
Resistance of Hepatitis C Virus to Inhibitors: Complexity and Clinical Implications


Abstract
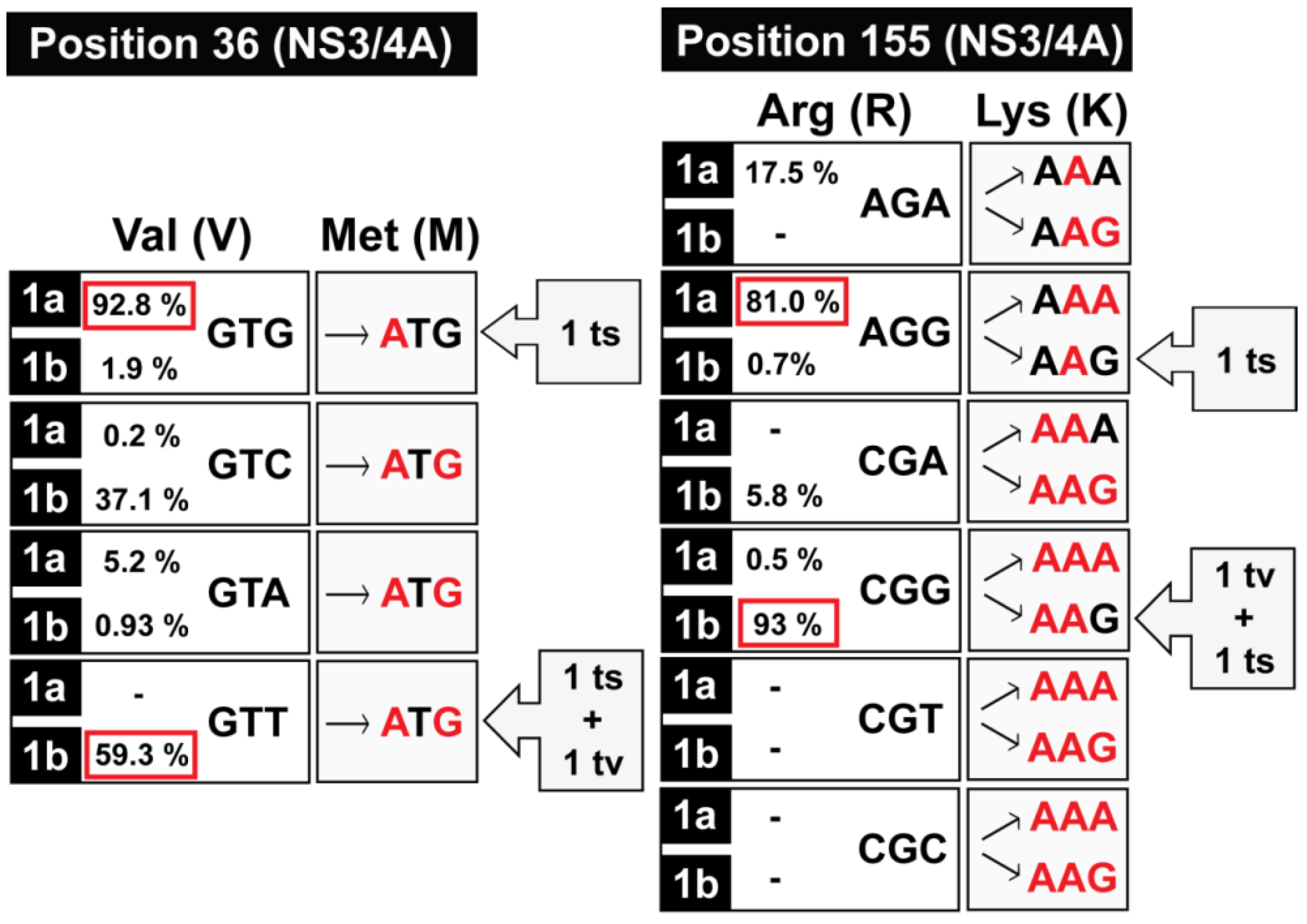
:1. Quasi-Species in the Interpretation of Virus Adaptability

{kind=link}
{kind=link}
| Observations | References |
|---|---|
| [5,9,19,20] |
| [5,9,19,20,21,22,23] |
| [5,6,7,8,10] |
| [9,24] |
| [25] |
2. Molecular Basis of Quasi-Species Dynamics in Connection with Antiviral Drug Resistance
3. Molecular and Population Aspects of drug Resistance
4. Resistance to Anti-HCV Inhibitors and Treatment Failure
4.1. Resistance to Interferon and Ribavirin
4.2. Resistance To Directly Acting Antiviral Agents and Host-Targeting Agents


4.3. HCV Inhibitor Resistance Independent of Specific Mutations
4.4. DAA Resistance Mutations in Naïve and Treatment-Experienced Patients: Effect of Natural and Selected Resistance Mutations on Treatment Response
5. Cell Culture Systems for the Study of Resistance Mutations
6. Concluding Remarks and Perspectives
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mas, A.; Lopez-Galindez, C.; Cacho, I.; Gomez, J.; Martinez, M.A. Unfinished stories on viral quasispecies and Darwinian views of evolution. J. Mol. Biol. 2010, 397, 865–877. [Google Scholar] [CrossRef] [PubMed]
- Ojosnegros, S.; Perales, C.; Mas, A.; Domingo, E. Quasispecies as a matter of fact: Viruses and beyond. Virus Res. 2011, 162, 203–215. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E. Virus as Populations; Academic Press: Waltham, MA, USA; Elsevier: Amsterdam, The Netherlands, 2015. [Google Scholar]
- Domingo, E.; Schuster, P. What is a quasispecies? Historical origins and current scope. Curr. Top. Microbiol. Immunol. 2015, in press. [Google Scholar]
- Bartolini, B.; Lionetti, R.; Giombini, E.; Sias, C.; Taibi, C.; Montalbano, M.; Gianpiero, D.; McPhee, F.; Hughes, E.A.; Zhou, N.; et al. Dynamics of HCV genotype 4 resistance-associated variants during virologic escape with pIFN/RBV + daclatasvir: A case study using ultra deep pyrosequencing. J. Clin. Virol. 2015, 66, 38–43. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.; Kozak, R.A.; Biondi, M.J.; Pilon, R.; Vallee, D.; Liang, B.B.; La, D.; Kim, J.; van Domselaar, G.; Leonard, L.; et al. Next generation sequencing of the hepatitis C virus NS5B gene reveals potential novel S282 drug resistance mutations. Virology 2015, 477, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Ogishi, M.; Yotsuyanagi, H.; Tsutsumi, T.; Gatanaga, H.; Ode, H.; Sugiura, W.; Moriya, K.; Oka, S.; Kimura, S.; Koike, K. Deconvoluting the composition of low-frequency hepatitis C viral quasispecies: Comparison of genotypes and NS3 resistance-associated variants between HCV/HIV coinfected hemophiliacs and HCV monoinfected patients in Japan. PLoS ONE 2015, 10, e0119145. [Google Scholar] [CrossRef] [PubMed]
- Hirotsu, Y.; Kanda, T.; Matsumura, H.; Moriyama, M.; Yokosuka, O.; Omata, M. HCV NS5A resistance-associated variants in a group of real-world Japanese patients chronically infected with HCV genotype 1b. Hepatol. Int. 2015, 9, 424–430. [Google Scholar] [CrossRef] [PubMed]
- Thys, K.; Verhasselt, P.; Reumers, J.; Verbist, B.M.; Maes, B.; Aerssens, J. Performance assessment of the Illumina massively parallel sequencing platform for deep sequencing analysis of viral minority variants. J. Virol. Methods 2015, 221, 29–38. [Google Scholar] [CrossRef] [PubMed]
- Simen, B.B.; Braverman, M.S.; Abbate, I.; Aerssens, J.; Bidet, Y.; Bouchez, O.; Gabriel, C.; Izopet, J.; Kessler, H.H.; Stelzl, E.; et al. An international multicenter study on HIV-1 drug resistance testing by 454 ultra-deep pyrosequencing. J. Virol. Methods 2014, 204, 31–37. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Li, Y.; Li, S.; Hu, N.; He, Y.; Pong, R.; Lin, D.; Lu, L.; Law, M. Comparison of next-generation sequencing systems. J. Biomed. Biotechnol. 2012, 2012. [Google Scholar] [CrossRef] [PubMed]
- Arias, A.; Lázaro, E.; Escarmís, C.; Domingo, E. Molecular intermediates of fitness gain of an RNA virus: Characterization of a mutant spectrum by biological and molecular cloning. J. Gen. Virol. 2001, 82, 1049–1060. [Google Scholar] [CrossRef] [PubMed]
- Domingo, E.; Sheldon, J.; Perales, C. Viral quasispecies evolution. Microbiol. Mol. Biol. Rev. 2012, 76, 159–216. [Google Scholar] [CrossRef] [PubMed]
- Palmer, S.; Kearney, M.; Maldarelli, F.; Halvas, E.K.; Bixby, C.J.; Bazmi, H.; Rock, D.; Falloon, J.; Davey, R.T., Jr.; Dewar, R.L.; et al. Multiple, linked human immunodeficiency virus type 1 drug resistance mutations in treatment-experienced patients are missed by standard genotype analysis. J. Clin. Microbiol. 2005, 43, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Bao, R.; Huang, L.; Andrade, J.; Tan, W.; Kibbe, W.A.; Jiang, H.; Feng, G. Review of current methods, applications, and data management for the bioinformatics analysis of whole exome sequencing. Cancer Inform. 2014, 13, 67–82. [Google Scholar] [PubMed]
- Pabinger, S.; Dander, A.; Fischer, M.; Snajder, R.; Sperk, M.; Efremova, M.; Krabichler, B.; Speicher, M.R.; Zschocke, J.; Trajanoski, Z. A survey of tools for variant analysis of next-generation genome sequencing data. Brief. Bioinform. 2014, 15, 256–278. [Google Scholar] [CrossRef] [PubMed]
