
A Suggested New Bacteriophage Genus, “Kp34likevirus”, within the Autographivirinae Subfamily of Podoviridae

Abstract
:1. Introduction
2. Materials and Methods
2.1. Biological Materials

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
2.2. Electron Microscopy
2.3. Burst Size Experiments
2.4. Sensitivity of Phage Particles to Temperature, Chloroform and pH
2.5. Determination of Phage Bacterial Host Range
2.6. Phage Structural Protein Analysis
2.7. DNA Purification
2.8. DNA Sequencing
2.9. In Silico Analysis and Annotation of Genomes
2.10. Comparative Genomics
2.11. Analysis of Lysis Cassette
3. Results
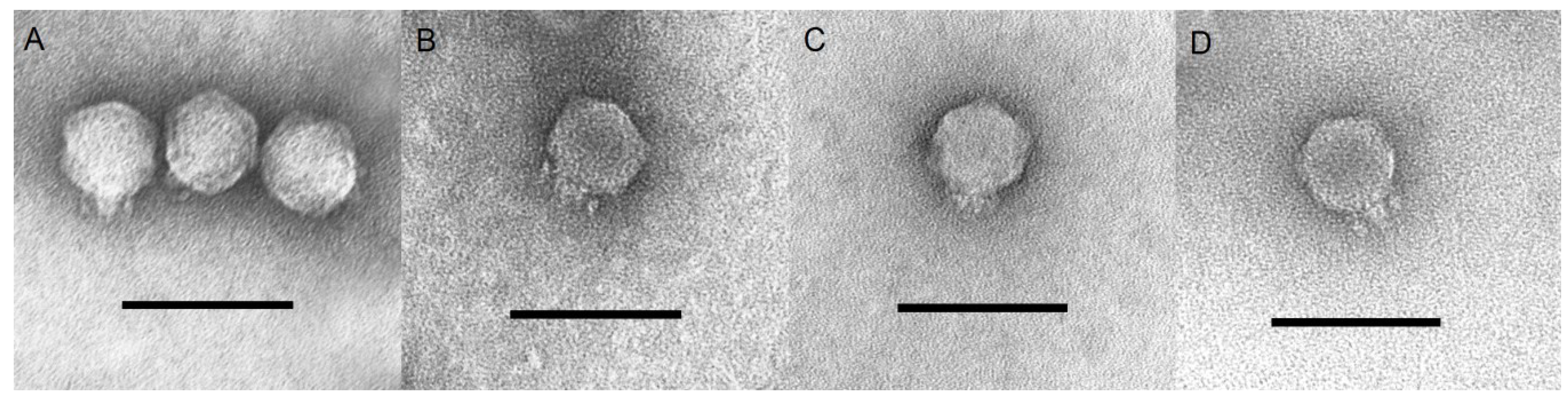
3.1. Phage Isolation and Morphological Characteristics
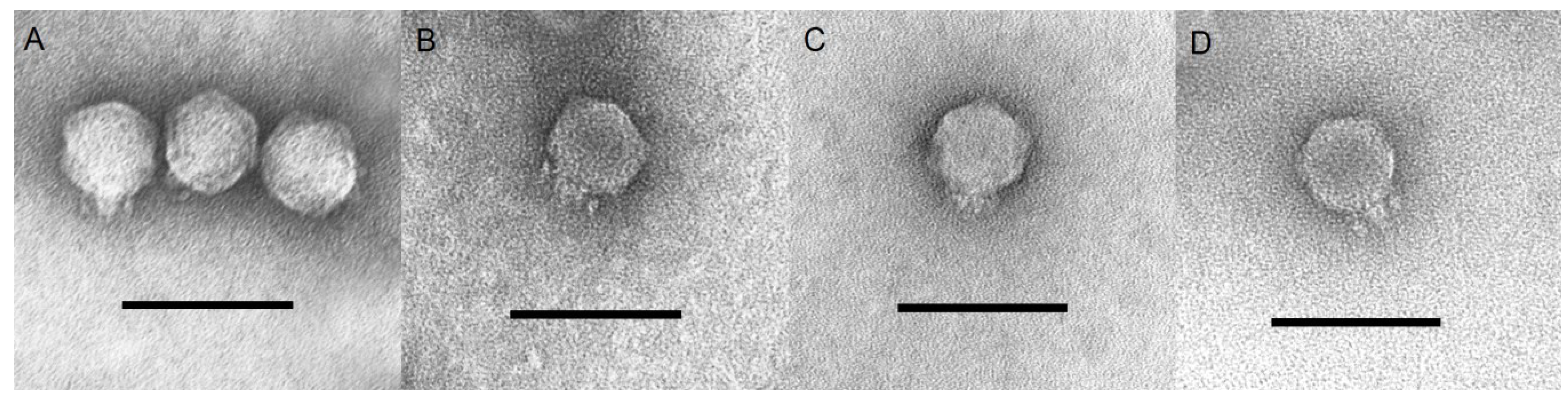
3.2. Physicochemical Properties
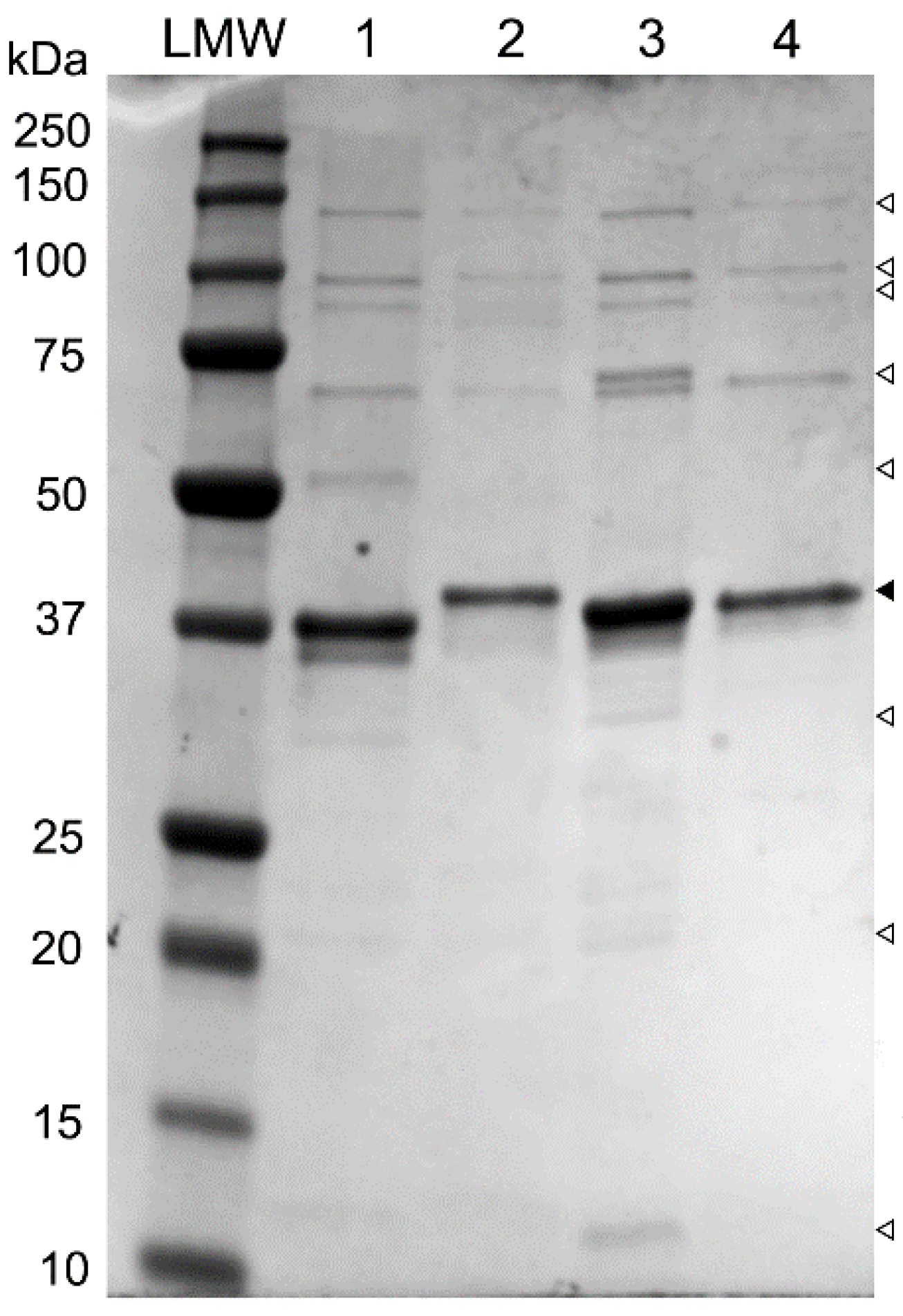
3.3. Analysis of Structural Proteins

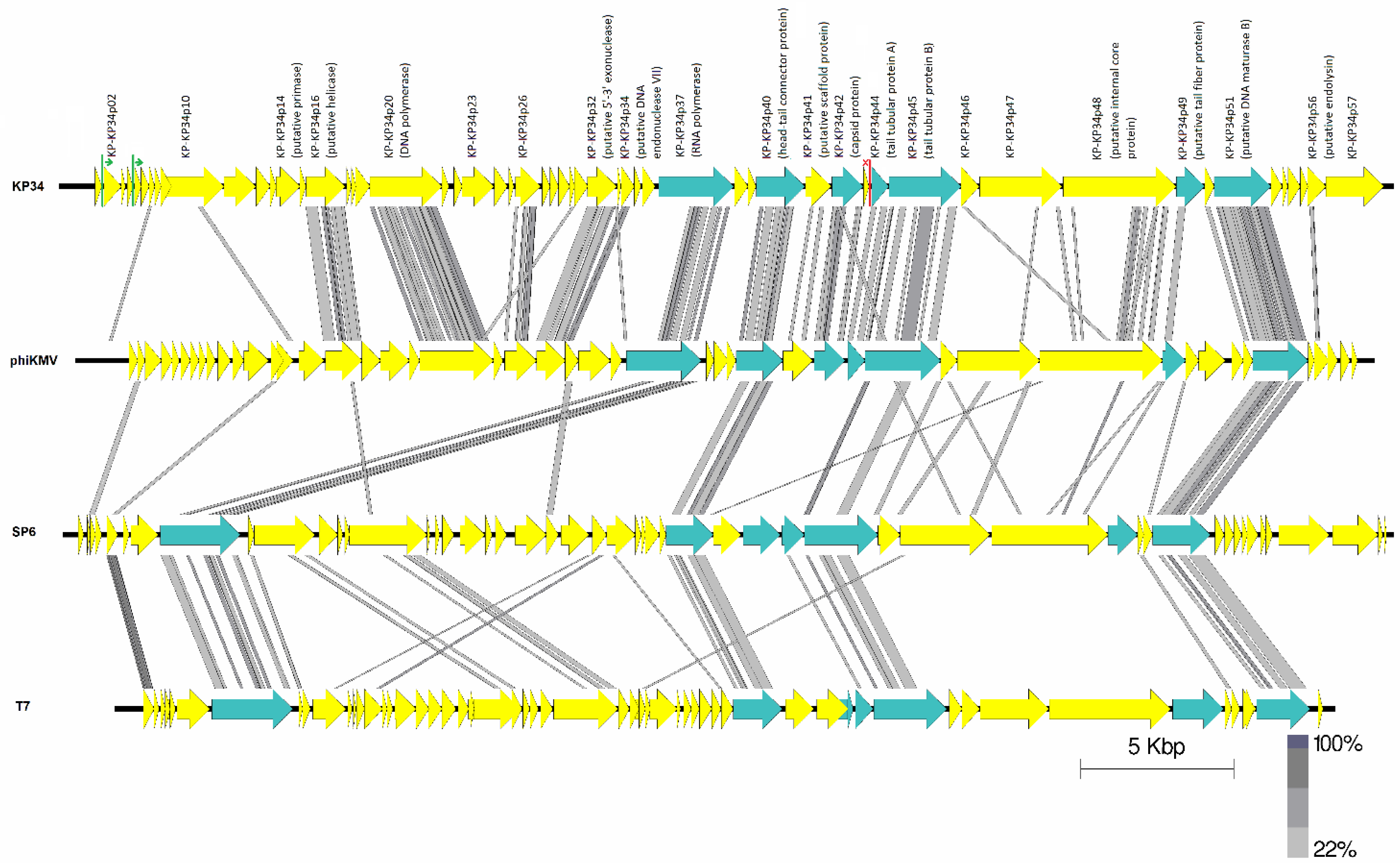
3.4. Genome Analysis
| Phage | Size (bp) | G+C content, % | CDS | Nucleotide pairwise identity, % | ||||
|---|---|---|---|---|---|---|---|---|
| KP34 | SU503 | SU552A | NTUH-K2044-K1-1 | F19 | ||||
| KP34 | 43,809 | 54.1 | 57 | - | 78.6 | 79.3 | 77.5 | 77.5 |
| SU503 | 43,809 | 53.7 | 55 | 78.6 | - | 75.1 | 78.1 | 76.8 |
| SU552A | 43,594 | 54.2 | 56 | 79.3 | 75.1 | - | 76.4 | 76.6 |
| NTUH-K2044-K1-1 | 43,871 | 54.2 | 54 | 77.5 | 78.1 | 76.4 | - | 76.1 |
| F19 | 43,766 | 53.8 | 52 | 77.5 | 76.8 | 76.6 | 76.1 | - |
| Host Promoter | ||
|---|---|---|
| Phage | location | nucleotide sequence |
| KP34 | 931..959 | TTGACACCGCGAAGAACATAAG |
| SU503 | 1040..1068 | TTGACACCGCGAAGGACATAAGCTAGATT |
| 1169..1196 | TTAAAATAAACGCTTGACAAGTTATGAT | |
| 1182..1210 | TTGACAAGTTATGATTCACTGAGTAACTT | |
| SU552A | 683..710 | TTGCCCTGCTTACCATTTTTGCTATAAG |
| 874..902 | TTGACACCGCGAAGAACATAAGCTAGATT | |
| 943..971 | TTGACACCGCGAAGAACATAAGCTAGATT | |
| NTUH-K2044-K1-1 | 953..981 | TTGACACCGCGAAGGACATAAGCTAGATT |
| 1022..1050 | TTGACAAGTTCTGATTCACTGAGTAACTT | |
| 1251..1280 | CTCACAGGTTAGCAGTCCTGAGCCGATAAG | |
| F19 | 968..996 | TTGACACCGCGAAGGACATAAGCTAGATT |
| 1051..1079 | TTGACAAGTTCCGATTCACTGAGTAACTT | |
| 1192..1220 | ACGACAAACGGCGGGTGCGCTTAGATGAT | |
