
Identification of a Novel Human Rhinovirus C Type by Antibody Capture VIDISCA-454


 ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Clinical Samples
2.2. Ethical Approval
2.3. Antibody Capture
2.4. VIDISCA and Roche Titanium-454 Sequencing
2.5. Xcompare2 Pipeline
2.6. Sequence Analysis
2.7. Full-Length Genome Sequencing
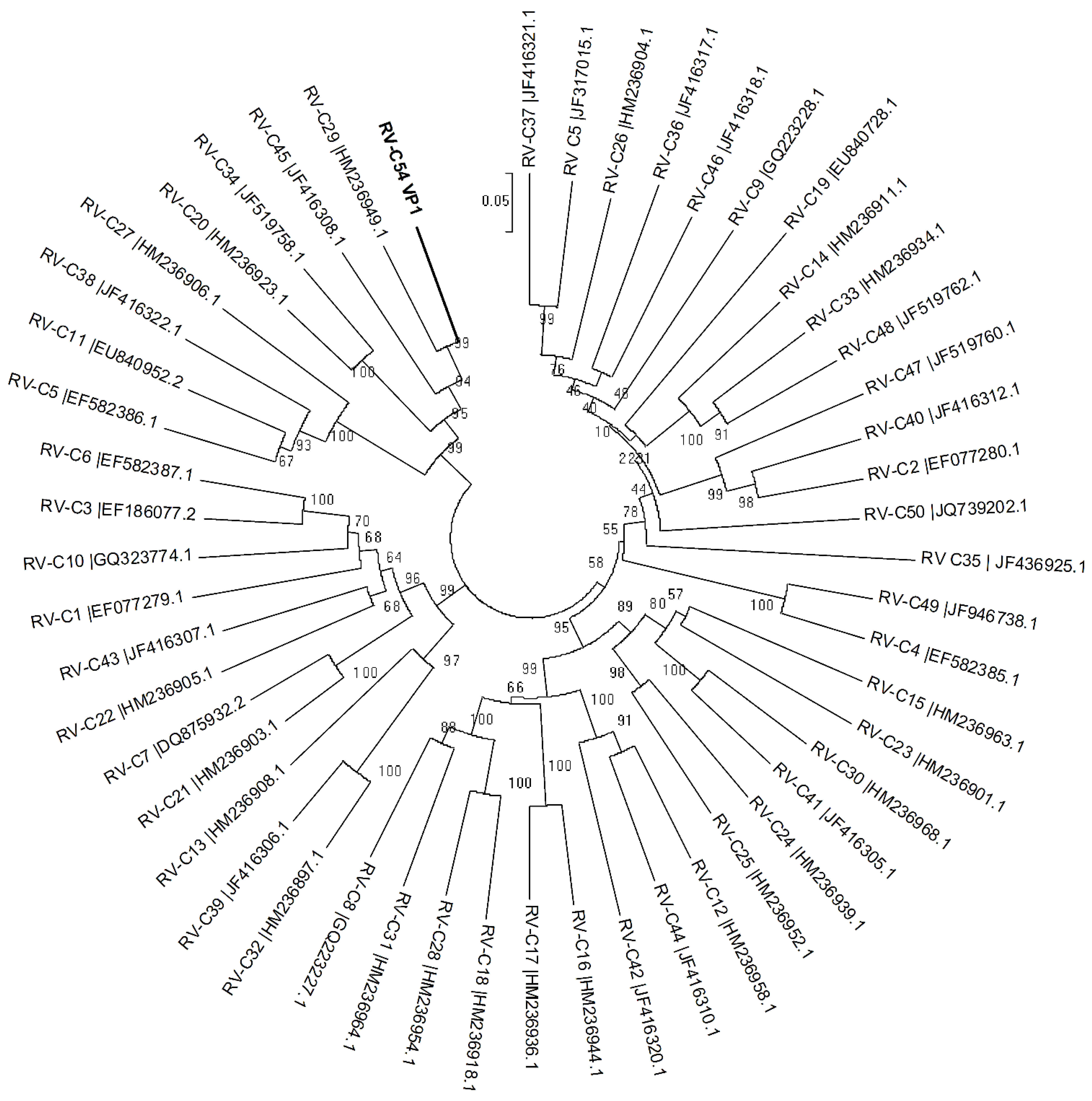
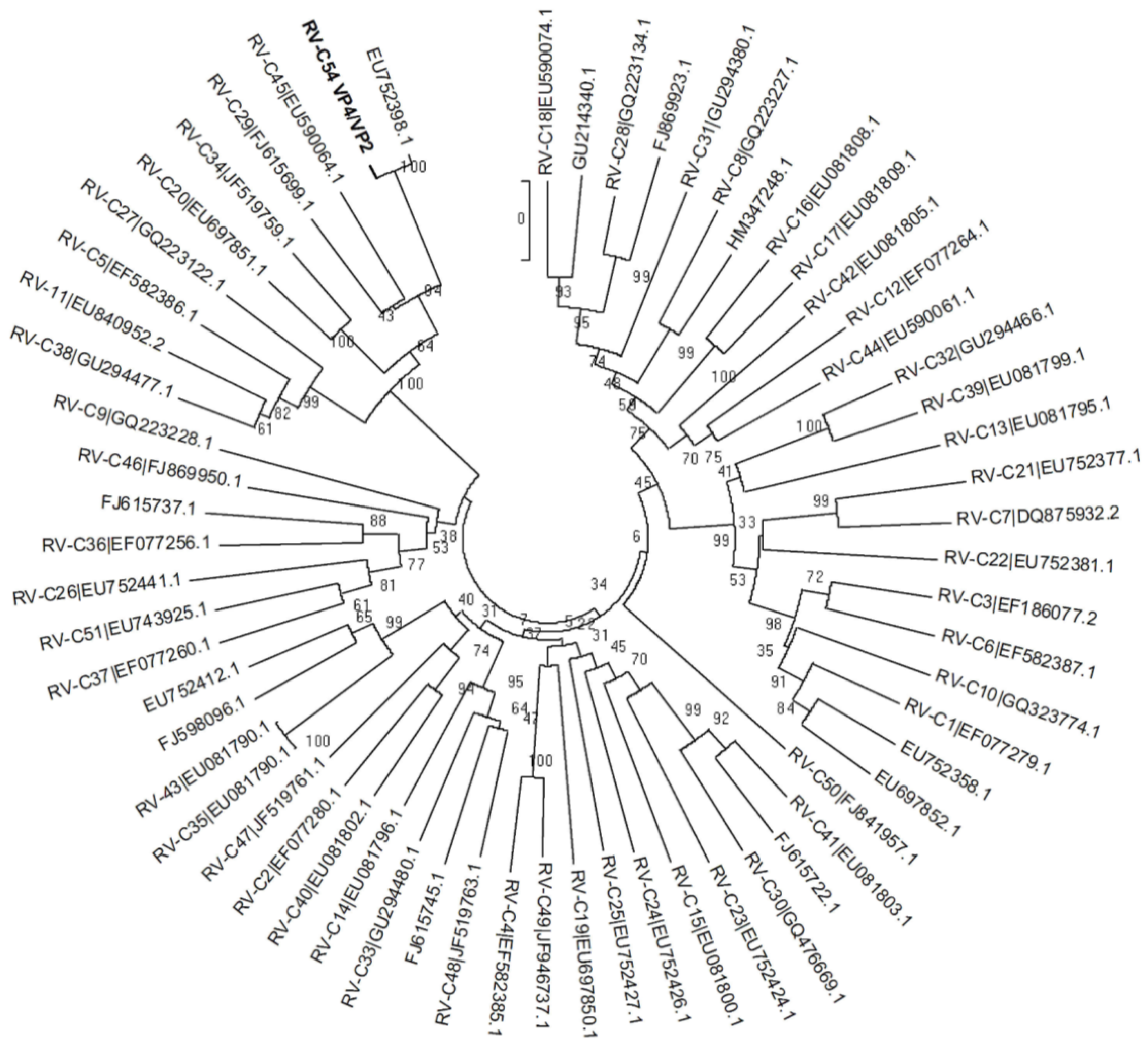
2.8. Phylogenetic Analysis
2.9. Virus Genome Characterization
3. Results
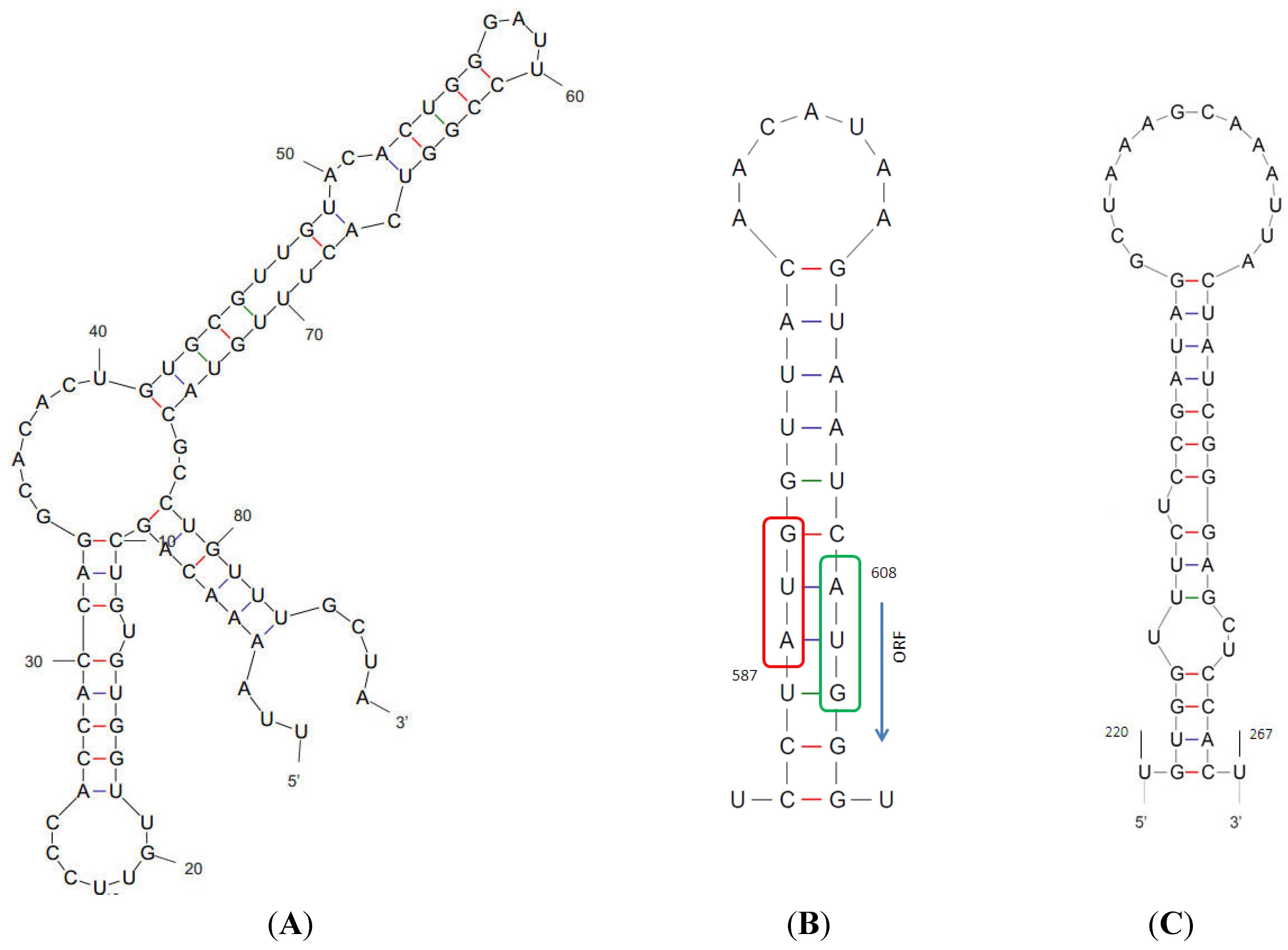

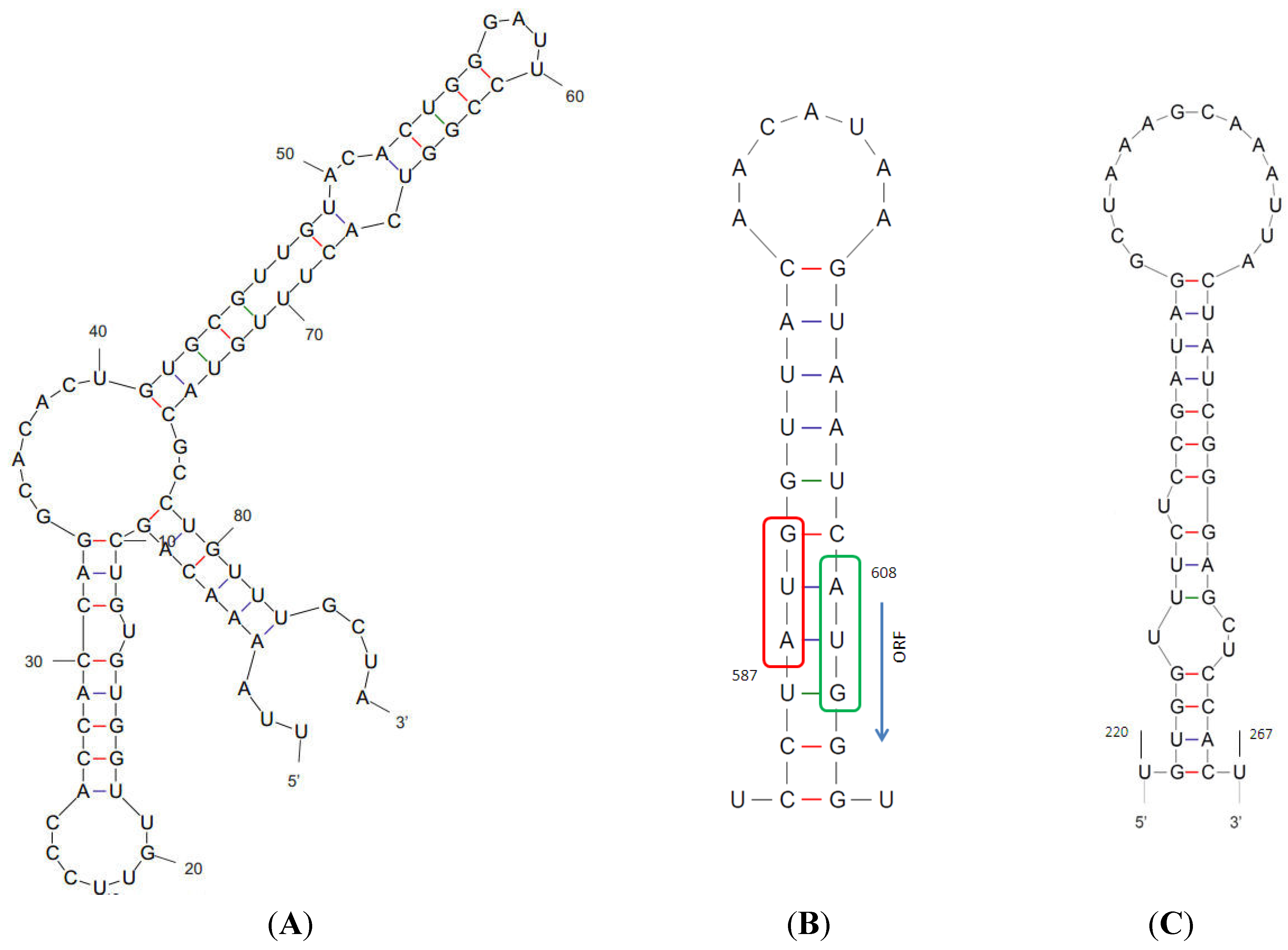{kind=link}
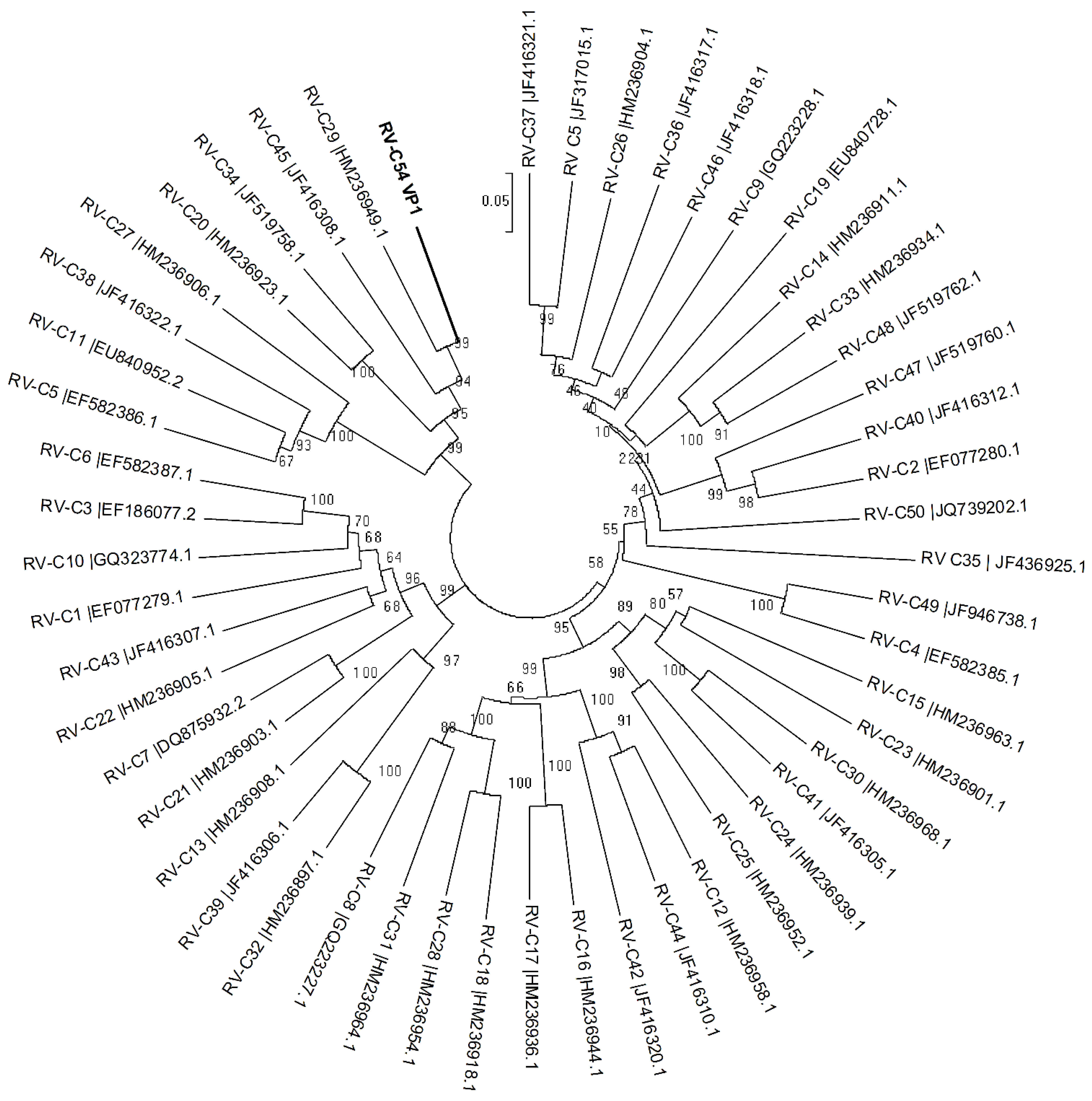{kind=link}
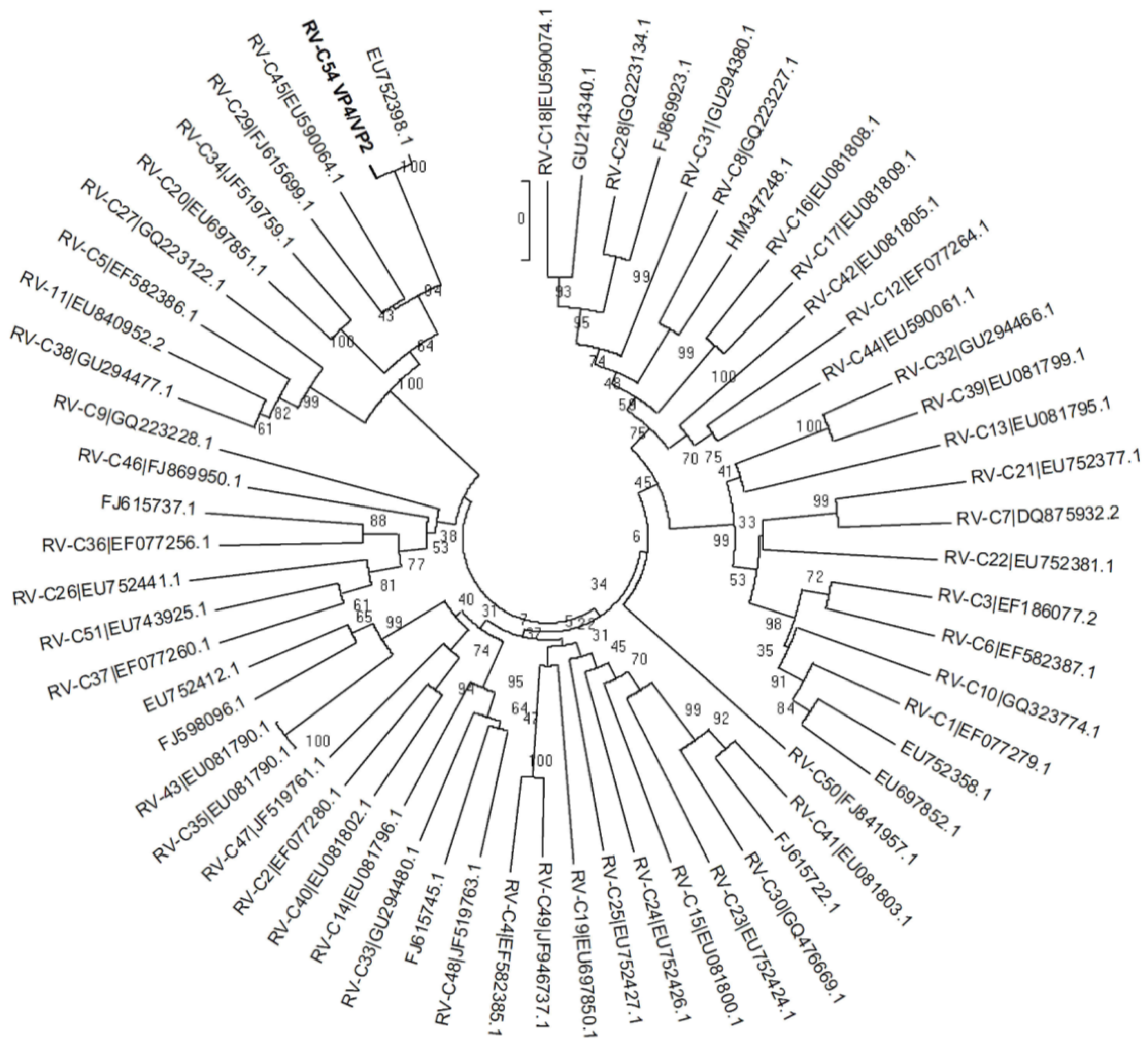{kind=link}
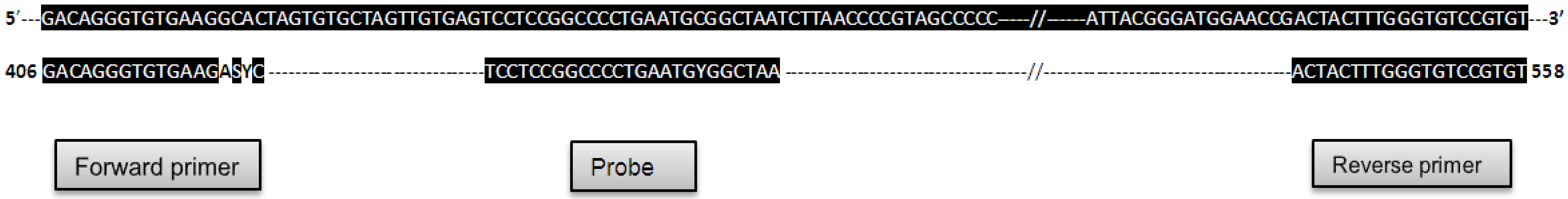
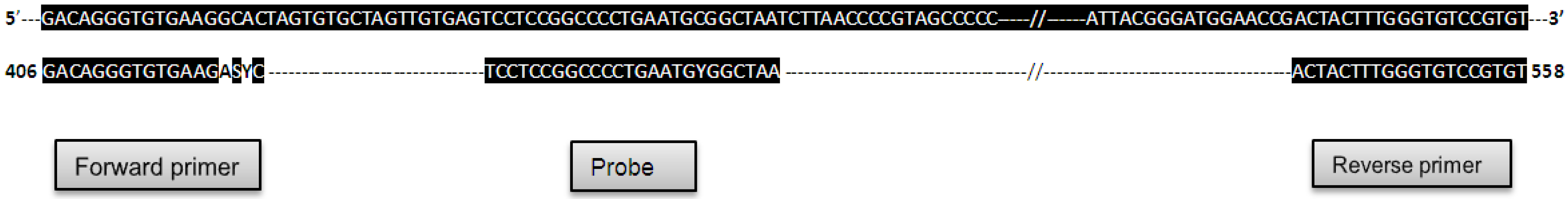{kind=link}
| Position | Protease | Sequence |
|---|---|---|
| 841 | 2A | DSIKTA * GPSDL |
| 330 | 3C | SNRTQ * GLPV |
| 537 | 3C | VQSGQ * GAIL |
| 983 | 3C | LATTQ * GPIT |
| 1329 | 3C | LVIRQ * GFKT |
