
Activation of the Mitochondrial Apoptotic Signaling Platform during Rubella Virus Infection


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
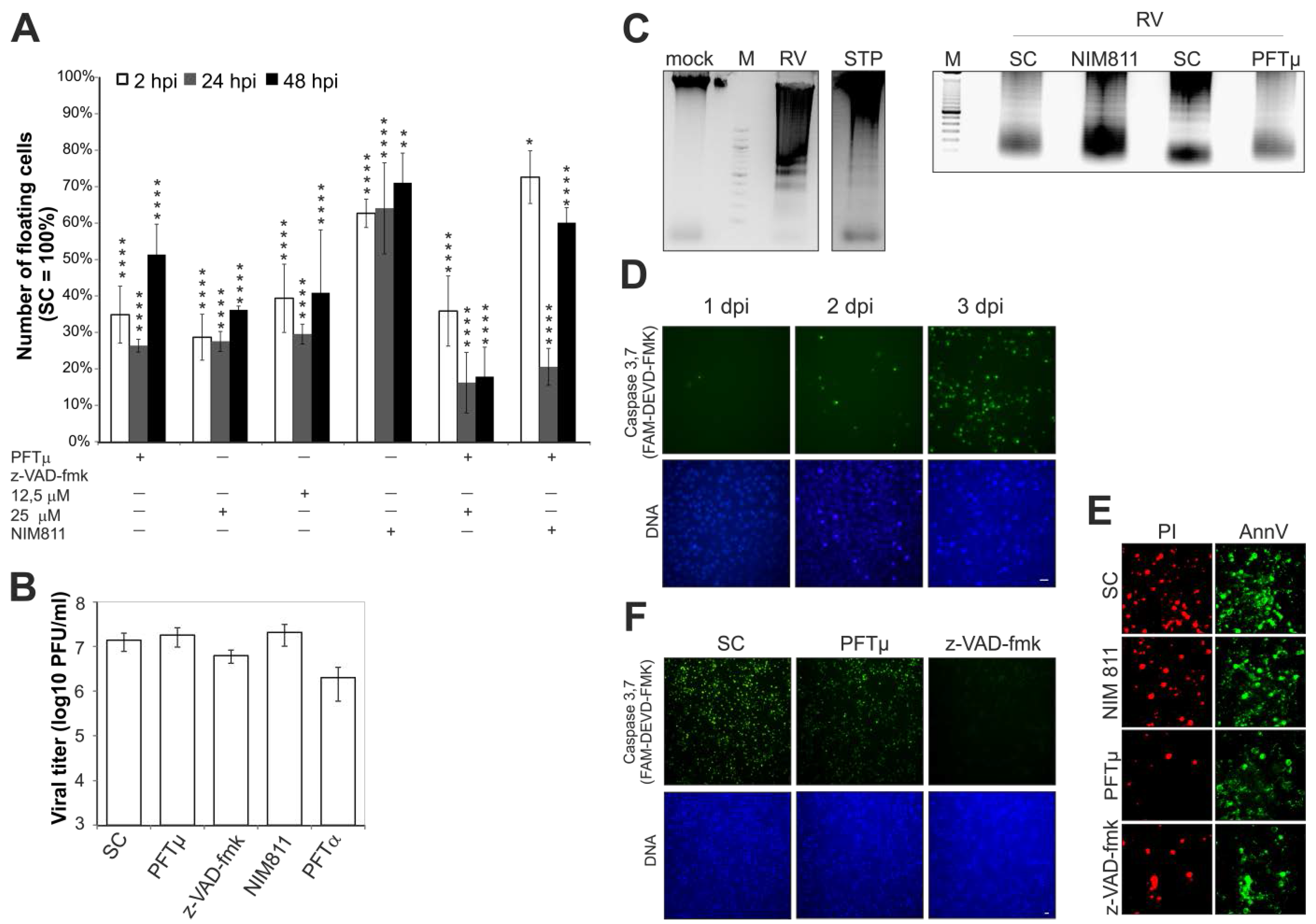
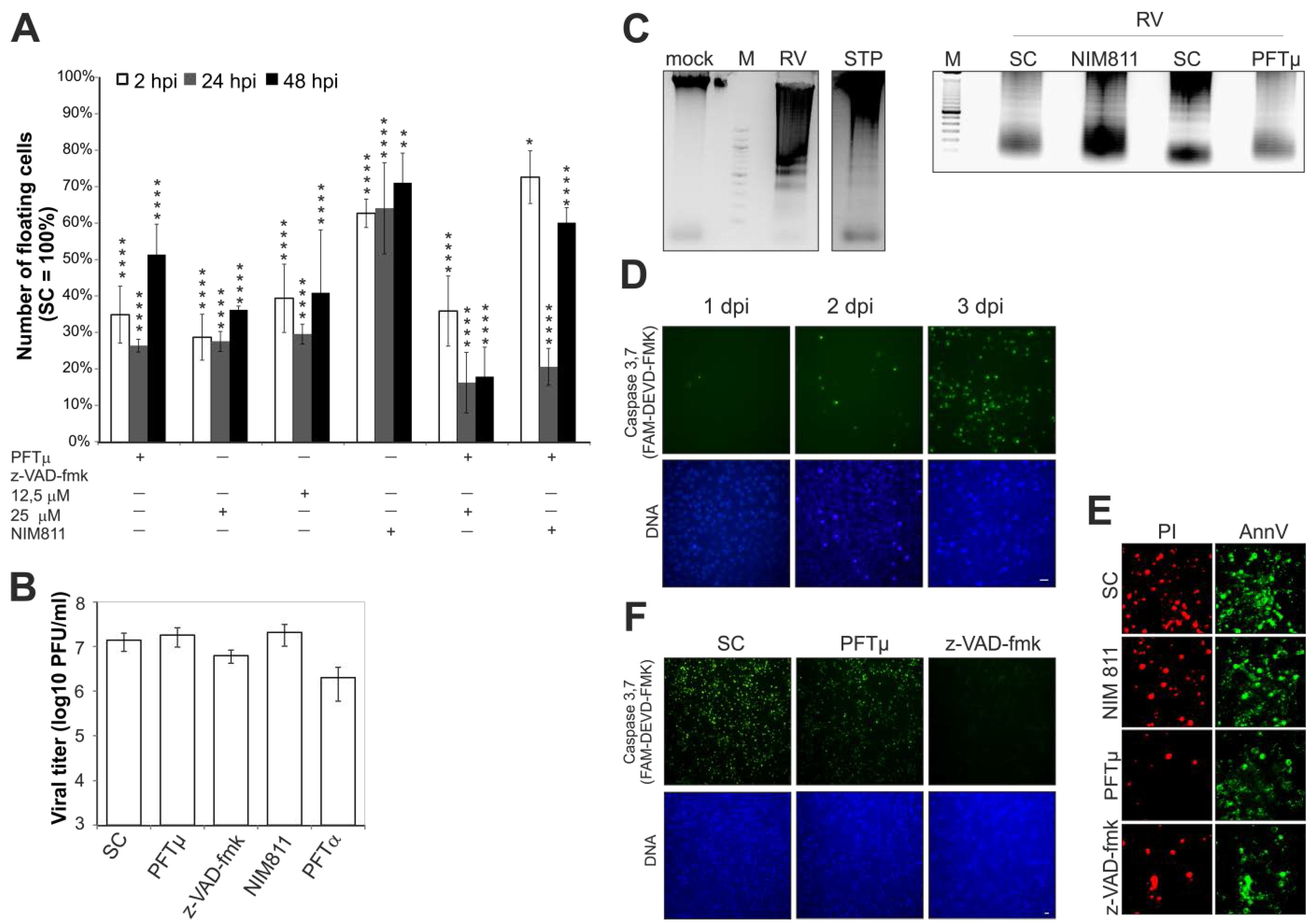
2.1. Effect of Pharmacological Inhibitors of Apoptotic Signaling Pathways on Rubella Virus-Induced Cell Death
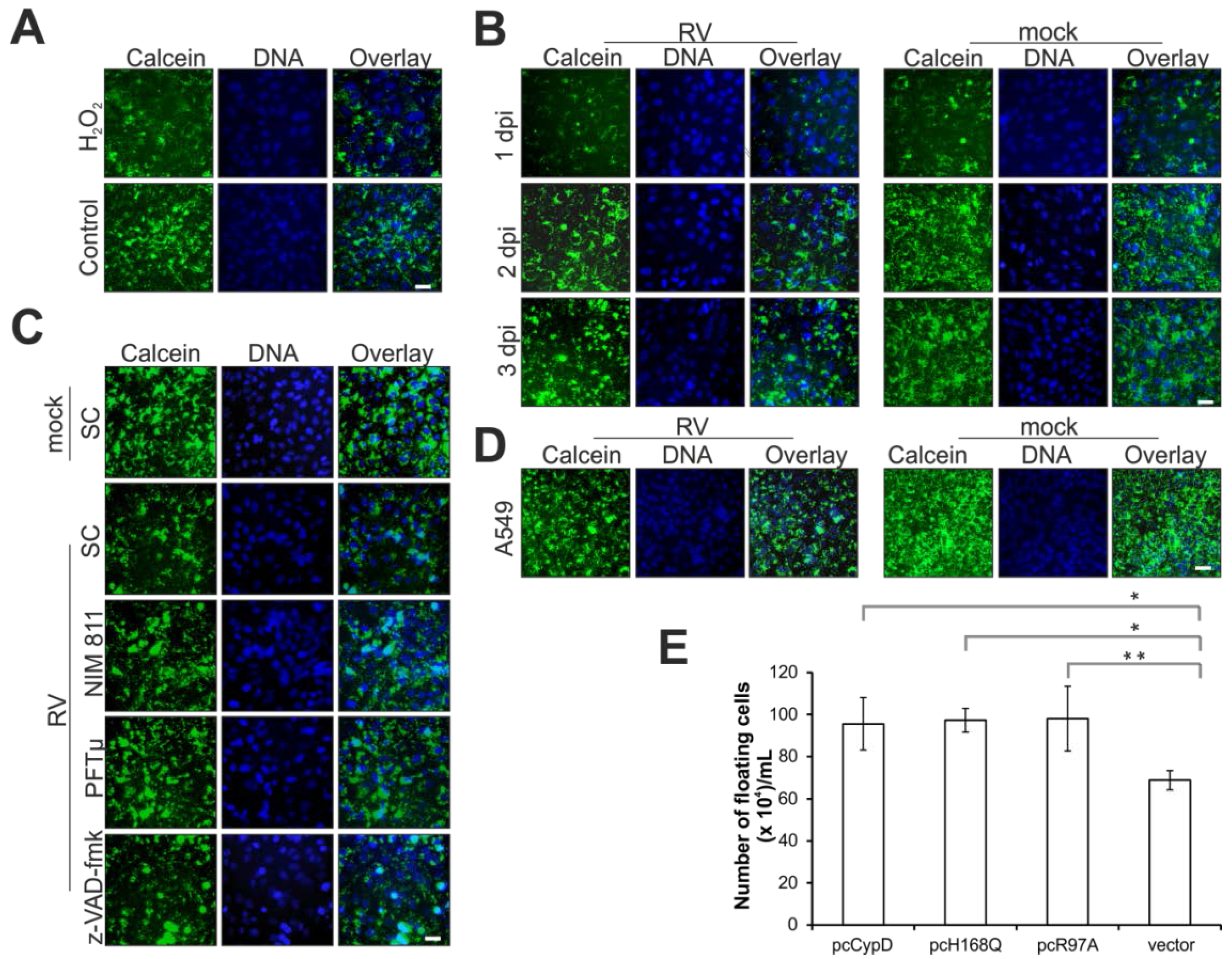
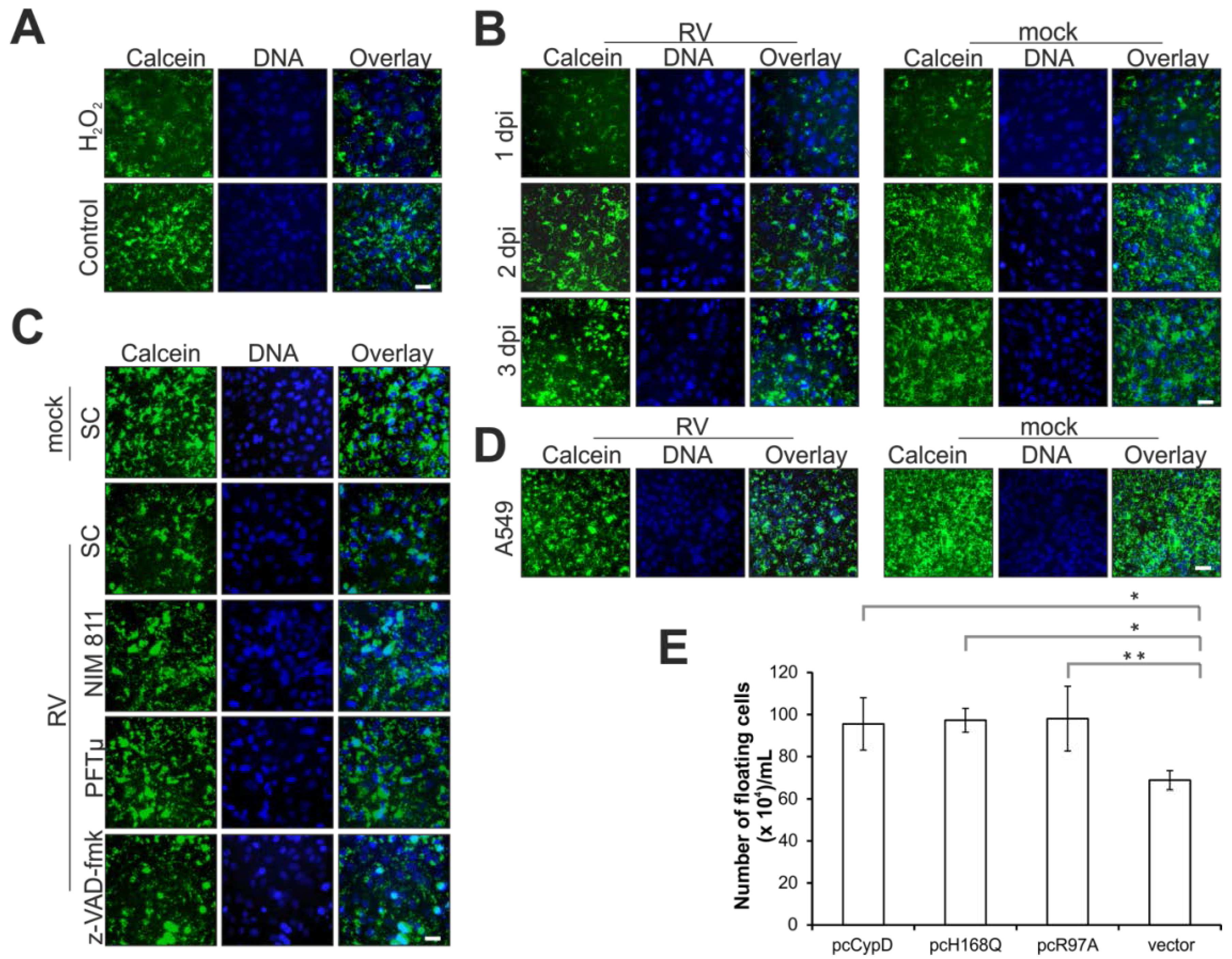
2.2. Rubella Virus Infection Results in Partial Opening of the Mitochondrial Permeability Transition Pore
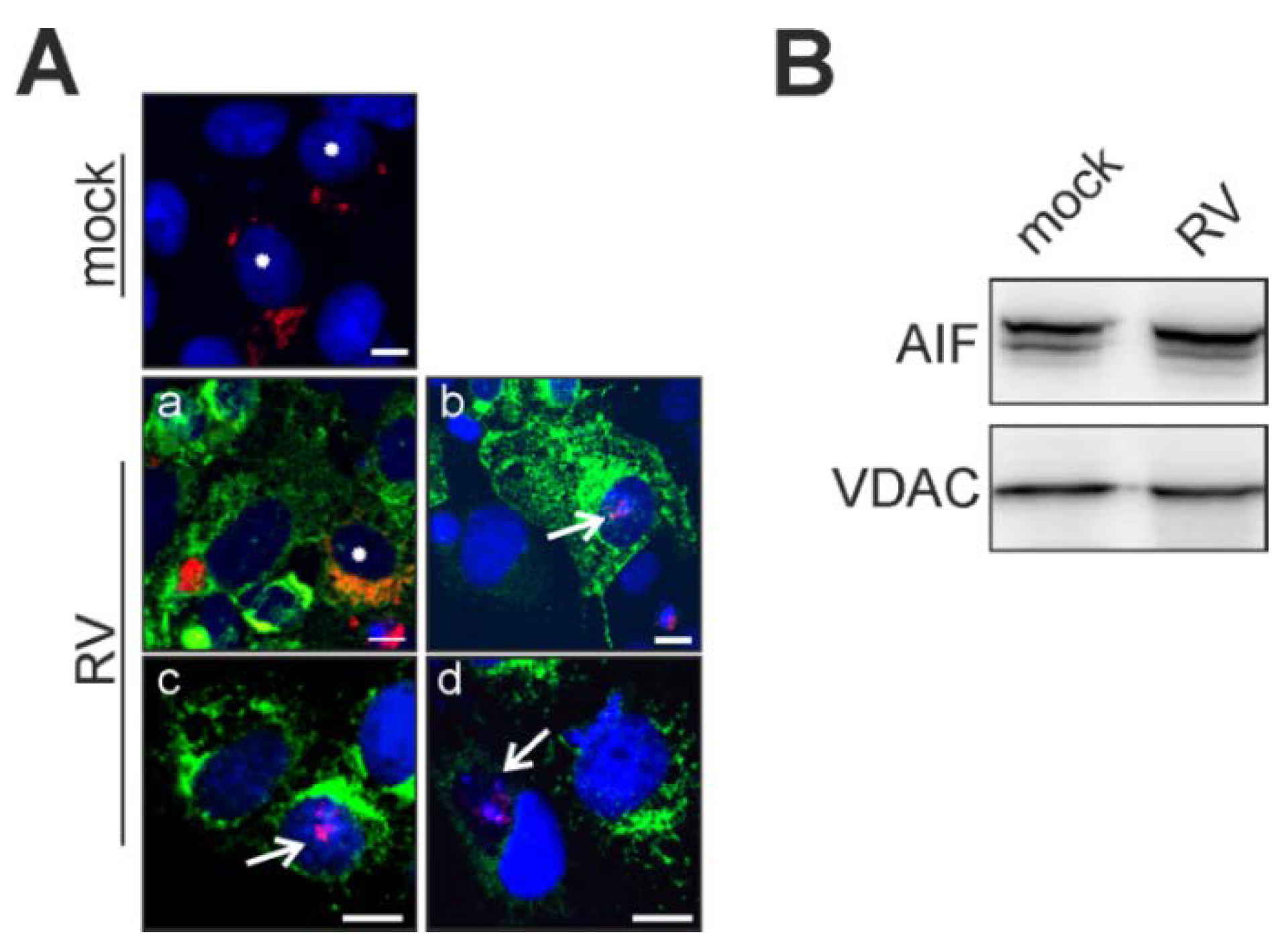
2.3. Rubella Virus Induces Nuclear Translocation of the Pro-Apoptotic Mitochondrial Protein Apoptosis-Inducing Factor (AIF)

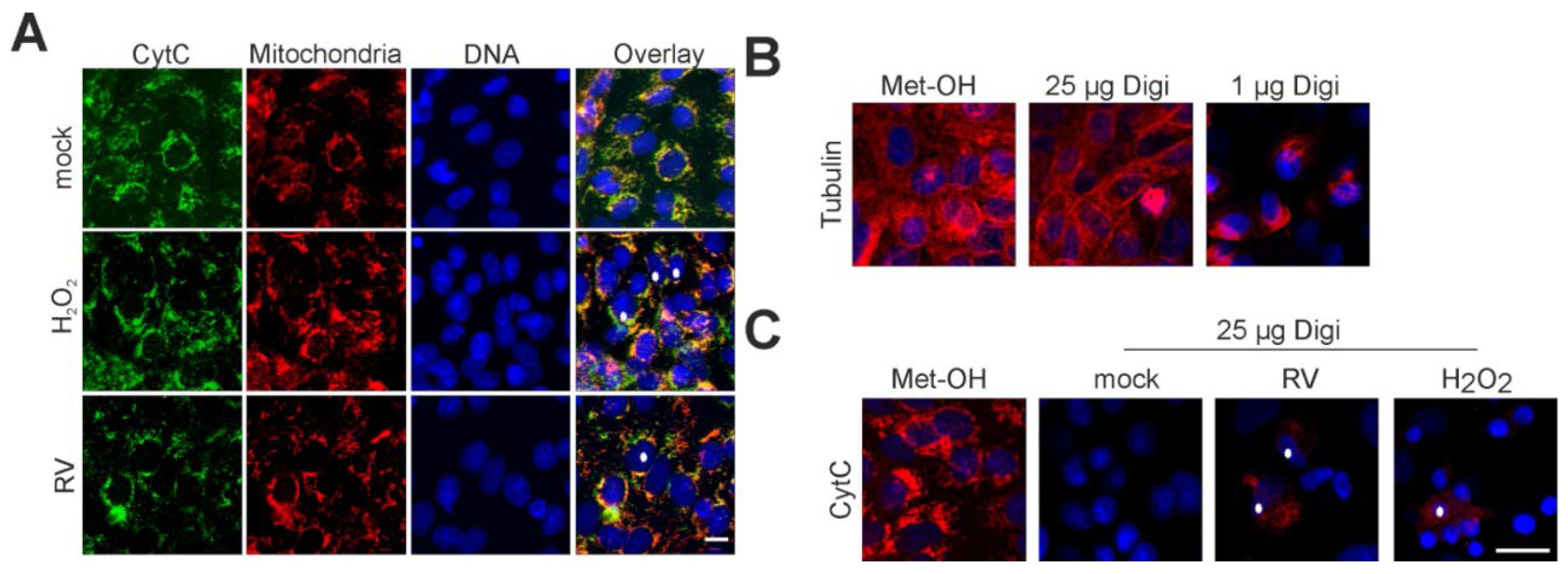
2.4. Point of No Return: Release of the Apoptosis-Promoting Protein Cytochrome C from Mitochondria

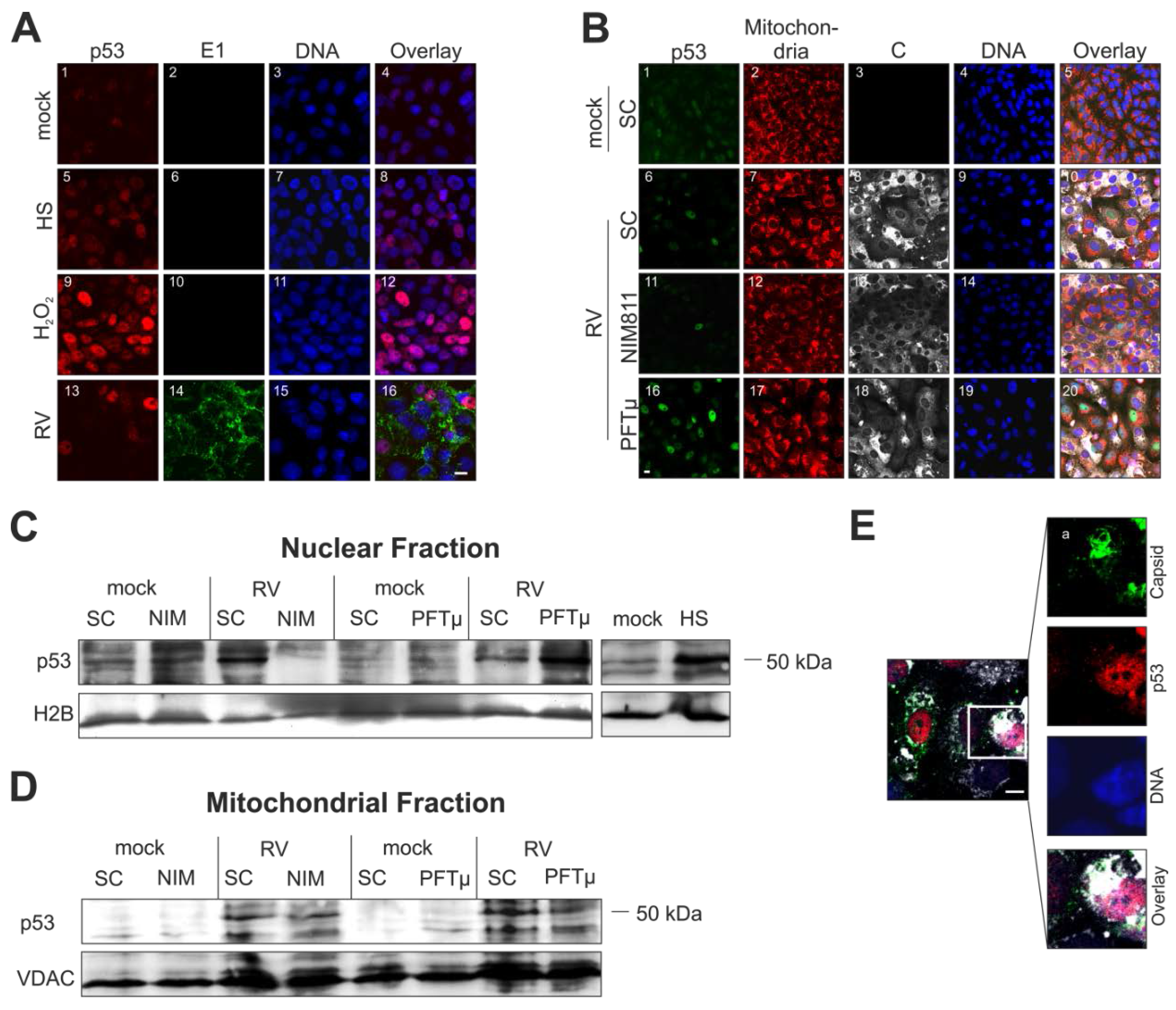
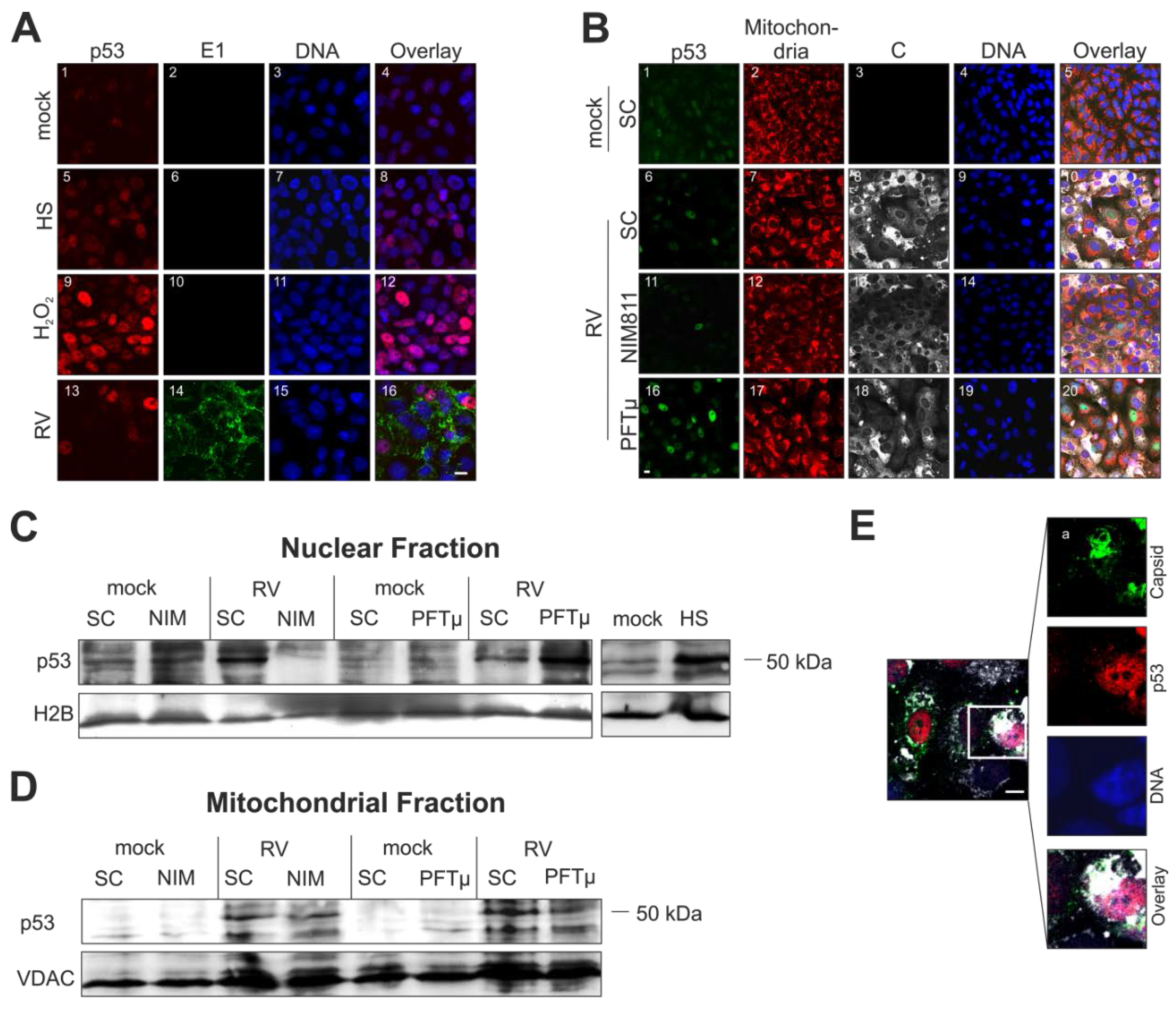
2.5. Cellular Distribution of p53 Is Altered during Rubella Virus Infection
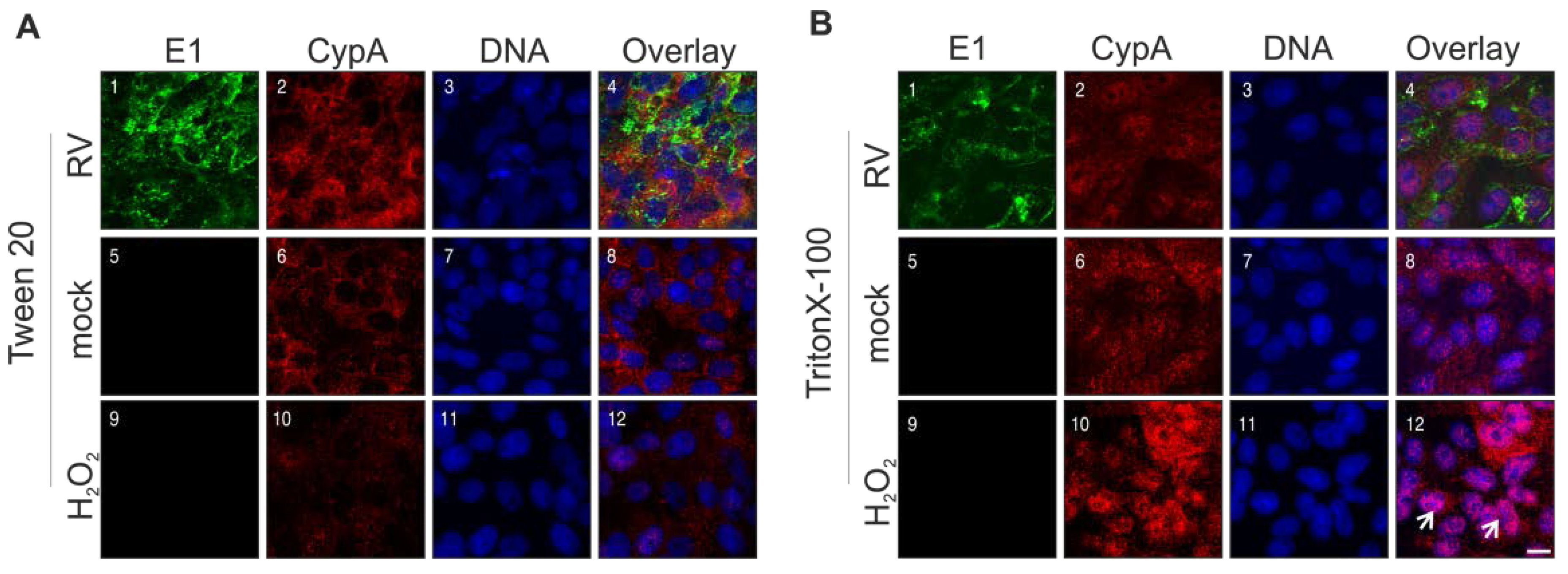
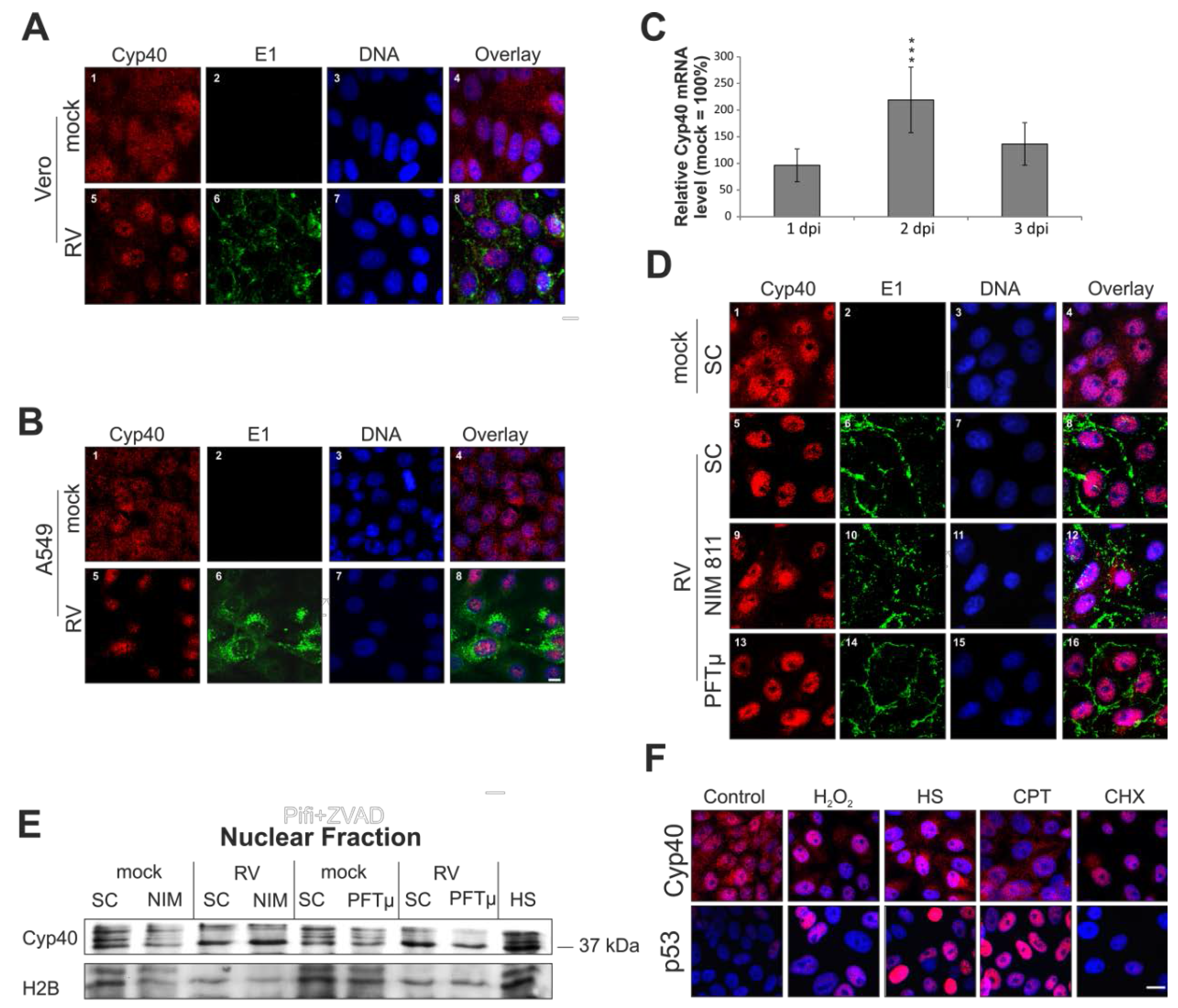
2.6. Rubella Virus Induces Nuclear Translocation of Cyclophilin 40
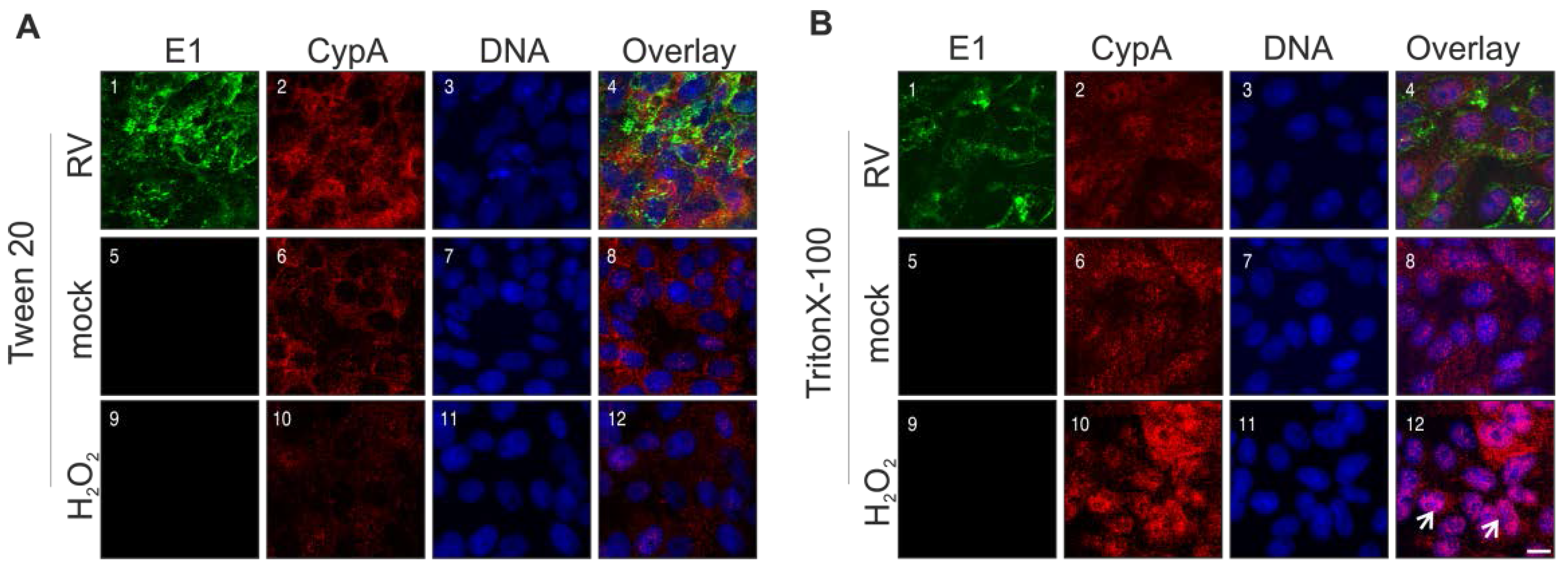
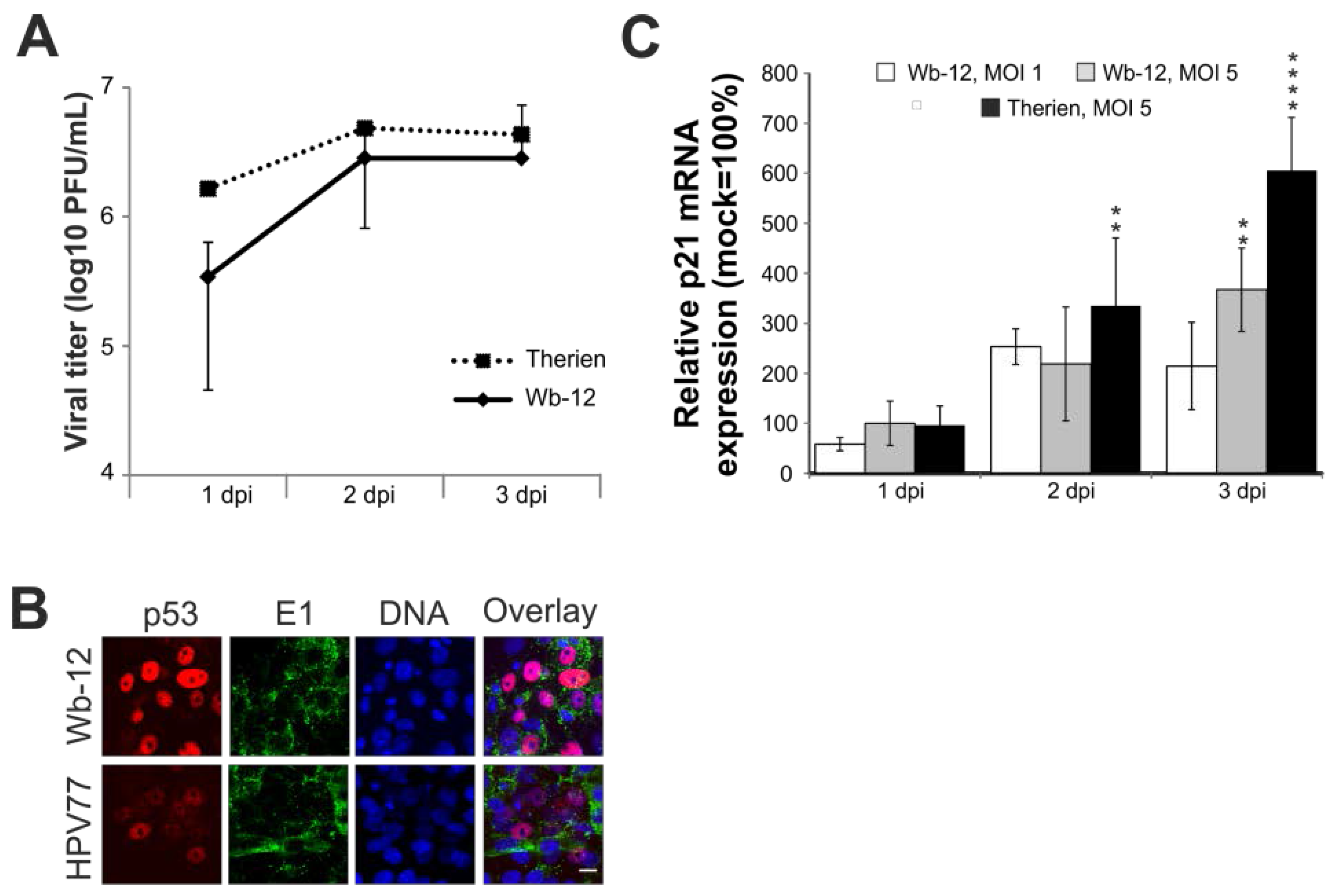
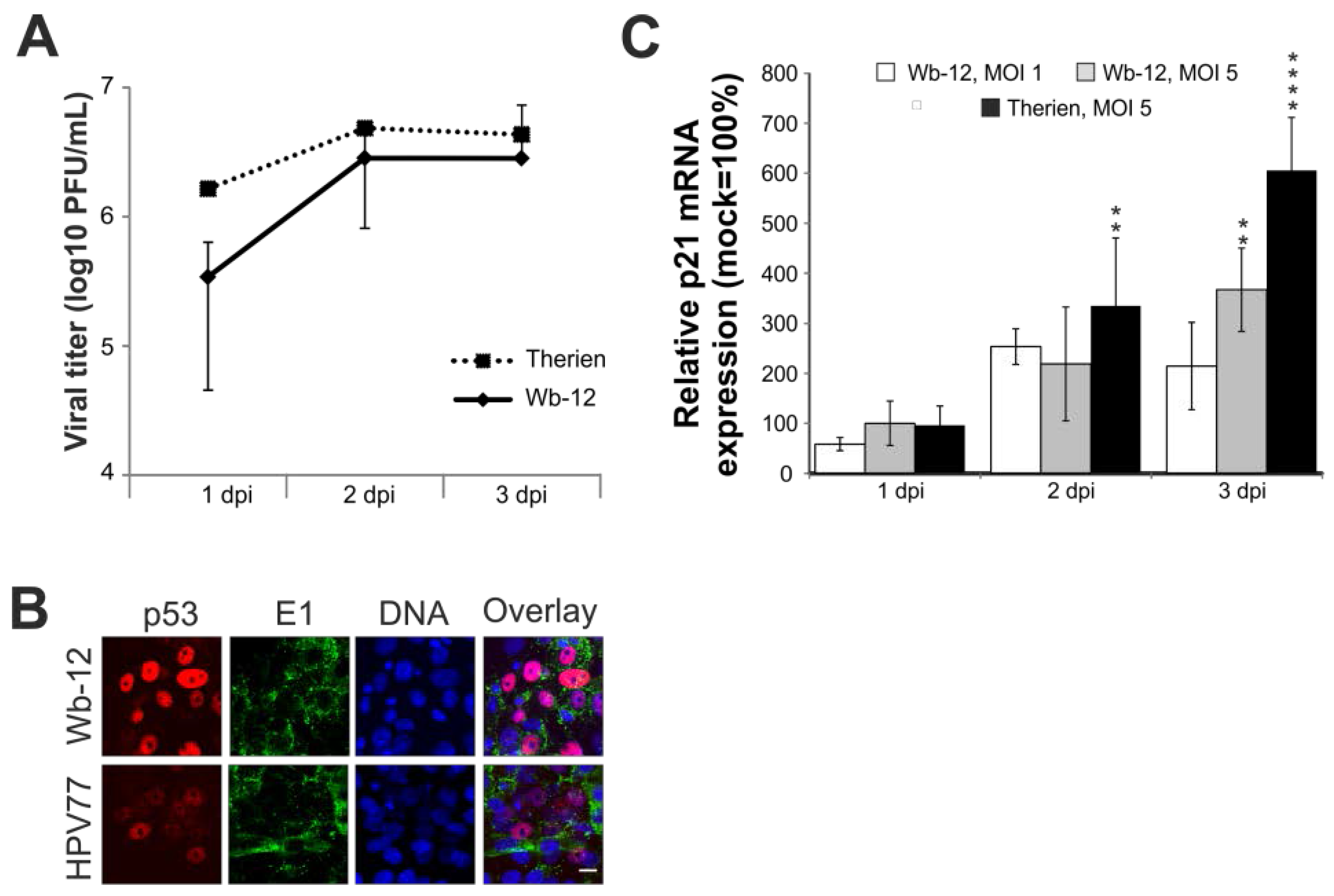
2.7. Rubella Virus Reveals Strain-Specific Effects in Its Mode of Apoptosis Induction
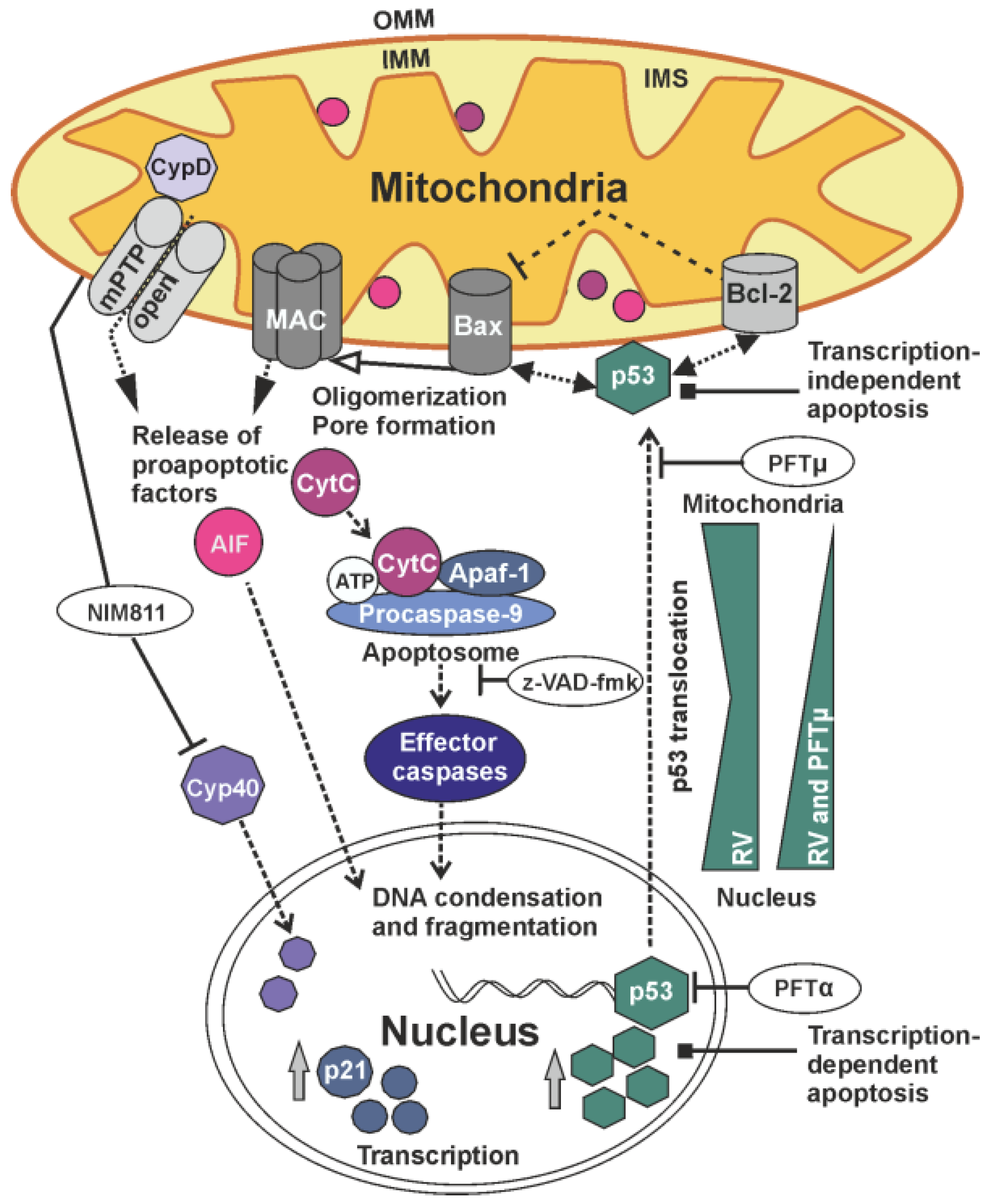
3. Discussion

4. Materials and Methods
4.1. Antibodies and Pharmacological Reagents
4.2. Vero Cell Cultivation, Drug Treatment and Virus Strains
4.3. Plasmids and Transient Transfection
4.4. Characterization of Floaters: DNA Fragmentation Assay