| E. coli consensus sequence | TTGACA-(N15-18)-TATAAT | |
| Putative phage promoters | ||
| Phage | location | nucleotide sequence |
| KP34 | 1487..1538 | CACTAATTACAGCCTATAGCATCCTACGGGGTGCTATGTGAAGTAATTACCT |
| 2508..2559 | TTTAGTAGCAAGCCTATAGCGTCCTATGGGGCGCTATGTGAATGCAACTGGC | |
| SU503 | 2010..2061 | CGTTAATTACAGCCTATAGCATCCTACGGGGTGCTATGTGAAGTAATTACCT |
| 3192..3243 | TCCAGTAGCAAGCCTATAGCGTCCTACGGGGCGCTATGTGAATGCAACTGGC | |
| SU552A | 1449..1500 | CGTTAATTACAGCCTATAGCATCCTACGGGGTGCTATGTGAAGTAATTACCT |
| 2225..2276 | TATAGTAGCAAGCCTATAGCGTCCTACGGGGCGCTATGTGAATGCAACTAGC | |
| NTUH-K2044-K1-1 | 1752..1803 | TACTAATTACAGCCTATAGCATCCTATGGGGTGCTATGTGAAGTAATTACCT |
| 2618..2669 | TCTAGTAGCAAGCCTATAGCGTCCTACGGGGCGCTATGTGAATGCAACCGGC | |
| F19 | 1795..1846 | CGTTAATTACAGCCTATAGCATCCTATGGGGTGCTATGTGAAGTAATTACAT |
| 2571..2622 | TATAGTAGCAAGCCTATAGCGTCCGACTGGGCGCTATGTGAATGCAACTAGC | |
| Phage promoter consensus | AGCCTATAGCGTCCTACGGGGCGCTATGTGAA | |
| Rho-independent terminators | ||
| Phage | location | nucleotide sequence |
| KP34 | 26623..26660 | GCCCCTGGTGCCTTCGGGTGCCAGGGGCTTTTTTTTTT |
| SU503 | 26936..26972 | GCCCCTGGTGCCTTCGGGTGCCAGGGGCTTTTTTTTT |
| SU552A | 25384..25420 | GCCCCTGGTGCCTTCGGGTGCCAGGGGCTTTTTTTTT |
| NTUH-K2044-K1-1 | 25925..25961 | GCCCCTGGTGCCTTCTGGTGCCGGGGGCTTTTTTTTT |
| F19 | 26363..26400 | GCCCCTGGTGCCTTCGGGTGCCAGGGGCTTTTTTTTTT |
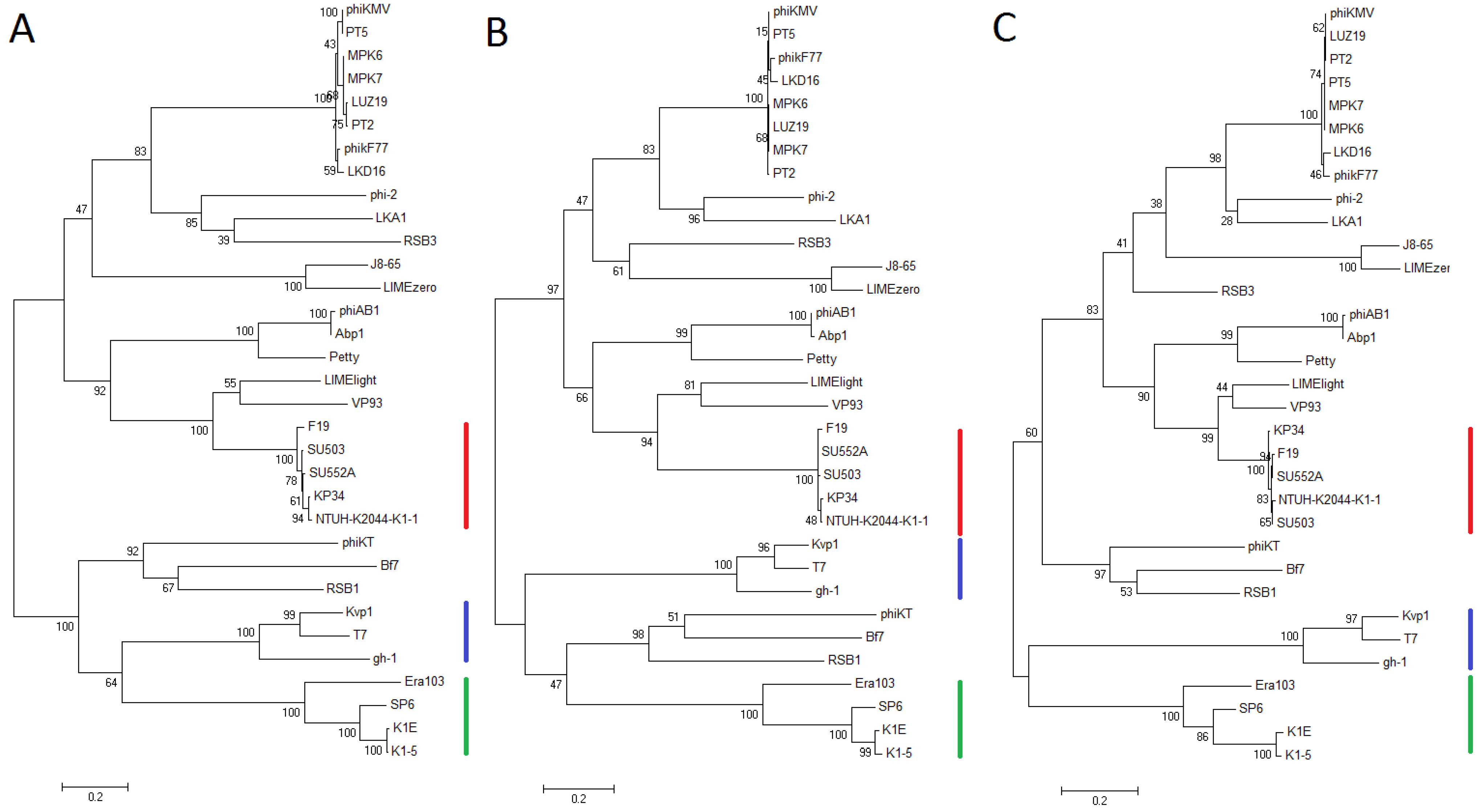
3.5. Comparative Genomics

| No. | Description | KP34 Locus_Tag | Accession no. | % Pairwise AA identity to KP34 | |||
|---|---|---|---|---|---|---|---|
| SU503 | SU552A | K2044 | F19 | ||||
| 1 | hypothetical protein | KP-KP34p02 | GI:282554636 | 96.8 | 97.9 | 94.1 | 97.9 |
| 2 | hypothetical protein | KP-KP34p05 | GI:294661414 | 63.0 | 65.8 | 64.4 | 65.8 |
| 3 | hypothetical protein | KP-KP34p10 | GI:282554643 | 84.0 a | 89.0 a | 88.3 a | 87.9 a |
| 4 | putative peptidase | KP-KP34p11 | GI:282554645 | 87.4 | 97.1 | 98.0 | 98.0 |
| 5 | hypothetical protein | KP-KP34p12 | GI:282554646 | 68.8 | 70.5 | 62.7 | 67.1 |
| 6 | putative DNA primase | KP-KP34p14 | GI:282554648 | 93.6 | 95.8 | 95.0 | 96.5 |
| 7 | putative DNA helicase | KP-KP34p16 | GI:294661415 | 96.0 b | 98.6 | 97.7 | 97.7 b |
