
Rapid Detection of Listeria by Bacteriophage Amplification and SERS-Lateral Flow Immunochromatography


Abstract
:1. Introduction
2. Materials and Methods
2.1. A511 Propagation
2.2. Production and Purification of Anti-A511 Antibodies
2.3. Nanoparticle Reporter and Control Particle Preparation
2.4. LFI Device Fabrication
2.5. Determination of LFI Limit of Detection
2.6. Phage Amplification and LFI Analysis
3. Results and Discussion
3.1. SERS-LFI Device Optimization

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Parameter Investigated | Optimized Conditions |
|---|---|
| Conjugation Buffer | 10 mM MOPS, pH 7.15 |
| Running Buffer | 0.1 M sodium borate, 3%, 1% Tween 20, pH 8 |
| Antibody:SERS NP ratio | 350:1 |
| Thiol blocker | N-ethylmaleimide |
| Capture antibody concentration | 2 mg/mL |
| Membrane Flow Rate | 180 mm/4 s |
| SERS reporter release pad concentration | 0.02% solids |
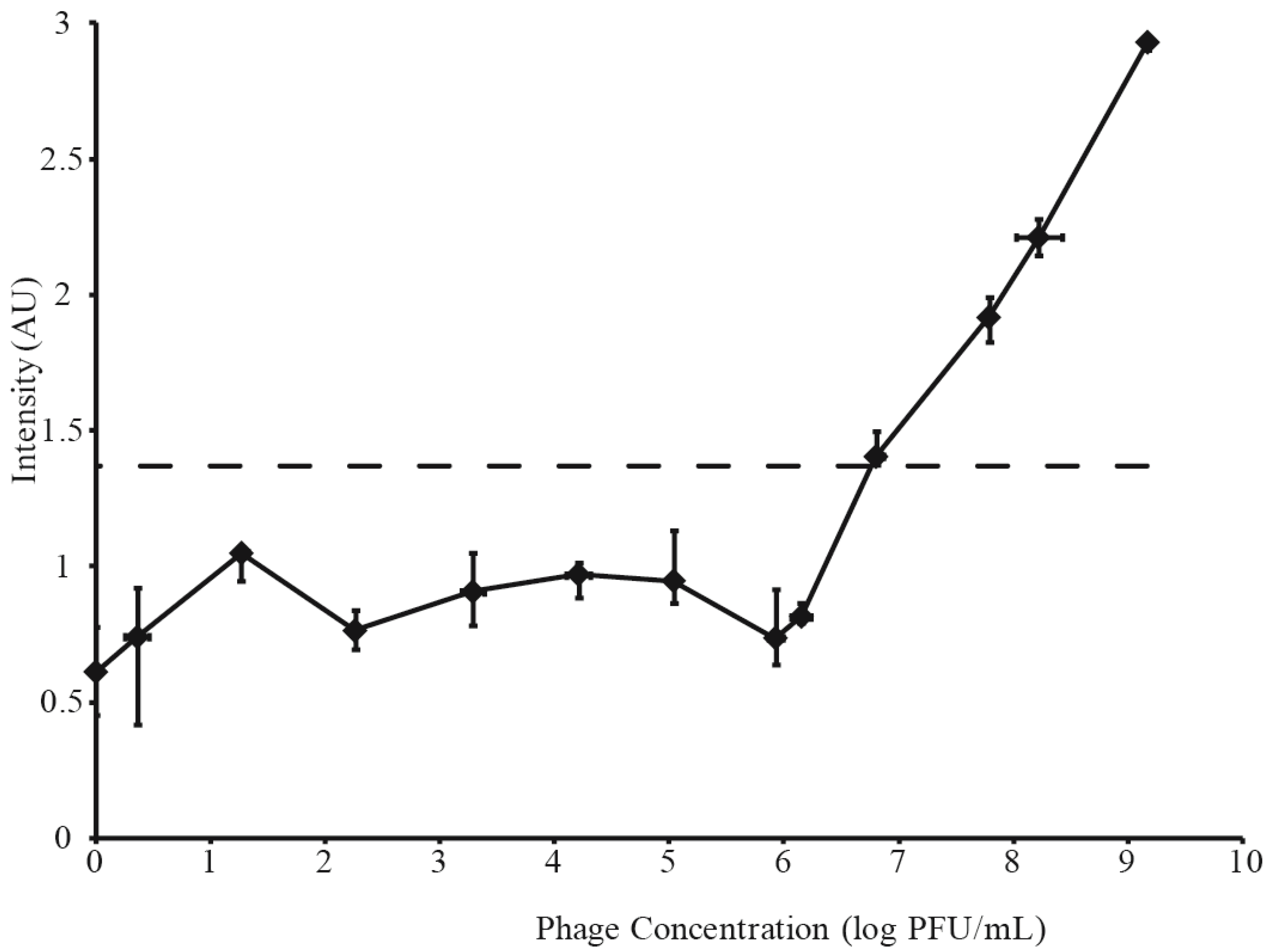
3.2. Determination of LFI Limit of Detection

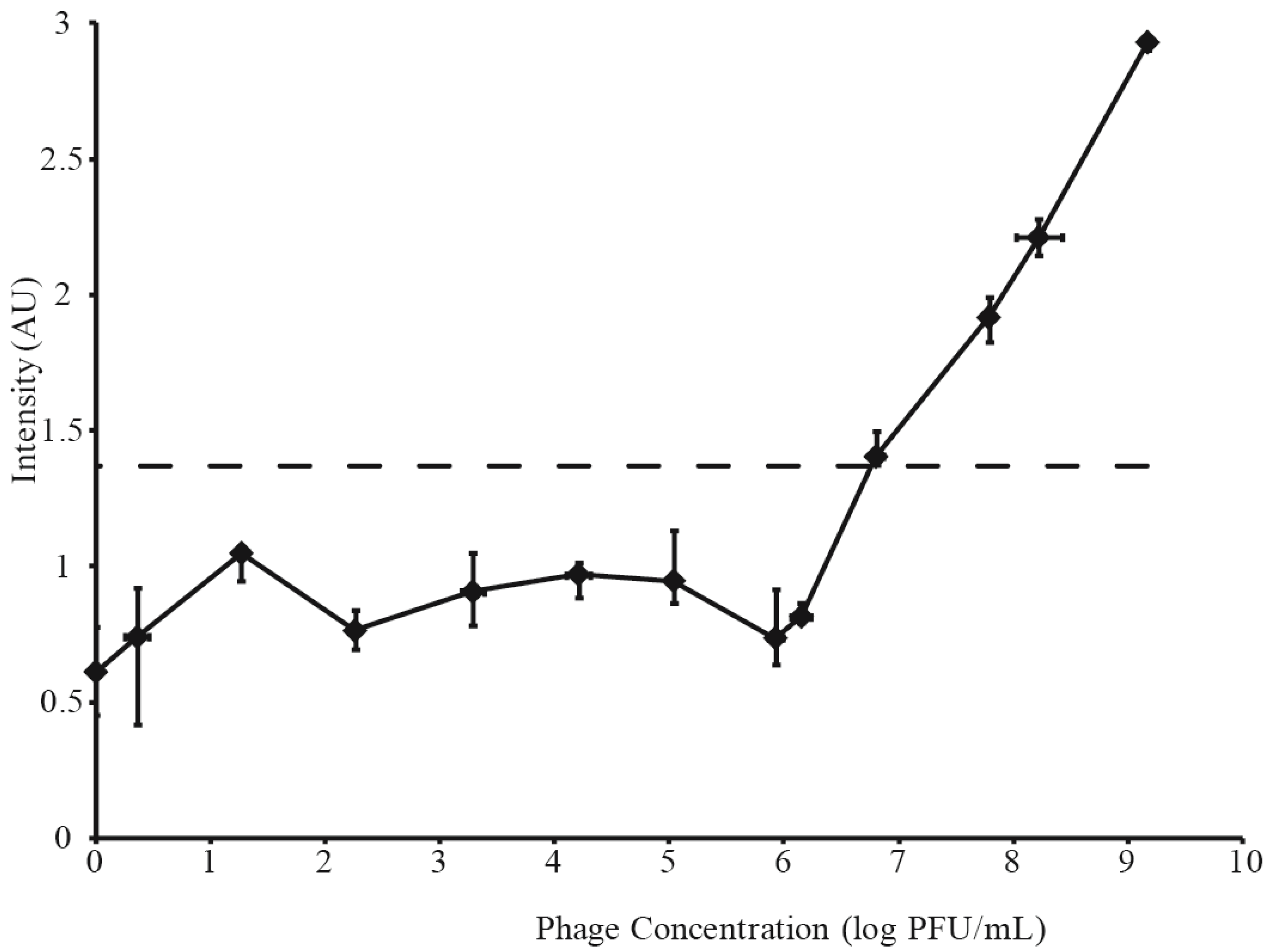
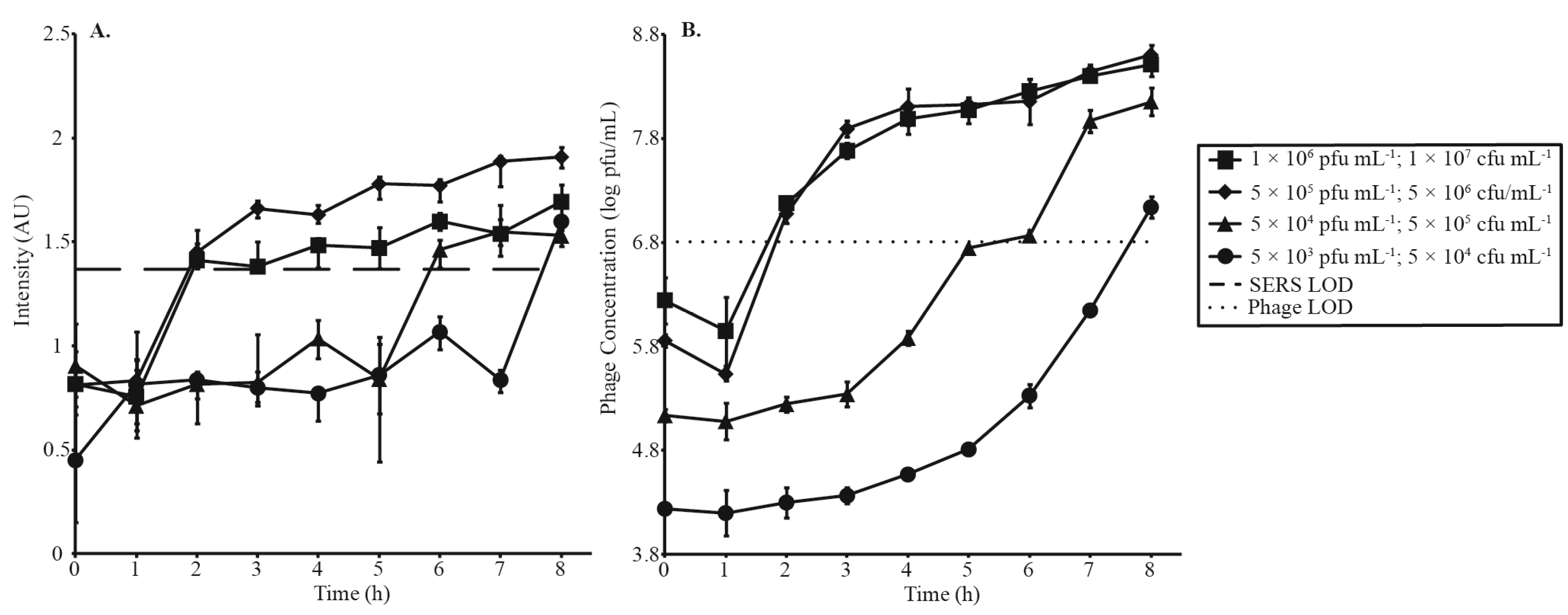
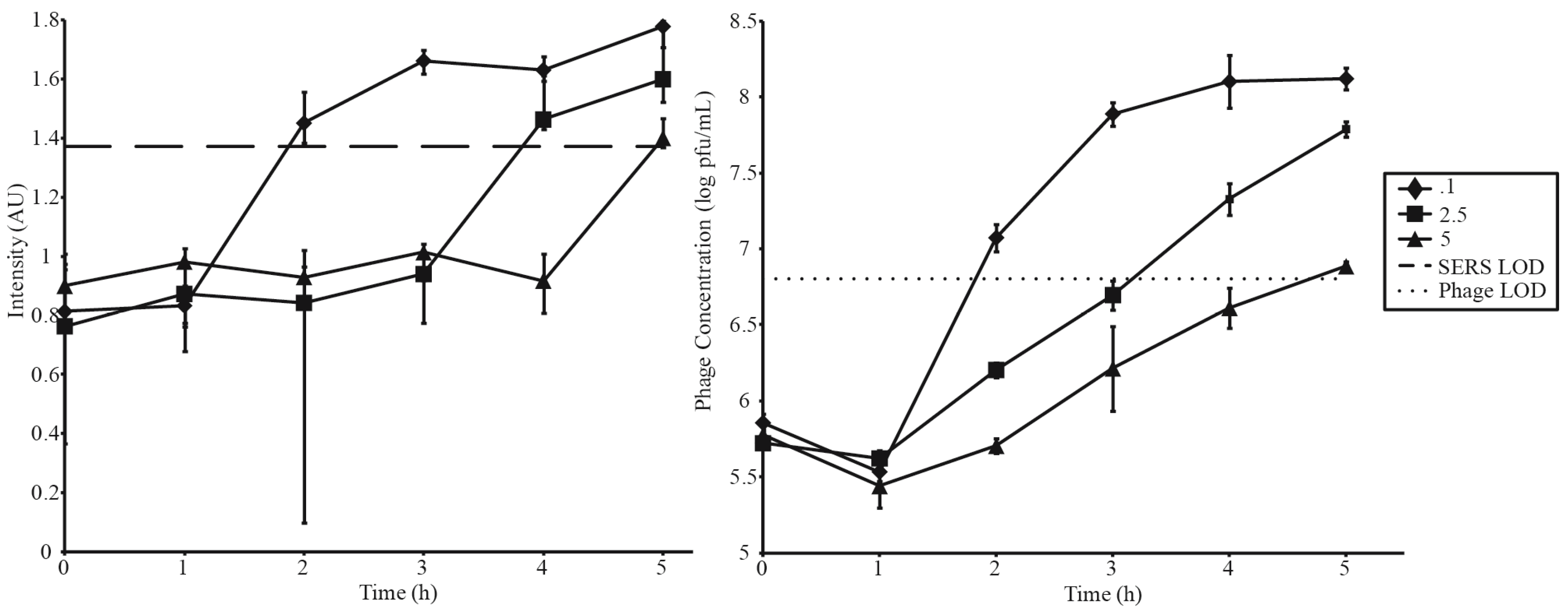
