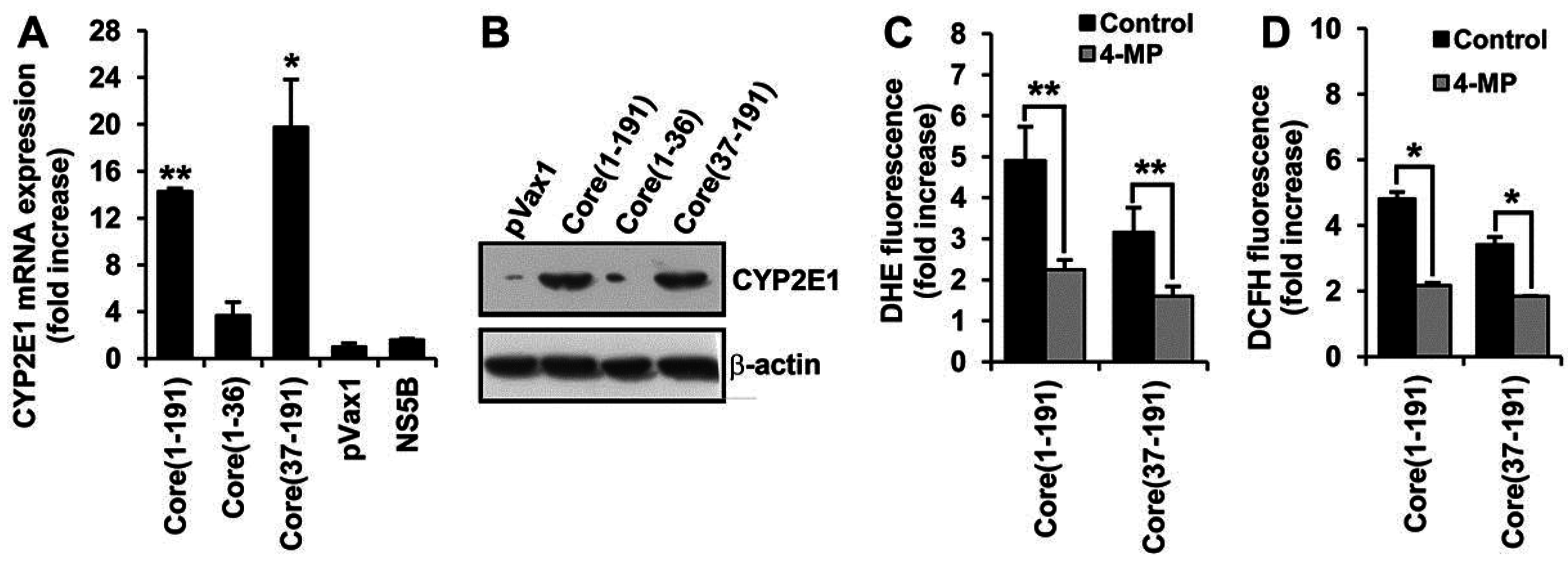
3.5. Core Amino Acids 37-191 Trigger the Production of ROS by Inducing the Expression of Cytochrome P450 2E1 and Ero1α
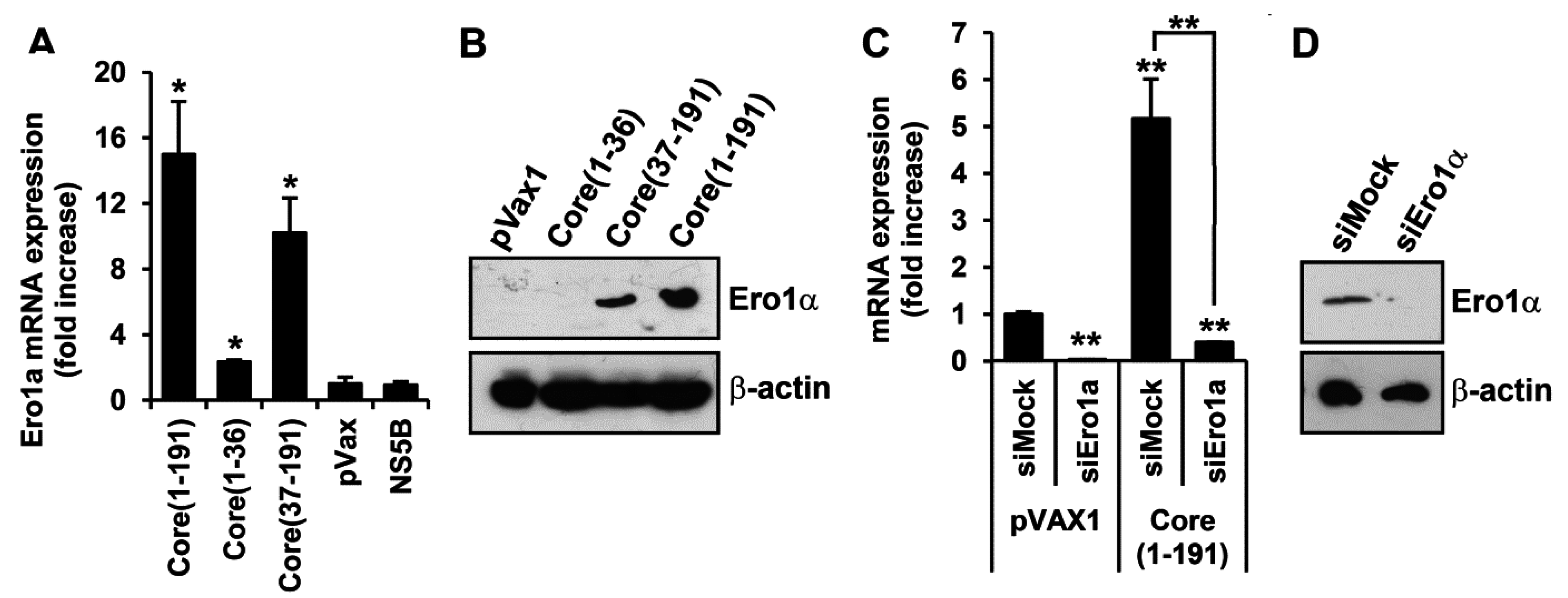
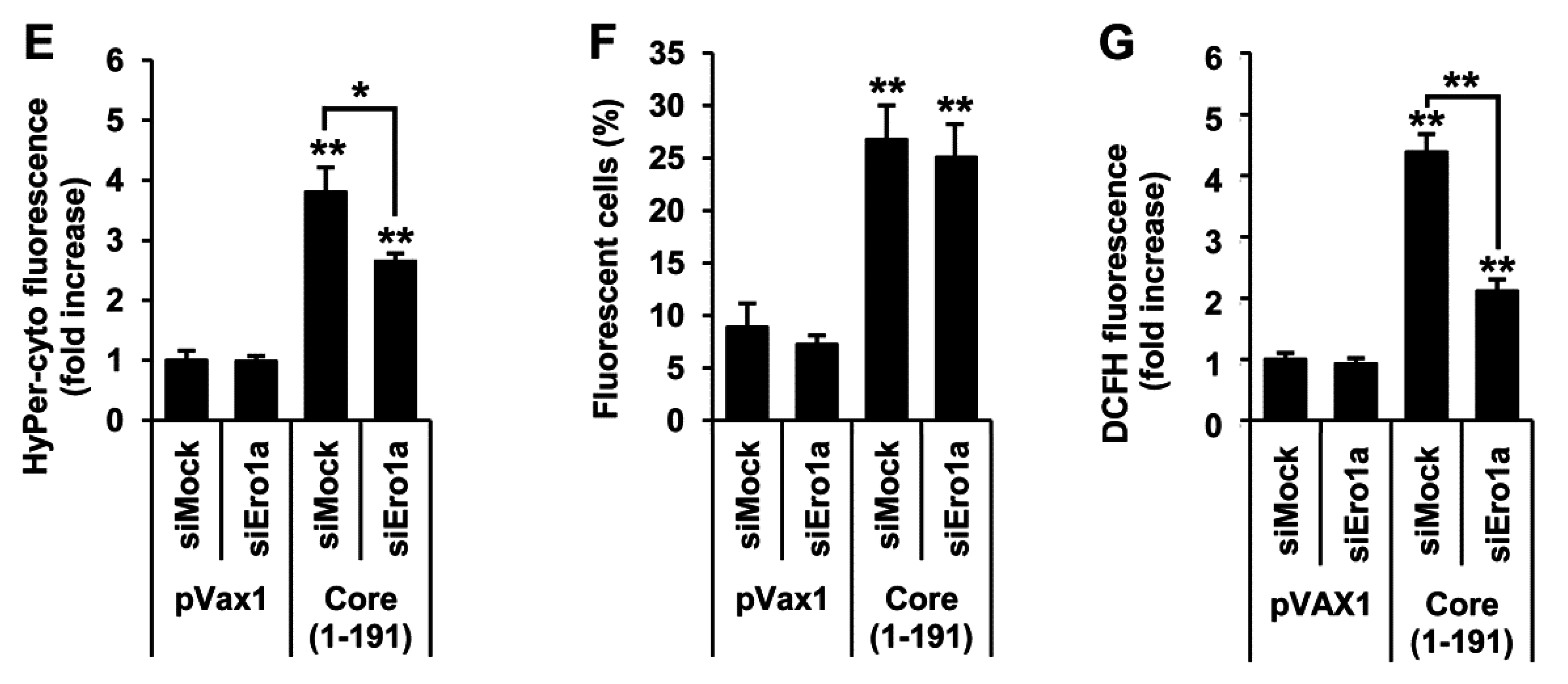
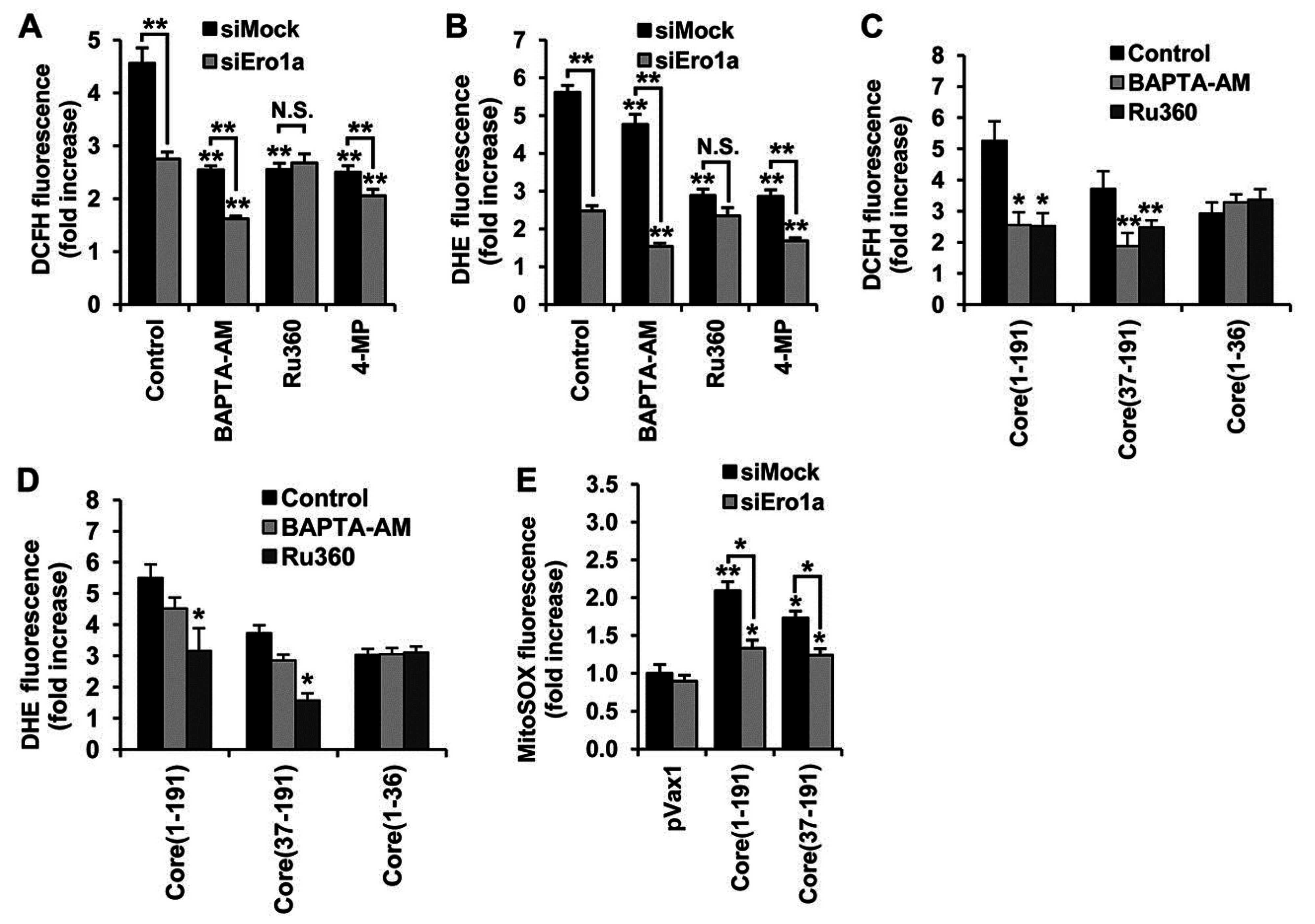
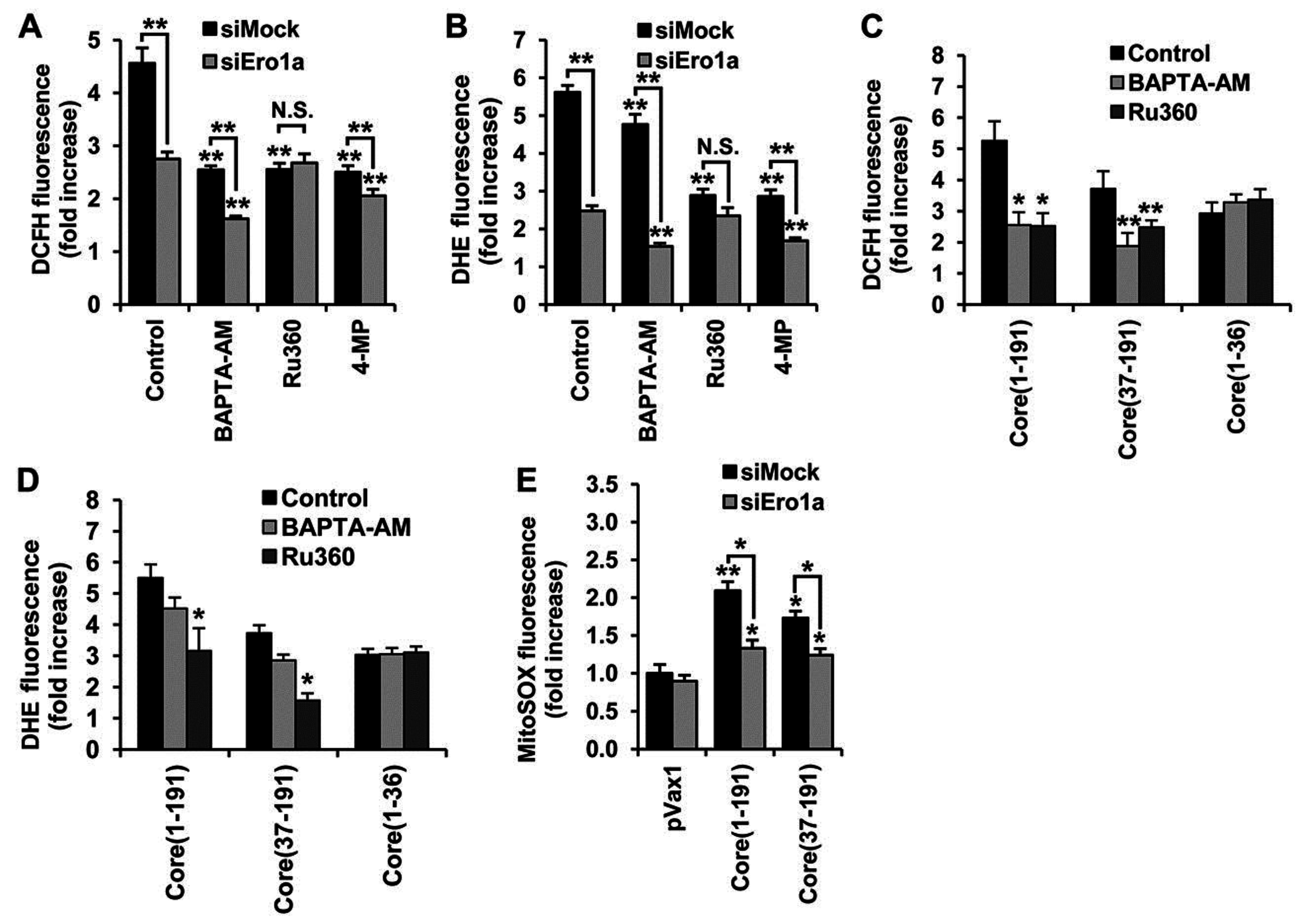
3.6. Core aa 37–191 Fragment Triggers the Production of Superoxide Anion by Ero1α-Mediated Efflux of Calcium Ions
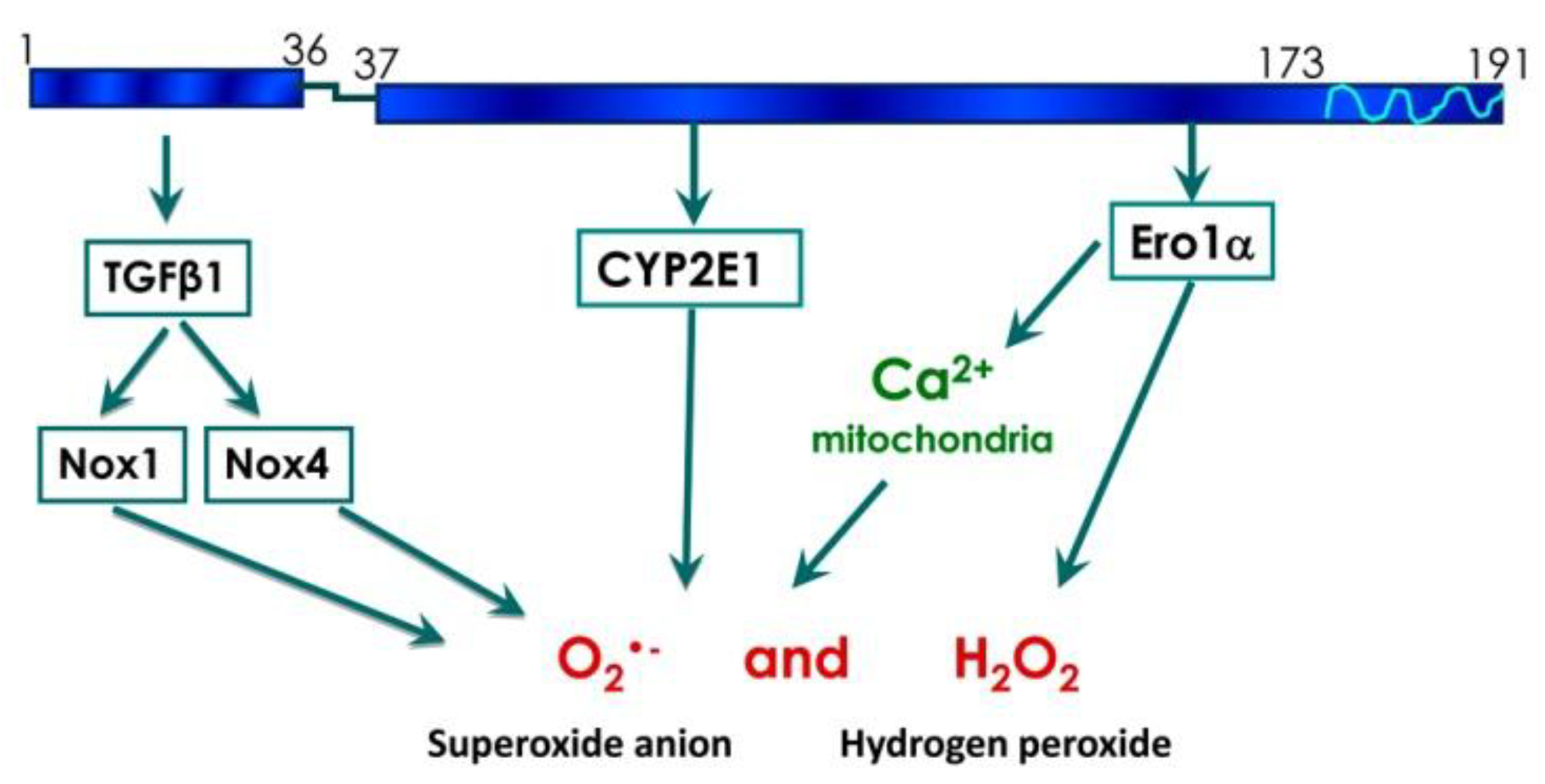
4. Discussion
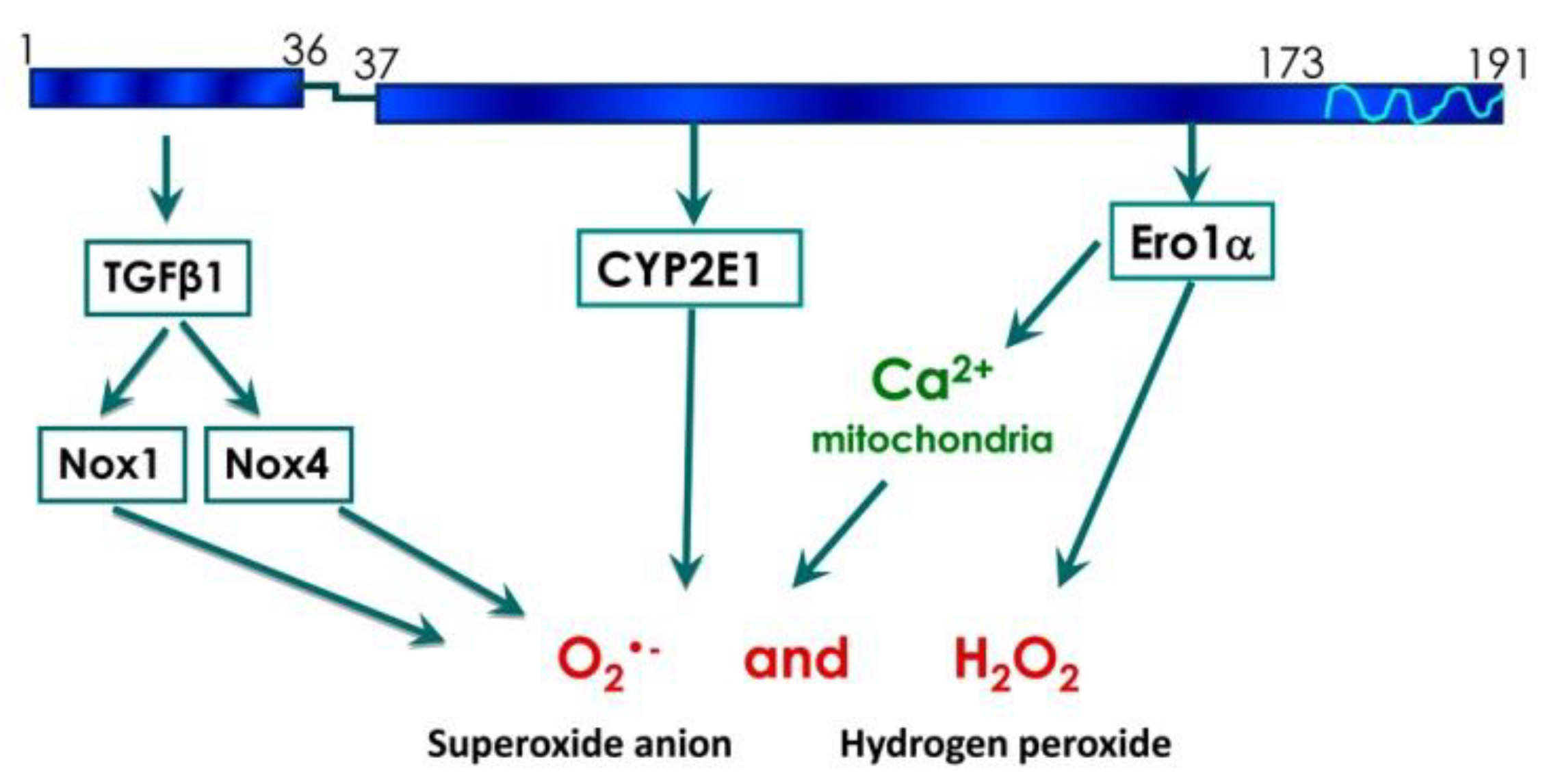
Conclusions
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Levrero, M. Viral hepatitis and liver cancer: The case of hepatitis C. Oncogene 2006, 25, 3834–3847. [Google Scholar] [CrossRef] [PubMed]
- Jin, D.Y. Molecular pathogenesis of hepatitis C virus-associated hepatocellular carcinoma. Front. Biosci. 2007, 12, 222–233. [Google Scholar] [CrossRef] [PubMed]
- Clement, S.; Pascarella, S.; Negro, F. Hepatitis C virus infection: Molecular pathways to steatosis, insulin resistance and oxidative stress. Viruses 2009, 1, 126–143. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Bartosch, B.; Smirnova, O.A.; Isaguliants, M.G.; Kochetkov, S.N. HCV and oxidative stress in the liver. Viruses 2013, 5, 439–469. [Google Scholar] [CrossRef] [PubMed]
- Smirnova, I.S.; Aksenov, N.D.; Kashuba, E.V.; Payakurel, P.; Grabovetsky, V.V.; Zaberezhny, A.D.; Vonsky, M.S.; Buchinska, L.; Biberfeld, P.; Hinkula, J.; et al. Hepatitis C virus core protein transforms murine fibroblasts by promoting genomic instability. Cell. Oncol. 2006, 28, 177–190. [Google Scholar] [PubMed]
- Moriya, K.; Nakagawa, K.; Santa, T.; Shintani, Y.; Fujie, H.; Miyoshi, H.; Tsutsumi, T.; Miyazawa, T.; Ishibashi, K.; Horie, T.; et al. Oxidative stress in the absence of inflammation in a mouse model for hepatitis C virus-associated hepatocarcinogenesis. Cancer Res. 2001, 61, 4365–4370. [Google Scholar] [PubMed]
- Benzoubir, N.; Lejamtel, C.; Battaglia, S.; Testoni, B.; Benassi, B.; Gondeau, C.; Perrin-Cocon, L.; Desterke, C.; Thiers, V.; Samuel, D.; et al. HCV core-mediated activation of latent tgf-beta via thrombospondin drives the crosstalk between hepatocytes and stromal environment. J. Hepatol. 2013, 59, 1160–1168. [Google Scholar] [CrossRef] [PubMed]
- Waris, G.; Felmlee, D.J.; Negro, F.; Siddiqui, A. Hepatitis C virus induces proteolytic cleavage of sterol regulatory element binding proteins and stimulates their phosphorylation via oxidative stress. J. Virol. 2007, 81, 8122–8130. [Google Scholar] [CrossRef] [PubMed]