| 8 | DNA polymerase | KP-KP34p20 | GI:282554654 | 81.5 | 84.1 | 81.8 | 80.8 |
| 9 | hypothetical protein | KP-KP34p23 | GI:282554657 | 88.1 | 81.9 | 85.8 | 72.9 |
| 10 | hypothetical protein | KP-KP34p26 | GI:282554660 | 96.5 | 98.7 | 96.5 | 95.8 |
| 11 | hypothetical protein | KP-KP34p29 | GI:282554662 | 78.5 | 83.5 | 91.8 | 86.0 |
| 12 | putative 5'-3' exonuclease | KP-KP34p32 | GI:282554665 | 93.5 | 92.2 | 91.9 | 92.5 |
| 13 | putative DNA endo- nuclease VII | KP-KP34p34 | GI:294661421 | 99.3 | 94.3 c | 51.4 c | 98.6 |
| 14 | DNA-dependent RNA polymerase | KP-KP34p37 | GI:282554612 | 96.8 | 96.4 | 98.4 | 94.6 |
| 15 | hypothetical protein | KP-KP34p38 | GI:282554613 | 97.9 | 90.4 | 89.7 | 91.1 |
| 16 | head-tail connector protein | KP-KP34p40 | GI:294661422 | 98.9 | 98.9 | 99.2 | 98.3 |
| 17 | putative scaffolding protein | KP-KP34p41 | GI:282554617 | 98.2 | 98.2 | 97.9 | 97.5 |
| 18 | capsid protein | KP-KP34p42 | GI:282554619 | 92.9 | 93 | 93.5 | 92.7 |
| 19 | tail tubular protein A | KP-KP34p44 | GI:282554621 | 87.9 | 91.9 | 97.7 | 92.2 |
| 20 | tail tubular protein B | KP-KP34p45 | GI:282554622 | 99.0 | 98.0 | 96.2 | 88.4 |
| 21 | putative internal virion protein B | KP-KP34p46 | GI:282554623 | 75.4 | 98.5 | 75.9 | 99.0 |
| 22 | hypothetical protein | KP-KP34p47 | GI:282554624 | 96.1 | 95.0 | 92.6 | 88.0 |
| 23 | putative internal core protein | KP-KP34p48 | GI:282554625 | 97.4 | 97.9 | 89.0 | 70.2 |
| 24 | putative tail fiber protein | KP-KP34p49 | GI:282554626 | 93.8 | 65.1 | 90.3 | 92.5 |
| 25 | putative DNA maturase A | KP-KP34p50 | GI:282554627 | 97.0 | 100 | 100 | 100 |
| 26 | putative DNA maturase B | KP-KP34p51 | GI:282554628 | 98.7 | 98.9 | 98.1 | 98.9 |
| 27 | hypothetical protein | KP-KP34p52 | GI:282554629 | 97.4 | 98.4 | 98.4 | 97.6 |
| 28 | hypothetical protein (spanin) | KP-KP34p54 | GI:282554631 | 98.5 | 97.8 | 97.0 | 96.3 |
| 29 | putative endolysin | KP-KP34p56 | GI:282554633 | 90.6 | 96.0 | 93.3 | 89.8 |
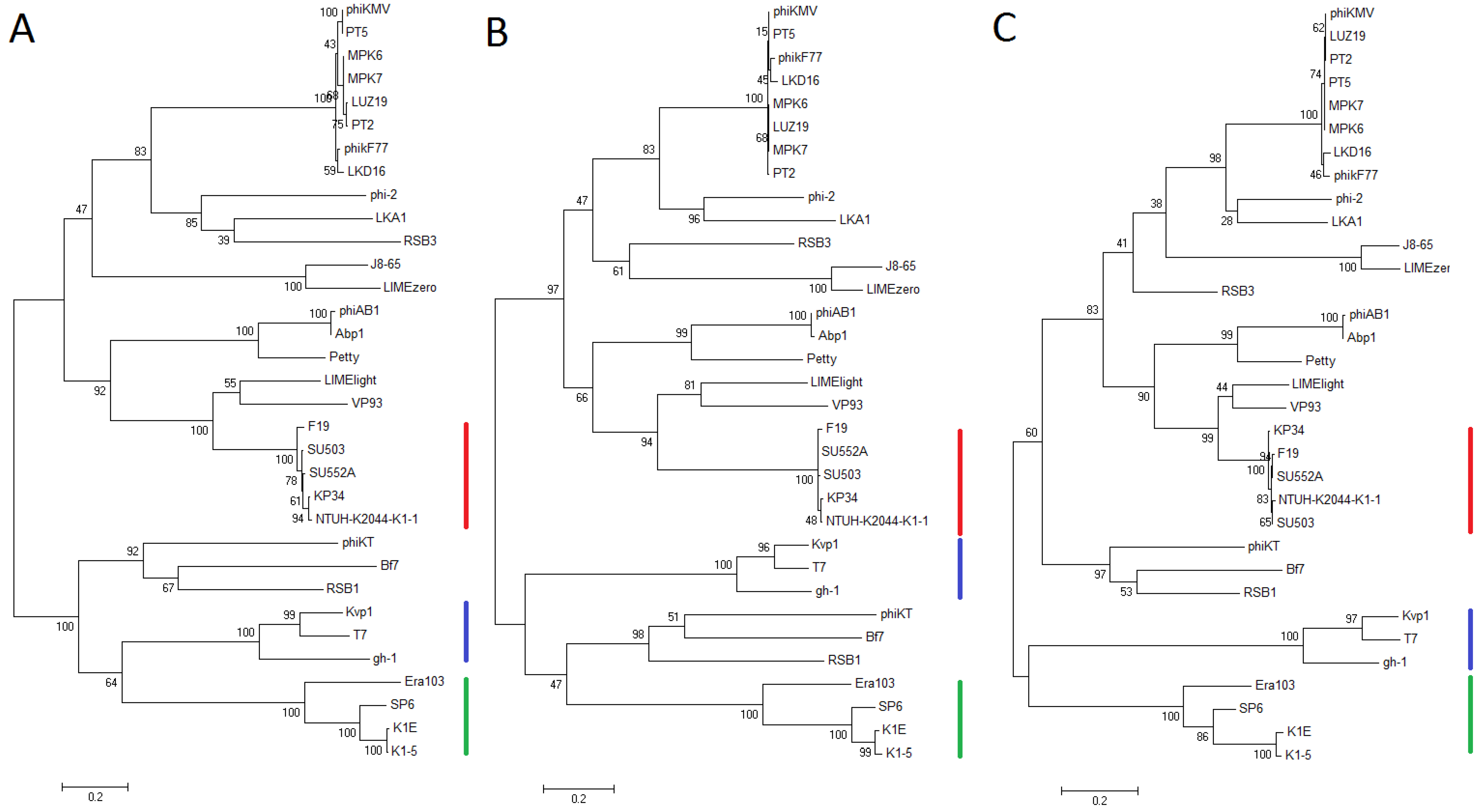
3.6. RNA Polymerase
| Phage | Recognition loop | Specificity loop |
|---|---|---|
| φKMV | HQEAKAAKPAAKL | EEVRVRLRAEAVEYVTLYEAK-DEL |
| KP34 | MRNVKAPGIGGKY | EEVRVRIDCMNLSAVLVHNRDFKTC |