- El-Metwally, S.; Hamza, T.; Zakaria, M.; Helmy, M. Next-generation sequence assembly: Four stages of data processing and computational challenges. PLoS Comput. Biol. 2013, 9, e1003345. [Google Scholar] [CrossRef] [PubMed]
- Magi, A.; Benelli, M.; Gozzini, A.; Girolami, F.; Torricelli, F.; Brandi, M.L. Bioinformatics for next generation sequencing data. Genes 2010, 1, 294–307. [Google Scholar] [CrossRef] [PubMed]
- Gregori, J.; Esteban, J.I.; Cubero, M.; Garcia-Cehic, D.; Perales, C.; Casillas, R.; Alvarez-Tejado, M.; Rodriguez-Frias, F.; Guardia, J.; Domingo, E.; et al. Ultra-Deep Pyrosequencing (UDPS) Data Treatment to Study Amplicon HCV Minor Variants. PLoS ONE 2013, 8, e83361. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, C.; Gregori, J.; Buti, M.; Tabernero, D.; Camos, S.; Casillas, R.; Quer, J.; Esteban, R.; Homs, M.; Rodriguez-Frias, F. A comparative study of ultra-deep pyrosequencing and cloning to quantitatively analyze the viral quasispecies using hepatitis B virus infection as a model. Antivir. Res. 2013, 98, 273–283. [Google Scholar] [CrossRef] [PubMed]
- Li, M.; Stoneking, M. A new approach for detecting low-level mutations in next-generation sequence data. Genome Biol. 2012, 13. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, K.; Oshima, T.; Morimoto, T.; Ikeda, S.; Yoshikawa, H.; Shiwa, Y.; Ishikawa, S.; Linak, M.C.; Hirai, A.; Takahashi, H.; et al. Sequence-specific error profile of Illumina sequencers. Nucl. Acids Res. 2011, 39. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, P.; Ma, J.; Pei, D.; Obert, C.; Cheng, C.; Geiger, T.L. Identification of errors introduced during high throughput sequencing of the T cell receptor repertoire. BMC Genom. 2011, 12. [Google Scholar] [CrossRef] [PubMed]
- De Leeneer, K.; de Schrijver, J.; Clement, L.; Baetens, M.; Lefever, S.; de Keulenaer, S.; van Criekinge, W.; Deforce, D.; van Nieuwerburgh, F.; Bekaert, S.; et al. Practical tools to implement massive parallel pyrosequencing of PCR products in next generation molecular diagnostics. PLoS ONE 2011, 6, e25531. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giallonardo, F.D.; Topfer, A.; Rey, M.; Prabhakaran, S.; Duport, Y.; Leemann, C.; Schmutz, S.; Campbell, N.K.; Joos, B.; Lecca, M.R.; et al. Full-length haplotype reconstruction to infer the structure of heterogeneous virus populations. Nucl. Acids Res. 2014, 42. [Google Scholar] [CrossRef] [PubMed]
- Acevedo, A.; Brodsky, L.; Andino, R. Mutational and fitness landscapes of an RNA virus revealed through population sequencing. Nature 2014, 505, 686–690. [Google Scholar] [CrossRef] [PubMed]
- Eigen, M. Self-organization of matter and the evolution of biological macromolecules. Naturwissenschaften 1971, 58, 465–523. [Google Scholar] [CrossRef] [PubMed]
- Eigen, M.; Schuster, P. The Hypercycle. A Principle of Natural Self-Organization; Springer: Berlin, Germany, 1979. [Google Scholar]
- Page, K.M.; Nowak, M.A. Unifying evolutionary dynamics. J. Theor. Biol. 2002, 219, 93–98. [Google Scholar] [CrossRef]
- Ferrer-Orta, C.; Agudo, R.; Domingo, E.; Verdaguer, N. Structural insights into replication initiation and elongation processes by the FMDV RNA-dependent RNA polymerase. Curr. Opin. Struct. Biol. 2009, 19, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Campagnola, G.; McDonald, S.; Beaucourt, S.; Vignuzzi, M.; Peersen, O.B. Structure-function relationships underlying the replication fidelity of viral RNA-dependent RNA polymerases. J. Virol. 2015, 89, 275–286. [Google Scholar] [CrossRef] [PubMed]
- Friedberg, E.C.; Walker, G.C.; Siede, W.; Wood, R.D.; Schultz, R.A.; Ellenberger, T. DNA Repair and Mutagenesis; American Society for Microbiology: Washington, DC, USA, 2006. [Google Scholar]
- Eigen, M.; Biebricher, C.K. Sequence space and quasispecies distribution. In RNA Genetics; Domingo, E., Ahlquist, P., Holland, J.J., Eds.; CRC Press: Boca Raton, FL, USA, 1988; Volume 3, pp. 211–245. [Google Scholar]
- Lauring, A.S.; Andino, R. Quasispecies theory and the behavior of RNA viruses. PLoS Pathog. 2010, 6, e1001005. [Google Scholar] [CrossRef] [PubMed]
- Schuster, P. Quasispecies on fitness landscapes. Curr. Top. Microbiol. Immunol. 2015, in press. [Google Scholar]
- Bordería, A.V.; Rozen-Gagnon, K.; Vignuzzi, M. Fidelity variants and RNA quasispecies. Curr. Top. Microbiol. Immunol. 2015, in press. [Google Scholar]
- Ke, R.; Loverdo, C.; Qi, H.; Sun, R.; Lloyd-Smith, J.O. Rational Design and Adaptive Management of Combination Therapies for Hepatitis C Virus Infection. PLoS Comput. Biol. 2015, 11, e1004040. [Google Scholar] [CrossRef] [PubMed]
- Eggers, H.J.; Tamm, I. Coxsackie A9 virus: Mutation from drug dependence to drug independence. Science 1965, 148, 97–98. [Google Scholar] [CrossRef] [PubMed]
- Melnick, J.L.; Crowther, D.; Barrera-Oro, J. Rapid development of drug-resistant mutants of poliovirus. Science 1961, 134. [Google Scholar] [CrossRef]
- Domingo, E. RNA virus evolution and the control of viral disease. Prog. Drug Res. 1989, 33, 93–133. [Google Scholar] [PubMed]
- Richman, D.D. (Ed.) Antiviral Drug Resistance; John Wiley and Sons Inc.: New York, NY, USA, 1996.