- Miura, K.; Taura, K.; Kodama, Y.; Schnabl, B.; Brenner, D.A. Hepatitis c virus-induced oxidative stress suppresses hepcidin expression through increased histone deacetylase activity. Hepatology 2008, 48, 1420–1429. [Google Scholar] [CrossRef] [PubMed]
- Choi, J. Oxidative stress, endogenous antioxidants, alcohol, and hepatitis C: Pathogenic interactions and therapeutic considerations. Free Radic. Biol. Med. 2012, 52, 1135–1150. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, A.; Tazuma, S.; Nishioka, T.; Ohishi, W.; Hyogo, H.; Nomura, S.; Chayama, K. Hepatitis C virus core protein modulates fatty acid metabolism and thereby causes lipid accumulation in the liver. Dig. Dis. Sci. 2005, 50, 1361–1371. [Google Scholar] [CrossRef] [PubMed]
- Sharma, P.; Jha, A.B.; Dubey, R.S.; Pessarakli, M. Reactive oxygen species, oxidative damage, and antioxidative defense mechanism in plants under stressful conditions. J. Bot. 2012, 2012, e217037. [Google Scholar] [CrossRef]
- Brault, C.; Levy, P.L.; Bartosch, B. Hepatitis C virus-induced mitochondrial dysfunctions. Viruses 2013, 5, 954–980. [Google Scholar] [CrossRef] [PubMed]
- Gong, G.; Waris, G.; Tanveer, R.; Siddiqui, A. Human hepatitis C virus ns5a protein alters intracellular calcium levels, induces oxidative stress, and activates stat-3 and nf-kappa b. Proc. Natl. Acad. Sci. USA 2001, 98, 9599–9604. [Google Scholar] [PubMed]
- Ivanov, A.V.; Smirnova, O.A.; Ivanova, O.N.; Masalova, O.V.; Kochetkov, S.N.; Isaguliants, M.G. Hepatitis C virus proteins activate nrf2/are pathway by distinct ros-dependent and independent mechanisms in huh7 cells. PLoS ONE 2011, 6, e24957. [Google Scholar] [CrossRef] [PubMed]
- Korenaga, M.; Wang, T.; Li, Y.; Showalter, L.A.; Chan, T.; Sun, J.; Weinman, S.A. Hepatitis C virus core protein inhibits mitochondrial electron transport and increases reactive oxygen species (ros) production. J. Biol. Chem. 2005, 280, 37481–37488. [Google Scholar] [CrossRef] [PubMed]
- Seronello, S.; Montanez, J.; Presleigh, K.; Barlow, M.; Park, S.B.; Choi, J. Ethanol and reactive species increase basal sequence heterogeneity of hepatitis C virus and produce variants with reduced susceptibility to antivirals. PLoS ONE 2011, 6, e27436. [Google Scholar] [CrossRef] [PubMed]
- Casero, R.A.; Pegg, A.E. Polyamine catabolism and disease. Biochem. J. 2009, 421, 323–338. [Google Scholar] [CrossRef] [PubMed]