| K2044 | MRNVKAPGIGGKY | EEVRVRIDCMNLSAVLVHNRDFKTC |
| F19 | MRNVKAPGIGGKY | EEVRVRIDCMNLTIMRVHNRDFKTC |
| SU503 | MRNVKAPGIGGKY | EEVRVRIDCMNLTIMRVHNRDFKTC |
| SU552A | MRNVKAPGIGGKY | EEVRVRIDCMNLTIMRVHNRDFKTC |
| LIMElight | IKAEKAPGVGGKY | EEKRVNIRSMGLTQVVAYNRNYDLN |
| VP93 | LKASKTRGVGAKY | HETRVKVRSMGINQVVLYNFDYERN |
3.7. Lysis Cassette

4. Discussion
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Lavigne, R.; Seto, D.; Mahadevan, P.; Ackermann, H.W.; Kropinski, A.M. Unifying classical and molecular taxonomic classification: Analysis of the Podoviridae using blastp-based tools. Res. Microbiol. 2008, 159, 406–414. [Google Scholar] [CrossRef] [PubMed]
- Dunn, J.J.; Studier, F.W. Complete nucleotide sequence of bacteriophage T7 DNA and the locations of T7 genetic elements. J. Mol. Biol. 1983, 166, 477–535. [Google Scholar] [CrossRef] [PubMed]
- Dobbins, A.T.; George, M., Jr.; Basham, D.A.; Ford, M.E.; Houtz, J.M.; Pedulla, M.L.; Lawrence, J.G.; Hatfull, G.F.; Hendrix, R.W. Complete genomic sequence of the virulent Salmonella bacteriophage SP6. J. Bacteriol. 2004, 186, 1933–1944. [Google Scholar] [CrossRef] [PubMed]
- Ceyssens, P.J.; Lavigne, R.; Mattheus, W.; Chibeu, A.; Hertveldt, K.; Mast, J.; Robben, J.; Volckaert, G. Genomic analysis of Pseudomonas aeruginosa phages LKD16 and LKA1: Establishment of the φKMV subgroup within the T7 supergroup. J. Bacteriol. 2006, 188, 6924–6931. [Google Scholar] [CrossRef] [PubMed]
- Drulis-Kawa, Z.; Mackiewicz, P.; Kesik-Szeloch, A.; Maciaszczyk-Dziubinska, E.; Weber-Dabrowska, B.; Dorotkiewicz-Jach, A.; Augustyniak, D.; Majkowska-Skrobek, G.; Bocer, T.; Empel, J.; et al. Isolation and characterisation of KP34—A novel φKMV-like bacteriophage for Klebsiella pneumoniae. Appl. Microbiol. Biotechnol. 2011, 90, 1333–1345. [Google Scholar] [CrossRef] [PubMed]
- Lin, T.L.; Hsieh, P.F.; Huang, Y.T.; Lee, W.C.; Tsai, Y.T.; Su, P.A.; Pan, Y.J.; Hsu, C.R.; Wu, M.C.; Wang, J.T. Isolation of a bacteriophage and its depolymerase specific for K1 capsule of Klebsiella pneumoniae: Implication in typing and treatment. J. Infect. Dis. 2014, 210, 1734–1744. [Google Scholar] [CrossRef] [PubMed]