- Lam, N.P.; Neumann, A.U.; Gretch, D.R.; Wiley, T.E.; Perelson, A.S.; Layden, T.J. Dose-dependent acute clearance of hepatitis C genotype 1 virus with interferon α. Hepatology 1997, 26, 226–231. [Google Scholar] [CrossRef] [PubMed]
- Zeuzem, S.; Schmidt, J.M.; Lee, J.H.; Ruster, B.; Roth, W.K. Effect of interferon α on the dynamics of hepatitis C virus turnover in vivo. Hepatology 1996, 23, 366–371. [Google Scholar] [CrossRef] [PubMed]
- Perales, C.; Beach, N.M.; Gallego, I.; Soria, M.E.; Quer, J.; Esteban, J.I.; Rice, C.; Domingo, E.; Sheldon, J. Response of hepatitis C virus to long-term passage in the presence of α interferon: Multiple mutations and a common phenotype. J. Virol. 2013, 87, 7593–7607. [Google Scholar] [CrossRef] [PubMed]
- Davis, G.L.; Esteban-Mur, R.; Rustgi, V.; Hoefs, J.; Gordon, S.C.; Trepo, C.; Shiffman, M.L.; Zeuzem, S.; Craxi, A.; Ling, M.H.; et al. Interferon α-2b alone or in combination with ribavirin for the treatment of relapse of chronic hepatitis C. International Hepatitis Interventional Therapy Group. N. Engl. J. Med. 1998, 339, 1493–1499. [Google Scholar] [CrossRef] [PubMed]
- Poynard, T.; Marcellin, P.; Lee, S.S.; Niederau, C.; Minuk, G.S.; Ideo, G.; Bain, V.; Heathcote, J.; Zeuzem, S.; Trepo, C.; et al. Randomised trial of interferon alpha2b plus ribavirin for 48 weeks or for 24 weeks versus interferon α2b plus placebo for 48 weeks for treatment of chronic infection with hepatitis C virus. International Hepatitis Interventional Therapy Group (IHIT). Lancet 1998, 352, 1426–1432. [Google Scholar] [CrossRef]
- Manns, M.P.; McHutchison, J.G.; Gordon, S.C.; Rustgi, V.K.; Shiffman, M.; Reindollar, R.; Goodman, Z.D.; Koury, K.; Ling, M.; Albrecht, J.K. Peginterferon α-2b plus ribavirin compared with interferon α-2b plus ribavirin for initial treatment of chronic hepatitis C: A randomised trial. Lancet 2001, 358, 958–965. [Google Scholar] [CrossRef]
- Lindsay, K.L.; Trepo, C.; Heintges, T.; Shiffman, M.L.; Gordon, S.C.; Hoefs, J.C.; Schiff, E.R.; Goodman, Z.D.; Laughlin, M.; Yao, R.; et al. A randomized, double-blind trial comparing pegylated interferon α-2b to interferon alfa-2b as initial treatment for chronic hepatitis C. Hepatology 2001, 34, 395–403. [Google Scholar] [CrossRef] [PubMed]
- Hadziyannis, S.J.; Sette, H., Jr.; Morgan, T.R.; Balan, V.; Diago, M.; Marcellin, P.; Ramadori, G.; Bodenheimer, H., Jr.; Bernstein, D.; Rizzetto, M.; et al. Peginterferon-α2a and ribavirin combination therapy in chronic hepatitis C: A randomized study of treatment duration and ribavirin dose. Ann. Intern. Med. 2004, 140, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Zeuzem, S.; Hultcrantz, R.; Bourliere, M.; Goeser, T.; Marcellin, P.; Sanchez-Tapias, J.; Sarrazin, C.; Harvey, J.; Brass, C.; Albrecht, J. Peginterferon α-2b plus ribavirin for treatment of chronic hepatitis C in previously untreated patients infected with HCV genotypes 2 or 3. J. Hepatol. 2004, 40, 993–999. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.H.; Keeffe, E.B. Prevalence and treatment of hepatitis C virus genotypes 4, 5, and 6. Clin. Gastroenterol. Hepatol. 2005, 3, S97–S101. [Google Scholar] [CrossRef]
- Antaki, N.; Hermes, A.; Hadad, M.; Ftayeh, M.; Antaki, F.; Abdo, N.; Kebbewar, K. Efficacy of interferon plus ribavirin in the treatment of hepatitis C virus genotype 5. J. Viral Hepat. 2008, 15, 383–386. [Google Scholar] [CrossRef] [PubMed]
- Kamal, S.M.; el Tawil, A.A.; Nakano, T.; He, Q.; Rasenack, J.; Hakam, S.A.; Saleh, W.A.; Ismail, A.; Aziz, A.A.; Madwar, M.A. Peginterferon α-2b and ribavirin therapy in chronic hepatitis C genotype 4: impact of treatment duration and viral kinetics on sustained virological response. Gut 2005, 54, 858–866. [Google Scholar] [CrossRef] [PubMed]
- Sostegni, R.; Ghisetti, V.; Pittaluga, F.; Marchiaro, G.; Rocca, G.; Borghesio, E.; Rizzetto, M.; Saracco, G. Sequential versus concomitant administration of ribavirin and interferon α-n3 in patients with chronic hepatitis C not responding to interferon alone: Results of a randomized, controlled trial. Hepatology 1998, 28, 341–346. [Google Scholar] [CrossRef] [PubMed]
- Asahina, Y.; Izumi, N.; Enomoto, N.; Uchihara, M.; Kurosaki, M.; Onuki, Y.; Nishimura, Y.; Ueda, K.; Tsuchiya, K.; Nakanishi, H.; et al. Mutagenic effects of ribavirin and response to interferon/ribavirin combination therapy in chronic hepatitis C. J. Hepatol. 2005, 43, 623–629. [Google Scholar] [CrossRef] [PubMed]
- Cuevas, J.M.; Gonzalez-Candelas, F.; Moya, A.; Sanjuan, R. Effect of ribavirin on the mutation rate and spectrum of hepatitis C virus in vivo. J. Virol. 2009, 83, 5760–5764. [Google Scholar] [CrossRef] [PubMed]
- Dietz, J.; Schelhorn, S.E.; Fitting, D.; Mihm, U.; Susser, S.; Welker, M.W.; Fuller, C.; Daumer, M.; Teuber, G.; Wedemeyer, H.; et al. Deep sequencing reveals mutagenic effects of ribavirin during monotherapy of hepatitis C virus genotype 1-infected patients. J. Virol. 2013, 87, 6172–6181. [Google Scholar] [CrossRef] [PubMed]
