
Development of a Recombination System for the Generation of Occlusion Positive Genetically Modified Anticarsia Gemmatalis Multiple Nucleopolyhedrovirus

Abstract
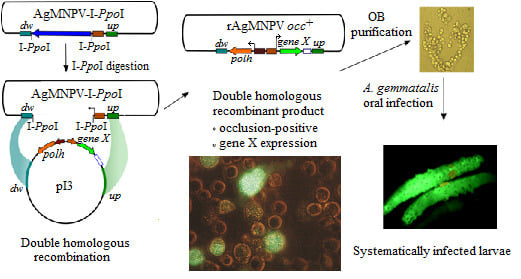
:
1. Introduction
2. Materials and Methods
2.1. Virus, Cells and Insects
2.2. Recombinant DNA Methods

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Primer name | Primer sequence |
|---|---|
| Upr10-NdeI | GCCCATATGCACAGTCAACGCCGGCC |
| Lpr10-SgfI | GCCCGCGATCGCGACGATATTGAAATGGTTGAAATAAATATAC |
| Uprom-NdeI | GCCCCATATGAAGTTGCAGCTCAAGCAGGATTGT |
| Ppolhrev-NotI | CATTGCGGCCGCAATTCAAGCTTAGTTATAGCAAATTTTACTAC |
| Uup-RsrII | CCCCGGTCCGATGACCGAATTGAGCAACGCG |
| Lup-SfiI | CTAGTTGGCCGCCTCGGCCTGCTGACTAAGCGTAGACC |
| Lred-SfiI | CGCTTAGT GGCCGAGGCGGCCAACTAGAATGCAGTGAAAAAAATG |
| SV40/CcdB-XmaI | ATGGACCACCCCGGGTTCCTGTAGCGGCCGCG |
| Polhi-SgfI | AAATTTGCGATCGCTATGCCAGATTATACG |
| Ldw-BglII | GGAAAGATCTATACACACGTTAGGCGAGCGCCG |
| eGFP/Up-EcoRI | TCCATCGAATTCATGGTGAGCAAGGGC |
| eGFP/Dw-XhoI | CTGATAAGCTTCTCGAGTCGCGGCCG |
| Primer name | Primer sequence |
|---|---|
| Polhi-SgfI | AAATTTGCGATCGCTATGCCAGATTATACG |
| AgDwrec | AACCCGTAAAGCCGCCGTTG |
| AgUpsrec | GGCGCGAGTTAAATAGTCTG |
| SV40/CcdB-XmaI | ATGGACCACCCCGGGTTCCTGTAGCGGCCGCG |
| LacZ | TGGATCTGCAACATGTCCCAGGTGA |
| ppolhAg600UpHindIII | TGTACAAAGCTTCTAATTGCGTAAAAATG |
| pie1Agfw | TATAAGATCTCAGGGTACAATTG |
| pie1Agrev | CATGAAGATCTATTTATACC |
2.3. Construction of the AgMNPV-I-PpoI Recombinant