3.3. Phage Amplification and SERS-LFI Analysis
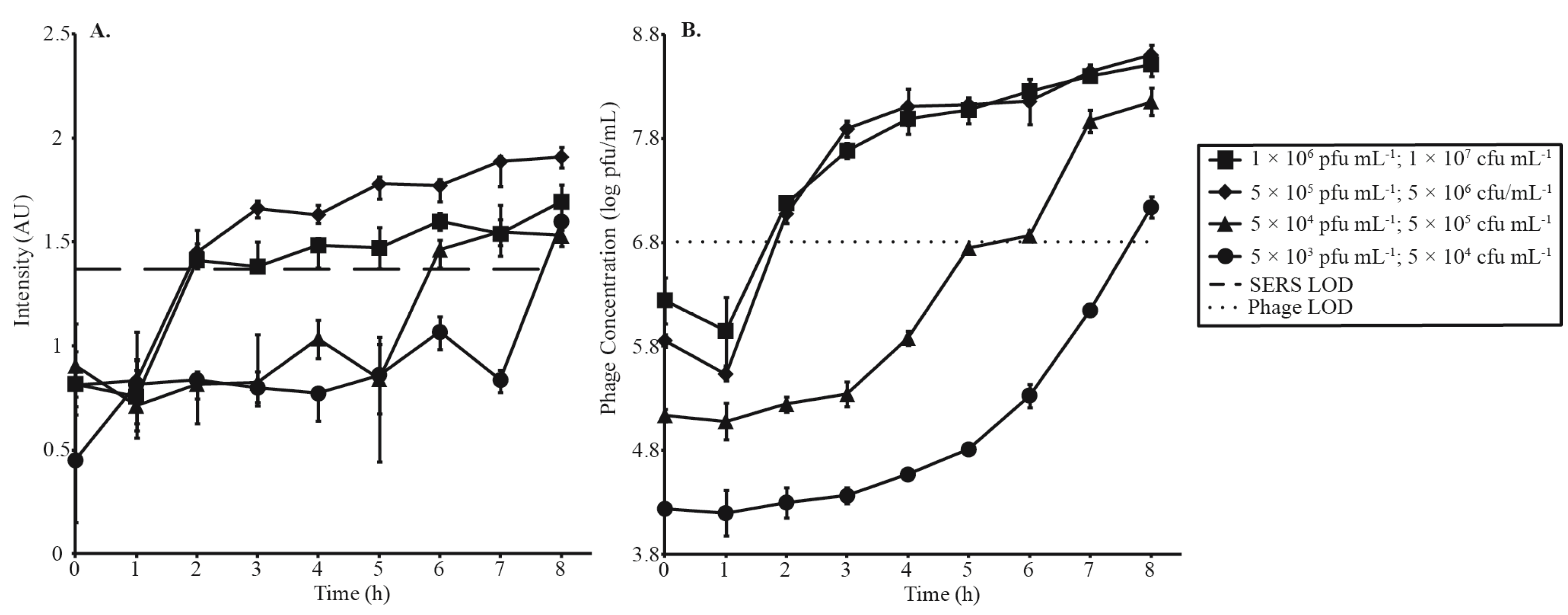
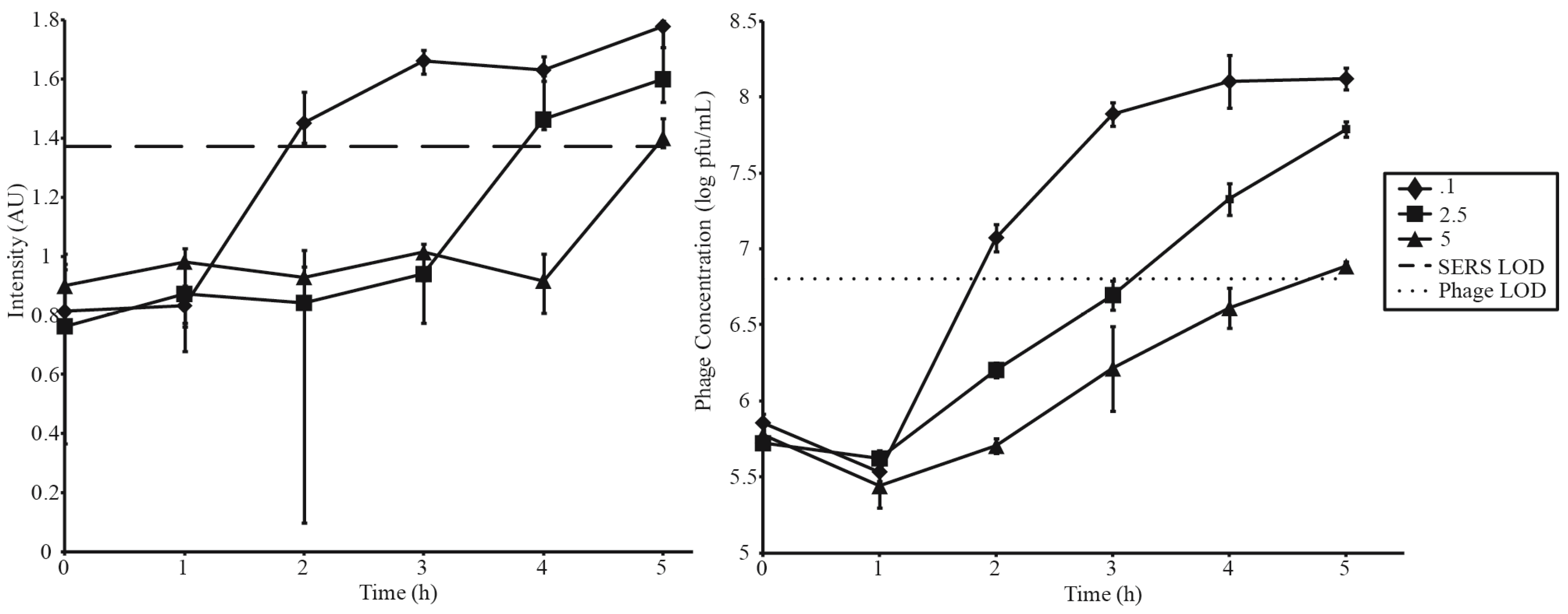
| Phage Amplification Series 1 | |||
| Phage Concentration (pfu·mL−1) | Bacterial Concentration (cfu·mL−1) | MOI | SERS-LFI Detection Time (h) |
| 1 × 106 | 1 × 107 | 0.1 | 2 |
| 5 × 105 | 5 × 106 | 0.1 | 2 |
| 5 × 104 | 5 × 105 | 0.1 | 6 |
| 5 × 103 | 5 × 104 | 0.1 | 8 |
| Phage Amplification Series 2 | |||
| Phage Concentration (pfu·mL−1) | Bacterial Concentration (cfu·mL−1) | MOI | SERS-LFI Detection Time (h) |
| 5 × 105 | 1 × 106 | 0.1 | 2 |