| 1407 | 3C | NAIFQ * GLGS |
| 1482 | 3C | LCMTQ * GAYT |
| 1504 | 3C | RAVVQ * GPQH |
| 1687 | 3C | FVESQ * GEII |
4. Discussion
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Regamey, N.; Kaiser, L.; Roiha, H.L.; Deffernez, C.; Kuehni, C.E.; Latzin, P.; Aebi, C.; Frey, U. Viral etiology of acute respiratory infections with cough in infancy—A community-based birth cohort study. Pediatr. Infect. Dis. J. 2008, 27, 100–105. [Google Scholar] [PubMed]
- Tsuchiya, L.R.R.V.; Costa, L.M.D.; Raboni, S.M.; Nogueira, M.B.; Pereira, L.A.; Rotta, I.; Takahashi, G.R.A.; Coelho, M.; Siqueira, M.M. Viral respiratory infection in Curitiba, Southern Brazil. J. Infect. 2005, 51, 401–407. [Google Scholar] [CrossRef] [PubMed]
- Fowlkes, A.; Giorgi, A.; Erdman, D.; Temte, J.; Goodin, K.; Di, L.S.; Sun, Y.; Martin, K.; Feist, M.; Linz, R.; et al. Viruses Associated With Acute Respiratory Infections and Influenza-like Illness Among Outpatients From the Influenza Incidence Surveillance Project, 2010–2011. J. Infect. Dis. 2014. [Google Scholar] [CrossRef]
