
Phosphorylation of Single Stranded RNA Virus Proteins and Potential for Novel Therapeutic Strategies

{kind=link}
Abstract
:1. Introduction
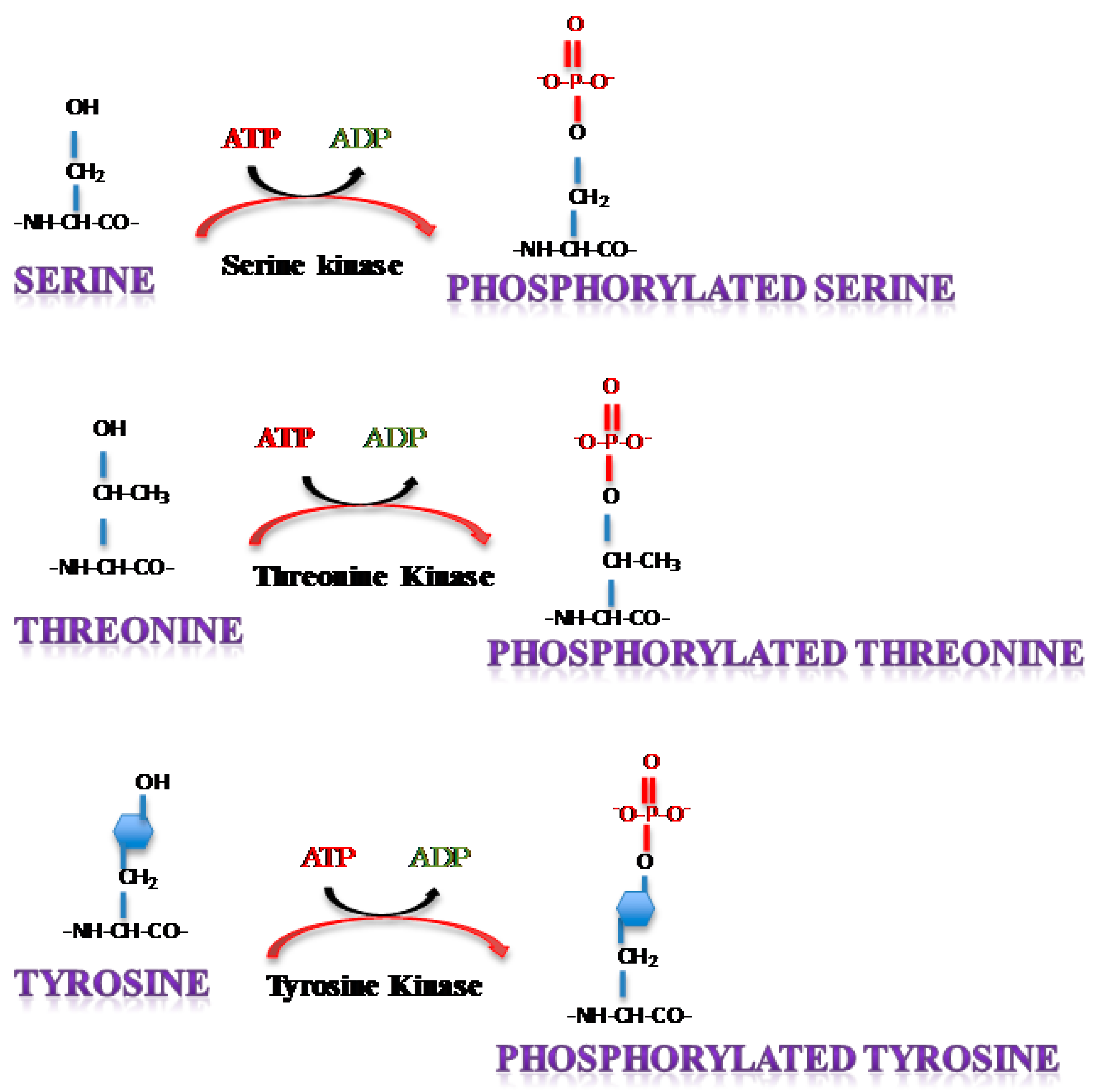
2. Protein Phosphorylation
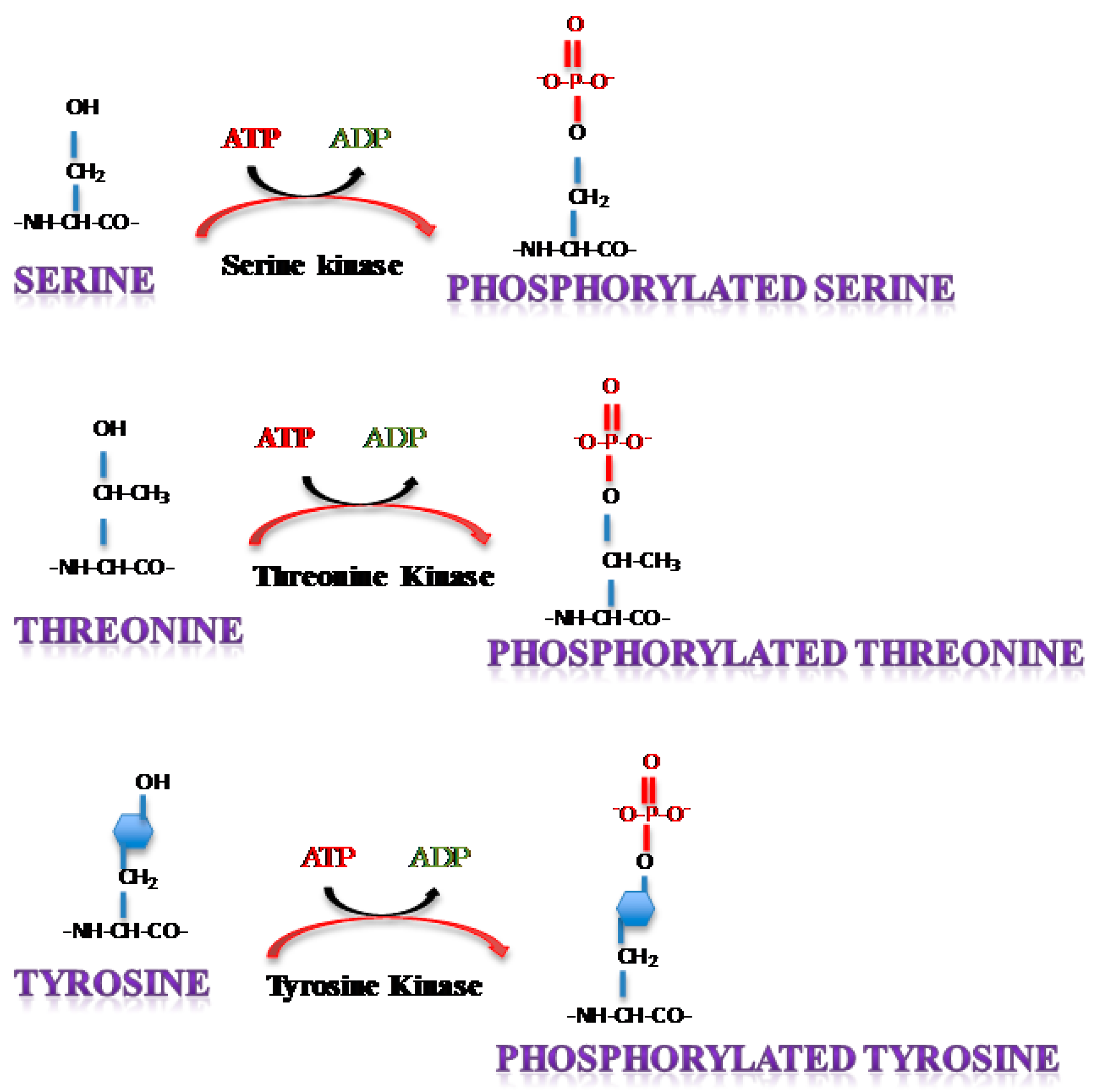
3. Phosphorylation of Single Stranded RNA Virus Proteins
3.1. Positive Sense ssRNA Viruses
3.1.1. Flaviviruses
3.1.2. Alphaviruses
3.1.3. Rubivirus
3.2. Negative Sense ssRNA Viruses
3.2.1. Filovirus
3.2.2. Orthomyxovirus
4. Viral Protein Phosphorylation Using Host Enzymes—Strength or Weakness?
5. Repurposing of FDA Approved Therapeutics
6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Walsh, C.T.; Garneau-Tsodikova, S.; Gatto, G.J. Protein Posttranslational Modifications: The Chemistry of Proteome Diversifications. Angew. Chem. Int. Ed. 2005, 44, 7342–7372. [Google Scholar] [CrossRef] [PubMed]
- Bretaña, N.A.; Lu, C.-T.; Chiang, C.-Y.; Su, M.-G.; Huang, K.-Y.; Lee, T.-Y.; Weng, S.-L. Identifying Protein Phosphorylation Sites with Kinase Substrate Specificity on Human Viruses. PLoS ONE 2012, 7, e40694. [Google Scholar] [CrossRef] [PubMed]
- Schweiger, R.; Linial, M. Cooperativity within proximal phosphorylation sites is revealed from large-scale proteomics data. Biol. Direct 2010, 5, 6. [Google Scholar] [CrossRef] [PubMed]
- Mann, M.; Ong, S.-E.; Grønborg, M.; Steen, H.; Jensen, O.N.; Pandey, A. Analysis of protein phosphorylation using mass spectrometry: Deciphering the phosphoproteome. Trends Biotechnol. 2002, 20, 261–268. [Google Scholar] [CrossRef]
- Manning, G.; Whyte, D.B.; Martinez, R.; Hunter, T.; Sudarsanam, S. The protein kinase complement of the human genome. Science 2002, 298, 1912–1934. [Google Scholar] [CrossRef] [PubMed]
- Rothwarf, D.M.; Karin, M. The NF-κB Activation Pathway: A Paradigm in Information Transfer from Membrane to Nucleus. Sci. Signal. 1999, 1999, re1. [Google Scholar] [CrossRef] [PubMed]
- Gustin, M.C.; Albertyn, J.; Alexander, M.; Davenport, K. MAP Kinase Pathways in the Yeast Saccharomyces cerevisiae. Microbiol. Mol. Biol. Rev. 1998, 62, 1264–1300. [Google Scholar] [PubMed]
- Holmberg, C.I.; Hietakangas, V.; Mikhailov, A.; Rantanen, J.O.; Kallio, M.; Meinander, A.; Hellman, J.; Morrice, N.; MacKintosh, C.; Morimoto, R.I.; et al. Phosphorylation of serine 230 promotes inducible transcriptional activity of heat shock factor 1. EMBO J. 2001, 20, 3800–3810. [Google Scholar] [CrossRef] [PubMed]
- Appella, E.; Anderson, C.W. Post-translational modifications and activation of p53 by genotoxic stresses. Eur. J. Biochem. FEBS 2001, 268, 2764–2772. [Google Scholar] [CrossRef]