| 5 × 105 | 2 × 105 | 2.5 | 4 |
| 5 × 105 | 1 × 105 | 5 | 5 |
Supplementary Files
Supplementary File 1Acknowledgments
Author Contributions
Conflicts of Interest
References
- Swaminathan, B.; Gerner-Smidt, P. The epidemiology of human listeriosis. Microbes Infect. 2007, 9, 1236–1243. [Google Scholar] [CrossRef] [PubMed]
- Scallan, E.; Hoekstra, R.M.; Angulo, F.J.; Tauxe, R.V.; Widdowson, M.-A.; Roy, S.L.; Jones, J.L.; Griffin, P.M. Foodborne illness acquired in the United States—Major pathogens. Emerg. Infect. Dis. 2011, 17. [Google Scholar] [CrossRef]
- Todd, E.; Notermans, S. Surveillance of listeriosis and its causative pathogen, Listeria monocytogenes. Food Control 2011, 22, 1484–1490. [Google Scholar] [CrossRef]
- Farber, J.; Peterkin, P. Listeria monocytogenes, a food-borne pathogen. Microbiol. Rev. 1991, 55, 476–511. [Google Scholar] [PubMed]
- Center for Disease Control and Prevention. Multistate Outbreak of Listeriosis Linked to Whole Cantaloupes from Jensen Farms, Colorado. Available online: http://www.cdc.gov/listeria/outbreaks/cantaloupes-jensen-farms/index.html (accessed on 9 February 2015).