2.4. Transfection of Linearized AgMNPV-I-PpoI DNA vs. Circular Undigested Genome
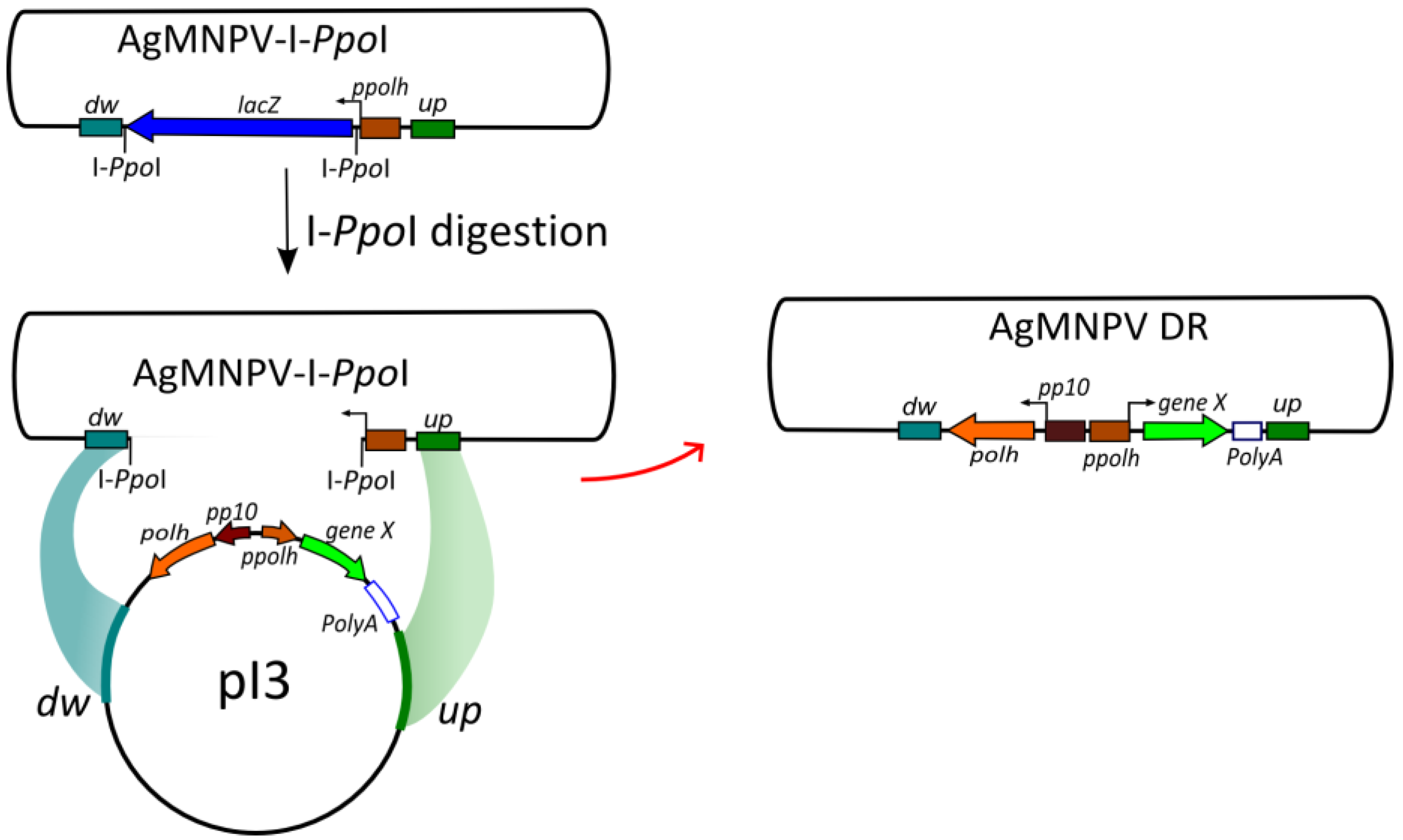
2.5. Generation of the Transfer Vector pI3
2.6. Construction of Recombinant AgMNPV-GFP
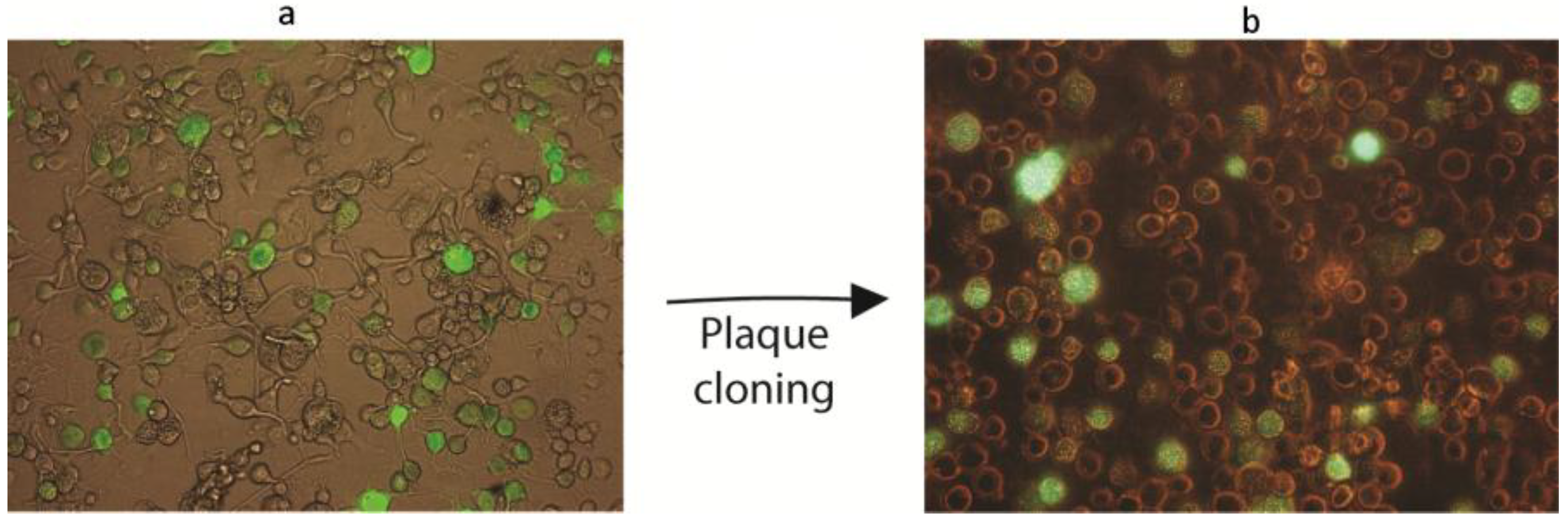
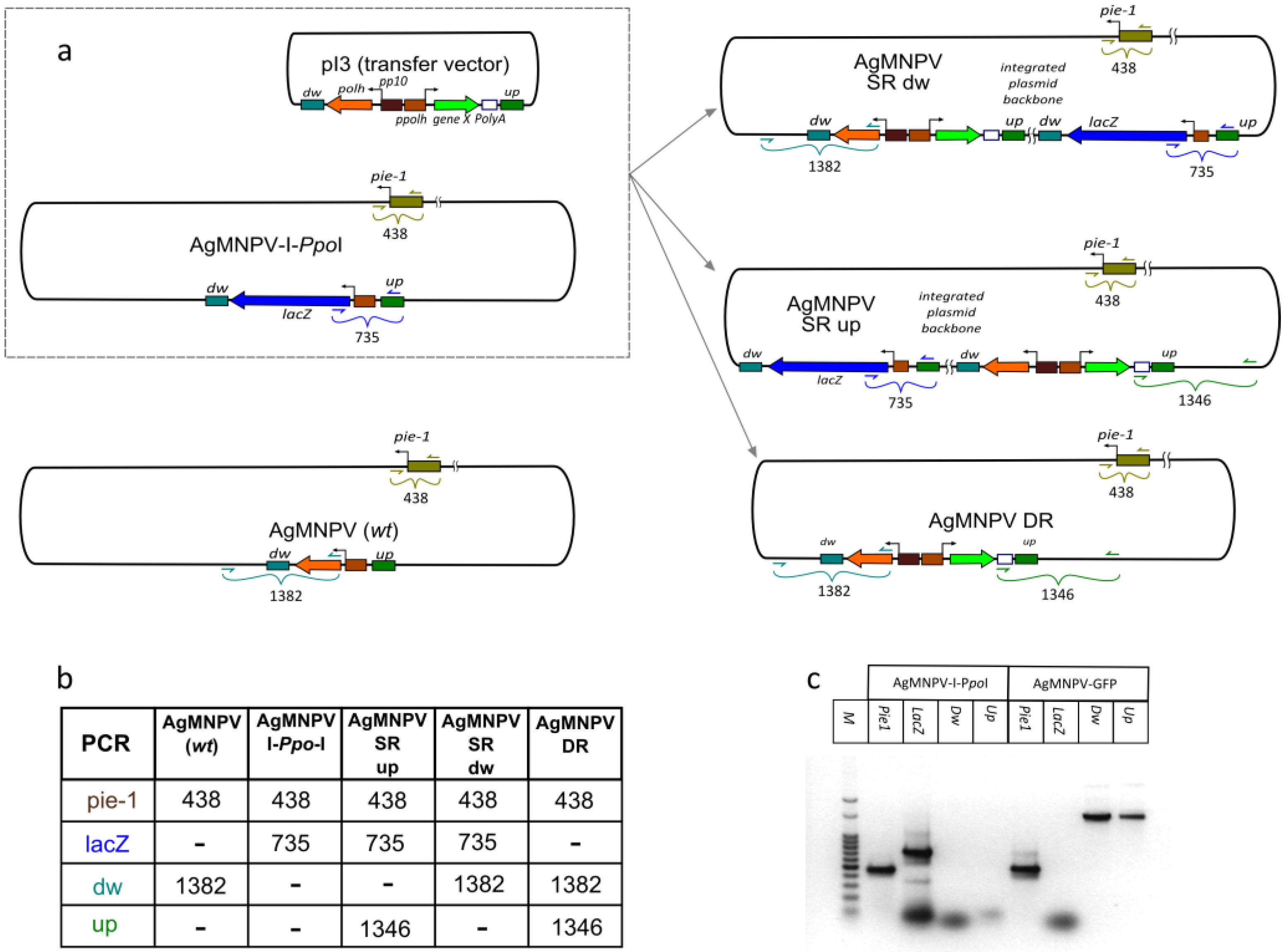
2.7. Characterization of AgMNPV-GFP
2.8. Per os Infection of A. gemmatalis Larvae with Recombinant AgMNPV
3. Results
3.1. Construction of a Linearizable AgMNPV Genome
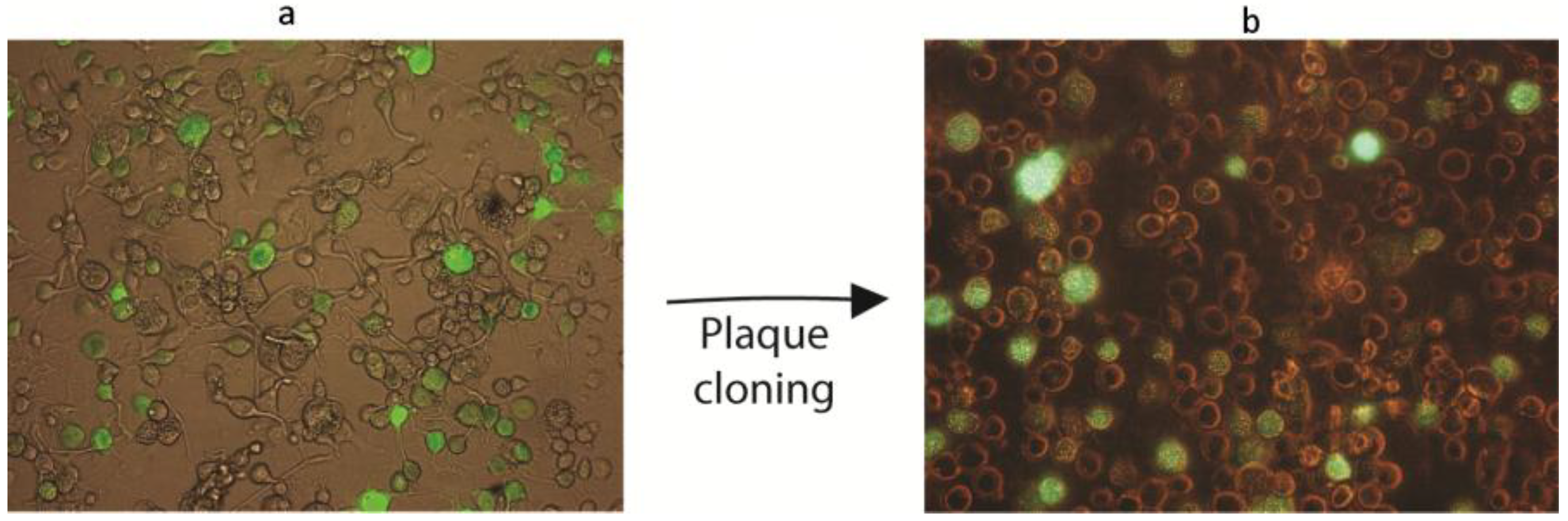
3.2. Transfection of Insect Cells with Circular vs. Linear AgMNPV Genomic DNA
3.3. Construction of a Transfer Vector for the Generation of Occlusion-Positive Recombinant AgMNPV
3.4. Co-Transfection of Viral DNA and Transfer Plasmid
3.5. The Recombinant AgMNPV-GFP Infects Cells Producing Polyhedra, which Are Orally Infective to A. gemmatalis Larvae
4. Discussion
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Szewczyk, B.; Hoyos-Carvajal, L.; Paluszek, M.; Skrzecz, I.; Lobo de Souza, M. Baculoviruses—Re-emerging biopesticides. Biotechnol. Adv. 2006, 24, 143–160. [Google Scholar] [CrossRef] [PubMed]
- Allen, G.E.; Knell, J.D. A nuclear polyhedrosis virus of Anticarsia gemmatalis: I. Ultrastructure, replication and pathogenicity. Fla. Entomol. 1977, 60, 233–240. [Google Scholar] [CrossRef]