- Schmitz, M.L.; Mattioli, I.; Buss, H.; Kracht, M. NF-kappaB: A multifaceted transcription factor regulated at several levels. Chembiochem Eur. J. Chem. Biol. 2004, 5, 1348–1358. [Google Scholar] [CrossRef] [PubMed]
- Vanden Berghe, W.; Vermeulen, L.; Delerive, P.; de Bosscher, K.; Staels, B.; Haegeman, G. A paradigm for gene regulation: Inflammation, NF-kappaB and PPAR. Adv. Exp. Med. Biol. 2003, 544, 181–196. [Google Scholar] [PubMed]
- Vermeulen, L.; de Wilde, G.; Notebaert, S.; Vanden Berghe, W.; Haegeman, G. Regulation of the transcriptional activity of the nuclear factor-kappaB p65 subunit. Biochem. Pharmacol. 2002, 64, 963–970. [Google Scholar] [CrossRef]
- Okazaki, T.; Sakon, S.; Sasazuki, T.; Sakurai, H.; Doi, T.; Yagita, H.; Okumura, K.; Nakano, H. Phosphorylation of serine 276 is essential for p65 NF-κB subunit-dependent cellular responses. Biochem. Biophys. Res. Commun. 2003, 300, 807–812. [Google Scholar] [PubMed]
- Jacob, T.; Van den Broeke, C.; Favoreel, H.W. Viral Serine/Threonine Protein Kinases. J. Virol. 2011, 85, 1158–1173. [Google Scholar] [CrossRef] [PubMed]
- Condit, R.C.; Motyczka, A.; Spizz, G. Isolation, characterization, and physical mapping of temperature-sensitive mutants of vaccinia virus. Virology 1983, 128, 429–443. [Google Scholar] [CrossRef]
- Kovacs, G.R.; Vasilakis, N.; Moss, B. Regulation of Viral Intermediate Gene Expression by the Vaccinia Virus B1 Protein Kinase. J. Virol. 2001, 75, 4048–4055. [Google Scholar] [CrossRef] [PubMed]
- Lenard, J. Host cell protein kinases in nonsegmented negative-strand virus (mononegavirales) infection. Pharmacol. Ther. 1999, 83, 39–48. [Google Scholar] [CrossRef]
- Afrikanova, I.; Miozzo, M.C.; Giambiagi, S.; Burrone, O. Phosphorylation generates different forms of rotavirus NSP5. J. Gen. Virol. 1996, 77, 2059–2065. [Google Scholar] [CrossRef] [PubMed]
- Krishnan, M.N.; Garcia-Blanco, M.A. Targeting host factors to treat West Nile and dengue viral infections. Viruses 2014, 6, 683–708. [Google Scholar] [CrossRef] [PubMed]
- Best, S.M.; Morris, K.L.; Shannon, J.G.; Robertson, S.J.; Mitzel, D.N.; Park, G.S.; Boer, E.; Wolfinbarger, J.B.; Bloom, M.E. Inhibition of Interferon-Stimulated JAK-STAT Signaling by a Tick-Borne Flavivirus and Identification of NS5 as an Interferon Antagonist. J. Virol. 2005, 79, 12828–12839. [Google Scholar] [CrossRef]
- Lin, R.-J.; Chang, B.-L.; Yu, H.-P.; Liao, C.-L.; Lin, Y.-L. Blocking of interferon-induced Jak-Stat signaling by Japanese encephalitis virus NS5 through a protein tyrosine phosphatase-mediated mechanism. J. Virol. 2006, 80, 5908–5918. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, D.; Mayuri; Best, S.M.; Perera, R.; Kuhn, R.J.; Striker, R. Protein Kinase G Phosphorylates Mosquito-Borne Flavivirus NS5. J. Virol. 2009, 83, 9195–9205. [Google Scholar] [CrossRef] [PubMed]
