The Role of Ubiquitin and Ubiquitin-Like Modification Systems in Papillomavirus Biology

Department of Microbial Pathogenesis and Immunology, College of Medicine, Texas A&M Health Science Center, 8447 HWY 47, Bryan, TX 77807, USA
Viruses 2014, 6(9), 3584-3611; https://doi.org/10.3390/v6093584
Submission received: 22 August 2014
/
Revised: 17 September 2014
/
Accepted: 18 September 2014
/
Published: 24 September 2014
(This article belongs to the Special Issue Viruses and the Ubiquitin/Proteasome System)
{kind=link}
{kind=link}
Abstract
:Human papillomaviruses (HPVs) are small DNA viruses that are important etiological agents of a spectrum of human skin lesions from benign to malignant. Because of their limited genome coding capacity they express only a small number of proteins, only one of which has enzymatic activity. Additionally, the HPV productive life cycle is intimately tied to the epithelial differentiation program and they must replicate in what are normally non-replicative cells, thus, these viruses must reprogram the cellular environment to achieve viral reproduction. Because of these limitations and needs, the viral proteins have evolved to co-opt cellular processes primarily through protein-protein interactions with critical host proteins. The ubiquitin post-translational modification system and the related ubiquitin-like modifiers constitute a widespread cellular regulatory network that controls the levels and functions of thousands of proteins, making these systems an attractive target for viral manipulation. This review describes the interactions between HPVs and the ubiquitin family of modifiers, both to regulate the viral proteins themselves and to remodel the host cell to facilitate viral survival and reproduction.
1. Introduction –— Human Papillomaviruses
The human papillomaviruses (HPVs) are important pathogens with clinical manifestations ranging from benign to malignant lesions [1,2]. Over 170 different types have been sequenced with many more isolates identified but not yet classified [3]. The existing HPVs are classified into five genera, Alphapapillomaviruses, Betapapillomaviruses, Gammapapillomaviruses, Mupapillomaviruses, and Nupapillomaviruses, with the majority falling into genus alpha and genus beta. Most of the alpha group viruses infect mucosal epithelium while the beta types exhibit tropism primarily for cutaneous epithelium. Among the mucosal types a functional distinction exists between types that are highly associated with the development of cervical cancer, the so-called high-risk types, and the remaining nononcogenic, low-risk types. Because of their contribution to the development of cervical cancer, research has often focused heavily on the high risk types, particularly the most prevalent types, 16 and 18. More recently, a growing body of evidence linking cutaneous beta type HPVs to skin cancers [4] has led to increased examination of the biology and molecular biology of the beta HPVs. What has arisen from studies across multiple HPV types is that there are both common and type-specific features reflecting the great diversity of types and their biological niches [5,6,7].
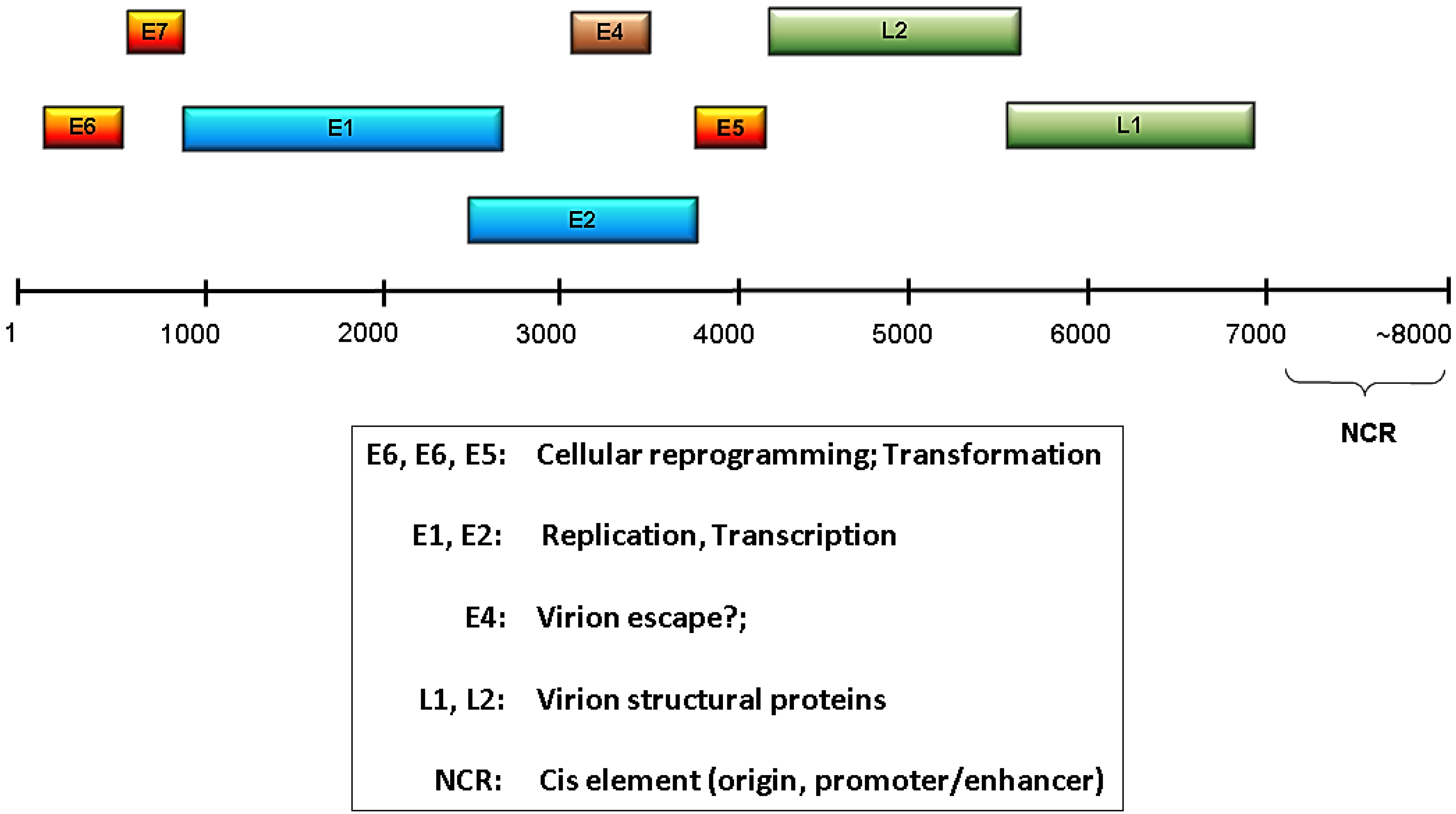
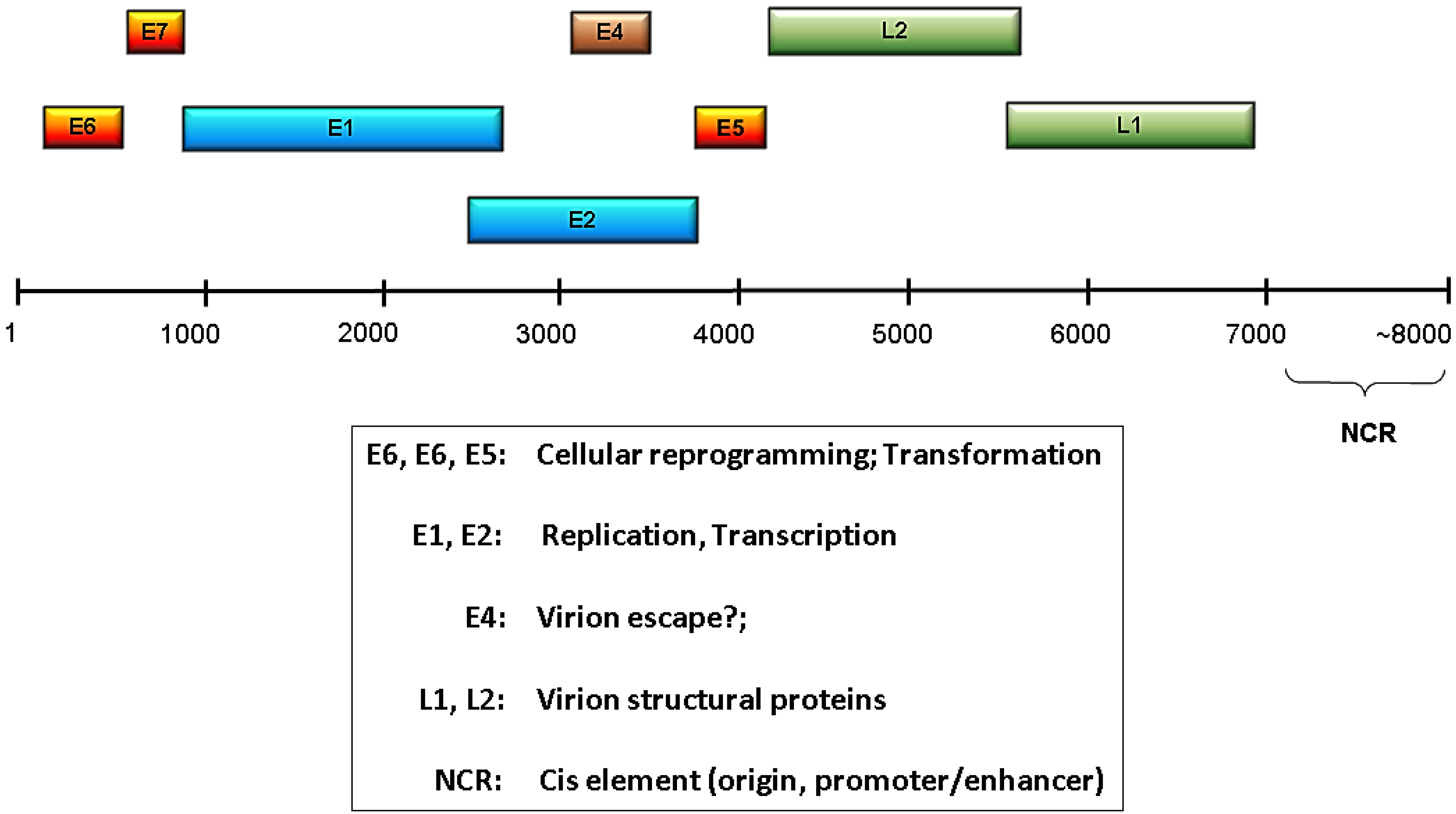
A common feature of all papillomaviruses is a small, double-stranded, circular genome of about 8000 base pairs containing a single origin of replication [8]. These small genomes encode a limited number of proteins that are classified as either early or late depending on their time of expression. The early region of the genome contains open reading frames (ORFs) E1, E2, E4, E5, E6, and E7, and the early protein functions include viral genome replication, transcriptional regulation, and modulation of the host cell environment to favor viral replication and/or persistence (Figure 1). E6 and E7 gene products are particularly important for reprogramming the host cell, and for the high risk HPVs their E6 and E7 products constitute the predominant oncoproteins. There are only two late proteins, L1 and L2, and these are the viral capsid structural proteins [9,10]. Given the very limited repertoire of viral proteins, the HPVs have evolved to be highly dependent upon the host cells to supply needed functions and enzymatic activities. Much more detailed information about HPV proteins and their functions can be found in several recent reviews [11,12,13,14,15,16].