- Moscardi, F. Assessment of the application of baculoviruses for control of Lepidoptera. Annu. Rev. Entomol. 1999, 44, 257–289. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, J.V.; Wolff, J.L.; Garcia-Maruniak, A.; Ribeiro, B.M.; de Castro, M.E.; de Souza, M.L.; Moscardi, F.; Maruniak, J.E.; Zanotto, P.M. Genome of the most widely used viral biopesticide: Anticarsia gemmatalis multiple nucleopolyhedrovirus. J. Gen. Virol. 2006, 87, 3233–3250. [Google Scholar] [CrossRef] [PubMed]
- Bonning, B.C.; Hammock, B.D. Development of recombinant baculoviruses for insect control. Annu. Rev. Entomol. 1996, 41, 191–210. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S. Increased insecticidal effect by a recombinant baculovirus carrying a synthetic diuretic hormone gene. Biochem. Biophys. Res. Commun. 1989, 165, 1177–1183. [Google Scholar] [CrossRef] [PubMed]
- Haase, S.; Ferrelli, M.L.; Pidre, M.L.; Romanowski, V. Genetic Engineering of Baculoviruses. In Current Issues in Molecular Virology—Viral Genetics and Biotechnological Applications; Romanowski, V., Ed.; InTech: Rijeka, Croatia, 2013. [Google Scholar] [CrossRef]
- Kitts, P.A.; Ayres, M.D.; Possee, R.D. Linearization of baculovirus DNA enhances the recovery of recombinant virus expression vectors. Nucleic Acids Res. 1990, 18, 5667–5672. [Google Scholar] [CrossRef] [PubMed]
- Kitts, P.A.; Possee, R.D. A method for producing recombinant baculovirus expression vectors at high frequency. BioTechniques 1993, 14, 810–817. [Google Scholar] [PubMed]