- Forwood, J.K.; Brooks, A.; Briggs, L.J.; Xiao, C.-Y.; Jans, D.A.; Vasudevan, S.G. The 37-Amino-Acid Interdomain of Dengue Virus NS5 Protein Contains a Functional NLS and Inhibitory CK2 Site. Biochem. Biophys. Res. Commun. 1999, 257, 731–737. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, M.; Zhang, L.; Ramachandra, M.; Kusukawa, J.; Ebner, K.E.; Padmanabhan, R. Association between NS3 and NS5 Proteins of Dengue Virus Type 2 in the Putative RNA Replicase is Linked to Differential Phosphorylation of NS5. J. Biol. Chem. 1995, 270, 19100–19106. [Google Scholar] [PubMed]
- Mackenzie, J.M.; Kenney, M.T.; Westaway, E.G. West Nile virus strain Kunjin NS5 polymerase is a phosphoprotein localized at the cytoplasmic site of viral RNA synthesis. J. Gen. Virol. 2007, 88, 1163–1168. [Google Scholar] [CrossRef] [PubMed]
- Bhattacharya, D.; Hoover, S.; Falk, S.P.; Weisblum, B.; Vestling, M.; Striker, R. Phosphorylation of yellow fever virus NS5 alters methyltransferase activity. Virology 2008, 380, 276–284. [Google Scholar] [CrossRef] [PubMed]
- Yamauchi, S.; Takeuchi, K.; Chihara, K.; Sun, X.; Honjoh, C.; Yoshiki, H.; Hotta, H.; Sada, K. Hepatitis C Virus Particle Assembly Involves Phosphorylation of NS5A by the c-Abl Tyrosine Kinase. J. Biol. Chem. 2015, 290, 21857–21864. [Google Scholar] [CrossRef] [PubMed]
- Morozova, O.V.; Tsekhanovskaya, N.A.; Maksimova, T.G.; Bachvalova, V.N.; Matveeva, V.A.; Kit, Y.Y. Phosphorylation of tick-borne encephalitis virus NS5 protein. Virus Res. 1997, 49, 9–15. [Google Scholar] [CrossRef]
- Jose, J.; Snyder, J.E.; Kuhn, R.J. A structural and functional perspective of alphavirus replication and assembly. Future Microbiol. 2009, 4, 837–856. [Google Scholar] [CrossRef] [PubMed]
- Steele, K.E.; Twenhafel, N.A. REVIEW PAPER: Pathology of animal models of alphavirus encephalitis. Vet. Pathol. 2010, 47, 790–805. [Google Scholar] [CrossRef] [PubMed]
- Tretyakova, I.; Lukashevich, I.S.; Glass, P.; Wang, E.; Weaver, S.; Pushko, P. Novel vaccine against Venezuelan equine encephalitis combines advantages of DNA immunization and a live attenuated vaccine. Vaccine 2013, 31, 1019–1025. [Google Scholar] [CrossRef]
- Weaver, S.C.; Ferro, C.; Barrera, R.; Boshell, J.; Navarro, J.-C. Venezuelan Equine Encephalitis. Annu. Rev. Entomol. 2004, 49, 141–174. [Google Scholar] [CrossRef]
- Atasheva, S.; Fish, A.; Fornerod, M.; Frolova, E.I. Venezuelan equine Encephalitis virus capsid protein forms a tetrameric complex with CRM1 and importin alpha/beta that obstructs nuclear pore complex function. J. Virol. 2010, 84, 4158–4171. [Google Scholar] [CrossRef] [PubMed]