Figure 1.
HPV genome organization and function. Shown is a linearized HPV genome with the relative genomic locations of the early (E) and late (L) ORFs indicated. The NCR is the noncoding region that contains the viral replication origin and transcriptional regulatory elements. Functions associated with the various ORFs are listed in the box below the genome.
Figure 1.
HPV genome organization and function. Shown is a linearized HPV genome with the relative genomic locations of the early (E) and late (L) ORFs indicated. The NCR is the noncoding region that contains the viral replication origin and transcriptional regulatory elements. Functions associated with the various ORFs are listed in the box below the genome.
HPVs have a complex lifecycle that is intimately connected to the differentiation program of skin [17]. Establishment of infection requires viral entry into the replicative basal cells where the viral genome can persist episomally in the host cell nucleus [18]. Subsequent stages of the productive lifecycle occur in the subrabasal layers as differentiation progresses. Normally, these differentiating cells are nondividing and would not provide the replicative machinery and S phase environment required by the HPVs for their DNA replication, thus, a requirement of these viruses is to push the cells back into a proliferative state [19]. Additionally, HPVs must ensure appropriate expression of the viral proteins at different differentiation stages, effective assembly of new virions, and avoidance of host defense mechanisms [20]. Surprisingly, these viruses have only one enzymatically active protein, E1, which has helicase activity for viral genome replication [21,22,23]. Consequently, the actions of the other viral proteins are mediated entirely through protein-protein interaction, and this review will focus on how the HPV proteins interact with the ubiquitin and related ubiquitin-like modification systems.
2. Overview of the Ubiquitin Superfamily
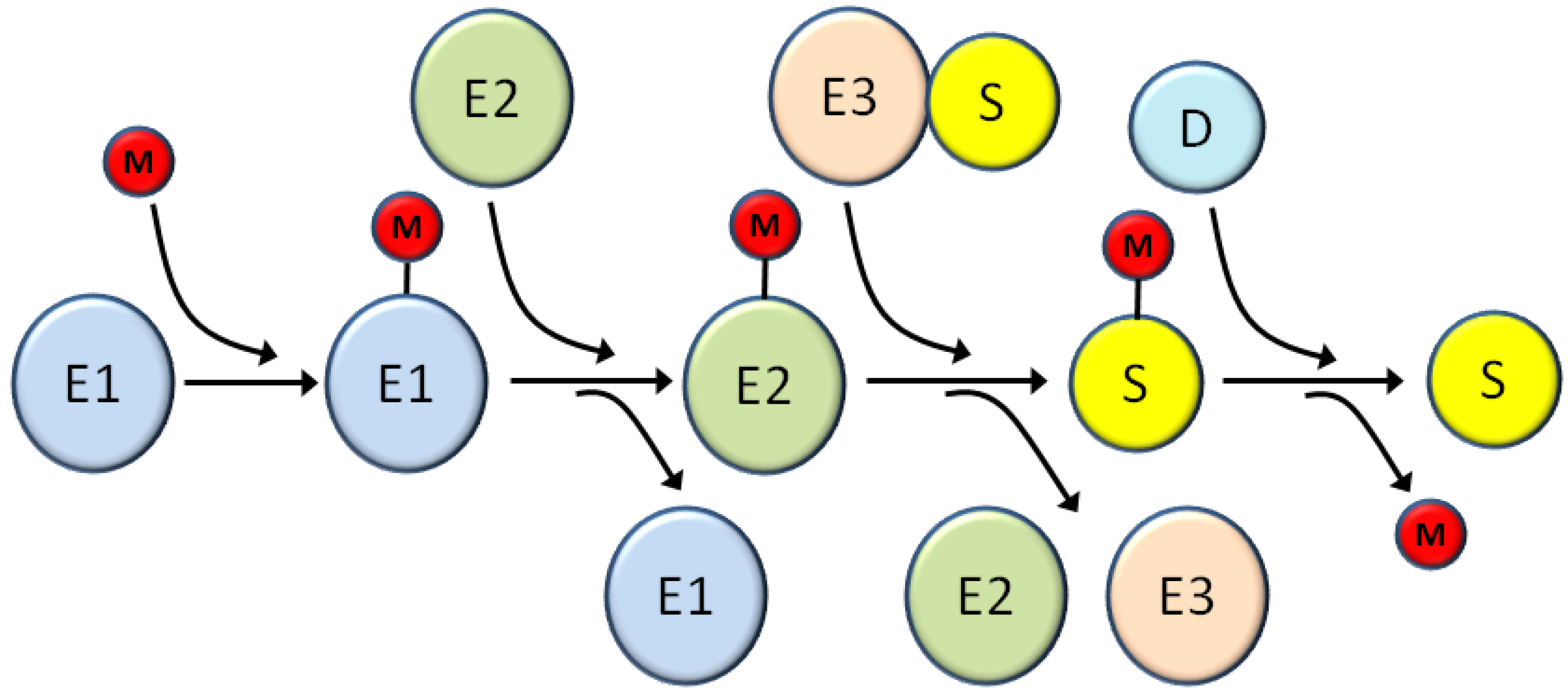
All viruses have common missions that they must accomplish in order to successfully reproduce, including optimizing viral gene expression, promoting viral genome replication, and avoiding or abrogating host defense systems. At each of these steps viruses typically have evolved numerous complementary and/or redundant mechanisms to co-opt cellular proteins and machinery to support the viral lifecycle. Because of this dependence on host cells, viruses often target cellular systems with broad and important regulatory functions such as the ubiquitin super family [24]. This superfamily is characterized by small protein modifiers that are covalently attached to proteins substrates through a series of biochemically similar steps (Figure 2). These modifiers include ubiquitin and a series of ubiquitin-like proteins (Ubls), such as SUMOs [25], ISG15 [26], NEDD8 [27], FAT10 [28], and others [29]. The first step in the conjugation process is activation of the modifier through an ATP-dependent reaction that forms a thioester linkage between the modifier and the activation enzyme (E1) [30]. Subsequently the modifier is transferred to the conjugation enzyme (E2), again through a thioester linkage [31]. Finally, the modifier is transferred to the substrate using an E3 ligase that provides substrate specificity [32]. This final conjugation of the modifier to the substrate occurs through an isopeptide bond linking the C-terminus of the modifier with the epsilon amino group of a lysine residue in the target protein. Depending on the substrate, more than one lysine may be modified and more than one type of modifier may be utilized; in some cases the different modifications occur on separate lysine residues while in other cases different modifiers compete for the same lysine, often with opposing functional effects [33,34]. Additionally, depending on the modifier used both mono- and poly-chains of the modifier can be formed resulting in different outcomes for the substrate [35,36].
The prototype of this superfamily is ubiquitin, a 76 amino acids protein. Polyubiquitination targets proteins for proteosomal degradation and is a key cellular mechanism for removing damaged proteins and for regulating protein activity [37]. Ubiquitin chains can be removed by deubiquitinating enzymes (DUBs), thus, the balance between the modification and demodification reactions can be important for controlling substrate levels, including viral proteins [24,38]. In addition to ubiquitin, the SUMOs [39,40] and ISG15 [41] are the other two Ubls that have been implicated as modifiers involved in HPV viral processes. There are four human SUMO genes encoding SUMOs 1-4 [42], though SUMO 4 appears to have limited tissue expression [43] and has been much less studied than the other three. The processed forms of SUMOs 2 and 3 are 97% identical, while both are approximately 50% identical to SUMO1. While highly similar and often treated interchangeably in many studies, functional and biological differences have emerged for the three predominant SUMO types [44,45,46,47]. Consequently, the possibility of type-specific effects on substrates should be kept in mind when interpreting sumoylation studies. Like ubiquitin, SUMO moieties can be removed from substrates by a set of desumoylating enzymes known as SENPs (sentrin proteases) [48]. Lastly, mature ISG15 is a 157 amino acid ubiquitin-like modifier whose expression is highly stimulated by type I interferon [26]. While ISG15 modification is now known to impact a variety of cellular processes, its original discovery as an interferon-stimulated gene has focused a great deal of work in this field towards its role in viral infections where it has an antiviral effect [41].