- Hitchins, A.D.; Jinneman, K. Bacteriologial Analytical Manual Chapter 10 Detection and Enumeration of Listeria monocytogenes in Foods. Available online: http://www.fda.gov/Food/FoodScienceResearch/LaboratoryMethods/ucm071400.htm (accessed on 9 February 2015).
- U.S. Department of Agriculture. Isolation and Identification of Listeria monocytogenes from Red Meat, Poultry and Egg Products, and Environmental Samples. Available online: http://www.fsis.usda.gov/wps/wcm/connect/1710bee8-76b9-4e6c-92fc-fdc290dbfa92/MLG-8.pdf?MOD=AJPERES (accessed on 9 Feburary 2015).
- Al-Soud, W.A.; Rådström, P. Capacity of nine thermostable DNA polymerases to mediate DNA amplification in the presence of PCR-inhibiting samples. Appl. Environ. Microbiol. 1998, 64, 3748–3753. [Google Scholar] [PubMed]
- Schmelcher, M.; Loessner, M.J. Application of bacteriophages for detection of foodborne pathogens. Bacteriophage 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Guenther, S.; Huwyler, D.; Richard, S.; Loessner, M.J. Virulent bacteriophage for efficient biocontrol of Listeria monocytogenes in ready-to-eat foods. Appl. Environ. Microbiol. 2009, 75, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Loessner, M.J.; Rees, C.E.D.; Stewart, G.; Scherer, S. Construction of luciferase reporter bacteriophage A511::luxAB for rapid and sensitive detection of viable Listeria cells. Appl. Environ. Microbiol. 1996, 62, 1133–1140. [Google Scholar] [PubMed]
- Klumpp, J.; Dorscht, J.; Lurz, R.; Bielmann, R.; Wieland, M.; Zimmer, M.; Calendar, R.; Loessner, M.J. The terminally redundant, nonpermuted genome of Listeria bacteriophage A511: A model for the SPO1-like myoviruses of gram-positive bacteria. J. Bacteriol. 2008, 190, 5753–5765. [Google Scholar] [CrossRef] [PubMed]
- U.S. Food and Drug Adminstration. Guidance for Industry: Control of Listeria monocytogenes in Refrigerated or Frozen Ready-to-Eat Foods. Draft Guidance. Available online: http://www.fda.gov/Food/GuidanceRegulation/GuidanceDocumentsRegulatoryInformation/FoodProcessingHACCP/ucm073110.htm#ftn1 (accessed on 24 October 2015).
- Gasanov, U.; Hughes, D.; Hansbro, P.M. Methods for the isolation and identification of Listeria spp. and Listeria monocytogenes: A review. FEMS Microbiol. Rev. 2005, 29, 851–875. [Google Scholar] [CrossRef] [PubMed]
- Loessner, M.J.; Rudolf, M.; Scherer, S. Evaluation of luciferase reporter bacteriophage A511::luxAB for detection of Listeria monocytogenes in contaminated foods. Appl. Environ. Microbiol. 1997, 63, 2961–2965. [Google Scholar] [PubMed]
- Oliveira, I.; Almeida, R.; Hofer, E.; Almeida, P. Bacteriophage amplification assay for detection of Listeria spp. using virucidal laser treatment. Braz. J. Microbiol. 2012, 43, 1128–1136. [Google Scholar] [CrossRef] [PubMed]
- Hagens, S.; de Wouters, T.; Vollenweider, P.; Loessner, M.J. Reporter bacteriophage A511::celB transduces a hyperthermostable glycosidase from Pyrococcus furiosus for rapid and simple detection of viable Listeria cells. Bacteriophage 2011, 1, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Cherry, W.B.; Davis, B.R.; Edwards, P.R.; Hogan, R.B. A simple procedure for the identification of the genus Salmonella by means of a specific bacteriophage. J. Lab. Clin. Med. 1954, 44, 51–55. [Google Scholar] [PubMed]
- Fernandes, E.; Martins, V.; Nóbrega, C.; Carvalho, C.; Cardoso, F.; Cardoso, S.; Dias, J.; Deng, D.; Kluskens, L.; Freitas, P. A bacteriophage detection tool for viability assessment of Salmonella cells. Biosens. Bioelectron. 2014, 52, 239–246. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balasubramanian, S.; Sorokulova, I.B.; Vodyanoy, V.J.; Simonian, A.L. Lytic phage as a specific and selective probe for detection of Staphylococcus aureus—A surface plasmon resonance spectroscopic study. Biosens. Bioelectron. 2007, 22, 948–955. [Google Scholar] [CrossRef] [PubMed]
- Madonna, A.J.; Cuyk, S.V.; Voorhees, K.J. Detection of Escherichia coli using immunomagnetic separation and bacteriophage amplification coupled with matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 2003, 17, 257–263. [Google Scholar] [CrossRef] [PubMed]