- Pyrc, K.; Berkhout, B.; van der Hoek, L. Identification of new human coronaviruses. Expert Rev. Anti Infect. Ther. 2007, 5, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Tan le, V.; van Doorn, H.R.; Nghia, H.D.; Chau, T.T.; Tu le, T.P.; de Vries, M.; Canuti, M.; Deijs, M.; Jebbink, M.F.; Baker, S.; et al. Identification of a new cyclovirus in cerebrospinal fluid of patients with acute central nervous system infections. MBio 2013, 4. [Google Scholar] [CrossRef]
- Canuti, M.; Deijs, M.; Jazaeri Farsani, S.M.; Holwerda, M.; Jebbink, M.F.; de Vries, M.; van Vugt, S.; Brugman, C.; Verheij, T.; Lammens, C.; et al. Metagenomic analysis of a sample from a patient with respiratory tract infection reveals the presence of a gamma-papillomavirus. Front. Microbiol. 2014, 5, 347. [Google Scholar] [CrossRef] [PubMed]
- Canuti, M.; Eis-Huebinger, A.M.; Deijs, M.; de Vries, M.; Drexler, J.F.; Oppong, S.K.; Muller, M.A.; Klose, S.M.; Wellinghausen, N.; Cottontail, V.M.; et al. Two novel parvoviruses in frugivorous New and Old World bats. PLoS One 2011, 6, e29140. [Google Scholar] [CrossRef] [PubMed]
- De Vries, M.; Pyrc, K.; Berkhout, R.; Vermeulen-Oost, W.; Dijkman, R.; Jebbink, M.F.; Bruisten, S.; Berkhout, B.; van der Hoek, L. Human parechovirus type 1, 3, 4, 5, and 6 detection in picornavirus cultures. J. Clin. Microbiol. 2008, 46, 759–762. [Google Scholar] [CrossRef] [PubMed]
- De Vries, M.; Deijs, M.; Canuti, M.; van Schaik, B.D.; Faria, N.R.; van de Garde, M.D.; Jachimowski, L.C.; Jebbink, M.F.; Jakobs, M.; Luyf, A.C.; et al. A sensitive assay for virus discovery in respiratory clinical samples. PLoS One 2011, 6, e16118. [Google Scholar]
- Jazaeri Farsani, S.M.; Jebbink, M.F.; Deijs, M.; Canuti, M.; van Dort, K.A.; Bakker, M.; Grady, B.P.; Prins, M.; van Hemert, F.J.; Kootstra, N.A.; et al. Identification of a new genotype of Torque Teno Mini virus. Virol. J. 2013, 10, 323. [Google Scholar] [CrossRef] [PubMed]