Figure 2.
Schematic of the general cascade for modification by ubiquitin and ubiquitin-like (Ubl) modifiers. E1, E2, and E3 are the activating enzyme, conjugating enzyme, and ligase, respectively for the various Ubl pathways; the number of distinct enzymes at each step various for the particular modifier system. M represents ubiquitin or any other Ubl. S is the substrate and D is the demodifying enzyme that removes the modifier and returns the substrate to the unmodified form. See the text for more details about each step in the modification/demodification process.
Figure 2.
Schematic of the general cascade for modification by ubiquitin and ubiquitin-like (Ubl) modifiers. E1, E2, and E3 are the activating enzyme, conjugating enzyme, and ligase, respectively for the various Ubl pathways; the number of distinct enzymes at each step various for the particular modifier system. M represents ubiquitin or any other Ubl. S is the substrate and D is the demodifying enzyme that removes the modifier and returns the substrate to the unmodified form. See the text for more details about each step in the modification/demodification process.

3. Modification of HPV Viral Proteins by Ubiquitin and Ubiquitin-Like Modifiers (Ubls)
As for cellular gene products, the expression levels of viral proteins need to be highly regulated to coordinate their individual protein activities with the functional stages of the viral life cycle. This regulation typically occurs at both transcriptional and post-transcriptional levels. For HPVs, the viral early proteins tend to be expressed at relatively low levels and have short half-lives (Section 3.1., Section 3.2., Section 3.3., Section 3.4., Section 3.5. and Section 3.6.), while the L1 [49] and L2 [50] late proteins are much more stable (Section 3.7.). Proteasome inhibitor studies showed that the instability of these viral early proteins is proteasome dependent, and studies on individual HPV early proteins have shown that ubiquitination is a key element in targeting these proteins to the proteasome [51,52,53,54]. In combination, these results indicate that the ubiquitin-proteasome system is a key mechanism used by HPVs to regulate the intracellular levels of the viral gene products at the post-translational level. In addition to modification by ubiquitination, several HPV proteins are modified by sumoylation [39]. Sumoylation does not directly lead to degradation of the viral substrates, but does modulate biological functions. Both the ubiquitin and SUMO systems are utilized by the host cell to regulate aspects of cell cycle [55,56,57] and differentiation [57,58], and HPVs presumably take advantage of these systems to coordinate the levels of viral proteins with critical aspects of host cell cycle and epithelial cell differentiation. Alternatively, in some cases, these Ubl modifications may be antiviral in nature as the host cell attempts to reduce the level or activities of viral proteins as a protective measure. More specific details concerning the modification of the individual early proteins by ubiquitin and SUMO are presented below.
3.1. The E1 Proteins
E1 proteins are replication helicases that nucleate the formation of viral genome replication complexes and are essential for papillomavirus replication [13]. An early study revealed that the bovine papillomavirus (BPV) E1 protein was an unstable protein that was ubiquitinated and degraded via proteasomes [51]. In these studies, E1 was stabilized by association with the cyclin E/Cdk2 complex, apparently simply through complex formation, as Cdk2 kinase activity and E1 phosphorylation were not required for the protection from degradation. Under conditions which supported BPV origin-dependent DNA replication, E1 again became rapidly degraded, possibly as a consequence of dissociation from the cyclin E/Cdk2 complex. The elongation phase of DNA synthesis was required for E1 degradation, suggesting that E1 may dissociate from the cyclin E/Cdk2 complex at the completion of each round of genome replication. Subsequently, the anaphase-promoting complex (APC) was shown to be a primary E3 ligase for E1, specifically the APCCdh1 complex [59]. E1 interaction with, and degradation mediated by, the APC complex was dependent upon a KEN box and a D box within E1. As these motifs are highly conserved among HPV E1 proteins this suggests that the APC complex may be a universal regulator of E1 stability. However, comparable studies have not been directly done for HPV E1 proteins, thus, virtually nothing is known about the specific components of the ubiquitin-proteasome system that promote degradation of the human papillomavirus types.
In addition to modification by ubiquitin, E1 proteins are also substrates for SUMO, first shown for BPV E1 [60] and then for HPV 11 [61]. E1 sumoylation requires direct interaction with the SUMO conjugase, Ubc9 [60], and the interaction appears to be enhanced by E1 oligomerization [62]. Sumoylation of E1 is also stimulated by three SUMO ligases of the PIAS (protein inhibitor of activated STAT) family, suggesting that some or all of the members of this family may be the authentic in vivo ligases for E1 proteins [61]. While the available evidence strongly supports that E1 proteins can be sumoylated as a consequence of their interaction with Ubc9, the extent of E1 sumoylation is limited and the functional consequences of this modification are unclear. Sumoylation was originally reported to be required for nuclear localization of E1 [63], however, subsequent studies did not confirm this phenotype [62,64]. Intriguingly, all E1 mutants defective for Ubc9 binding are also impaired for DNA replication [62]. This could indicate that E1 sumoylation is required for some step in viral genome replication, but could also reflect requisite sumoylation of some other replication factor, such as PCNA (proliferating cell nuclear antigen) [65]. In this latter case E1 could be recruiting Ubc9 to the replication complex to redirect its activity to a host substrate critical for the viral replication process.
3.2. The E2 Proteins
E2 is a central regulatory factor for papillomaviruses and its expression is tightly regulated at multiple levels, including protein turn over. Ubiquitination and proteosomal degradation leading to a short half-life was first demonstrated for bovine papillomavirus E2 [66]. Similar ubiquitination and proteosomal degradation leading to short half-lives has been shown for E2 proteins from both high risk (types 16, 18, and 31) and low risk (types 6 and 11) HPVs, and their rapid degradation is dependent upon sequences within the amino terminal transactivation domain [52,67,68]. Importantly, E2 protein levels are cell cycle regulated with degradation occurring specifically at the end of G1 phase, and this degradation is mediated at least in part via interaction with the SCFSkp2 ubiquitin ligase [69]. High risk E2 proteins also associate with the Mdm2 ligase [70] and the APC/C ubiquitin ligase [71], though E2 does not appear to be a direct substrate for either ligase. For APC/C, E2 interacts with the activators, Cdc209 and Cdh1, and inhibits APC/C activity leading to the stabilization of several substrates involved in cell cycle control and chromosomal instability, including Skp2 [71]. This suggests a possible feedback mechanism to control E2 levels in cycling basal keratinocytes whereby E2 acts on APC/C to increase Skp2 which subsequently leads to E2 degradation via the SCFSkp2 ligase [69]. In contrast, in differentiated keratinocytes where Skp2 is not expressed this feedback would absent thus contributing to the observed increase in E2 levels in the upper layers of the epithelium [72].
While HPV 18E2 was shown to interact with the SCFSkp2 ubiquitin ligase that contains cullin1, HPV 16E2 associated with cullin3 but only weakly if at all with cullin1 [73]. Inhibition of cullin3-based E3 ligases with a dominant-negative CUL3 led to reduced ubiquitination and a significantly increased half-life for 16E2, suggesting that 16E2 degradation is mediated via a cullin-3 containing ubiquitin ligase. Whether or not 18E2 and 16E2 are actually ubiquitinated via different E3 ligases, or if there is redundancy in which ligases can target E2 proteins, remains to be determined. Interestingly, Brd4, an activator of E2 transcriptional activity, blocks the interaction between E2 and cullin-3 resulting in increased stability of E2, presumably through reduced ubiquitination [73]. Similar stabilization by Brd4 has been reported for the E2 proteins from bovine papillomavirus [74], HPV 11 [68,75], and HPV 31 [68]. Brd4 directly binds to transactivation domain (TAD) of E2 which suggests that Brd4 may be a universal regulator of E2 stability through competition with E3 ligase complexes that ubiquitinate the TAD domain [68]. Alternatively, a recent report suggests that E2 is primarily ubiquitinated in the cytoplasm and that Brd4 stabilizes E2 by sequestering it in the nucleus, where it is not accessible for degradation [76].
In addition to Brd4, several other proteins have been shown to increase E2 half-life by preventing proteosomal degradation, including two cellular proteins, Tax1BP1 [77] and NRIP [78], and two HPV proteins, E1 [79] and E1^E4 [80]. None of these proteins had any significant effect on E2 transcript levels, thus, they appear to be acting at the protein level. For the two viral proteins, E1 and E1^E4, no mechanism was explored. Tax1BP1 is a subunit of an ubiquitin-editing enzyme complex [81] and binds E2 through the TAD region, consistent with Tax1BP1 potentially acting to protect E2 by deubiquitinating this domain. However, no reduction of E2 poly-ubiquitination was observed in the presence of Tax1BP1, so the mechanism of E2 stabilization remains undefined. Like Tax1BP1 and Brd4, NRIP also interacts with E2 through the TAD domain [78]. Unlike Tax1BP1, NRIP was able to reduce ubiquitination of 16E2 and this was related to dephosphorylation of E2 mediated via recruitment of calcineurin by NRIP. Exactly how phosphorylation promotes or contributes to ubiquitination remains unclear, especially as other reports suggest that phosphorylation may stabilize 16E2 [82] and 8E2 [83]. An intriguing suggestion is that phosphorylation may regulate chromatin association leading to bound E2 that is protected from degradation, possibly by sequestration from the ubiquitination machinery, and is consequently much more stable than unbound E2 [83]. In this model phosphorylation could be a determinant of either stability or degradation depending on how it affected chromatin binding by a particular E2 type and during a particular cell cycle stage.