- Foy, N.J.; Akhrymuk, M.; Akhrymuk, I.; Atasheva, S.; Bopda-Waffo, A.; Frolov, I.; Frolova, E.I. Hypervariable domains of nsP3 proteins of New World and Old World alphaviruses mediate formation of distinct, virus-specific protein complexes. J. Virol. 2013, 87, 1997–2010. [Google Scholar] [CrossRef] [PubMed]
- Vihinen, H.; Ahola, T.; Tuittila, M.; Merits, A.; Kääriäinen, L. Elimination of Phosphorylation Sites of Semliki Forest Virus Replicase Protein nsP3. J. Biol. Chem. 2001, 276, 5745–5752. [Google Scholar] [CrossRef] [PubMed]
- Foy, N.J.; Akhrymuk, M.; Shustov, A.V.; Frolova, E.I.; Frolov, I. Hypervariable domain of nonstructural protein nsP3 of Venezuelan equine encephalitis virus determines cell-specific mode of virus replication. J. Virol. 2013, 87, 7569–7584. [Google Scholar] [CrossRef] [PubMed]
- Li, G.P.; La Starza, M.W.; Hardy, W.R.; Strauss, J.H.; Rice, C.M. Phosphorylation of Sindbis virus nsP3 in vivo and in vitro. Virology 1990, 179, 416–427. [Google Scholar] [CrossRef]
- Dé, I.; Fata-Hartley, C.; Sawicki, S.G.; Sawicki, D.L. Functional Analysis of nsP3 Phosphoprotein Mutants of Sindbis Virus. J. Virol. 2003, 77, 13106–13116. [Google Scholar] [CrossRef] [PubMed]
- Vihinen, H.; Saarinen, J. Phosphorylation Site Analysis of Semliki Forest Virus Nonstructural Protein 3. J. Biol. Chem. 2000, 275, 27775–27783. [Google Scholar] [CrossRef] [PubMed]
- Amaya, M.; Voss, K.; Sampey, G.; Senina, S.; de la Fuente, C.; Mueller, C.; Calvert, V.; Kehn-Hall, K.; Carpenter, C.; Kashanchi, F.; et al. The role of IKKβ in Venezuelan equine encephalitis virus infection. PLoS ONE 2014, 9, e86745. [Google Scholar] [CrossRef] [PubMed]
- Saxton-Shaw, K.D.; Ledermann, J.P.; Borland, E.M.; Stovall, J.L.; Mossel, E.C.; Singh, A.J.; Wilusz, J.; Powers, A.M. O’nyong nyong Virus Molecular Determinants of Unique Vector Specificity Reside in Non-Structural Protein 3. PLoS Negl. Trop. Dis. 2013, 7, e1931. [Google Scholar] [CrossRef] [PubMed]
- Galbraith, S.E.; Sheahan, B.J.; Atkins, G.J. Deletions in the hypervariable domain of the nsP3 gene attenuate Semliki Forest virus virulence. J. Gen. Virol. 2006, 87, 937–947. [Google Scholar] [CrossRef] [PubMed]
- Park, E.; Griffin, D.E. The nsP3 macro domain is important for Sindbis virus replication in neurons and neurovirulence in mice. Virology 2009, 388, 305–314. [Google Scholar] [CrossRef] [PubMed]
- Liu, N.; Brown, D.T. Phosphorylation and dephosphorylation events play critical roles in Sindbis virus maturation. Virology 1993, 196, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.N.; Lee, H.; Hernandez, R.; Brown, D.T. Mutations in the endo domain of Sindbis virus glycoprotein E2 block phosphorylation, reorientation of the endo domain, and nucleocapsid binding. Virology 1996, 222, 236–246. [Google Scholar] [CrossRef] [PubMed]