4.5. Cell Death Assays
4.6. Calcein Release Assay for Assessment of Mitochondrial Permeability Transition Pore (mPTP) Opening
4.7. Immunofluorescence Analysis
4.8. Cell Fractionation and Western Blot Analysis
4.9. RNA Preparation and Real-Time Quantitative PCR
4.10. Statistics
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Frey, T.K. Molecular biology of rubella virus. Adv. Virus Res. 1994, 44, 69–160. [Google Scholar] [PubMed]
- Duncan, R.; Muller, J.; Lee, N.; Esmaili, A.; Nakhasi, H.L. Rubella virus-induced apoptosis varies among cell lines and is modulated by Bcl-xl and caspase inhibitors. Virology 1999, 255, 117–128. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, J.; Pletz, M.W.; Liebert, U.G. Rubella virus-induced cytopathic effect in vitro is caused by apoptosis. J. Gen. Virol. 1999, 80, 1657–1664. [Google Scholar] [CrossRef] [PubMed]
- Domegan, L.M.; Atkins, G.J. Apoptosis induction by the therien and vaccine RA27/3 strains of rubella virus causes depletion of oligodendrocytes from rat neural cell cultures. J. Gen. Virol. 2002, 83, 2135–2143. [Google Scholar] [CrossRef] [PubMed]
- Megyeri, K.; Berencsi, K.; Halazonetis, T.D.; Prendergast, G.C.; Gri, G.; Plotkin, S.A.; Rovera, G.; Gonczol, E. Involvement of a p53-dependent pathway in rubella virus-induced apoptosis. Virology 1999, 259, 74–84. [Google Scholar] [CrossRef] [PubMed]
- Cooray, S.; Jin, L.; Best, J.M. The involvement of survival signaling pathways in rubella-virus induced apoptosis. Virol. J. 2005, 2, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ilkow, C.S.; Goping, I.S.; Hobman, T.C. The rubella virus capsid is an anti-apoptotic protein that attenuates the pore-forming ability of bax. PLoS Pathog. 2011, 7, e1001291. [Google Scholar] [CrossRef] [PubMed]