Lastly, there is evidence that E2 degradation is subject to crosstalk between the ubiquitin system and the sumoylation system [84]. BPV, HPV 11, HPV 16, and HPV 18E2 proteins have all been shown to bind Ubc9, the SUMO conjugating enzyme, and the 16 and 18E2 proteins could be sumoylated in vitro and in vivo [85]. Type 16E2 was tested for functional effects, and sumoylation was found to contribute to both transactivation and repressive activity. In addition to these functional effects, increasing overall intracellular sumoylation levels led to a dramatic stabilization of 16 and 18E2 proteins via prevention of proteosomal degradation [86]. Surprisingly, the stabilization mediated by increasing overall sumoylation did not occur through direct sumoylation of the E2 proteins themselves as sumoylation defective E2 mutants were stabilized as effectively as the wild-type protein. This indirect effect on E2 degradation suggests that increasing sumoylation regulates some component of the ubiquitination/deubiquitination pathway to prevent proteosomal degradation. Biologically this may contribute to enhanced E2 expression in suprabasal layers of the skin where sumoylation is increased [87]. In summary, the overall levels of E2 reflect a complex interplay between numerous factors that can interact with E2 to facilitate or inhibit degradation, and there appear to be type specific nuances that further confound our current understanding.
3.3. The E4 Proteins
The E4 ORF is located within the E2 gene and is expressed primarily as an E1^E4 product [16]. The E1^E4 protein is expressed at relatively high levels during productive viral replication and accumulates in the mid to upper epithelial layers, suggesting that it is a fairly stable protein [88,89]. While modification of E1^E4 by kinases [90,91,92] and proteases [93,94] is known, modification by the ubiquitin superfamily members has not been reported.
3.4. The E5 Proteins
The E5 protein is the major oncoprotein for BPV and a contributor to the oncogenic process in HPVs [14]. It is a very small (44 amino acids for BPV and approximately 80 amino acids for HPVs), hydrophobic, and membrane associated protein that is expressed at very low levels. Like E4 proteins, the turnover and degradative mechanisms have not been explored in any detail, thus, whether or not it is modified by ubiquitin for proteosomal targeting is unknown.
3.5. The E6 Proteins
E6 proteins from high-risk HPV types are moderately short-lived proteins [95,96] that are degraded via the proteasome [53,97]. The degradative mechanism for other E6 proteins is less certain as an initial study reported that two low-risk genital type E6 proteins (6a and 11) were not stabilized by a proteasome inhibitor and two cutaneous HPV E6 types (5 and 8) showed only slight stabilization by the inhibitor [53]. However, a subsequent study found that type 11E6 was ubiquitinated and degraded via the proteasome, suggesting that all E6 types may share a common degradative pathway [97]. Differences in epitope tagging or cell type for the expression experiments may account for these conflicting results, and further studies are needed to clarify possible differences in degradative mechanisms between various E6 types. In contrast, both studies agreed that this degradative process does not require interaction with E6AP, a known ubiquitin E3 ligase that binds E6 proteins [98]. Interestingly, a latter study found that the presence E6AP actually stabilizes both HPV 16 and 18E6, though the E6AP enzymatic activity is unnecessary for this protective effect [99], and similar results were later found for cutaneous HPV E6 proteins [100]. The combined results infer that E6AP is not the E3 ligase responsible for ubiquitinating E6 itself, that E6AP is acting through some other mechanism to affect E6 turnover, and that this mechanism may be common to all E6 types. The true E3 ligase(s) for E6 proteins remains undetermined, but other possible candidates have been identified through recent proteomic approaches. HERC2, a putative E3 ligase of the HECT-domain type, was associated with HPV 16E6 [101], a HECT-domain E3 ligase, called EDD, binds strongly to HPV 18E6 and weakly to type 16 and 11 E6 proteins [102], and some E6 proteins from genus beta interact with the Ccr4-Not complex which has known ubiquitin ligase activity [6]. However, none of these candidates has been functionally tested for ability to ubiquitinate E6, thus, all may simply be E3 ligases recruited to E6 to facilitate modification of other substrates rather than E6 itself. For example, EDD enhances ubiquitination of E6AP and reduces E6AP levels which indirectly reduces E6 levels, but does not appear to affect E6 directly [102]. Likewise, HERC2 is known to stimulate the ligase activity of E6AP [103], so could be acting to enhance the activity of the E6-E6AP complex rather than targeting E6 itself. No such HERC2 enhancing effect was observed for E6-mediated p53 degradation in HeLa cells [104], but this does not rule out a possible HERC2 effect on other E6 targeted substrates.
Lastly, in addition to binding E3 ligases, HPV 16E6 binds a deubiquitinating enzyme, USP15, and both 16 and18E6 proteins are stabilized by catalytically active, but not an inactive, mutant of USP15 [101]. These results suggest that USP15 is protecting E6 proteins by removing ubiquitin chains and are consistent with a typical ubiquitin-proteasome pathway for degradation of at least the high-risk HPV E6 proteins. Somewhat surprisingly, HPV 16E6 is also stabilized by association with PDZ domain proteins [105]. As PDZ proteins are themselves targets for E6-E6AP mediated degradation [106,107], this observation suggests that the complex network of interactions between E6 and its associated proteins can influence E6 stability and proteolysis, similar to what has been observed for E2 proteins.
3.6. The E7 Proteins
Like its partner oncoprotein, E6, the HPV E7 proteins are also short-lived with HPV 18E7, initially shown to have a half-life of less than 15 minutes in HeLa cells [108]. Subsequent studies of HPV 16E7 in Cos 7 cells [54] and CaSki cells [109] both showed that E7 was ubiquitinated and that its short half-life was due to proteosomal-dependent degradation. The 16E7 half-life in Cos7 cells was 30-40 minutes, consistent with the high-risk E7 proteins, generally being short-lived proteins. Interestingly, mutational interrogation revealed that E7 degradation required the N-terminal eleven amino acids of E7 which directed N-terminal poly-ubiquitination of E7; neither of the two internal lysines in E7 was involved [54]. Tandem mass spectrometry (MS/MS) analysis of ubiquitinated HPV58 E7 confirmed N-terminal conjugation of the ubiquitin moiety suggesting that this may be a general mechanism for E7 ubiquitination [110].
While ubiquitin-mediated, proteosomal degradation of E7 proteins is well established, less certain are the components of the ubiquitination machinery that are specifically involved in E7 modification as several candidates have been identified. The first potential candidate identified was SOCS1 which binds E7 in vitro, co-localizes with E7 in vivo, stimulates E7 ubiquitination, and promotes proteosomal degradation of E7 [111]. SOCS1 is a component of the VCB-type ubiquitin E3 ligase that appears to function as an adapter protein to target this ligase to substrates [112], implying that SOCS1 is part of an E3 ligase complex that controls E7 degradation. However, it was noted the E7 was still partially ubiquitinated in the presence of an inactive SOCS-box defective mutant of SOCS1, suggesting that additional E3 ligases could be involved [111]. Consistent with this observation, Oh et al. demonstrated that the Cull1-Skp2 E3 ligase could also bind E7, function to ubiquitinate E7, and mediate E7 proteosomal degradation [113]. Like the SOCS1 study, E7 degradation was only partially blocked in the absence of Skp2, thus, at least two E3 ligase pathways for E7 proteolysis seem plausible. Additionally, Oh et al. identified UbcH7 as a functional E2 ligase for E7 while seven other E2 ligases (UbcH2, UbcH3, Ubc5A, Ubc5B, Ubc5C, UbcH6, and UbcH10) were ineffective. Unfortunately, later studies have not demonstrated an association of E7 with cullin 1 [5,114], thus, the authentic E3 ligase complexes that ubiquitinate E7 remains uncertain.
In addition to the dual E3 ligases that have been reported to facilitate E7 degradation, there is one report of a deubiquitinating enzyme (DUB) specific for E7, USP11 [115]. Both in vitro and in vivo, USP11 binds E7 and reduces the extent of poly-ubiquitination. Over expression of USP11 increases the half-life of E7 while siRNA knockdown of USP11 enhanced E7 degradation with a concomitant loss of E7 transforming activity in CaSki cells. While these studies do not rule out the existence of other DUBs that could contribute to the overall balance of ubiquitinated versus unmodified E7, they do strongly implicate a role for USP11 in at least some cell types.
3.7. L1 and L2 Proteins
The L1 and L2 capsid proteins are relatively stable and there are no reports of ubiquitination for either protein [9,10]. However, HPV16 L2 can be sumoylated at lysine 35 with a strong preference for SUMO2/3 over SUMO1 [50,116], and HPV16 L1 can be modified with ISG15 [117]. ISG15 modified L1 was incorporated less effectively into pseudovirion particles and the resulting modified pseudovirions had significantly reduced infectively, demonstrating that this modification could be regulating virion production during an authentic infection. Similarly, sumoylation of L2 prevents binding to L1, thus, sumoylation may also contribute to regulation of capsid assembly [50]. Sumoylation of L2 did not prevent binding to E2, but the effects of this modification on other known interactions of L2 were not examined. Interestingly, a lysine 35 to arginine mutant L2 protein had reduced stability compared to the wild type protein. However, the study did not distinguish whether sumoylation at lysine 35 actually protects L2 from degradation or that the arginine at position 35 simply makes L2 intrinsically less stable. Additionally, neither sumoylation nor ISGylation has been examined in the context of normal keratinocytes, so the presence and potential function of these modifications during the normal viral life cycle has not been verified.