- Fros, J.J.; Liu, W.J.; Prow, N.A.; Geertsema, C.; Ligtenberg, M.; Vanlandingham, D.L.; Schnettler, E.; Vlak, J.M.; Suhrbier, A.; Khromykh, A.A.; et al. Chikungunya virus nonstructural protein 2 inhibits type I/II interferon-stimulated JAK-STAT signaling. J. Virol. 2010, 84, 10877–10887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhrymuk, I.; Kulemzin, S.V.; Frolova, E.I. Evasion of the innate immune response: The Old World alphavirus nsP2 protein induces rapid degradation of Rpb1, a catalytic subunit of RNA polymerase II. J. Virol. 2012, 86, 7180–7191. [Google Scholar] [CrossRef] [PubMed]
- Rathore, A.P.S.; Ng, M.-L.; Vasudevan, S.G. Differential unfolded protein response during Chikungunya and Sindbis virus infection: CHIKV nsP4 suppresses eIF2α phosphorylation. Virol. J. 2013, 10, 36. [Google Scholar] [CrossRef] [PubMed]
- Fros, J.J.; Major, L.D.; Scholte, F.E.M.; Gardner, J.; van Hemert, M.J.; Suhrbier, A.; Pijlman, G.P. Chikungunya virus non-structural protein 2-mediated host shut-off disables the unfolded protein response. J. Gen. Virol. 2015, 96, 580–589. [Google Scholar] [CrossRef] [PubMed]
- Thaa, B.; Biasiotto, R.; Eng, K.; Neuvonen, M.; Götte, B.; Rheinemann, L.; Mutso, M.; Utt, A.; Varghese, F.; Balistreri, G.; et al. Differential PI3K-Akt-mTOR activation by Semliki Forest and chikungunya virus, dependent on nsP3 and connected to replication complex internalisation. J. Virol. 2015. [Google Scholar] [CrossRef] [PubMed]
- Law, L.M.J.; Everitt, J.C.; Beatch, M.D.; Holmes, C.F.B.; Hobman, T.C. Phosphorylation of Rubella Virus Capsid Regulates Its RNA Binding Activity and Virus Replication. J. Virol. 2003, 77, 1764–1771. [Google Scholar] [CrossRef] [PubMed]
- Claus, C.; Tzeng, W.-P.; Liebert, U.G.; Frey, T.K. Rubella virus-like replicon particles: Analysis of encapsidation determinants and non-structural roles of capsid protein in early post-entry replication. J. Gen. Virol. 2012, 93, 516–525. [Google Scholar] [CrossRef] [PubMed]
- Law, L.J.; Ilkow, C.S.; Tzeng, W.-P.; Rawluk, M.; Stuart, D.T.; Frey, T.K.; Hobman, T.C. Analyses of Phosphorylation Events in the Rubella Virus Capsid Protein: Role in Early Replication Events. J. Virol. 2006, 80, 6917–6925. [Google Scholar] [CrossRef] [PubMed]
- Biedenkopf, N.; Hartlieb, B.; Hoenen, T.; Becker, S. Phosphorylation of Ebola virus VP30 influences the composition of the viral nucleocapsid complex: Impact on viral transcription and replication. J. Biol. Chem. 2013, 288, 11165–11174. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.J.; Volchkova, V.A.; Raoul, H.; Alazard-Dany, N.; Reynard, O.; Volchkov, V.E. Role of VP30 phosphorylation in the Ebola virus replication cycle. J. Infect. Dis. 2011, 204 (Suppl. 3), S934–S940. [Google Scholar] [CrossRef] [PubMed]