- Tzeng, W.P.; Xu, J.; Frey, T.K. Characterization of cell lines stably transfected with rubella virus replicons. Virology 2012, 429, 29–36. [Google Scholar] [CrossRef] [PubMed]
- Beatch, M.D.; Hobman, T.C. Rubella virus capsid associates with host cell protein p32 and localizes to mitochondria. J. Virol. 2000, 74, 5569–5576. [Google Scholar] [CrossRef] [PubMed]
- Beatch, M.D.; Everitt, J.C.; Law, L.J.; Hobman, T.C. Interactions between rubella virus capsid and host protein p32 are important for virus replication. J. Virol. 2005, 79, 10807–10820. [Google Scholar] [CrossRef] [PubMed]
- Claus, C.; Chey, S.; Heinrich, S.; Reins, M.; Richardt, B.; Pinkert, S.; Fechner, H.; Gaunitz, F.; Schafer, I.; Seibel, P.; et al. Involvement of p32 and microtubules in alteration of mitochondrial functions by rubella virus. J. Virol. 2011, 85, 3881–3892. [Google Scholar] [CrossRef] [PubMed]
- Claus, C.; Schonefeld, K.; Hubner, D.; Chey, S.; Reibetanz, U.; Liebert, U.G. Activity increase in respiratory chain complexes by rubella virus with marginal induction of oxidative stress. J. Virol. 2013, 87, 8481–8492. [Google Scholar] [CrossRef] [PubMed]
- Buchakjian, M.R.; Kornbluth, S. The engine driving the ship: Metabolic steering of cell proliferation and death. Nat. Rev. Mol. Cell. Biol. 2010, 11, 715–727. [Google Scholar] [CrossRef] [PubMed]