- Dixit, N.M.; Layden-Almer, J.E.; Layden, T.J.; Perelson, A.S. Modelling how ribavirin improves interferon response rates in hepatitis C virus infection. Nature 2004, 432, 922–924. [Google Scholar] [CrossRef] [PubMed]
- Lutchman, G.; Danehower, S.; Song, B.C.; Liang, T.J.; Hoofnagle, J.H.; Thomson, M.; Ghany, M.G. Mutation rate of the hepatitis C virus NS5B in patients undergoing treatment with ribavirin monotherapy. Gastroenterology 2007, 132, 1757–1766. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Prieto, A.M.; Sheldon, J.; Grande-Perez, A.; Tejero, H.; Gregori, J.; Quer, J.; Esteban, J.I.; Domingo, E.; Perales, C. Extinction of hepatitis C virus by ribavirin in hepatoma cells involves lethal mutagenesis. PLoS ONE 2013, 8, e71039. [Google Scholar] [CrossRef] [PubMed]
- Young, K.C.; Lindsay, K.L.; Lee, K.J.; Liu, W.C.; He, J.W.; Milstein, S.L.; Lai, M.M. Identification of a ribavirin-resistant NS5B mutation of hepatitis C virus during ribavirin monotherapy. Hepatology 2003, 38, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Ibarra, K.D.; Pfeiffer, J.K. Reduced ribavirin antiviral efficacy via nucleoside transporter-mediated drug resistance. J. Virol. 2009, 83, 4538–4547. [Google Scholar] [CrossRef] [PubMed]
- Pfeiffer, J.K.; Kirkegaard, K. Ribavirin resistance in hepatitis C virus replicon-containing cell lines conferred by changes in the cell line or mutations in the replicon RNA. J. Virol. 2005, 79, 2346–2355. [Google Scholar] [CrossRef] [PubMed]
- Hmwe, S.S.; Aizaki, H.; Date, T.; Murakami, K.; Ishii, K.; Miyamura, T.; Koike, K.; Wakita, T.; Suzuki, T. Identification of hepatitis C virus genotype 2a replicon variants with reduced susceptibility to ribavirin. Antivir. Res. 2010, 85, 520–524. [Google Scholar] [CrossRef] [PubMed]
- Feigelstock, D.A.; Mihalik, K.B.; Feinstone, S.M. Selection of hepatitis C virus resistant to ribavirin. Virol. J. 2011, 8. [Google Scholar] [CrossRef] [PubMed]
- Barth, H. Hepatitis C virus: Is it time to say goodbye yet? Perspectives and challenges for the next decade. World J. Hepatol. 2015, 7, 725–737. [Google Scholar] [CrossRef] [PubMed]
- Horner, S.M.; Naggie, S. Successes and Challenges on the Road to Cure Hepatitis C. PLoS Pathog. 2015, 11, e1004854. [Google Scholar] [CrossRef] [PubMed]
- Pawlotsky, J.M. Hepatitis C treatment: The data flood goes on-an update from the liver meeting 2014. Gastroenterology 2015, 148, 468–479. [Google Scholar] [CrossRef] [PubMed]
- Conteduca, V.; Sansonno, D.; Russi, S.; Pavone, F.; Dammacco, F. Therapy of chronic hepatitis C virus infection in the era of direct-acting and host-targeting antiviral agents. J. Infect. 2014, 68, 1–20. [Google Scholar] [CrossRef] [PubMed]
- DeLemos, A.S.; Chung, R.T. Hepatitis C treatment: An incipient therapeutic revolution. Trends Mol. Med. 2014, 20, 315–321. [Google Scholar] [CrossRef] [PubMed]
- Feld, J.J. The beginning of the end: What is the future of interferon therapy for chronic hepatitis C? Antivir. Res. 2014, 105, 32–38. [Google Scholar] [CrossRef] [PubMed]
- Hayes, C.N.; Chayama, K. Emerging treatments for chronic hepatitis C. J. Formos. Med. Assoc. 2015, 114, 204–215. [Google Scholar] [CrossRef] [PubMed]
- Solund, C.; Krarup, H.; Ramirez, S.; Thielsen, P.; Roge, B.T.; Lunding, S.; Barfod, T.S.; Madsen, L.G.; Tarp, B.; Christensen, P.B.; et al. Nationwide experience of treatment with protease inhibitors in chronic hepatitis C patients in denmark: Identification of viral resistance mutations. PLoS ONE 2014, 9, e113034. [Google Scholar] [CrossRef] [PubMed]
- Vermehren, J.; Sarrazin, C. The role of resistance in HCV treatment. Best Pract. Res. Clin. Gastroenterol. 2012, 26, 487–503. [Google Scholar] [CrossRef] [PubMed]
- Lontok, E.; Harrington, P.; Howe, A.; Kieffer, T.; Lennerstrand, J.; Lenz, O.; McPhee, F.; Mo, H.; Parkin, N.; Pilot-Matias, T.; et al. Hepatitis C virus drug resistance-associated substitutions: State of the art summary. Hepatology 2015, 62, 1623–32. [Google Scholar] [CrossRef] [PubMed]
- Lange, C.M.; Zeuzem, S. Perspectives and challenges of interferon-free therapy for chronic hepatitis C. J. Hepatol. 2013, 58, 583–592. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.J.; Locarnini, S.A.; Beard, M.R. Resistance to anti-HCV protease inhibitors. Curr. Opin. Virol. 2011, 1, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Sarrazin, C.; Zeuzem, S. Resistance to direct antiviral agents in patients with hepatitis C virus infection. Gastroenterology 2010, 138, 447–462. [Google Scholar] [CrossRef] [PubMed]
- Wyles, D.L. Antiviral resistance and the future landscape of hepatitis C virus infection therapy. J. Infect. Dis. 2013, 207, S33–S39. [Google Scholar] [CrossRef] [PubMed]
- Gao, M. Antiviral activity and resistance of HCV NS5A replication complex inhibitors. Curr. Opin. Virol. 2013, 3, 514–520. [Google Scholar] [CrossRef] [PubMed]