- Zanotto, P.M.; Sampaio, M.J.; Johnson, D.W.; Rocha, T.L.; Maruniak, J.E. The Anticarsia gemmatalis nuclear polyhedrosis virus polyhedrin gene region: Sequence analysis, gene product and structural comparisons. J. Gen. Virol. 1992, 73, 1049–1056. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann-Campo, C.B.; de Oliveira, E.B.; Moscardi, F. Criação Massal da Lagarta da Soja Anticarsia gemmatalis; Documentos 10; EMBRAPA-CNPSo: Londrina, Brazil, 1985; p. 23. [Google Scholar]
- Sambrook, J.; Fritsch, E.F.; Maniatis, T. Molecular Cloning: A Laboratory Manual, 2nd ed.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 1989. [Google Scholar]
- McCarthy, C.B.; Romanowski, V. Digestion of I-PpoI recognition sites in unfavorable sequence contexts achieved by changing the reaction conditions. Biochem. Genet. 2006, 44, 61–68. [Google Scholar] [CrossRef] [PubMed]
- Arana, E.I.; Albariño, C.G.; O’Reilly, D.; Ghiringhelli, P.D.; Romanowski, V. Generation of a recombinant Anticarsia gemmatalis multicapsid nucleopolyhedrovirus expressing a foreign gene under the control of a very late promoter. Virus Genes 2001, 22, 363–372. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, C.B.; Romanowski, V. A simplified method for the extraction of baculoviral DNA for PCR analysis: A practical application. J. Virol. Methods 2008, 148, 286–290. [Google Scholar] [CrossRef] [PubMed]
- O’Reilly, D.R.; Miller, L.K.; Luckow, V.A. Baculovirus Expression Vectors: A Laboratory Manual; Oxford University Press: New York, NY, USA, 1994. [Google Scholar]
- Murhammer, D.W. Baculovirus and Insect Cell Expression Protocols. Methods Mol. Biol. 2007, 388, 1–404. [Google Scholar]