- Laurindo, F.R.; Pescatore, L.A.; Fernandes Dde, C. Protein disulfide isomerase in redox cell signaling and homeostasis. Free Radic. Biol. Med. 2012, 52, 1954–1969. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Campbell, R.V.; Yi, M.K.; Lemon, S.M.; Weinman, S.A. Role of hepatitis C virus core protein in viral-induced mitochondrial dysfunction. J. Viral Hepat. 2010, 17, 784–793. [Google Scholar] [CrossRef] [PubMed]
- De Mochel, N.S.; Seronello, S.; Wang, S.H.; Ito, C.; Zheng, J.X.; Liang, T.J.; Lambeth, J.D.; Choi, J. Hepatocyte nad(p)h oxidases as an endogenous source of reactive oxygen species during hepatitis C virus infection. Hepatology 2010, 52, 47–59. [Google Scholar] [CrossRef] [PubMed]
- Boudreau, H.E.; Emerson, S.U.; Korzeniowska, A.; Jendrysik, M.A.; Leto, T.L. Hepatitis C virus (HCV) proteins induce nadph oxidase 4 expression in a transforming growth factor beta-dependent manner: A new contributor to HCV-induced oxidative stress. J. Virol. 2009, 83, 12934–12946. [Google Scholar] [CrossRef] [PubMed]
- Benali-Furet, N.L.; Chami, M.; Houel, L.; de Giorgi, F.; Vernejoul, F.; Lagorce, D.; Buscail, L.; Bartenschlager, R.; Ichas, F.; Rizzuto, R.; et al. Hepatitis c virus core triggers apoptosis in liver cells by inducing er stress and er calcium depletion. Oncogene 2005, 24, 4921–4933. [Google Scholar] [CrossRef] [PubMed]
- McLauchlan, J. Properties of the hepatitis C virus core protein: A structural protein that modulates cellular processes. J. Viral Hepat. 2000, 7, 2–14. [Google Scholar] [CrossRef] [PubMed]
- Boulant, S.; Montserret, R.; Hope, R.G.; Ratinier, M.; Targett-Adams, P.; Lavergne, J.P.; Penin, F.; McLauchlan, J. Structural determinants that target the hepatitis C virus core protein to lipid droplets. J. Biol. Chem. 2006, 281, 22236–22247. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, R.; Sakamoto, S.; Tsutsumi, T.; Rikimaru, A.; Tanaka, K.; Shimoike, T.; Moriishi, K.; Iwasaki, T.; Mizumoto, K.; Matsuura, Y.; et al. Molecular determinants for subcellular localization of hepatitis C virus core protein. J. Virol. 2005, 79, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Boehning, D.F.; Qian, T.; Popov, V.L.; Weinman, S.A. Hepatitis C virus core protein increases mitochondrial ROS production by stimulation of Ca2+ uniporter activity. FASEB J. 2007, 21, 2474–2485. [Google Scholar] [CrossRef] [PubMed]
- Isaguliants, M.G.; Iakimtchouk, K.; Petrakova, N.V.; Yermalovich, M.A.; Zuber, A.K.; Kashuba, V.I.; Belikov, S.V.; Andersson, S.; Kochetkov, S.N.; Klinman, D.M.; et al. Gene immunization may induce secondary antibodies reacting with DNA. Vaccine 2004, 22, 1576–1585. [Google Scholar] [CrossRef] [PubMed]