- Podschun, R.; Ullmann, U. Klebsiella spp. As nosocomial pathogens: Epidemiology, taxonomy, typing methods, and pathogenicity factors. Clin. Microbiol. Rev. 1998, 11, 589–603. [Google Scholar] [PubMed]
- Pieroni, P.; Rennie, R.P.; Ziola, B.; Deneer, H.G. The use of bacteriophages to differentiate serologically cross-reactive isolates of Klebsiella pneumoniae. J. Med. Microbiol. 1994, 41, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Brolund, A.; Haeggman, S.; Edquist, P.J.; Gezelius, L.; Olsson-Liljequist, B.; Wisell, K.T.; Giske, C.G. The diversilab system versus pulsed-field gel electrophoresis: Characterisation of extended spectrum beta-lactamase producing Escherichia coli and Klebsiella pneumoniae. J. Microbiol. Methods 2010, 83, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Russell, D.W. Molecular Cloning—A Laboratory Manual, 3rd ed.; Cold Spring Harbor Laboratory Press: New York, NY, USA, 2001. [Google Scholar]
- Klebsiella Phage F19, Complete Genome. Available online: http://www.ncbi.nlm.nih.gov/nuccore/NC_023567.2 (accessed on 2 April 2015).
- Pajunen, M.; Kiljunen, S.; Skurnik, M. Bacteriophage φyeo3-12, specific for Yersinia enterocolitica serotype O:3, is related to coliphages T3 and T7. J. Bacteriol. 2000, 182, 5114–5120. [Google Scholar] [CrossRef] [PubMed]
- Kesik-Szeloch, A.; Drulis-Kawa, Z.; Weber-Dabrowska, B.; Kassner, J.; Majkowska-Skrobek, G.; Augustyniak, D.; Lusiak-Szelachowska, M.; Zaczek, M.; Gorski, A.; Kropinski, A.M. Characterising the biology of novel lytic bacteriophages infecting multidrug resistant Klebsiella pneumoniae. Virol. J. 2013, 10, e100. [Google Scholar] [CrossRef]
- Kutter, E. Phage host range and efficiency of plating. In Bacteriophages: Methods and Protocols Volume 1—Isolation, Characterization and Interactions, 1st ed.; Martha, R.J.C., Andrew, K., Eds.; Humana Press: New York, NY, USA, 2009; pp. 141–149. [Google Scholar]
- Boulanger, P. Purification of bacteriophages and SDS-PAGE analysis of phage structural proteins from ghost particles. Methods Mol. Biol. 2009, 502, 227–238. [Google Scholar] [PubMed]