- Hughes, P.R.; Wood, H.A. A synchronous peroral technique for the bioassay of insect viruses. J. Invertebr. Pathol. 1981, 37, 154–159. [Google Scholar] [CrossRef]
- Kunimi, Y.; Fuxa, J.R. Volumes ingested by four species of noctuids with reference to peroral droplet bioassay of baculoviruses. J. Invertebr. Pathol. 1996, 68, 310–311. [Google Scholar] [CrossRef] [PubMed]
- Abot, A.R.; Moscardi, F.; Fuxa, J.R.; Sosa-Gómez, D.R.; Richter, A.R. Development of Resistance by Anticarsia gemmatalis from Brazil and the United States to a Nuclear Polyhedrosis Virus under Laboratory Selection Pressure. Biol. Control 1996, 7, 126–130. [Google Scholar] [CrossRef]
- Lima, A.A.; Aragao, C.W.; de Castro, M.E.; Oliveira, J.V.; Sosa-Gómez, D.R.; Ribeiro, B.M. A Recombinant Anticarsia gemmatalis MNPV Harboring chiA and v-cath genes from Choristoneura fumiferana defective NPV induce host liquefaction and increased insecticidal activity. PLoS ONE 2013, 8, e74592. [Google Scholar] [CrossRef]
- Pinedo, F.J.R.; Moscardi, F.; Luque, T.; Olszewski, J.A.; Ribeiro, B.M. Inactivation of the ecdysteroid UDP-glucosyltransferase (egt) gene of Anticarsia gemmatalis nucleopolyhedrovirus (AgMNPV) improves its virulence towards its insect host. Biol. Control 2003, 27, 336–344. [Google Scholar] [CrossRef]
- Ribeiro, B.M.; Gatti, C.D.; Costa, M.H.; Moscardi, F.; Maruniak, J.E.; Possee, R.D.; Zanotto, P.M. Construction of a recombinant Anticarsia gemmatalis nucleopolyhedrovirus (AgMNPV-2D) harbouring the beta-galactosidase gene. Arch. Virol. 2001, 146, 1355–1367. [Google Scholar] [CrossRef] [PubMed]