- Alekseeva, E.; Sominskaya, I.; Skrastina, D.; Egorova, I.; Starodubova, E.; Kushners, E.; Mihailova, M.; Petrakova, N.; Bruvere, R.; Kozlovskaya, T.; et al. Enhancement of the expression of HCV core gene does not enhance core-specific immune response in DNA immunization: Advantages of the heterologous DNA prime, protein boost immunization regimen. Genet. Vaccines Ther. 2009, 7, e7. [Google Scholar] [CrossRef] [PubMed]
- Zielonka, J.; Vasquez-Vivar, J.; Kalyanaraman, B. Detection of 2-hydroxyethidium in cellular systems: A unique marker product of superoxide and hydroethidine. Nat. Protoc. 2008, 3, 8–21. [Google Scholar] [CrossRef] [PubMed]
- Mitkevich, V.A.; Petrushanko, I.Y.; Kretova, O.V.; Zelenikhin, P.V.; Prassolov, V.S.; Tchurikov, N.A.; Ilinskaya, O.N.; Makarov, A.A. Oncogenic c-kit transcript is a target for binase. Cell Cycle 2010, 9, 2674–2678. [Google Scholar] [CrossRef] [PubMed]
- Anelli, T.; Bergamelli, L.; Margittai, E.; Rimessi, A.; Fagioli, C.; Malgaroli, A.; Pinton, P.; Ripamonti, M.; Rizzuto, R.; Sitia, R. Ero1alpha regulates Ca(2+) fluxes at the endoplasmic reticulum-mitochondria interface (mam). Antioxid. Redox Signal. 2012, 16, 1077–1087. [Google Scholar] [CrossRef] [PubMed]
- Sancho, P.; Martin-Sanz, P.; Fabregat, I. Reciprocal regulation of nadph oxidases and the cyclooxygenase-2 pathway. Free Radic. Biol. Med. 2011, 51, 1789–1798. [Google Scholar] [CrossRef] [PubMed]
- Cerutti, A.; Maillard, P.; Minisini, R.; Vidalain, P.O.; Roohvand, F.; Pecheur, E.I.; Pirisi, M.; Budkowska, A. Identification of a functional, crm-1-dependent nuclear export signal in hepatitis C virus core protein. PLoS ONE 2011, 6, e25854. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.M.; You, L.R.; Hwang, L.H.; Lee, Y.H. Direct interaction of hepatitis C virus core protein with the cellular lymphotoxin-beta receptor modulates the signal pathway of the lymphotoxin-beta receptor. J. Virol. 1997, 71, 9417–9426. [Google Scholar] [PubMed]
- Lin, W.; Kim, S.S.; Yeung, E.; Kamegaya, Y.; Blackard, J.T.; Kim, K.A.; Holtzman, M.J.; Chung, R.T. Hepatitis C virus core protein blocks interferon signaling by interaction with the stat1 sh2 domain. J. Virol. 2006, 80, 9226–9235. [Google Scholar] [CrossRef] [PubMed]
- Sun, C.; Pager, C.T.; Luo, G.; Sarnow, P.; Cate, J.H. Hepatitis c virus core-derived peptides inhibit genotype 1b viral genome replication via interaction with ddx3x. PLoS ONE 2010, 5, e12826. [Google Scholar] [CrossRef] [PubMed]