- Halfon, P.; Sarrazin, C. Future treatment of chronic hepatitis C with direct acting antivirals: Is resistance important? Liver Int. 2012, 32, 79–87. [Google Scholar] [CrossRef] [PubMed]
- Nakamoto, S.; Kanda, T.; Wu, S.; Shirasawa, H.; Yokosuka, O. Hepatitis C virus NS5A inhibitors and drug resistance mutations. World J. Gastroenterol. 2014, 20, 2902–2912. [Google Scholar] [CrossRef] [PubMed]
- Nettles, R.E.; Gao, M.; Bifano, M.; Chung, E.; Persson, A.; Marbury, T.C.; Goldwater, R.; DeMicco, M.P.; Rodriguez-Torres, M.; Vutikullird, A.; et al. Multiple ascending dose study of BMS-790052, a nonstructural protein 5A replication complex inhibitor, in patients infected with hepatitis C virus genotype 1. Hepatology 2011, 54, 1956–1965. [Google Scholar] [CrossRef] [PubMed]
- Lam, A.M.; Espiritu, C.; Bansal, S.; Micolochick Steuer, H.M.; Niu, C.; Zennou, V.; Keilman, M.; Zhu, Y.; Lan, S.; Otto, M.J.; et al. Genotype and subtype profiling of PSI-7977 as a nucleotide inhibitor of hepatitis C virus. Antimicrob. Agents Chemother. 2012, 56, 3359–3368. [Google Scholar] [CrossRef] [PubMed]
- Donaldson, E.F.; Harrington, P.R.; O'Rear, J.J.; Naeger, L.K. Clinical evidence and bioinformatics characterization of potential hepatitis C virus resistance pathways for sofosbuvir. Hepatology 2015, 61, 56–65. [Google Scholar] [CrossRef] [PubMed]
- Tong, X.; le Pogam, S.; Li, L.; Haines, K.; Piso, K.; Baronas, V.; Yan, J.M.; So, S.S.; Klumpp, K.; Najera, I. In vivo emergence of a novel mutant L159F/L320F in the NS5B polymerase confers low-level resistance to the HCV polymerase inhibitors mericitabine and sofosbuvir. J. Infect. Dis. 2014, 209, 668–675. [Google Scholar] [CrossRef] [PubMed]
- Childs-Kean, L.M.; Hand, E.O. Simeprevir and sofosbuvir for treatment of chronic hepatitis C infection. Clin. Ther. 2015, 37, 243–267. [Google Scholar] [CrossRef] [PubMed]
- Gane, E.J.; Stedman, C.A.; Hyland, R.H.; Ding, X.; Svarovskaia, E.; Symonds, W.T.; Hindes, R.G.; Berrey, M.M. Nucleotide polymerase inhibitor sofosbuvir plus ribavirin for hepatitis C. N. Engl. J. Med. 2013, 368, 34–44. [Google Scholar] [CrossRef] [PubMed]
- Hedskog, C.; Dvory-Sobol, H.; Gontcharova, V.; Martin, R.; Ouyang, W.; Han, B.; Gane, E.J.; Brainard, D.; Hyland, R.H.; Miller, M.D.; et al. Evolution of the HCV viral population from a patient with S282T detected at relapse after sofosbuvir monotherapy. J. Viral Hepat. 2015, 22, 871–881. [Google Scholar] [CrossRef] [PubMed]
- Svarovskaia, E.S.; Dvory-Sobol, H.; Parkin, N.; Hebner, C.; Gontcharova, V.; Martin, R.; Ouyang, W.; Han, B.; Xu, S.; Ku, K.; et al. Infrequent development of resistance in genotype 1–6 hepatitis C virus-infected subjects treated with sofosbuvir in phase 2 and 3 clinical trials. Clin. Infect. Dis. 2014, 59, 1666–1674. [Google Scholar] [CrossRef] [PubMed]
- Osinusi, A.; Meissner, E.G.; Lee, Y.J.; Bon, D.; Heytens, L.; Nelson, A.; Sneller, M.; Kohli, A.; Barrett, L.; Proschan, M.; et al. Sofosbuvir and ribavirin for hepatitis C genotype 1 in patients with unfavorable treatment characteristics: A randomized clinical trial. JAMA 2013, 310, 804–811. [Google Scholar] [CrossRef] [PubMed]
- Lawitz, E.; Poordad, F.F.; Pang, P.S.; Hyland, R.H.; Ding, X.; Mo, H.; Symonds, W.T.; McHutchison, J.G.; Membreno, F.E. Sofosbuvir and ledipasvir fixed-dose combination with and without ribavirin in treatment-naive and previously treated patients with genotype 1 hepatitis C virus infection (LONESTAR): An open-label, randomised, phase 2 trial. Lancet 2014, 383, 515–523. [Google Scholar] [CrossRef]
- Maimone, S.; Tripodi, G.; Musolino, C.; Cacciola, I.; Pollicino, T.; Raimondo, G. Lack of the NS5B S282T mutation in HCV isolates from liver tissue of treatment-naive patients with HCV genotype-1b infection. Antivir. Ther. 2015, 20, 245–247. [Google Scholar] [CrossRef] [PubMed]
- Smith, D.B.; Bukh, J.; Kuiken, C.; Muerhoff, A.S.; Rice, C.M.; Stapleton, J.T.; Simmonds, P. Expanded classification of hepatitis C virus into 7 genotypes and 67 subtypes: Updated criteria and genotype assignment web resource. Hepatology 2014, 59, 318–327. [Google Scholar] [CrossRef] [PubMed]
- McCown, M.F.; Rajyaguru, S.; Kular, S.; Cammack, N.; Najera, I. GT-1a or GT-1b subtype-specific resistance profiles for hepatitis C virus inhibitors telaprevir and HCV-796. Antimicrob. Agents Chemother. 2009, 53, 2129–2132. [Google Scholar] [CrossRef] [PubMed]
- Quer, J.; Gregori, J.; Rodriguez-Frias, F.; Buti, M.; Madejon, A.; Perez-del-Pulgar, S.; Garcia-Cehic, D.; Casillas, R.; Blasi, M.; Homs, M.; et al. High-resolution hepatitis C virus subtyping using NS5B deep sequencing and phylogeny, an alternative to current methods. J. Clin. Microbiol. 2015, 53, 219–226. [Google Scholar] [CrossRef] [PubMed]
- Los Alamos database. Available online: https://hcv.lanl.gov/content/sequence/HCV/ToolsOutline.html (accessed on 19 February 2015).