- Ilinykh, P.A.; Tigabu, B.; Ivanov, A.; Ammosova, T.; Obukhov, Y.; Garron, T.; Kumari, N.; Kovalskyy, D.; Platonov, M.O.; Naumchik, V.S.; et al. Role of protein phosphatase 1 in dephosphorylation of Ebola virus VP30 protein and its targeting for the inhibition of viral transcription. J. Biol. Chem. 2014, 289, 22723–22738. [Google Scholar] [CrossRef] [PubMed]
- Modrof, J.; Mühlberger, E.; Klenk, H.-D.; Becker, S. Phosphorylation of VP30 impairs ebola virus transcription. J. Biol. Chem. 2002, 277, 33099–33104. [Google Scholar] [CrossRef] [PubMed]
- García, M.; Cooper, A.; Shi, W.; Bornmann, W.; Carrion, R.; Kalman, D.; Nabel, G.J. Productive replication of Ebola virus is regulated by the c-Abl1 tyrosine kinase. Sci. Transl. Med. 2012, 4, 123ra24. [Google Scholar] [CrossRef] [PubMed]
- Hale, B.G.; Randall, R.E.; Ortín, J.; Jackson, D. The multifunctional NS1 protein of influenza A viruses. J. Gen. Virol. 2008, 89, 2359–2376. [Google Scholar] [CrossRef] [PubMed]
- Hsiang, T.-Y.; Zhou, L.; Krug, R.M. Roles of the Phosphorylation of Specific Serines and Threonines in the NS1 Protein of Human Influenza A Viruses. J. Virol. 2012, 86, 10370–10376. [Google Scholar] [CrossRef]
- Hale, B.G.; Knebel, A.; Botting, C.H.; Galloway, C.S.; Precious, B.L.; Jackson, D.; Elliott, R.M.; Randall, R.E. CDK/ERK-mediated phosphorylation of the human influenza A virus NS1 protein at threonine-215. Virology 2009, 383, 6–11. [Google Scholar] [CrossRef] [PubMed]
- Hirata, N.; Suizu, F.; Matsuda-Lennikov, M.; Edamura, T.; Bala, J.; Noguchi, M. Inhibition of Akt kinase activity suppresses entry and replication of influenza virus. Biochem. Biophys. Res. Commun. 2014, 450, 891–898. [Google Scholar] [CrossRef] [PubMed]
- Halder, U.C.; Bhowmick, R.; Roy Mukherjee, T.; Nayak, M.K.; Chawla-Sarkar, M. Phosphorylation Drives an Apoptotic Protein to Activate Antiapoptotic Genes. J. Biol. Chem. 2013, 288, 14554–14568. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.; Zhao, Z.; Bi, Y.; Sun, L.; Liu, X.; Liu, W. Tyrosine 132 phosphorylation of influenza A virus M1 protein is crucial for virus replication by controlling the nuclear import of M1. J. Virol. 2013, 87, 6182–6191. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, E.C.; Denham, E.M.; Thomas, B.; Trudgian, D.C.; Hester, S.S.; Ridlova, G.; York, A.; Turrell, L.; Fodor, E. Mapping the Phosphoproteome of Influenza A and B Viruses by Mass Spectrometry. PLoS Pathog 2012, 8, e1002993. [Google Scholar] [CrossRef] [PubMed]