- Cox, C.R.; Saichek, N.R.; Schweizer, H.P.; Voorhees, K.J. Rapid Burkholderia pseudomallei identification and antibiotic resistance determination by bacteriophage amplification and MALDI-TOF MS. Bacteriophage 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Rees, J.C.; Voorhees, K.J. Simultaneous detection of two bacterial pathogens using bacteriophage amplification coupled with matrix-assisted laser desorption/ionization time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 2005, 19, 2757–2761. [Google Scholar] [CrossRef] [PubMed]
- Bhowmick, T.; Mirrett, S.; Reller, L.; Price, C.; Qi, C.; Weinstein, M.; Kirn, T. Controlled multicenter evaluation of a bacteriophage-based method for rapid detection of Staphylococcus aureus in positive blood cultures. J. Clin. Microbiol. 2013, 51, 1226–1230. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, K.V.; Turner, N.N.; Roundtree, S.S.; McGowan, K.L. Rapid detection of methicillin-resistant Staphylococcus aureus (MRSA) and methicillin-susceptible Staphylococcus aureus (MSSA) using KeyPath MRSA/MSSA blood culture test and the BacT/ALERT system in a pediatric population. Arch. Pathol. Lab. Med. 2013, 137, 1103–1105. [Google Scholar] [CrossRef] [PubMed]
- Cox, C.R.; Jensen, K.R.; Mondesire, R.R.; Voorhees, K.J. Rapid detection of Bacillus anthracis by γ phage amplification and lateral flow immunochromatography. J. Microbiol. Methods 2015, 118, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Khreich, N.; Lamourette, P.; Boutal, H.; Devilliers, K.; Créminon, C.; Volland, H. Detection of Staphylococcus enterotoxin B using fluorescent immunoliposomes as label for immunochromatographic testing. Anal. Biochem. 2008, 377, 182–188. [Google Scholar] [CrossRef] [PubMed]
- Doering, W.E.; Piotti, M.E.; Natan, M.J.; Freeman, R.G. SERS as a foundation for nanoscale, optically detected biological labels. Adv. Mater. 2007, 19, 3100–3108. [Google Scholar] [CrossRef]
- Parolo, C.; de la Escosura-Muñiz, A.; Merkoçi, A. Enhanced lateral flow immunoassay using gold nanoparticles loaded with enzymes. Biosens. Bioelectron. 2013, 40, 412–416. [Google Scholar] [CrossRef] [PubMed]
- Noble, J.; Attree, S.; Horgan, A.; Knight, A.; Kumarswami, N.; Porter, R.; Worsley, G. Optical scattering artifacts observed in the development of multiplexed surface enhanced Raman spectroscopy nanotag immunoassays. Anal. Chem. 2012, 84, 8246–8252. [Google Scholar] [CrossRef] [PubMed]
- Mulvaney, S.P.; Musick, M.D.; Keating, C.D.; Natan, M.J. Glass-coated, analyte-tagged nanoparticles: A new tagging system based on detection with surface-enhanced Raman scattering. Langmuir 2003, 19, 4784–4790. [Google Scholar] [CrossRef]
- Doering, W.E.; Nie, S. Spectroscopic tags using dye-embedded nanoparticles and surface-enhanced Raman scattering. Anal. Chem. 2003, 75, 6171–6176. [Google Scholar] [CrossRef] [PubMed]
- Zavaleta, C.L.; Smith, B.R.; Walton, I.; Doering, W.; Davis, G.; Shojaei, B.; Natan, M.J.; Gambhir, S.S. Multiplexed imaging of surface enhanced Raman scattering nanotags in living mice using noninvasive Raman spectroscopy. Proc. Natl. Acad. Sci. USA 2009, 106, 13511–13516. [Google Scholar] [CrossRef] [PubMed]
- Sambrook, J.; Russell, D.W. Molecular Cloning—A Laboratory Manual; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2001; Volume 3. [Google Scholar]
- Carlson, K. Working with bacteriophages: Common techniques and methological approaches. In Bacteriophages Biology and Applications; Kutter, E., Sulakvelidze, A., Eds.; CRC Press: New York, NY, USA, 2005; pp. 437–494. [Google Scholar]
- Wang, Y.W.; Khan, A.; Som, M.; Wang, D.; Chen, Y.; Leigh, S.Y.; Meza, D.; McVeigh, P.Z.; Wilson, B.C.; Liu, J.T. Rapid ratiometric biomarker detection with topically applied SERS nanoparticles. Technolgy 2014, 2, 118–132. [Google Scholar] [CrossRef] [PubMed]
- Doering, W.E.; University of California-Davis, Davis, CA, USA. Personal communication, 2014.