- Perales, C.; Beach, N.M.; Sheldon, J.; Domingo, E. Molecular basis of interferon resistance in hepatitis C virus. Curr. Opin. Virol. 2014, 8C, 38–44. [Google Scholar] [CrossRef] [PubMed]
- Geller, R.; Vignuzzi, M.; Andino, R.; Frydman, J. Evolutionary constraints on chaperone-mediated folding provide an antiviral approach refractory to development of drug resistance. Genes Dev. 2007, 21, 195–205. [Google Scholar] [CrossRef] [PubMed]
- Hopkins, S.; Scorneaux, B.; Huang, Z.; Murray, M.G.; Wring, S.; Smitley, C.; Harris, R.; Erdmann, F.; Fischer, G.; Ribeill, Y. SCY-635, a novel nonimmunosuppressive analog of cyclosporine that exhibits potent inhibition of hepatitis C virus RNA replication in vitro. Antimicrob. Agents Chemother. 2010, 54, 660–672. [Google Scholar] [CrossRef] [PubMed]
- Garbelli, A.; Radi, M.; Falchi, F.; Beermann, S.; Zanoli, S.; Manetti, F.; Dietrich, U.; Botta, M.; Maga, G. Targeting the human DEAD-box polypeptide 3 (DDX3) RNA helicase as a novel strategy to inhibit viral replication. Curr. Med. Chem. 2011, 18, 3015–3027. [Google Scholar] [CrossRef] [PubMed]
- Kumar, N.; Liang, Y.; Parslow, T.G.; Liang, Y. Receptor tyrosine kinase inhibitors block multiple steps of influenza a virus replication. J. Virol. 2011, 85, 2818–2827. [Google Scholar] [CrossRef] [PubMed]
- Vidalain, P.O.; Lucas-Hourani, M.; Helynck, O.; Tangy, F.; Munier-Lehmann, H. Stimulation of the antiviral innate immune response by pyrimidine biosynthesis inhibitors: A surprise of phenotypic screening. Med. Sci. 2015, 31, 98–104. [Google Scholar]
- Pawlotsky, J.M. What are the pros and cons of the use of host-targeted agents against hepatitis C? Antivir. Res. 2014, 105, 22–25. [Google Scholar] [CrossRef] [PubMed]
- Chatterji, U.; Lim, P.; Bobardt, M.D.; Wieland, S.; Cordek, D.G.; Vuagniaux, G.; Chisari, F.; Cameron, C.E.; Targett-Adams, P.; Parkinson, T.; et al. HCV resistance to cyclosporin A does not correlate with a resistance of the NS5A-cyclophilin A interaction to cyclophilin inhibitors. J. Hepatol. 2010, 53, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Delang, L.; Vliegen, I.; Froeyen, M.; Neyts, J. Comparative Study of the Genetic Barriers and Pathways towards Resistance of Selective Inhibitors of Hepatitis C Virus Replication. Antimicrob. Agents Chemother. 2011, 55, 4103–4113. [Google Scholar] [CrossRef] [PubMed]
- Sheldon, J.; Beach, N.M.; Moreno, E.; Gallego, I.; Pineiro, D.; Martinez-Salas, E.; Gregori, J.; Quer, J.; Esteban, J.I.; Rice, C.M.; et al. Increased replicative fitness can lead to decreased drug sensitivity of hepatitis C virus. J. Virol. 2014, 88, 12098–12111. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, J.C.; de Meyer, S.; Bartels, D.J.; Dierynck, I.; Zhang, E.Z.; Spanks, J.; Tigges, A.M.; Ghys, A.; Dorrian, J.; Adda, N.; et al. Evolution of treatment-emergent resistant variants in telaprevir phase 3 clinical trials. Clin. Infect. Dis. 2013, 57, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Sato, M.; Maekawa, S.; Komatsu, N.; Tatsumi, A.; Miura, M.; Muraoka, M.; Suzuki, Y.; Amemiya, F.; Takano, S.; Fukasawa, M.; et al. Deep sequencing and phylogenetic analysis of variants resistant to interferon-based protease inhibitor therapy in chronic hepatitis induced by genotype 1b hepatitis C virus. J. Virol. 2015, 89, 6105–6116. [Google Scholar] [CrossRef] [PubMed]
- Paolucci, S.; Fiorina, L.; Mariani, B.; Gulminetti, R.; Novati, S.; Barbarini, G.; Bruno, R.; Baldanti, F. Naturally occurring resistance mutations to inhibitors of HCV NS5A region and NS5B polymerase in DAA treatment-naive patients. Virol. J. 2013, 10. [Google Scholar] [CrossRef] [PubMed]
- Suzuki, F.; Sezaki, H.; Akuta, N.; Suzuki, Y.; Seko, Y.; Kawamura, Y.; Hosaka, T.; Kobayashi, M.; Saito, S.; Arase, Y.; et al. Prevalence of hepatitis C virus variants resistant to NS3 protease inhibitors or the NS5A inhibitor (BMS-790052) in hepatitis patients with genotype 1b. J. Clin. Virol. 2012, 54, 352–354. [Google Scholar] [CrossRef] [PubMed]
- Margeridon-Thermet, S.; le Pogam, S.; Li, L.; Liu, T.F.; Shulman, N.; Shafer, R.W.; Najera, I. Similar prevalence of low-abundance drug-resistant variants in treatment-naive patients with genotype 1a and 1b hepatitis C virus infections as determined by ultradeep pyrosequencing. PLoS ONE 2014, 9, e105569. [Google Scholar] [CrossRef] [PubMed]
- Hernandez, D.; Zhou, N.; Ueland, J.; Monikowski, A.; McPhee, F. Natural prevalence of NS5A polymorphisms in subjects infected with hepatitis C virus genotype 3 and their effects on the antiviral activity of NS5A inhibitors. J. Clin. Virol. 2013, 57, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Alves, R.; Queiroz, A.T.; Pessoa, M.G.; da Silva, E.F.; Mazo, D.F.; Carrilho, F.J.; Carvalho-Filho, R.J.; de Carvalho, I.M. The presence of resistance mutations to protease and polymerase inhibitors in Hepatitis C virus sequences from the Los Alamos databank. J. Viral Hepat. 2013, 20, 414–421. [Google Scholar] [CrossRef] [PubMed]
- Trimoulet, P.; Pinson, P.; Papuchon, J.; Foucher, J.; Vergniol, J.; Chermak, F.; Wittkop, L.; Castaing, N.; Merrouche, W.; Reigadas, S.; et al. Dynamic and rapid changes in viral quasispecies by UDPS in chronic hepatitis C patients receiving telaprevir-based therapy. Antivir. Ther. 2013, 18, 723–727. [Google Scholar] [CrossRef] [PubMed]
- Ruggiero, T.; Proietti, A.; Boglione, L.; Milia, M.G.; Allice, T.; Burdino, E.; Orofino, G.; Bonora, S.; di Perri, G.; Ghisetti, V. Predominance of hepatitis C virus Q80K among NS3 baseline-resistance-associated amino acid variants in direct-antiviral-agent-naive patients with chronic hepatitis: Single-centre experience. Arch. Virol. 2015, in press. [Google Scholar] [CrossRef] [PubMed]
- Sarrazin, C.; Lathouwers, E.; Peeters, M.; Daems, B.; Buelens, A.; Witek, J.; Wyckmans, Y.; Fevery, B.; Verbinnen, T.; Ghys, A.; et al. Prevalence of the hepatitis C virus NS3 polymorphism Q80K in genotype 1 patients in the European region. Antivir. Res. 2015, 116, 10–16. [Google Scholar] [CrossRef] [PubMed]