- Belizario, J.E.; Alves, J.; Occhiucci, J.M.; Garay-Malpartida, M.; Sesso, A. A mechanistic view of mitochondrial death decision pores. Braz. J. Med. Biol. Res. 2007, 40, 1011–1024. [Google Scholar] [CrossRef] [PubMed]
- Vaseva, A.V.; Moll, U.M. The mitochondrial p53 pathway. Biochim. Biophys. Acta Bioenerg. 2009, 1787, 414–420. [Google Scholar] [CrossRef] [PubMed]
- Wolff, S.; Erster, S.; Palacios, G.; Moll, U.M. P53’s mitochondrial translocation and MOMP action is independent of puma and bax and severely disrupts mitochondrial membrane integrity. Cell. Res. 2008, 18, 733–744. [Google Scholar] [CrossRef] [PubMed]
- Andreeva, L.; Heads, R.; Green, C.J. Cyclophilins and their possible role in the stress response. Int. J. Exp. Pathol. 1999, 80, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Pugachev, K.V.; Frey, T.K. Rubella virus induces apoptosis in culture cells. Virology 1998, 250, 359–370. [Google Scholar] [CrossRef] [PubMed]
- Strom, E.; Sathe, S.; Komarov, P.G.; Chernova, O.B.; Pavlovska, I.; Shyshynova, I.; Bosykh, D.A.; Burdelya, L.G.; Macklis, R.M.; Skaliter, R.; et al. Small-molecule inhibitor of p53 binding to mitochondria protects mice from gamma radiation. Nat. Chem. Biol. 2006, 2, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Komarov, P.G.; Komarova, E.A.; Kondratov, R.V.; Christov-Tselkov, K.; Coon, J.S.; Chernov, M.V.; Gudkov, A.V. A chemical inhibitor of p53 that protects mice from the side effects of cancer therapy. Science 1999, 285, 1733–1737. [Google Scholar] [CrossRef] [PubMed]