4. Modulation of the Host Environment Mediated Through HPV-Ubl Interactions
In addition to utilizing the ubiquitin-proteasome system, along with other Ubls, to regulate turnover of HPV polypeptides themselves, several HPV proteins have targeted these systems to control the levels and activities of critical cellular proteins. The ubiquitin superfamily, particularly ubiquitin and SUMO, modify hundreds to thousands of host proteins [118,119]. Redirecting these ubiquitous post‑translational modifications systems provides the virus with enormous opportunities for reprogramming the cellular environment to favor viral persistence and reproduction. For HPVs, none of the viral proteins has intrinsic E3 ligase or deubiquitinating activity, so manipulation of these Ubl systems requires interaction between HPV proteins and cellular components of the ubiquitin or SUMO pathways. Retargeting host E3 ubiquitin ligases to new substrates to facilitate degradation of selected host proteins is an important mechanism for some HPV early proteins, particularly E6 and E7 [120]. Alternatively, some early proteins interfere with E3 ligases or recruit DUBs to prevent degradation of host proteins. Examples of how HPV proteins enhance, inhibit, and redirect the Ubl systems are presented below.
4.1. The E1 Proteins
While little is known about the specific enzymes involved in HPV E1 ubiquitination and proteasomal degradation, a recent association with a deubiquitinating complex has been described [121,122,123]. E1 proteins from anogenital papilloma virus types, but not cutaneous types, interact with a host protein originally named p80 and, subsequently, called UAF1 [121]. UAF1 is a cytoplasmic protein that is recruited to the viral origin in an E1- and E2-dependent fashion and is necessary for wild-type levels of viral DNA replication [122]. However, UAF1 is not required for assembly of the E1-E2-ori complex so must be acting at a downstream point in the replication process. In addition to binding viral E1, UAF1 interacts with three deubiquitinating enzymes, USP1, USP12, and USP46, leading to formation of a ternary complex on the viral origin, consisting of E1, UAF1, and any one of the three DUBs [123]. The presence of a DUB in the complex is as critical for replication activity as is UAF1 itself, suggesting that the primary role of UAF1 is to recruit a DUB to the replication initiation complex. What is missing at this point is the mechanism by which a DUB facilitates replication and the relevant target(s) for the DUB. Removal of ubiquitin from E1 to increase E1 levels is one possible mechanism to enhance replication, but the UAF1-USP complex is known to target other substrates involved in DNA replication and chromatin structure [124,125,126], thus, experimental determination of the functional targets will be needed to fully resolve how E1 utilizes host DUBs to promote viral genome replication. If the target(s) is not E1 itself, this would be a new example of how certain HPVs have usurped the ubiquitin system to target host proteins in ways that facilitate the viral life cycle.
4.2. The E2 Proteins
E2 is the major transcriptional regulatory protein for papillomaviruses, and it can exert either positive and negative effects on viral promoters depending on the context of E2 binding sites [12]. Over 80 host cell proteins have been identified as partners of various E2 types, many of them involved in transcriptional regulation and chromatin modification/remodeling as expected for a viral transcriptional regulatory protein. Among this group of known E2 partners is the E3 ligase, Mdm2 [70]. Mdm2 binds to C-terminal DNA binding domain of E2, can complex with E2 on E2 binding sites, and activates E2-dependent transcription of the HPV 16 promoter. While transcriptional activation requires the E3 ligase activity of Mdm2, E2 protein levels are unaffected by Mdm2 suggesting that E2 is not a direct substrate for Mdm2. Thus the transcriptional effects appears to be mediated via another mechanism involving ubiquitination, though the specific target for the E2-Mdm2 complex is still unknown. Potentially Mdm2 could be targeting other proteins associated with the HPV 16 promoter for proteolysis or affecting protein-protein interactions at the promoter, as is well established for other transcriptional units [127].
Perhaps because of a focus on E2’s transcriptional function, there has been very limited investigation of E2 interactions with the ubiquitin-proteasome system outside the context of transcriptional control. As mentioned in Section 3.2., high-risk E2 proteins type 16 and 18, but not low-risk E2s, associate with Cdc20 and Cdh1 (also known as FZR1) which are activators of the Anaphase Promoting Complex (APC), an important E3 ligase regulating cell cycle [71]. E2 appears to sequester Cdh1 into insoluble cytoplasmic structures, thus preventing APC activation and allowing APC substrates, such as cyclin B, to escape degradation. This dysregulation of APC ultimately leads to genomic instability which may contribute to the oncogenic potential of high-risk HPVs.
The two above example confirm that E2 proteins can in some instances function as modulators of the ubiquitin-proteasome system, but until recently this mechanism seemed to be relatively limited for E2 proteins. However, more global and unbiased evaluations of large-scale protein-protein interaction networks for multiple E2 protein types have revealed a more widespread connection between E2 and the ubiquitin-proteasome system [128,129]. A yeast two-hybrid screen of 12 different E2 types from three clades against a human keratinocyte (HaCaT) cDNA library revealed that proteins in the ubiquitin functional family comprised a major target for E2 proteins with 26 partner proteins identified [129]. Among these partners were two members of the HECT domain family of E3 ligases (HUWE1, WWP2) and several adaptor proteins of cullin-based ubiquitin ligases (BTBD1, SPOP, CDC20, CDH1, FBX022). While some of these interactions may be involved in ubiquitination and turnover of E2 itself, it is likely that some reflect novel pathways, whereby E2 redirects the ligases to new targets or modulates the ligase activity on their normal targets. Additionally, the interactor list contained PIAS1 and PIAS4, E3 ligases in the sumoylation pathway [130]. As E2 is known to be sumoylated [85], these PIAS proteins could be acting as SUMO ligases for E2, or again E2 could be retargeting them to enhance sumoylation of E2 partners. Regardless of the outcome, these more global interaction studies suggest that the ability of E2 to manipulate members of the ubiquitin super family may be broader and more extensive than previously considered.
4.3. The E4 Proteins
E4 proteins are known to mediate keratin network disruption which is believed to comprise epithelial integrity and promote virion release [131]. There is one report that at least part of the mechanism for this E4-mediated disruption of the keratin network is through hyper-ubiquitination of keratin proteins that are associated with E1^E4 protein, which could lead to enhanced proteosomal degradation of the keratins [132]. This hyper-ubiquitination is likely to be an indirect effect triggered by E1^E4 induced phosphorylation of the keratins rather than direct recruitment of an ubiquitin E3 ligase by E1^E4.
4.4. The E5 Proteins
The HPV E5 oncoproteins are membrane bound and mediate their biological effects through association with several host cell proteins, including EGFR [133]. E5 enhances EGFR activity through multiple mechanisms, at least one of which appears to be interference with ubiquitin-proteasome degradation [134]. In this mechanism, HPV16 E5 reduced association of an E3 ligase, c-Cbl, with EGFR leading to a reduction in EGFR ubiquitination. The 16E5 effect on EGFR levels was similar to that seen by proteosomal inhibition, suggesting that interfering with the EGFR/c-Cbl interaction was an important contributor to E5 activation of EGFR. Similarly, 16E5 is reported to block the ubiquitin degradative pathway for keratinocyte growth factor receptor (KGFR), also by affecting the c-Cbl E3 ligase [135]. In this case E5 impairs phosphorylation at tyrosine 196 of the FRS2α docking protein that is constitutively associated with KGFR. Phosphorylation of this tyrosine is necessary to recruit the Grb2-Cbl ligase complex, so reduction of modification at this residue by E5 likely leads to reduced ability of the ligase to ubiquitinate the KGFR-KFS2 complex.
In contrast to the above studies showing an E5-mediated inhibition of proteosomal degradation for two growth factor receptors, there are now two reports that E5 proteins can also enhance degradation of certain host proteins. CD1d is an important major histocompatibility complex (MHC) type 1 glycoprotein that is down regulated at the protein level in HPV 6 or 16 E5 expressing cells; down regulation is abrogated by a proteasome inhibitor indicating a likely ubiquitin-proteasome degradative process [136]. In this case, E5 interacts with calnexin, an ER chaperone for CD1d, which likely results in misfolding of CD1d leading to its degradation. Decreased CD1d mediated by E5 results in abrogation of IL-12, a major cytokine important in both innate and adaptive immunity to pathogens, which would like aid in HPV evasion of the host immune response. While not a direct interaction between E5 and components of the ubiquitin-proteasome system, this example illustrates more subtle mechanisms by which the HPV proteins can influence the ubiquitin-proteasome system to the virus’ advantage. Another example of E5-mediated degradation has been reported for the pro-apoptotic protein, Bax [137]. Expression of HPV16 E5 increased ubiquitination of Bax leading to a decreased half-like that was rescued by a proteosomal inhibitor. E5 appeared to be acting through a pathway involving XOX-2, prostaglandin E2, and PKA, thus, again this is likely due to an indirect mechanism rather than a direct E5 interaction with components of the ubiquitin-proteasome system. As there are additional reports in the literature that E5 proteins can decrease the levels and/or half-lives of other cellular proteins [138,139], direct or indirect utilization of the ubiquitin-proteasome system by E5 may be a general feature of this viral protein.