- McCloskey, R.M.; Liang, R.H.; Joy, J.B.; Krajden, M.; Montaner, J.S.; Harrigan, P.R.; Poon, A.F. Global origin and transmission of hepatitis C virus nonstructural protein 3 Q80K polymorphism. J. Infect. Dis. 2015, 211, 1288–1295. [Google Scholar] [CrossRef] [PubMed]
- Lenz, O.; de Bruijne, J.; Vijgen, L.; Verbinnen, T.; Weegink, C.; van Marck, H.; Vandenbroucke, I.; Peeters, M.; Simmen, K.; Fanning, G.; Verloes, R.; Picchio, G.; Reesink, H. Efficacy of re-treatment with TMC435 as combination therapy in hepatitis C virus-infected patients following TMC435 monotherapy. Gastroenterology 2012, 143, 1176–1178. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, P.; Tripathi, R.; Schnell, G.; Reisch, T.; Beyer, J.; Irvin, M.; Xie, W.; Larsen, L.; Cohen, D.; Podsadecki, T.; et al. Resistance Analysis of Baseline and Treatment-Emergent Variants in the AVIATOR Study With Paritaprevir/r, Ombitasvir and Dasabuvir in HCV Genotype 1. Antimicrob. Agents Chemother. 2015, 59, 5445–5454. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Jarabo, C.M.; Arias, A.; Baranowski, E.; Escarmís, C.; Domingo, E. Memory in viral quasispecies. J. Virol. 2000, 74, 3543–3547. [Google Scholar] [CrossRef] [PubMed]
- Briones, C.; Domingo, E.; Molina-París, C. Memory in retroviral quasispecies: Experimental evidence and theoretical model for human immunodeficiency virus. J. Mol. Biol. 2003, 331, 213–229. [Google Scholar] [CrossRef]
- Briones, C.; Domingo, E. Minority report: Hidden memory genomes in HIV-1 quasispecies and possible clinical implications. AIDS Rev. 2008, 10, 93–109. [Google Scholar] [PubMed]
- Howe, A.Y.; Long, J.; Nickle, D.; Barnard, R.; Thompson, S.; Howe, J.; Alves, K.; Wahl, J. Long-term follow-up of patients receiving boceprevir for treatment of chronic hepatitis C. Antivir. Res. 2015, 113, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Susser, S.; Vermehren, J.; Forestier, N.; Welker, M.W.; Grigorian, N.; Fuller, C.; Perner, D.; Zeuzem, S.; Sarrazin, C. Analysis of long-term persistence of resistance mutations within the hepatitis C virus NS3 protease after treatment with telaprevir or boceprevir. J. Clin. Virol. 2011, 52, 321–327. [Google Scholar] [CrossRef] [PubMed]
- Yoshimi, S.; Imamura, M.; Murakami, E.; Hiraga, N.; Tsuge, M.; Kawakami, Y.; Aikata, H.; Abe, H.; Hayes, C.N.; Sasaki, T.; et al. Long term persistence of NS5A inhibitor-resistant hepatitis C virus in patients who failed daclatasvir and asunaprevir therapy. J. Med. Virol. 2015, 87, 1913–1920. [Google Scholar] [CrossRef] [PubMed]
- Ruiz-Jarabo, C.M.; Arias, A.; Molina-París, C.; Briones, C.; Baranowski, E.; Escarmís, C.; Domingo, E. Duration and fitness dependence of quasispecies memory. J. Mol. Biol. 2002, 315, 285–296. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, V.; Korner, F.; Koch, J.; Herian, U.; Theilmann, L.; Bartenschlager, R. Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 1999, 285, 110–113. [Google Scholar] [CrossRef] [PubMed]
- Lohmann, V.; Bartenschlager, R. On the history of hepatitis C virus cell culture systems. J. Med. Chem. 2014, 57, 1627–1642. [Google Scholar] [CrossRef] [PubMed]
- Steinmann, E.; Pietschmann, T. Cell culture systems for hepatitis C virus. Curr. Top. Microbiol. Immunol. 2013, 369, 17–48. [Google Scholar] [PubMed]
- Gao, M.; Nettles, R.E.; Belema, M.; Snyder, L.B.; Nguyen, V.N.; Fridell, R.A.; Serrano-Wu, M.H.; Langley, D.R.; Sun, J.H.; O’Boyle, D.R., II; et al. Chemical genetics strategy identifies an HCV NS5A inhibitor with a potent clinical effect. Nature 2010, 465, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Lindenbach, B.D.; Evans, M.J.; Syder, A.J.; Wolk, B.; Tellinghuisen, T.L.; Liu, C.C.; Maruyama, T.; Hynes, R.O.; Burton, D.R.; McKeating, J.A.; et al. Complete replication of hepatitis C virus in cell culture. Science 2005, 309, 623–626. [Google Scholar] [CrossRef] [PubMed]
- Wakita, T.; Pietschmann, T.; Kato, T.; Date, T.; Miyamoto, M.; Zhao, Z.; Murthy, K.; Habermann, A.; Krausslich, H.G.; Mizokami, M.; et al. Production of infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat. Med. 2005, 11, 791–796. [Google Scholar] [CrossRef] [PubMed]
- Zhong, J.; Gastaminza, P.; Cheng, G.; Kapadia, S.; Kato, T.; Burton, D.R.; Wieland, S.F.; Uprichard, S.L.; Wakita, T.; Chisari, F.V. Robust hepatitis C virus infection in vitro. Proc. Natl. Acad. Sci. USA 2005, 102, 9294–9299. [Google Scholar] [CrossRef] [PubMed]
- Scheel, T.K.; Gottwein, J.M.; Jensen, T.B.; Prentoe, J.C.; Hoegh, A.M.; Alter, H.J.; Eugen-Olsen, J.; Bukh, J. Development of JFH1-based cell culture systems for hepatitis C virus genotype 4a and evidence for cross-genotype neutralization. Proc. Natl. Acad. Sci. USA 2008, 105, 997–1002. [Google Scholar] [CrossRef] [PubMed]
- Jensen, T.B.; Gottwein, J.M.; Scheel, T.K.; Hoegh, A.M.; Eugen-Olsen, J.; Bukh, J. Highly efficient JFH1-based cell-culture system for hepatitis C virus genotype 5a: Failure of homologous neutralizing-antibody treatment to control infection. J. Infect. Dis. 2008, 198, 1756–1765. [Google Scholar] [CrossRef] [PubMed]
- Gottwein, J.M.; Scheel, T.K.; Jensen, T.B.; Lademann, J.B.; Prentoe, J.C.; Knudsen, M.L.; Hoegh, A.M.; Bukh, J. Development and characterization of hepatitis C virus genotype 1–7 cell culture systems: Role of CD81 and scavenger receptor class B type I and effect of antiviral drugs. Hepatology 2009, 49, 364–377. [Google Scholar] [CrossRef] [PubMed]
- Pietschmann, T.; Kaul, A.; Koutsoudakis, G.; Shavinskaya, A.; Kallis, S.; Steinmann, E.; Abid, K.; Negro, F.; Dreux, M.; Cosset, F.L.; et al. Construction and characterization of infectious intragenotypic and intergenotypic hepatitis C virus chimeras. Proc. Natl. Acad. Sci. USA 2006, 103, 7408–7413. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Ma, Y.; Yates, J.; Lemon, S.M. Compensatory mutations in E1, p7, NS2, and NS3 enhance yields of cell culture-infectious intergenotypic chimeric hepatitis C virus. J. Virol. 2007, 81, 629–638. [Google Scholar] [CrossRef] [PubMed]