- Arrese, M.; Portela, A. Serine 3 is critical for phosphorylation at the N-terminal end of the nucleoprotein of influenza virus A/Victoria/3/75. J. Virol. 1996, 70, 3385–3391. [Google Scholar] [PubMed]
- Chenavas, S.; Estrozi, L.F.; Slama-Schwok, A.; Delmas, B.; Di Primo, C.; Baudin, F.; Li, X.; Crépin, T.; Ruigrok, R.W.H. Monomeric Nucleoprotein of Influenza A Virus. PLoS Pathog. 2013, 9, e1003275. [Google Scholar] [CrossRef] [PubMed]
- Zheng, W.; Li, J.; Wang, S.; Cao, S.; Jiang, J.; Chen, C.; Ding, C.; Qin, C.; Ye, X.; Gao, G.F.; et al. Phosphorylation controls the nuclear-cytoplasmic shuttling of influenza a virus nucleoprotein. J. Virol. 2015, 89, 5822–5834. [Google Scholar] [CrossRef]
- Durrant, M.G.; Eggett, D.L.; Busath, D.D. Investigation of a recent rise of dual amantadine-resistance mutations in the influenza A M2 sequence. BMC Genet. 2015, 16 (Suppl. 2), S3. [Google Scholar] [CrossRef] [PubMed]
- Wade, R.C. “Flu” and structure-based drug design. Struct. Lond. Engl. 1993 1997, 5, 1139–1145. [Google Scholar] [CrossRef]
- Ison, M.G. Antivirals and resistance: Influenza virus. Curr. Opin. Virol. 2011, 1, 563–573. [Google Scholar] [CrossRef] [PubMed]
- Johansen, L.M.; DeWald, L.E.; Shoemaker, C.J.; Hoffstrom, B.G.; Lear-Rooney, C.M.; Stossel, A.; Nelson, E.; Delos, S.E.; Simmons, J.A.; Grenier, J.M.; et al. A screen of approved drugs and molecular probes identifies therapeutics with anti-Ebola virus activity. Sci. Transl. Med. 2015, 7, 290ra89. [Google Scholar] [PubMed]
- Madrid, P.B.; Chopra, S.; Manger, I.D.; Gilfillan, L.; Keepers, T.R.; Shurtleff, A.C.; Green, C.E.; Iyer, L.V.; Dilks, H.H.; Davey, R.A.; et al. A Systematic Screen of FDA-Approved Drugs for Inhibitors of Biological Threat Agents. PLoS ONE 2013, 8, e60579. [Google Scholar] [CrossRef] [PubMed]
- Kouznetsova, J.; Sun, W.; Martínez-Romero, C.; Tawa, G.; Shinn, P.; Chen, C.Z.; Schimmer, A.; Sanderson, P.; McKew, J.C.; Zheng, W.; et al. Identification of 53 compounds that block Ebola virus-like particle entry via a repurposing screen of approved drugs. Emerg. Microbes Infect. 2014, 3, e84. [Google Scholar] [CrossRef] [PubMed]
- Amaya, M.; Keck, F.; Lindquist, M.; Voss, K.; Scavone, L.; Kehn-Hall, K.; Roberts, B.; Bailey, C.; Schmaljohn, C.; Narayanan, A. The ubiquitin proteasome system plays a role in venezuelan equine encephalitis virus infection. PLoS ONE 2015, 10, e0124792. [Google Scholar] [CrossRef]
- Voss, K.; Amaya, M.; Mueller, C.; Roberts, B.; Kehn-Hall, K.; Bailey, C.; Petricoin, E.; Narayanan, A. Inhibition of host extracellular signal-regulated kinase (ERK) activation decreases new world alphavirus multiplication in infected cells. Virology 2014, 468–470, 490–503. [Google Scholar] [CrossRef] [PubMed]