4.5. The E6 Proteins
Historically, high risk E6 proteins are the prototype for HPV modulation of the host cell through the ubiquitin-proteasome system. Via their ability to bind the host ubiquitin ligase, E6AP, high risk E6 proteins retarget the ligase to modify E6 partners and promote the proteosomal degradation of these partners [11]. At least for the high risk E6 proteins, knocking down E6AP levels has nearly the same effect on global gene expression as knocking down E6 itself [140], indicating that a large portion of E6’s functional effects are mediated through E6AP, either utilizing its E3 ligase activity or a possible transcriptional activity [141]. Furthermore, E6AP is necessary for E6 to cause cervical cancer in a mouse model which strongly confirms that the oncogenic activity of the high risk E6 proteins is mediated through E6AP targets [142]. The initially described target of the E6-E6AP complex was the p53 tumor suppressor protein [143,144], and the ability of the high risk E6 proteins to promote p53 degradation remains a key element in the transforming activity of these proteins [145]. In addition to targeting p53, the high risk E6 proteins have a carboxy-terminal motif that can interact with cellular PDZ proteins and target their proteosomal degradation via E6AP [106,107,146,147,148]. The E6-targeted PDZ proteins that have been identified to date are membrane associated proteins involved in cell-to-cell adhesion and communication [149], thus, their dysregulation by E6-E6AP is likely to have significant effects on the host cells and may contribute to the oncogenic potential of the high-risk HPVs. However, the true extent to which these PDZ proteins are degraded in vivo during a natural infection and the actual biological consequences of such degradation remain largely unknown.
While most early studies on HPV E6 proteins focused on the high-risk types, due to their importance in human cancers, more recent studies have shown interesting similarities and differences among various E6 types. For example, interaction with E6AP has also been shown for low risk mucosal HPVs [7,150,151], and this conserved ability to interact with E6AP occurs through the LXXLL motif on E6AP as for high risk E6 proteins [152,153,154]. However, in contrast to high risk E6 proteins, the low risk mucosal types fail to interact with or significantly degrade p53 even though they can bind E6AP [144,151,155]. This observation at least partially explains their low oncogenic potential and suggests that there must be subtle differences in the resulting E6-E6AP complexes between low and high risk mucosal E6 proteins that can affect target discrimination. Similarly, the low risk E6 proteins lack the PDZ binding motif so these proteins are not degraded by low risk E6s. Nonetheless, since the E6-E6AP interaction is conserved between high and low risk E6 types, there are likely to be other E6-E6AP targets that are biologically important for all mucosal HPVs.
In support of a broader role for the E6-E6AP complex in the normal life cycle of HPVs, many other E6 binding partners have been identified both through targeted studies of individual proteins and more recently through proteomics approaches [6,7,102,156,157], and many of these partners are proteasomally degraded in an E6AP-dependent fashion. Discussion of all the numerous identified E6-E6AP targets is beyond the scope of this review, but more comprehensive discussions of individual targets can be found in several recent reviews [11,158,159,160,161]. Furthermore, the general importance of this E6-E6AP interaction has been extended by the recent demonstration that E6 proteins from cutaneous HPVs of the beta genus associate with E6AP [162]. However, there is considerable variation between individual E6 types and the extent of association with E6AP [100] and at least one report that several beta type E6 proteins do not bind E6AP [6], thus, the true generality of the E6-E6AP complex remains unresolved. Consistent with a possible general role for E6-E6AP, there is one common E6-E6AP targets for nearly all tested E6 proteins, Bak [163,164,165]. Bak is a pro-apoptotic protein [166], thus, down regulating its activity through degradation may be a critical requirement for all HPVs. In contrast, the more type-specific targets for E6-E6AP may reflect unique requirements for viral persistence and/or reproduction in different types of epithelium.
The well-established and widespread ability of E6 proteins to interact with and utilize E6AP as an E3 ligase to ubiquitinate host proteins for targeted degradation does not exclude interactions between E6 and other components of the ubiquitin-proteasome system. As described in Section 3.5., proteomics approaches have demonstrated associations of various E6 proteins with other E3 ligases, including HERC2 [101], EDD [102], and the Ccr4-Not complex [6]. What role these ligases play with E6 in the HPV life cycle, and what their potential targets might be, remain undefined. Additionally, there is accumulating evidence that E6 proteins can interact directly with proteosomal components. Two recent proteomics studies detected association of E6 proteins with multiple proteosomal subunits [6,7], and direct binding to multiple subunit proteins, including S2, S4, S6a and b, S7, S8, and S10 was demonstrated in vitro for 16E6, 11E6, and 18E6 [167]. E6 was also able to increase the ubiquitination of the S5a subunit through E6AP which increased S5a degradation [167]. How these proteosomal interactions may affect overall proteosomal function and/or degradation of E6 targets has not been examined, but does suggest that the utilization of the ubiquitin-proteasome system by E6 proteins is multi-faceted and complex.
One other intriguing aspect of post-translational regulation by E6 is cross-talk between the ubiquitin and SUMO systems. The initial observation was that 16E6 binds PIASy, a SUMO E3 ligase, and inhibits its activity [168]. This reduction of PIASy activity results in decreased sumoylation of specific PIASy substrates, including p53 and pRb. PIASy normally activates p53 and represses pRb, thus, the E6 mediated inhibition of PIASy would contribute to the reduced p53 activity and increased pRb activity in high risk HPV infected cells. Subsequently, a more global effect of E6 on the sumoylation system was demonstrated with the finding that the SUMO conjugating enzyme, Ubc9, is a substrate for E6AP-dependent degradation by high risk E6 proteins, leading to numerous changes in the cellular sumoylation profile [169]. Sumoylation is important in normal keratinocyte differentiation [87,170], thus, the ability of E6 to disrupt the sumoylation program broadly may contribute to the aberrant differentiation phenotype of high risk HPVs. Other more substrate specific possibilities exist, such as described in a recent study on hADA3, a transcriptional coactivator [171]. In the absence of E6, hADA3 is a normal substrate for E6AP, and the ubiquitination and subsequent degradation of hADA3 is enhanced by its increased sumoylation. Expression of high risk E6 further enhances sumoylation of hADA3 thus leading to its decreased stability. While enhanced sumoylation of hADA3 might seem to conflict with generally reduced sumoylation caused by E6 targeting of Ubc9, on an individual substrate basis there may be no conflict. For example, recruitment of E6 to the E6AP-hADA3 complex could retarget E6AP to cause degradation of an associated SUMO protease resulting in a net increase in hADA3 sumoylation. While speculative, such models illustrate the additional levels of complexity that arise through cross-talk between the Ubl systems. Significant further studies will be needed to untangle and understand the biological consequences of these processes.
4.6. The E7 Proteins
Like E6, the characterization of E7 proteins has focused heavily on the major oncogenic types, 16, 18, and 31, with many fewer studies on the low risk alpha types or the beta types [15]. The primary target for the high risk E7 proteins that is associated with their oncogenic potential is the host pRb protein which is degraded through the ubiquitin-proteasome pathway in an E7-dependent fashion [172,173,174,175]. MDM2, the natural E3 ligase for pRB is not involved in the E7-mediated degradation of pRB [176]. Instead, for HPV16 E7, a cullin 2 ubiquitin ligase associates with the E6-pRb complex, leading to pRB ubiquitination and degradation [114]. 16E6 preferentially associates with the activated NEDD8 modified form of cullin 2, and may induce neddylation and/or stabilize the neddylated form of cullin 2 [114]. However, 16E6 does not appear to bind directly to cullin 2, and association with cullin 2 requires the substrate specificity factor, ZER1 [5]. Intriguingly, ZER1 interacts only with 16E7 and not 18E7, and 18E7 does not directly bind to cullin 2 [114], thus, apparently, 18E7 utilizes a different E3 ligase to target pRb for degradation. Consistent with a requirement for another E7-associated E3 ligase, proteomics studies identify several other E3 ligase components, including KCMF1, UBR4, and cullin 3, that associate more generally with E7 proteins from alpha and beta HPV types [5]. What role these other potential E3 ligase complexes play in E7 modulation of the host environment is currently unknown, but the conservation of these interactions suggests that there are common functions across most, if not all, HPV E7 types that require the ubiquitin-proteasome system. One example of such a target is the pRB family member, p130, which is an important regulator of differentiation, cell growth, and senescence [177]. P130 is proteasomally degraded in an E7-dependent fashion by both high and low risk alpha HPVs [178,179,180]. While the specific E3 ligase components involved are undetermined, this critical regulatory protein could be a mandatory target for mucosal E7 proteins to facilitate a replicative environment in terminally differentiating keratinocytes; there is very little information about the role of p130 for the cutaneous types, though HPV1 has been reported to destabilize p130 in raft cultures [181].