- Gottwein, J.M.; Scheel, T.K.; Hoegh, A.M.; Lademann, J.B.; Eugen-Olsen, J.; Lisby, G.; Bukh, J. Robust hepatitis C genotype 3a cell culture releasing adapted intergenotypic 3a/2a (S52/JFH1) viruses. Gastroenterology 2007, 133, 1614–1626. [Google Scholar] [CrossRef] [PubMed]
- Scheel, T.K.; Gottwein, J.M.; Carlsen, T.H.; Li, Y.P.; Jensen, T.B.; Spengler, U.; Weis, N.; Bukh, J. Efficient culture adaptation of hepatitis C virus recombinants with genotype-specific core-NS2 by using previously identified mutations. J. Virol. 2011, 85, 2891–2906. [Google Scholar] [CrossRef] [PubMed]
- Prentoe, J.; Jensen, T.B.; Meuleman, P.; Serre, S.B.; Scheel, T.K.; Leroux-Roels, G.; Gottwein, J.M.; Bukh, J. Hypervariable region 1 differentially impacts viability of hepatitis C virus strains of genotypes 1 to 6 and impairs virus neutralization. J. Virol. 2011, 85, 2224–2234. [Google Scholar] [CrossRef] [PubMed]
- Griffin, S.; Stgelais, C.; Owsianka, A.M.; Patel, A.H.; Rowlands, D.; Harris, M. Genotype-dependent sensitivity of hepatitis C virus to inhibitors of the p7 ion channel. Hepatology 2008, 48, 1779–1790. [Google Scholar] [CrossRef] [PubMed]
- Gottwein, J.M.; Scheel, T.K.; Jensen, T.B.; Ghanem, L.; Bukh, J. Differential efficacy of protease inhibitors against HCV genotypes 2a, 3a, 5a, and 6a NS3/4A protease recombinant viruses. Gastroenterology 2011, 141, 1067–1079. [Google Scholar] [CrossRef] [PubMed]
- Scheel, T.K.; Gottwein, J.M.; Mikkelsen, L.S.; Jensen, T.B.; Bukh, J. Recombinant HCV variants with NS5A from genotypes 1–7 have different sensitivities to an NS5A inhibitor but not interferon-α. Gastroenterology 2011, 140, 1032–1042. [Google Scholar] [CrossRef] [PubMed]
- Scheel, T.K.; Prentoe, J.; Carlsen, T.H.; Mikkelsen, L.S.; Gottwein, J.M.; Bukh, J. Analysis of functional differences between hepatitis C virus NS5A of genotypes 1–7 in infectious cell culture systems. PLoS Pathog. 2012, 8, e1002696. [Google Scholar] [CrossRef] [PubMed]
- Galli, A.; Scheel, T.K.; Prentoe, J.C.; Mikkelsen, L.S.; Gottwein, J.M.; Bukh, J. Analysis of hepatitis C virus core/NS5A protein co-localization using novel cell culture systems expressing core-NS2 and NS5A of genotypes 1–7. J. Gen. Virol. 2013, 94, 2221–2235. [Google Scholar] [CrossRef] [PubMed]
- Gottwein, J.M.; Jensen, S.B.; Li, Y.P.; Ghanem, L.; Scheel, T.K.; Serre, S.B.; Mikkelsen, L.; Bukh, J. Combination treatment with hepatitis C virus protease and NS5A inhibitors is effective against recombinant genotype 1a, 2a, and 3a viruses. Antimicrob. Agents Chemother. 2013, 57, 1291–1303. [Google Scholar] [CrossRef] [PubMed]
- Gottwein, J.M.; Jensen, S.B.; Serre, S.B.; Ghanem, L.; Scheel, T.K.; Jensen, T.B.; Krarup, H.; Uzcategui, N.; Mikkelsen, L.S.; Bukh, J. Adapted J6/JFH1-based Hepatitis C virus recombinants with genotype-specific NS4A show similar efficacies against lead protease inhibitors, alpha interferon, and a putative NS4A inhibitor. Antimicrob. Agents Chemother. 2013, 57, 6034–6049. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.P.; Ramirez, S.; Humes, D.; Jensen, S.B.; Gottwein, J.M.; Bukh, J. Differential sensitivity of 5'UTR-NS5A recombinants of hepatitis C virus genotypes 1–6 to protease and NS5A inhibitors. Gastroenterology 2014, 146, 812–821. [Google Scholar] [CrossRef] [PubMed]
- Saeed, M.; Andreo, U.; Chung, H.Y.; Espiritu, C.; Branch, A.D.; Silva, J.M.; Rice, C.M. SEC14L2 enables pan-genotype HCV replication in cell culture. Nature 2015, 524, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Yi, M.; Villanueva, R.A.; Thomas, D.L.; Wakita, T.; Lemon, S.M. Production of infectious genotype 1a hepatitis C virus (Hutchinson strain) in cultured human hepatoma cells. Proc. Natl. Acad. Sci. USA 2006, 103, 2310–2315. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.P.; Ramirez, S.; Jensen, S.B.; Purcell, R.H.; Gottwein, J.M.; Bukh, J. Highly efficient full-length hepatitis C virus genotype 1 (strain TN) infectious culture system. Proc. Natl. Acad. Sci. USA 2012, 109, 19757–19762. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.P.; Ramirez, S.; Mikkelsen, L.; Bukh, J. Efficient Infectious Cell Culture Systems of the Hepatitis C Virus (HCV) Prototype Strains HCV-1 and H77. J. Virol. 2015, 89, 811–823. [Google Scholar] [CrossRef] [PubMed]
- Shimakami, T.; Welsch, C.; Yamane, D.; McGivern, D.; Yi, M.; Zeuzem, S.; Lemon, S.M. Protease Inhibitor-Resistant Hepatitis C Virus Mutants with Reduced Fitness from Impaired Production of Infectious Virus. Gastroenterology 2011, 140, 667–675. [Google Scholar] [CrossRef] [PubMed]
- Pietschmann, T.; Zayas, M.; Meuleman, P.; Long, G.; Appel, N.; Koutsoudakis, G.; Kallis, S.; Leroux-Roels, G.; Lohmann, V.; Bartenschlager, R. Production of infectious genotype 1b virus particles in cell culture and impairment by replication enhancing mutations. PLoS Pathog. 2009, 5, e1000475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Date, T.; Morikawa, K.; Tanaka, Y.; Tanaka-Kaneko, K.; Sata, T.; Mizokami, M.; Wakita, T. Replication and infectivity of a novel genotype 1b hepatitis C virus clone. Microbiol. Immunol. 2012, 56, 308–317. [Google Scholar] [CrossRef] [PubMed]
- Buti, M.; Riveiro-Barciela, M.; Esteban, R. Management of Direct Antiviral Agent Failures. J. Hepatol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Perales, C.; Iranzo, J.; Manrubia, S.C.; Domingo, E. The impact of quasispecies dynamics on the use of therapeutics. Trends Microbiol. 2012, 20, 595–603. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Perales, C.; Quer, J.; Gregori, J.; Esteban, J.I.; Domingo, E. Resistance of Hepatitis C Virus to Inhibitors: Complexity and Clinical Implications. Viruses 2015, 7, 5746-5766. https://doi.org/10.3390/v7112902
Perales C, Quer J, Gregori J, Esteban JI, Domingo E. Resistance of Hepatitis C Virus to Inhibitors: Complexity and Clinical Implications. Viruses. 2015; 7(11):5746-5766. https://doi.org/10.3390/v7112902
Chicago/Turabian StylePerales, Celia, Josep Quer, Josep Gregori, Juan Ignacio Esteban, and Esteban Domingo. 2015. "Resistance of Hepatitis C Virus to Inhibitors: Complexity and Clinical Implications" Viruses 7, no. 11: 5746-5766. https://doi.org/10.3390/v7112902