- Soares, J.S.; Ribeiro, B.M. Pathology of Anticarsia gemmatalis larvae infected by two recombinant A. gemmatalis multicapsid nucleopolyhedroviruses. Res. Microbiol. 2005, 156, 263–269. [Google Scholar]
- Pijlman, G.P.; van den Born, E.; Martens, D.E.; Vlak, J.M. Autographa californica baculoviruses with large genomic deletions are rapidly generated in infected insect cells. Virology 2001, 283, 132–138. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Haase, S.; McCarthy, C.B.; Ferrelli, M.L.; Pidre, M.L.; Sciocco-Cap, A.; Romanowski, V. Development of a Recombination System for the Generation of Occlusion Positive Genetically Modified Anticarsia Gemmatalis Multiple Nucleopolyhedrovirus. Viruses 2015, 7, 1599-1612. https://doi.org/10.3390/v7041599
Haase S, McCarthy CB, Ferrelli ML, Pidre ML, Sciocco-Cap A, Romanowski V. Development of a Recombination System for the Generation of Occlusion Positive Genetically Modified Anticarsia Gemmatalis Multiple Nucleopolyhedrovirus. Viruses. 2015; 7(4):1599-1612. https://doi.org/10.3390/v7041599
Chicago/Turabian StyleHaase, Santiago, Christina B. McCarthy, M. Leticia Ferrelli, Matias L. Pidre, Alicia Sciocco-Cap, and Victor Romanowski. 2015. "Development of a Recombination System for the Generation of Occlusion Positive Genetically Modified Anticarsia Gemmatalis Multiple Nucleopolyhedrovirus" Viruses 7, no. 4: 1599-1612. https://doi.org/10.3390/v7041599