- Chung, D.-H.; Jonsson, C.B.; Tower, N.A.; Chu, Y.-K.; Sahin, E.; Golden, J.E.; Noah, J.W.; Schroeder, C.E.; Sotsky, J.B.; Sosa, M.I.; et al. Discovery of a novel compound with anti-venezuelan equine encephalitis virus activity that targets the nonstructural protein 2. PLoS Pathog. 2014, 10, e1004213. [Google Scholar] [CrossRef] [PubMed]
- Panchal, R.G.; Kota, K.P.; Spurgers, K.B.; Ruthel, G.; Tran, J.P.; Boltz, R.C.D.; Bavari, S. Development of high-content imaging assays for lethal viral pathogens. J. Biomol. Screen. 2010, 15, 755–765. [Google Scholar] [CrossRef] [PubMed]
- Cheung, Y.Y.; Chen, K.C.; Chen, H.; Seng, E.K.; Chu, J.J.H. Antiviral activity of lanatoside C against dengue virus infection. Antiviral Res. 2014, 111, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Quintavalle, M.; Sambucini, S.; Summa, V.; Orsatti, L.; Talamo, F.; de Francesco, R.; Neddermann, P. Hepatitis C virus NS5A is a direct substrate of casein kinase I-alpha, a cellular kinase identified by inhibitor affinity chromatography using specific NS5A hyperphosphorylation inhibitors. J. Biol. Chem. 2007, 282, 5536–5544. [Google Scholar] [CrossRef] [PubMed]
- Thorne, C.A.; Hanson, A.J.; Schneider, J.; Tahinci, E.; Orton, D.; Cselenyi, C.S.; Jernigan, K.K.; Meyers, K.C.; Hang, B.I.; Waterson, A.G.; et al. Small-molecule inhibition of Wnt signaling through activation of casein kinase 1α. Nat. Chem. Biol. 2010, 6, 829–836. [Google Scholar] [CrossRef] [PubMed]
- Shum, D.; Smith, J.L.; Hirsch, A.J.; Bhinder, B.; Radu, C.; Stein, D.A.; Nelson, J.A.; Früh, K.; Djaballah, H. High-content assay to identify inhibitors of dengue virus infection. Assay Drug Dev. Technol. 2010, 8, 553–570. [Google Scholar] [CrossRef] [PubMed]
- Panchal, R.G.; Reid, S.P.; Tran, J.P.; Bergeron, A.A.; Wells, J.; Kota, K.P.; Aman, J.; Bavari, S. Identification of an antioxidant small-molecule with broad-spectrum antiviral activity. Antiviral Res. 2012, 93, 23–29. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Keck, F.; Ataey, P.; Amaya, M.; Bailey, C.; Narayanan, A. Phosphorylation of Single Stranded RNA Virus Proteins and Potential for Novel Therapeutic Strategies. Viruses 2015, 7, 5257-5273. https://doi.org/10.3390/v7102872
Keck F, Ataey P, Amaya M, Bailey C, Narayanan A. Phosphorylation of Single Stranded RNA Virus Proteins and Potential for Novel Therapeutic Strategies. Viruses. 2015; 7(10):5257-5273. https://doi.org/10.3390/v7102872
Chicago/Turabian StyleKeck, Forrest, Pouya Ataey, Moushimi Amaya, Charles Bailey, and Aarthi Narayanan. 2015. "Phosphorylation of Single Stranded RNA Virus Proteins and Potential for Novel Therapeutic Strategies" Viruses 7, no. 10: 5257-5273. https://doi.org/10.3390/v7102872