- Gainutdinov, T.; Molkentin, J.D.; Siemen, D.; Ziemer, M.; Debska-Vielhaber, G.; Vielhaber, S.; Gizatullina, Z.; Orynbayeva, Z.; Gellerich, F.N. Knockout of cyclophilin D in Ppif(-/-) mice increases stability of brain mitochondria against Ca2+ stress. Arch. Biochem. Biophy. 2015, 579, 40–46. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Aguilar, M.; Baines, C.P. Structural mechanisms of cyclophilin D-dependent control of the mitochondrial permeability transition pore. Biochim. Biophys. Acta 2015, 1850, 2041–2047. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, P. The mitochondrial permeability transition pore: A mystery solved? Front. Physiol. 2013, 4, 95. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Mei, Q.; Li, J.; He, H. Cyclophilin A and viral infections. Biochem. Biophys. Res. Commun. 2012, 424, 647–650. [Google Scholar] [CrossRef] [PubMed]
- Chey, S.; Claus, C.; Liebert, U.G. Validation and application of normalization factors for gene expression studies in rubella virus-infected cell lines with quantitative real-time PCR. J. Cell. Biochem. 2010, 110, 118–128. [Google Scholar] [CrossRef] [PubMed]
- Rumlova, M.; Krizova, I.; Keprova, A.; Hadravova, R.; Dolezal, M.; Strohalmova, K.; Pichova, I.; Hajek, M.; Ruml, T. HIV-1 protease-induced apoptosis. Retrovirology 2014, 11, 37. [Google Scholar] [PubMed]
- Galluzzi, L.; Morselli, E.; Kepp, O.; Vitale, I.; Pinti, M.; Kroemer, G. Mitochondrial liaisons of p53. Antioxid. Redox Signal. 2011, 15, 1691–1714. [Google Scholar] [CrossRef] [PubMed]
- Frausto, S.D.; Lee, E.; Tang, H.L. Cyclophilins as Modulators of Viral Replication. Viruses 2013, 5, 1684–1701. [Google Scholar] [CrossRef] [PubMed]
- Baugh, J.; Gallay, P. Cyclophilin involvement in the replication of hepatitis C virus and other viruses. Biol. Chem. 2012, 393, 579–587. [Google Scholar] [CrossRef] [PubMed]