- Jett, B.D.; Hatter, K.L.; Huycke, M.M.; Gilmore, M.S. Simplified agar plate method for quantifying viable bacteria. Biotechnolgy 1997, 23, 648–650. [Google Scholar]
- Smartt, A.E.; Ripp, S. Bacteriophage reporter technology for sensing and detecting microbial targets. Anal. Bioanal. Chem. 2011, 400, 991–1007. [Google Scholar] [CrossRef] [PubMed]
- Schofield, D.; Sharp, N.J.; Westwater, C. Phage-based platforms for the clinical detection of human bacterial pathogens. Bacteriophage 2012, 2, 105–121. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-C.; Yang, Y.-M.; Liao, P.-H.; Chen, D.-W.; Lin, H.-P.; Chang, H.-C. A filter-like AuNPs@ MS SERS substrate for Staphylococcus aureus detection. Biosens. Bioelectron. 2014, 53, 519–527. [Google Scholar] [CrossRef] [PubMed]
- Jarvis, R.M.; Goodacre, R. Characterisation and identification of bacteria using SERS. Chem. Soc. Rev. 2008, 37, 931–936. [Google Scholar] [CrossRef] [PubMed]
- Ngom, B.; Guo, Y.; Wang, X.; Bi, D. Development and application of lateral flow test strip technology for detection of infectious agents and chemical contaminants: A review. Anal. Bioanal. Chem. 2010, 397, 1113–1135. [Google Scholar] [CrossRef] [PubMed]
- Martelet, A.; L’Hostis, G.; Tavares, P.; Brasilès, S.; Fenaille, F.O.; Rozand, C.; Theretz, A.; Gervasi, G.; Tabet, J.-C.; Ezan, E. Bacterial detection using unlabeled phage amplification and mass spectrometry through structural and nonstructural phage markers. J. Proteome Res. 2014, 13, 1450–1465. [Google Scholar] [CrossRef] [PubMed]
- Martelet, A.; L’Hostis, G.; Nevers, M.-C.; Volland, H.; Junot, C.; Becher, F.; Muller, B.H. Phage amplification and immunomagnetic separation combined with targeted mass spectrometry for sensitive detection of viable bacteria in complex food matrices. Anal. Chem. 2015, 87, 5553–5560. [Google Scholar] [CrossRef] [PubMed]
- Pierce, C.L.; Rees, J.C.; Fernández, F.M.; Barr, J.R. Detection of Staphylococcus aureus using 15N-labeled bacteriophage amplification coupled with matrix-assisted laser desorption/ionization-time-of-flight mass spectrometry. Anal. Chem. 2011, 83, 2286–2293. [Google Scholar] [CrossRef] [PubMed]
- Hagens, S.; Loessner, M.J. Bacteriophage for biocontrol of foodborne pathogens: Calculations and considerations. Curr. Pharm. Biotechnol. 2010, 11, 58–68. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stambach, N.R.; Carr, S.A.; Cox, C.R.; Voorhees, K.J. Rapid Detection of Listeria by Bacteriophage Amplification and SERS-Lateral Flow Immunochromatography. Viruses 2015, 7, 6631-6641. https://doi.org/10.3390/v7122962
Stambach NR, Carr SA, Cox CR, Voorhees KJ. Rapid Detection of Listeria by Bacteriophage Amplification and SERS-Lateral Flow Immunochromatography. Viruses. 2015; 7(12):6631-6641. https://doi.org/10.3390/v7122962
Chicago/Turabian StyleStambach, Nicholas R., Stephanie A. Carr, Christopher R. Cox, and Kent J. Voorhees. 2015. "Rapid Detection of Listeria by Bacteriophage Amplification and SERS-Lateral Flow Immunochromatography" Viruses 7, no. 12: 6631-6641. https://doi.org/10.3390/v7122962