In addition to the direct targeting of host proteins through complex formation between E7, E3 ligases, and the substrate, E7 also indirectly influences the ubiquitin-proteasome system to modulate levels of some host proteins. Spardy et al. demonstrated that 16E7 accelerates claspin degradation which helps abrogate DNA damage checkpoint responses and facilitates mitotic entry [182]. Claspin is normally targeted for proteosomal degradation through ubiquitination by the SCFβ-TrCP type E3 ligase, and 16E7 up regulates several components of this complex presumably leading to increased ligase activity and enhanced modification/proteolysis of claspin. As the unregulated components all possess E2F-responsive promoter elements, it is likely that E2F activation resulting from E7-mediated degradation of pRb is the responsible for the increased expression of these components rather than a direct association of E7 with the SCF complex. A similar E2F-dependent mechanism appears to be involved in E7 regulation of the APC/C E3 ligase complex [183]. HPV16 E7 expression inhibits the degradation of APC/C substrates, such as cyclin A and cyclin B, during mitotic progression [184], and this inhibition is related to increased expression of EMI1, a negative regulator of APC/C [183]. At least part of the increased EMI1 levels results from increased transcription, again likely due to E2F activation following pRB degradation. Additionally though, EMI1 is also stabilized at the protein level by E7 though the precise mechanism has not been determined. This combination of transcriptional and post-transcriptional effects raises EMI1 levels sufficiently to abrogate normal APC/C function. Decreased APC/C activity has been proposed to be a general requirement to create a nuclear environment permission for viral replication [185], thus, targeting APC/C via E7 is likely to be functionally significant for high-risk HPVs. Together, these two examples clearly illustrate the E7 proteins can also manipulate the ubiquitin-proteasome system via indirect pathways that do not require direct interaction of E7 with the ubiquitination machinery.
Lastly, like E6, high risk 16E7 can associate with at least one proteosomal component, S4 [186]. This same subunit is also up regulated by E7 [187], though no information is available about mechanisms or the functional significance of E7-S4 interaction. Nonetheless, this ability to interact with a proteosomal component suggests additional complexity in the relationship between E7 and the ubiquitin-proteasome system that needs further exploration.
4.7. L1 and L2 Proteins
Modulation of the ubiquitin superfamily by L1 and L2 has not been reported, but two recent publications have identified intriguing interactions of L2 with the sumoylation system. Marušič et al. found the HPV16 L2 expression in U2OS cells caused an increase in nuclear SUMO2/3 accumulation that did not require sumoylation of L2 itself [50]. While the functional significance of this L2-mediated up regulation is unknown, sumoylation has been shown to increase and be necessary during normal keratinocyte differentiation [87] and to be decreased by high risk E6 proteins [169], so perhaps L2 is acting to modulate the E6 effect in the higher layers of the epithelium. In addition to modulating host sumoylation, HPV16 L2 has been shown to have a functional SUMO interaction motif (SIM) that directly binds SUMO2 more strongly than SUMO1 [116]. The functional SIM was localized to amino acids 284-289, and this sequence is highly conserved among the high-risk L2 proteins. Mutation of this SIM motif had no effect on capsid assembly, binding to host cells, or intracellular trafficking, but abrogated the normal association of the L2-DNA complex with PML bodies and greatly reduced infectivity of HPV 16 pseudoviruses. These results strongly suggest that the L2 protein has evolved a SIM motif to facilitate localization of the viral genome to PML bodies during in the initial infection process.
5. Conclusions
Human papillomaviruses have evolved an incredibly varied and complex set of interactions with the host cell ubiquitin-proteasome system and some of the related Ubl modifier systems. Since the discovery of the E6-E6AP complex and its role in p53 ubiquitination and degradation 20 years ago, there is now evidence that most of, if not all, the viral proteins are either modified by one or more of the Ubl systems or exert an effect on how these systems modify and regulate host proteins. While many details remain to be determined, it is clear that these protein modifier based post-translational systems are of central importance for HPVs to effectively co-opt the host cells for viral reproduction. Initial studies with high risk mucosal HPVs confirmed that targeted degradation of critical host cell growth regulatory proteins, such as p53 and pRB, was essential for the transformation process. Subsequent examinations of low risk mucosal types and cutaneous types have confirmed that utilization of the ubiquitin-proteasome system is a more general feature of HPVs to regulate levels of their viral proteins and to impact the host cell environment. Burgeoning studies on other Ubl systems, primarily the SUMO system, have revealed examples where HPVs modulate or utilize sumoylation to regulate viral activities. Additionally, there are novel examples of HPV proteins exhibiting cross-talk between the ubiquitin and sumoylation systems, which adds another level of complexity to the ability of HPVs to modulate the host cell growth and differentiation processes. Hopefully, the next 20 years will be as productive as the previous 20 years in fully unraveling the mechanisms by which HPVs utilize the ubiquitin family of modifiers to reprogram host cells.
Acknowledgments
The author would like to thank all the previous members of his laboratory who have contributed to studies discussed in this review. Additionally, the author would like to acknowledge all the members of the papillomavirus research community for their contributions to the works described here. The open access publishing fees for this article have been covered by the Texas A&M University Online Access to Knowledge (OAK) Fund, supported by the University Libraries and the Office of the Vice President for Research.
Conflicts of Interest
The authors declare no conflict of interest.
References and Notes
- Bhatia, N.; Lynde, C.; Vender, R.; Bourcier, M. Understanding genital warts: epidemiology, pathogenesis, and burden of disease of human papillomavirus. J. Cutan. Med. Surg. 2013, 17, S47–S54. [Google Scholar] [PubMed]
- Bzhalava, D.; Guan, P.; Franceschi, S.; Dillner, J.; Clifford, G. A systematic review of the prevalence of mucosal and cutaneous human papillomavirus types. Virology 2013, 445, 224–231. [Google Scholar] [CrossRef] [PubMed]
- Chouhy, D.; Bolatti, E.M.; Perez, G.R.; Giri, A.A. Analysis of the genetic diversity and phylogenetic relationships of putative human papillomavirus types. J. Gen. Virol. 2013, 94, 2480–2488. [Google Scholar] [CrossRef] [PubMed]
- Dubina, M.; Goldenberg, G. Viral-associated nonmelanoma skin cancers: A review. Am. J. Dermatopathol. 2009, 31, 561–573. [Google Scholar] [CrossRef] [PubMed]
- White, E.A.; Sowa, M.E.; Tan, M.J. A.; Jeudy, S.; Hayes, S.D.; Santha, S.; Munger, K.; Harper, J.W.; Howley, P.M. Systematic identification of interactions between host cell proteins and E7 oncoproteins from diverse human papillomaviruses. Proc. Natl. Acad. Sci. USA 2012, 109, E260–E267. [Google Scholar] [CrossRef] [PubMed]
- White, E.A.; Kramer, R.E.; Tan, M.J.; Hayes, S.D.; Harper, J.W.; Howley, P.M. Comprehensive analysis of host cellular interactions with human papillomavirus E6 proteins identifies new E6 binding partners and reflects viral diversity. J. Virol. 2012, 86, 13174–13186. [Google Scholar] [PubMed]
- Rozenblatt-Rosen, O.; Deo, R.C.; Padi, M.; Adelmant, G.; Calderwood, M.A.; Rolland, T.; Grace, M.; Dricot, A.; Askenazi, M.; Tavares, M.; et al. Interpreting cancer genomes using systematic host network perturbations by tumour virus proteins. Nature 2012, 487, 491–495. [Google Scholar] [PubMed]
- Bernard, H.U. Regulatory elements in the viral genome. Virology 2013, 445, 197–204. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.W.; Roden, R.B. L2, the minor capsid protein of papillomavirus. Virology 2013, 445, 175–186. [Google Scholar] [CrossRef] [PubMed]
- Buck, C.B.; Day, P.M.; Trus, B.L. The papillomavirus major capsid protein L1. Virology 2013, 445, 169–174. [Google Scholar] [CrossRef] [PubMed]
- Vande Pol, S.B.; Klingelhutz, A.J. Papillomavirus E6 oncoproteins. Virology 2013, 445, 115–137. [Google Scholar]
- McBride, A.A. The papillomavirus E2 proteins. Virology 2013, 445, 57–79. [Google Scholar] [CrossRef] [PubMed]
- Bergvall, M.; Melendy, T.; Archambault, J. The E1 proteins. Virology 2013, 445, 35–56. [Google Scholar] [CrossRef] [PubMed]
- DiMaio, D.; Petti, L.M. The E5 proteins. Virology 2013, 445, 99–114. [Google Scholar] [CrossRef] [PubMed]