- Gaither, L.A.; Borawski, J.; Anderson, L.J.; Balabanis, K.A.; Devay, P.; Joberty, G.; Rau, C.; Schirle, M.; Bouwmeester, T.; Mickanin, C.; et al. Multiple cyclophilins involved in different cellular pathways mediate HCV replication. Virology 2010, 397, 43–55. [Google Scholar] [CrossRef] [PubMed]
- Jandova, J.; Janda, J.; Sligh, J.E. Cyclophilin 40 alters UVA-induced apoptosis and mitochondrial ROS generation in keratinocytes. Exp. Cell. Res. 2013, 319, 750–760. [Google Scholar] [CrossRef] [PubMed]
- Mark, P.J.; Ward, B.K.; Kumar, P.; Lahooti, H.; Minchin, R.F.; Ratajczak, T. Human cyclophilin 40 is a heat shock protein that exhibits altered intracellular localization following heat shock. Cell. Stress Chaperones 2001, 6, 59–70. [Google Scholar] [CrossRef]
- Galigniana, M.D.; Harrell, J.M.; O’Hagen, H.M.; Ljungman, M.; Pratt, W.B. Hsp90-binding immunophilins link p53 to dynein during p53 transport to the nucleus. J. Biol. Chem. 2004, 279, 22483–22489. [Google Scholar] [CrossRef] [PubMed]
- Sevrioukova, I.F. Apoptosis-inducing factor: Structure, function, and redox regulation. Antioxid. Redox Signal. 2011, 14, 2545–2579. [Google Scholar] [CrossRef] [PubMed]
- Pastorino, J.G.; Hoek, J.B. Regulation of hexokinase binding to VDAC. J. Bioenerg. Biomembr. 2008, 40, 171–182. [Google Scholar] [CrossRef] [PubMed]
- Lin, D.T.; Lechleiter, J.D. Mitochondrial targeted cyclophilin D protects cells from cell death by peptidyl prolyl isomerization. J. Biol. Chem. 2002, 277, 31134–31141. [Google Scholar] [CrossRef] [PubMed]