- Kalyanaraman, B.; Darley-Usmar, V.; Davies, K.J.; Dennery, P.A.; Forman, H.J.; Grisham, M.B.; Mann, G.E.; Moore, K.; Roberts, L.J., 2nd; Ischiropoulos, H. Measuring reactive oxygen and nitrogen species with fluorescent probes: Challenges and limitations. Free Radic. Biol. Med. 2012, 52, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Markvicheva, K.N.; Bogdanova, E.A.; Staroverov, D.B.; Lukyanov, S.; Belousov, V.V. Imaging of intracellular hydrogen peroxide production with hyper upon stimulation of hela cells with epidermal growth factor. Methods Mol. Biol. 2009, 476, 76–83. [Google Scholar]
- Knockaert, L.; Fromenty, B.; Robin, M.A. Mechanisms of mitochondrial targeting of cytochrome p450 2e1: Physiopathological role in liver injury and obesity. FEBS J. 2011, 278, 4252–4260. [Google Scholar] [CrossRef] [PubMed]
- Lu, Y.; Cederbaum, A.I. Cyp2e1 and oxidative liver injury by alcohol. Free Radic. Biol. Med. 2008, 44, 723–738. [Google Scholar] [CrossRef] [PubMed]
- Wu, D.; Cederbaum, A.I. Ethanol cytotoxicity to a transfected hepg2 cell line expressing human cytochrome p4502e1. J. Biol. Chem. 1996, 271, 23914–23919. [Google Scholar] [PubMed]
- Li, G.; Mongillo, M.; Chin, K.T.; Harding, H.; Ron, D.; Marks, A.R.; Tabas, I. Role of ero1-alpha-mediated stimulation of inositol 1,4,5-triphosphate receptor activity in endoplasmic reticulum stress-induced apoptosis. J. Cell Biol. 2009, 186, 783–792. [Google Scholar] [CrossRef] [PubMed]
- Takac, I.; Schroder, K.; Zhang, L.; Lardy, B.; Anilkumar, N.; Lambeth, J.D.; Shah, A.M.; Morel, F.; Brandes, R.P. The e-loop is involved in hydrogen peroxide formation by the nadph oxidase nox4. J. Biol. Chem. 2011, 286, 13304–13313. [Google Scholar] [CrossRef] [PubMed]
- Spencer, N.Y.; Yan, Z.; Boudreau, R.L.; Zhang, Y.; Luo, M.; Li, Q.; Tian, X.; Shah, A.M.; Davisson, R.L.; Davidson, B.; et al. Control of hepatic nuclear superoxide production by glucose 6-phosphate dehydrogenase and nadph oxidase-4. J. Biol. Chem. 2011, 286, 8977–8987. [Google Scholar] [CrossRef] [PubMed]
- Nisimoto, Y.; Diebold, B.A.; Constentino-Gomes, D.; Lambeth, J.D. Nox4: A hydrogen peroxide-generating oxygen sensor. Biochemistry 2014, 53, 5111–5120. [Google Scholar] [CrossRef] [PubMed]
- Helmcke, I.; Heumuller, S.; Tikkanen, R.; Schroder, K.; Brandes, R.P. Identification of structural elements in nox1 and nox4 controlling localization and activity. Antioxid. Redox Signal. 2009, 11, 1279–1287. [Google Scholar] [CrossRef] [PubMed]