- Aziz, R.K.; Bartels, D.; Best, A.A.; DeJongh, M.; Disz, T.; Edwards, R.A.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST server: Rapid annotations using subsystems technology. BMC Genomics 2008, 9, e75. [Google Scholar] [CrossRef]
- Noguchi, H.; Taniguchi, T.; Itoh, T. MetaGeneAnnotator: Detecting species-specific patterns of ribosomal binding site for precise gene prediction in anonymous prokaryotic and phage genomes. DNA Res. 2008, 15, 387–396. [Google Scholar] [CrossRef] [PubMed]
- Palleja, A.; Harrington, E.D.; Bork, P. Large gene overlaps in prokaryotic genomes: Result of functional constraints or mispredictions? BMC Genomics 2008, 9, e335. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped blast and psi-blast: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Eddy, S.R. tRNAscan-se: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Gautheret, D.; Lambert, A. Direct RNA motif definition and identification from multiple sequence alignments using secondary structure profiles. J. Mol. Biol. 2001, 313, 1003–1011. [Google Scholar] [CrossRef] [PubMed]
- Macke, T.J.; Ecker, D.J.; Gutell, R.R.; Gautheret, D.; Case, D.A.; Sampath, R. RNAmotif, an RNA secondary structure definition and search algorithm. Nucleic Acids Res. 2001, 29, 4724–4735. [Google Scholar] [CrossRef] [PubMed]
- Bailey, T.L.; Elkan, C. Fitting a mixture model by expectation maximization to discover motifs in biopolymers. Proc. Int. Conf. Intell. Syst. Mol. Biol. 1994, 2, 28–36. [Google Scholar] [PubMed]
- Turner, D.; Reynolds, D.; Seto, D.; Mahadevan, P. CoreGenes3.5: A webserver for the determination of core genes from sets of viral and small bacterial genomes. BMC Res. Notes 2013, 6, 140. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef] [PubMed]
- Kearse, M.; Moir, R.; Wilson, A.; Stones-Havas, S.; Cheung, M.; Sturrock, S.; Buxton, S.; Cooper, A.; Markowitz, S.; Duran, C.; et al. Geneious basic: An integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 2012, 28, 1647–1649. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Whelan, S.; Goldman, N. A general empirical model of protein evolution derived from multiple protein families using a maximum-likelihood approach. Mol. Biol. Evol. 2001, 18, 691–699. [Google Scholar] [CrossRef] [PubMed]