- Scorrano, L.; Nicolli, A.; Basso, E.; Petronilli, V.; Bernardi, P. Two modes of activation of the permeability transition pore: The role of mitochondrial cyclophilin. Mol. Cell. Biochem. 1997, 174, 181–184. [Google Scholar] [CrossRef] [PubMed]
- Marcu, R.; Kotha, S.; Zhi, Z.; Qin, W.; Neeley, C.K.; Wang, R.K.; Zheng, Y.; Hawkins, B.J. The mitochondrial permeability transition pore regulates endothelial bioenergetics and angiogenesis. Circ. Res. 2015, 116, 1336–1345. [Google Scholar] [CrossRef] [PubMed]
- Raimondi, F.; Migliaro, F.; di Pietro, E.; Borgia, F.; Rapacciuolo, A.; Capasso, L. Nitric oxide-sensitive pulmonary hypertension in congenital rubella syndrome. Case Rep. Crit. Care 2015, 2015, 198570. [Google Scholar] [CrossRef] [PubMed]
- Frascaroli, G.; Sinzger, C. Distinct properties of human cytomegalovirus strains and the appropriate choice of strains for particular studies. Methods Mol. Biol. 2014, 1119, 29–46. [Google Scholar] [PubMed]
- Villenave, R.; O’Donoghue, D.; Thavagnanam, S.; Touzelet, O.; Skibinski, G.; Heaney, L.G.; McKaigue, J.P.; Coyle, P.V.; Shields, M.D.; Power, U.F. Differential cytopathogenesis of respiratory syncytial virus prototypic and clinical isolates in primary pediatric bronchial epithelial cells. Virol. J. 2011, 8, 43. [Google Scholar] [CrossRef] [PubMed]
- Favreau, D.J.; Meessen-Pinard, M.; Desforges, M.; Talbot, P.J. Human coronavirus-induced neuronal programmed cell death is cyclophilin d dependent and potentially caspase dispensable. J. Virol. 2012, 86, 81–93. [Google Scholar] [CrossRef] [PubMed]