- Jazaeri Farsani, S.M.; Oude Munnink, B.B.; Deijs, M.; Canuti, M.; van der Hoek, L. Metagenomics in virus discovery. VOXS 2013, 8, 193–194. [Google Scholar]
- Galmes, J.; Li, Y.; Rajoharison, A.; Ren, L.; Dollet, S.; Richard, N.; Vernet, G.; Javouhey, E.; Wang, J.; Telles, J.N.; et al. Potential implication of new torque teno mini viruses in parapneumonic empyema in children. Eur. Respir. J. 2013, 42, 470–479. [Google Scholar] [CrossRef] [PubMed]
- Mokili, J.L.; Dutilh, B.E.; Lim, Y.W.; Schneider, B.S.; Taylor, T.; Haynes, M.R.; Metzgar, D.; Myers, C.A.; Blair, P.J.; Nosrat, B.; et al. Identification of a novel human papillomavirus by metagenomic analysis of samples from patients with febrile respiratory illness. PLoS One 2013, 8, e58404. [Google Scholar] [CrossRef] [PubMed]
- Oude Munnink, B.B.; Jazaeri Farsani, S.M.; Deijs, M.; Jonkers, J.; Verhoeven, J.T.; Ieven, M.; Goossens, H.; de Jong, M.D.; Berkhout, B.; Loens, K.; et al. Autologous antibody capture to enrich immunogenic viruses for viral discovery. PLoS One 2013, 8, e78454. [Google Scholar] [CrossRef] [PubMed]
- Waman, V.P.; Kolekar, P.S.; Kale, M.M.; Kulkarni-Kale, U. Population structure and evolution of rhinoviruses. PLoS One 2014, 9, e88981. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.E.; Lamson, D.M.; St, G.K.; Walsh, T.J. Human rhinoviruses. Clin. Microbiol. Rev. 2013, 26, 135–162. [Google Scholar] [CrossRef] [PubMed]
- McIntyre, C.L.; Knowles, N.J.; Simmonds, P. Proposals for the classification of human rhinovirus species A, B and C into genotypically assigned types. J. Gen. Virol. 2013, 94, 1791–1806. [Google Scholar] [CrossRef] [PubMed]
- Bochkov, Y.A.; Palmenberg, A.C.; Lee, W.M.; Rathe, J.A.; Amineva, S.P.; Sun, X.; Pasic, T.R.; Jarjour, N.N.; Liggett, S.B.; Gern, J.E. Molecular modeling, organ culture and reverse genetics for a newly identified human rhinovirus C. Nat. Med. 2011, 17, 627–632. [Google Scholar] [CrossRef] [PubMed]
- Tapparel, C.; Sobo, K.; Constant, S.; Huang, S.; Van, B.S.; Kaiser, L. Growth and characterization of different human rhinovirus C types in three-dimensional human airway epithelia reconstituted in vitro. Virology 2013, 446, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Hao, W.; Bernard, K.; Patel, N.; Ulbrandt, N.; Feng, H.; Svabek, C.; Wilson, S.; Stracener, C.; Wang, K.; Suzich, J.; et al. Infection and propagation of human rhinovirus C in human airway epithelial cells. J. Virol. 2012, 86, 13524–13532. [Google Scholar] [CrossRef] [PubMed]
- Ashraf, S.; Brockman-Schneider, R.; Bochkov, Y.A.; Pasic, T.R.; Gern, J.E. Biological characteristics and propagation of human rhinovirus-C in differentiated sinus epithelial cells. Virology 2013, 436, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Picornaviridae Website. Available online: http://www.picornaviridae.com/enterovirus/rv-c/rv-c.htm (accessed on 21 March 2014).