- Finn, R.D.; Clements, J.; Eddy, S.R. HMMER web server: Interactive sequence similarity searching. Nucleic Acids Res. 2011, 39, W29–W37. [Google Scholar] [CrossRef] [PubMed]
- Bernsel, A.; Viklund, H.; Hennerdal, A.; Elofsson, A. TOPCONS: Consensus prediction of membrane protein topology. Nucleic Acids Res. 2009, 37, W465–W468. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting transmembrane protein topology with a hidden Markov model: Application to complete genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed]
- Adams, M.H.; Park, B.H. An enzyme produced by a phage-host cell system. Ii. The properties of the polysaccharide depolymerase. Virology 1956, 2, 719–736. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, H.W. Classification of bacteriophages. In The Bacteriophages, 2nd ed.; Calendar, R., Ed.; Oxford University Press: New York, NY, USA, 2006; pp. 8–16. [Google Scholar]
- Kulakov, L.A.; Ksenzenko, V.N.; Shlyapnikov, M.G.; Kochetkov, V.V.; del Casale, A.; Allen, C.C.; Larkin, M.J.; Ceyssens, P.J.; Lavigne, R. Genomes of “φKMV-like viruses” of Pseudomonas aeruginosa contain localized single-strand interruptions. Virology 2009, 391, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Cheetham, G.M.; Steitz, T.A. Structure of a transcribing T7 RNA polymerase initiation complex. Science 1999, 286, 2305–2309. [Google Scholar] [CrossRef] [PubMed]
- Cheetham, G.M.; Steitz, T.A. Insights into transcription: Structure and function of single-subunit DNA-dependent RNA polymerases. Curr. Opin. Struct. Biol. 2000, 10, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Wang, I.N.; Smith, D.L.; Young, R. Holins: The protein clocks of bacteriophage infections. Annu. Rev. Microbiol. 2000, 54, 799–825. [Google Scholar] [CrossRef] [PubMed]
- Briers, Y.; Peeters, L.M.; Volckaert, G.; Lavigne, R. The lysis cassette of bacteriophage φKMV encodes a signal-arrest-release endolysin and a pinholin. Bacteriophage 2011, 1, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Drulis-Kawa, Z.; Majkowska-Skrobek, G.; Maciejewska, B.; Delattre, A.S.; Lavigne, R. Learning from bacteriophages—Advantages and limitations of phage and phage-encoded protein applications. Curr. Protein Pept. Sci. 2012, 13, 699–722. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, H.; Azeredo, J.; Lavigne, R.; Kluskens, L.D. Bacteriophage endolysins as a response to emerging foodborne pathogens. Trends Food Sci. Technol. 2012, 28, 103–115. [Google Scholar] [CrossRef] [Green Version]