- Anilkumar, N.; San Jose, G.; Sawyer, I.; Santos, C.X.; Sand, C.; Brewer, A.C.; Warren, D.; Shah, A.M. A 28-kda splice variant of nadph oxidase-4 is nuclear-localized and involved in redox signaling in vascular cells. Arterioscler. Thromb. Vasc. Biol. 2013, 33, e104–e112. [Google Scholar] [CrossRef] [PubMed]
- Nakai, K.; Tanaka, H.; Hanada, K.; Ogata, H.; Suzuki, F.; Kumada, H.; Miyajima, A.; Ishida, S.; Sunouchi, M.; Habano, W.; et al. Decreased expression of cytochromes P450 1A2, 2E1, and 3A4 and drug transporters Na+-taurocholate-cotransporting polypeptide, organic cation transporter 1, and organic anion-transporting peptide-c correlates with the progression of liver fibrosis in chronic hepatitis C patients. Drug Metab. Dispos. 2008, 36, 1786–1793. [Google Scholar] [PubMed]
- Burdette, D.; Olivarez, M.; Waris, G. Activation of transcription factor Nrf2 by hepatitis C virus induces the cell-survival pathway. J. Gen. Virol. 2010, 91, 681–690. [Google Scholar] [CrossRef] [PubMed]
- Hayes, J.D.; Dinkova-Kostova, A.T. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem. Sci. 2014, 39, 199–218. [Google Scholar] [CrossRef] [PubMed]
- Miyamoto, N.; Izumi, H.; Miyamoto, R.; Kondo, H.; Tawara, A.; Sasaguri, Y.; Kohno, K. Quercetin induces the expression of peroxiredoxins 3 and 5 via the Nrf2/NRF1 transcription pathway. Investig. Ophthalmol. Vis. Sci. 2011, 52, 1055–1063. [Google Scholar] [CrossRef] [PubMed]
- Diamond, D.L.; Syder, A.J.; Jacobs, J.M.; Sorensen, C.M.; Walters, K.A.; Proll, S.C.; McDermott, J.E.; Gritsenko, M.A.; Zhang, Q.; Zhao, R.; et al. Temporal proteome and lipidome profiles reveal hepatitis C virus-associated reprogramming of hepatocellular metabolism and bioenergetics. PLoS Pathog. 2010, 6, e1000719. [Google Scholar] [CrossRef] [PubMed]
- Brault, C.; Levy, P.; Duponchel, S.; Michelet, M.; Salle, A.; Pecheur, E.I.; Plissonnier, M.L.; Parent, R.; Vericel, E.; Ivanov, A.V.; et al. Glutathione peroxidase 4 is reversibly induced by HCV to control lipid peroxidation and to increase virion infectivity. Gut 2014. [Google Scholar] [CrossRef]
- Choi, J.; Corder, N.L.; Koduru, B.; Wang, Y. Oxidative stress and hepatic nox proteins in chronic hepatitis C and hepatocellular carcinoma. Free Radic. Biol. Med. 2014, 72, 267–284. [Google Scholar] [CrossRef] [PubMed]