- Zlateva, K.T.; de Vries, J.J.; Coenjaerts, F.E.; van Loon, A.M.; Verheij, T.; Little, P.; Butler, C.C.; Goossens, H.; Ieven, M.; Claas, E.C. Prolonged shedding of rhinovirus and re-infection in adults with respiratory tract illness. Eur. Respir. J. 2014, 44, 169–177. [Google Scholar] [CrossRef] [PubMed]
- Loens, K.; van Loon, A.M.; Coenjaerts, F.; van, A.Y.; Goossens, H.; Wallace, P.; Claas, E.J.; Ieven, M. Performance of different mono- and multiplex nucleic acid amplification tests on a multipathogen external quality assessment panel. J. Clin. Microbiol. 2012, 50, 977–987. [Google Scholar] [CrossRef] [PubMed]
- Boom, R.; Sol, C.J.; Salimans, M.M.; Jansen, C.L.; Wertheim-Van Dillen, P.M.; van der Noordaa, J. Rapid and simple method for purification of nucleic acids. J. Clin. Microbiol. 1990, 28, 495–503. [Google Scholar] [PubMed]
- Endoh, D.; Mizutani, T.; Kirisawa, R.; Maki, Y.; Saito, H.; Kon, Y.; Morikawa, S.; Hayashi, M. Species-independent detection of RNA virus by representational difference analysis using non-ribosomal hexanucleotides for reverse transcription. Nucleic Acids Res. 2005, 33, e65. [Google Scholar] [CrossRef] [PubMed]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinform. 2009, 10, 421. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed]
- Benson, D.A.; Cavanaugh, M.; Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2013, 41, D36–D42. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Huson, D.H.; Mitra, S.; Ruscheweyh, H.J.; Weber, N.; Schuster, S.C. Integrative analysis of environmental sequences using MEGAN4. Genome Res. 2011, 21, 1552–1560. [Google Scholar] [CrossRef] [PubMed]
- Cotten, M.; Oude Munnink, B.B.; Canuti, M.; Deijs, M.; Watson, S.J.; Kellam, P.; van der Hoek, L. Full genome virus detection in fecal samples using sensitive nucleic Acid preparation, deep sequencing, and a novel iterative sequence classification algorithm. PLoS One 2014, 9, e93269. [Google Scholar] [CrossRef] [PubMed]
- Tamura, K.; Stecher, G.; Peterson, D.; Filipski, A.; Kumar, S. MEGA6: Molecular Evolutionary Genetics Analysis version 6.0. Mol. Biol. Evol. 2013, 30, 2725–2729. [Google Scholar] [CrossRef] [PubMed]
- Blom, N.; Hansen, J.; Blaas, D.; Brunak, S. Cleavage site analysis in picornaviral polyproteins: Discovering cellular targets by neural networks. Protein Sci. 1996, 5, 2203–2216. [Google Scholar] [CrossRef] [PubMed]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef] [PubMed]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [PubMed]
- Martin, D.; Rybicki, E. RDP: Detection of recombination amongst aligned sequences. Bioinformatics 2000, 16, 562–563. [Google Scholar] [CrossRef] [PubMed]
- GRACE Website. Available online: http://www.grace-lrti.org (accessed on 24 November 2014).
- Palmenberg, A.C.; Spiro, D.; Kuzmickas, R.; Wang, S.; Djikeng, A.; Rathe, J.A.; Fraser-Liggett, C.M.; Liggett, S.B. Sequencing and analyses of all known human rhinovirus genomes reveal structure and evolution. Science 2009, 324, 55–59. [Google Scholar] [CrossRef] [PubMed]
- Bochkov, Y.A.; Gern, J.E. Clinical and molecular features of human rhinovirus C. Microbes Infect. 2012, 14, 485–494. [Google Scholar] [CrossRef] [PubMed]
- Palmenberg, A.C.; Rathe, J.A.; Liggett, S.B. Analysis of the complete genome sequences of human rhinovirus. J. Allergy Clin. Immunol. 2010, 125, 1190–1199. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jazaeri Farsani, S.M.; Oude Munnink, B.B.; Canuti, M.; Deijs, M.; Cotten, M.; Jebbink, M.F.; Verhoeven, J.; Kellam, P.; Loens, K.; Goossens, H.; et al. Identification of a Novel Human Rhinovirus C Type by Antibody Capture VIDISCA-454. Viruses 2015, 7, 239-251. https://doi.org/10.3390/v7010239
Jazaeri Farsani SM, Oude Munnink BB, Canuti M, Deijs M, Cotten M, Jebbink MF, Verhoeven J, Kellam P, Loens K, Goossens H, et al. Identification of a Novel Human Rhinovirus C Type by Antibody Capture VIDISCA-454. Viruses. 2015; 7(1):239-251. https://doi.org/10.3390/v7010239
Chicago/Turabian StyleJazaeri Farsani, Seyed Mohammad, Bas B. Oude Munnink, Marta Canuti, Martin Deijs, Matthew Cotten, Maarten F. Jebbink, Joost Verhoeven, Paul Kellam, Katherine Loens, Herman Goossens, and et al. 2015. "Identification of a Novel Human Rhinovirus C Type by Antibody Capture VIDISCA-454" Viruses 7, no. 1: 239-251. https://doi.org/10.3390/v7010239