- Pang, T.; Fleming, T.C.; Pogliano, K.; Young, R. Visualization of pinholin lesions in vivo. Proc. Natl. Acad. Sci. USA 2013, 110, E2054–E2063. [Google Scholar] [CrossRef] [PubMed]
- Berry, J.D.; Rajaure, M.; Young, R. Spanin function requires subunit homodimerization through intermolecular disulfide bonds. Mol. Microbiol. 2013, 88, 35–47. [Google Scholar] [CrossRef] [PubMed]
- Young, R. Phage lysis: Three steps, three choices, one outcome. J. Microbiol. 2014, 52, 243–258. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, E.M.; Ceyssens, P.J.; Dunon, V.; Ackermann, H.W.; van Vaerenbergh, J.; Maes, M.; de Proft, M.; Lavigne, R. Bacteriophages LIMElight and LIMEzero of Pantoea agglomerans, belonging to the “φKMV-like viruses”. Appl. Environ. Microbiol. 2011, 77, 3443–3450. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Eriksson, H.; Maciejewska, B.; Latka, A.; Majkowska-Skrobek, G.; Hellstrand, M.; Melefors, Ö.; Wang, J.-T.; Kropinski, A.M.; Drulis-Kawa, Z.; Nilsson, A.S. A Suggested New Bacteriophage Genus, “Kp34likevirus”, within the Autographivirinae Subfamily of Podoviridae. Viruses 2015, 7, 1804-1822. https://doi.org/10.3390/v7041804
Eriksson H, Maciejewska B, Latka A, Majkowska-Skrobek G, Hellstrand M, Melefors Ö, Wang J-T, Kropinski AM, Drulis-Kawa Z, Nilsson AS. A Suggested New Bacteriophage Genus, “Kp34likevirus”, within the Autographivirinae Subfamily of Podoviridae. Viruses. 2015; 7(4):1804-1822. https://doi.org/10.3390/v7041804
Chicago/Turabian StyleEriksson, Harald, Barbara Maciejewska, Agnieszka Latka, Grazyna Majkowska-Skrobek, Marios Hellstrand, Öjar Melefors, Jin-Town Wang, Andrew M. Kropinski, Zuzanna Drulis-Kawa, and Anders S. Nilsson. 2015. "A Suggested New Bacteriophage Genus, “Kp34likevirus”, within the Autographivirinae Subfamily of Podoviridae" Viruses 7, no. 4: 1804-1822. https://doi.org/10.3390/v7041804