- Sancho, P.; Mainez, J.; Crosas-Molist, E.; Roncero, C.; Fernandez-Rodriguez, C.M.; Pinedo, F.; Huber, H.; Eferl, R.; Mikulits, W.; Fabregat, I. Nadph oxidase nox4 mediates stellate cell activation and hepatocyte cell death during liver fibrosis development. PLoS ONE 2012, 7, e45285. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.X.; Chen, X.; Serizawa, N.; Szyndralewiez, C.; Page, P.; Schroder, K.; Brandes, R.P.; Devaraj, S.; Torok, N.J. Liver fibrosis and hepatocyte apoptosis are attenuated by GKT137831, a novel NOX4/NOX1 inhibitor in vivo. Free Radic. Biol. Med. 2012, 53, 289–296. [Google Scholar] [CrossRef] [PubMed]
- Otani, K.; Korenaga, M.; Beard, M.R.; Li, K.; Qian, T.; Showalter, L.A.; Singh, A.K.; Wang, T.; Weinman, S.A. Hepatitis c virus core protein, cytochrome P450 2e1, and alcohol produce combined mitochondrial injury and cytotoxicity in hepatoma cells. Gastroenterology 2005, 128, 96–107. [Google Scholar] [CrossRef] [PubMed]
- Nieto, N.; Friedman, S.L.; Cederbaum, A.I. Stimulation and proliferation of primary rat hepatic stellate cells by cytochrome P450 2e1-derived reactive oxygen species. Hepatology 2002, 35, 62–73. [Google Scholar] [CrossRef] [PubMed]
- Nieto, N.; Cederbaum, A.I. Increased Sp1-dependent transactivation of the lamgamma 1 promoter in hepatic stellate cells co-cultured with hepg2 cells overexpressing cytochrome p450 2e1. J. Biol. Chem. 2003, 278, 15360–15372. [Google Scholar] [CrossRef] [PubMed]
- Haufroid, V.; Ligocka, D.; Buysschaert, M.; Horsmans, Y.; Lison, D. Cytochrome p4502e1 (cyp2e1) expression in peripheral blood lymphocytes: Evaluation in hepatitis C and diabetes. Eur. J. Clin. Pharmacol. 2003, 59, 29–33. [Google Scholar] [PubMed]
- Haufroid, V.; Ligocka, D.; Wallemacq, P.; Lison, D.; Horsmans, Y. Comparison of cytochrome p4502e1 (cyp2e1) activity and hepatic and lymphocyte mrna expression in patients with chronic hepatitis C. Toxicol. Lett. 2005, 155, 171–177. [Google Scholar] [CrossRef] [PubMed]
- Gochee, P.A.; Jonsson, J.R.; Clouston, A.D.; Pandeya, N.; Purdie, D.M.; Powell, E.E. Steatosis in chronic hepatitis C: Association with increased messenger rna expression of collagen I, tumor necrosis factor-alpha and cytochrome p450 2e1. J. Gastroenterol. Hepatol. 2003, 18, 386–392. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Wang, P.; Cong, M.; Xu, Y.; Jia, J.; You, H. The CYP2E1 inhibitor DDC up-regulates MMP-1 expression in hepatic stellate cells via an ERK1/2- and Akt-dependent mechanism. Biosci. Rep. 2013, 33, e00041. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, R.P.; Kolli, D.; Casola, A. Respiratory syncytial virus infection: Mechanisms of redox control and novel therapeutic opportunities. Antioxid. Redox Signal. 2013, 18, 186–217. [Google Scholar] [CrossRef] [PubMed]