- Adamo, P.; Asis, L.; Silveyra, P.; Cuffini, C.; Pedranti, M.; Zapata, M. Rubella virus does not induce apoptosis in primary human embryo fibroblast cultures: A possible way of viral persistence in congenital infection. Viral Immunol. 2004, 17, 87–100. [Google Scholar] [CrossRef] [PubMed]
- Diaz, M.O.; Ziemin, S.; le Beau, M.M.; Pitha, P.; Smith, S.D.; Chilcote, R.R.; Rowley, J.D. Homozygous deletion of the α- and β 1-interferon genes in human leukemia and derived cell lines. Proc. Natl. Acad. Sci. USA 1988, 85, 5259–5263. [Google Scholar] [CrossRef] [PubMed]
- Neumann, S.; el Maadidi, S.; Faletti, L.; Haun, F.; Labib, S.; Schejtman, A.; Maurer, U.; Borner, C. How do viruses control mitochondria-mediated apoptosis? Virus Res. 2015, 209, 45–55. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Claus, C.; Manssen, L.; Hübner, D.; Roßmark, S.; Bothe, V.; Petzold, A.; Große, C.; Reins, M.; Mankertz, A.; Frey, T.K.; et al. Activation of the Mitochondrial Apoptotic Signaling Platform during Rubella Virus Infection. Viruses 2015, 7, 6108-6126. https://doi.org/10.3390/v7122928
Claus C, Manssen L, Hübner D, Roßmark S, Bothe V, Petzold A, Große C, Reins M, Mankertz A, Frey TK, et al. Activation of the Mitochondrial Apoptotic Signaling Platform during Rubella Virus Infection. Viruses. 2015; 7(12):6108-6126. https://doi.org/10.3390/v7122928
Chicago/Turabian StyleClaus, Claudia, Lena Manssen, Denise Hübner, Sarah Roßmark, Viktoria Bothe, Alice Petzold, Claudia Große, Mareen Reins, Annette Mankertz, Teryl K. Frey, and et al. 2015. "Activation of the Mitochondrial Apoptotic Signaling Platform during Rubella Virus Infection" Viruses 7, no. 12: 6108-6126. https://doi.org/10.3390/v7122928