- Grandvaux, N.; Soucy-Faulkner, A.; Fink, K. Innate host defense: Nox and Duox on phox’s tail. Biochimie 2007, 89, 1113–1122. [Google Scholar] [CrossRef] [PubMed]
- Kutomi, G.; Tamura, Y.; Tanaka, T.; Kajiwara, T.; Kukita, K.; Ohmura, T.; Shima, H.; Takamaru, T.; Satomi, F.; Suzuki, Y.; et al. Human endoplasmic reticulum oxidoreductin 1-alpha is a novel predictor for poor prognosis of breast cancer. Cancer Sci. 2013, 104, 1091–1096. [Google Scholar] [CrossRef] [PubMed]
- Reshi, M.L.; Su, Y.C.; Hong, J.R. RNA viruses: Ros-mediated cell death. Int. J. Cell Biol. 2014, 2014, e467452. [Google Scholar] [CrossRef] [PubMed]
- Olinski, R.; Styczynski, J.; Olinska, E.; Gackowski, D. Viral infection-oxidative stress/DNA damage-aberrant DNA methylation: Separate or interrelated events responsible for genetic instability and childhood all development? Biochim. Biophys. Acta 2014, 1846, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Tubbs, E.; Theurey, P.; Vial, G.; Bendridi, N.; Bravard, A.; Chauvin, M.A.; Ji-Cao, J.; Zoulim, F.; Bartosch, B.; Ovize, M.; et al. Mitochondria-associated endoplasmic reticulum membrane (mam) integrity is required for insulin signaling and is implicated in hepatic insulin resistance. Diabetes 2014, 63, 3279–3294. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ivanov, A.V.; Smirnova, O.A.; Petrushanko, I.Y.; Ivanova, O.N.; Karpenko, I.L.; Alekseeva, E.; Sominskaya, I.; Makarov, A.A.; Bartosch, B.; Kochetkov, S.N.; et al. HCV Core Protein Uses Multiple Mechanisms to Induce Oxidative Stress in Human Hepatoma Huh7 Cells. Viruses 2015, 7, 2745-2770. https://doi.org/10.3390/v7062745
Ivanov AV, Smirnova OA, Petrushanko IY, Ivanova ON, Karpenko IL, Alekseeva E, Sominskaya I, Makarov AA, Bartosch B, Kochetkov SN, et al. HCV Core Protein Uses Multiple Mechanisms to Induce Oxidative Stress in Human Hepatoma Huh7 Cells. Viruses. 2015; 7(6):2745-2770. https://doi.org/10.3390/v7062745
Chicago/Turabian StyleIvanov, Alexander V., Olga A. Smirnova, Irina Y. Petrushanko, Olga N. Ivanova, Inna L. Karpenko, Ekaterina Alekseeva, Irina Sominskaya, Alexander A. Makarov, Birke Bartosch, Sergey N. Kochetkov, and et al. 2015. "HCV Core Protein Uses Multiple Mechanisms to Induce Oxidative Stress in Human Hepatoma Huh7 Cells" Viruses 7, no. 6: 2745-2770. https://doi.org/10.3390/v7062745