- Roman, A.; Munger, K. The papillomavirus E7 proteins. Virology 2013, 445, 138–168. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J. The E4 protein; structure, function and patterns of expression. Virology 2013, 445, 80–98. [Google Scholar] [PubMed]
- Moody, C.A.; Laimins, L.A. Human papillomavirus oncoproteins: Pathways to transformation. Nature Rev. Cancer 2010, 10, 550–560. [Google Scholar] [CrossRef]
- Kines, R.C.; Thompson, C.D.; Lowy, D.R.; Schiller, J.T.; Day, P.M. The initial steps leading to papillomavirus infection occur on the basement membrane prior to cell surface binding. Proc. Natl. Acad. Sci. USA 2009, 106, 20458–20463. [Google Scholar]
- Munger, K.; Baldwin, A.; Edwards, K.M.; Hayakawa, H.; Nguyen, C.L.; Owens, M.; Grace, M.; Huh, K. Mechanisms of human papillomavirus-induced oncogenesis. J. Virol. 2004, 78, 11451–11460. [Google Scholar] [CrossRef] [PubMed]
- Frazer, I.H. Interaction of human papillomaviruses with the host immune system: A well evolved relationship. Virology 2009, 384, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Hughes, F.J.; Romanos, M.A. E1 protein of human papillomavirus is a DNA helicase/ATPase. Nucleic Acids Res. 1993, 21, 5817–5823. [Google Scholar] [CrossRef] [PubMed]
- Seo, Y.S.; Müller, F.; Lusky, M.; Hurwitz, J. Bovine papilloma virus (BPV)-encoded E1 protein contains multiple activities required for BPV DNA replication. Proc. Natl. Acad. Sci. USA 1993, 90, 702–706. [Google Scholar] [CrossRef] [PubMed]
- Jenkins, O.; Earnshaw, D.; Sarginson, G.; Delvecchio, A.; Tsai, J.; Kallender, H.; Amegadzie, B.; Browne, M. Characterization of the helicase and atpase activity of human papillomavirus type 6b E1 protein. J. Gen. Virol. 1996, 77, 1805–1809. [Google Scholar] [CrossRef]
- Calistri, A.; Munegato, D.; Carli, I.; Parolin, C.; Palu, G. The ubiquitin-conjugating system: Multiple roles in viral replication and infection. Cells 2014, 3, 386–417. [Google Scholar] [CrossRef] [PubMed]
- Hannoun, Z.; Greenhough, S.; Jaffray, E.; Hay, R.T.; Hay, D.C. Post-translational modification by SUMO. Toxicology 2010, 278, 288–293. [Google Scholar] [PubMed]
- Zhang, D.; Zhang, D.E. Interferon-stimulated gene 15 and the protein ISGylation system. J. Interferon Cytokine Res. 2011, 31, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Watson, I.R.; Irwin, M.S.; Ohh, M. NEDD8 pathways in cancer, Sine Quibus Non. Cancer Cell 2011, 19, 168–176. [Google Scholar] [CrossRef] [PubMed]
- Schmidtke, G.; Aichem, A.; Groettrup, M. FAT10ylation as a signal for proteasomal degradation. Biochim Biophys Acta 2014, 1843, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Van der Veen, A.G.; Ploegh, H.L. Ubiquitin-like proteins. Annu. Rev. Biochem. 2012, 81, 323–357. [Google Scholar] [CrossRef] [PubMed]
- Kerscher, O.; Felberbaum, R.; Hochstrasser, M. Modification of proteins by ubiquitin and ubiquitin-like proteins. Ann. Rev. Cell Develop. Biol. 2006, 22, 159–180. [Google Scholar]
- Wenzel, D.M.; Stoll, K.E.; Klevit, R.E. E2s: Structurally economical and functionally replete. Biochem. J. 2011, 433, 31–42. [Google Scholar] [CrossRef] [PubMed]
- Berndsen, C.E.; Wolberger, C. New insights into ubiquitin E3 ligase mechanism. Nat. Struct. Mol. Biol. 2014, 21, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Desterro, J.M.; Rodriguez, M.S.; Hay, R.T. SUMO-1 modification of IkappaBalpha inhibits NF‑kappaB activation. Mol. Cell 1998, 2, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Klenk, C.; Humrich, J.; Quitterer, U.; Lohse, M.J. SUMO-1 controls the protein stability and the biological function of phosducin. J. Biol. Chem. 2006, 281, 8357–8364. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, C.; Wang, E.; Wang, J. PolyUbiquitin chain linkage topology selects the functions from the underlying binding landscape. PLoS Comput. Biol. 2014, 10, e1003691. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.H.; Wang, H.; Wang, S.; Quon, D.; Liu, Y.W.; Cordell, B. Positive and negative regulation of APP amyloidogenesis by sumoylation. Proc. Natl. Acad. Sci. USA 2003, 100, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Myung, J.; Kim, K.B.; Crews, C.M. The ubiquitin-proteasome pathway and proteasome inhibitors. Medicinal Res. Revv 2001, 21, 245–273. [Google Scholar] [CrossRef]
- Isaacson, M.K.; Ploegh, H.L. Ubiquitination, ubiquitin-like modifiers, and deubiquitination in viral infection. Cell Host Microbe 2009, 5, 559–570. [Google Scholar] [PubMed]
- Wilson, V.G. SUMOylation at the host-pathogen interface. Biomolecules 2012, 2, 203–227. [Google Scholar] [CrossRef] [PubMed]
- Wimmer, P.; Schreiner, S.; Dobner, T. Human pathogens and the host cell SUMOylation system. J. Virol. 2012, 86, 642–654. [Google Scholar] [CrossRef] [PubMed]
- Lenschow, D.J. Antiviral properties of ISG15. Viruses 2010, 2, 2154–2168. [Google Scholar] [CrossRef] [PubMed]
- Geiss-Friedlander, R.; Melchior, F. Concepts in sumoylation: A decade on. Nat. Rev. Mol. Cell Biol. 2007, 8, 947–956. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Li, M.; Zhang, Y.; Yang, P.; Eckenrode, S.; Hopkins, D.; Zheng, W.; Purohit, S.; Podolsky, R.H.; Muir, A.; et al. A functional variant of SUMO4, a new I kappa B alpha modifier, is associated with type 1 diabetes. Nat. Genet. 2004, 36, 837–841. [Google Scholar] [CrossRef] [PubMed]
- Chang, P.C.; Cheng, C.Y.; Campbell, M.; Yang, Y.C.; Hsu, H.W.; Chang, T.Y.; Chu, C.H.; Lee, Y.W.; Hung, C.L.; Lai, S.M.; et al. The chromatin modification by SUMO-2/3 but not SUMO-1 prevents the epigenetic activation of key immune-related genes during Kaposi's sarcoma associated herpesvirus reactivation. BMC Genomics 2013, 14, 824. [Google Scholar] [CrossRef] [PubMed]
- Vertegaal, A.C.O.; Andersen, J.S.; Ogg, S.C.; Hay, R.T.; Mann, M.; Lamond, A.I. Distinct and overlapping sets of SUMO-1 and SUMO-2 target proteins revealed by quantitative proteomics. Mol. Cell. Proteomics 2006, 5, 2298–2310. [Google Scholar] [CrossRef] [PubMed]
- Ayaydin, F.; Dasso, M. Distinct in vivo dynamics of vertebrate SUMO paralogues. Mol. Biol. Cell. 2004, 15, 5208–5218. [Google Scholar] [CrossRef] [PubMed]
- Azuma, Y.; Arnaoutov, A.; Dasso, M. SUMO-2/3 regulates topoisomerase II in mitosis. J. Cell. Biol. 2003, 163, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Yeh, E.T.H. SUMOylation and de-SUMOylation: Wrestling with life's processes. J. Biol. Chem. 2009, 284, 8223–8227. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, J.; Zhao, W.M.; Zhao, K.N. Expression of HPV 58 long and short L1 capsid proteins in primary mouse keratinocyte cultures. Protein Pept. Lett. 2009, 16, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Marusic, M.B.; Mencin, N.; Licen, M.; Banks, L.; Grm, H.S.T. Modification of human papillomavirus minor capsid protein L2 by sumoylation. J. Virol. 2010, 84, 11585–11589. [Google Scholar] [CrossRef] [PubMed]
- Malcles, M.H.; Cueille, N.; Mechali, F.; Coux, O.; Bonne-Andrea, C. Regulation of bovine papillomavirus replicative helicase E1 by the ubiquitin-proteasome pathway. J. Virol. 2002, 76, 11350–11358. [Google Scholar] [CrossRef] [PubMed]
- Bellanger, S.; Demeret, C.; Goyat, S.; Thierry, F. Stability of the human papillomavirus type 18 E2 protein is regulated by a proteasome degradation pathway through its amino-terminal transactivation domain. J. Virol. 2001, 75, 7244–7251. [Google Scholar] [CrossRef] [PubMed]
- Kehmeier, E.; Ruhl, H.; Voland, B.; Stoppler, M.C.; Androphy, E.; Stoppler, H. Cellular steady-state levels of "high risk" but not "low risk" human papillomavirus (HPV) E6 proteins are increased by inhibition of proteasome-dependent degradation independent of their p53-and E6AP-binding capabilities. Virology 2002, 299, 72–87. [Google Scholar] [CrossRef] [PubMed]
- Reinstein, E.; Scheffner, M.; Oren, M.; Ciechanover, A.; Schwartz, A. Degradation of the E7 human papillomavirus oncoprotein by the ubiquitin-proteasome system: Targeting via ubiquitination of the N-terminal residue. Oncogene 2000, 19, 5944–5950. [Google Scholar] [CrossRef] [PubMed]
- Bassermann, F.; Eichner, R.; Pagano, M. The ubiquitin proteasome system—Implications for cell cycle control and the targeted treatment of cancer. Biochim. Biophys. Acta 2014, 1843, 150–162. [Google Scholar] [CrossRef] [PubMed]
- Teixeira, L.K.; Reed, S.I. Ubiquitin ligases and cell cycle control. Annu. Rev. Biochem. 2013, 82, 387–414. [Google Scholar] [CrossRef] [PubMed]
- Strikoudis, A.; Guillamot, M.; Aifantis, I. Regulation of stem cell function by protein ubiquitylation. EMBO Rep. 2014, 15, 365–382. [Google Scholar] [CrossRef] [PubMed]
- Lomeli, H.; Vazquez, M. Emerging roles of the SUMO pathway in development. Cell. Mol. Life Sci. 2011, 68, 4045–4064. [Google Scholar] [CrossRef]
- Mechali, F.; Hsu, C.Y.; Castro, A.; Lorca, T.; Bonne-Andrea, C. Bovine papillomavirus replicative helicase E1 is a target of the ubiquitin ligase APC. J. Virol. 2004, 78, 2615–2619. [Google Scholar] [CrossRef] [PubMed]
- Rangasamy, D.; Wilson, V.G. Bovine papillomavirus E1 protein is sumoylated by the host cell Ubc9 protein. J. Biol. Chem. 2000, 275, 30487–30495. [Google Scholar] [CrossRef]
- Rosas-Acosta, G.; Langereis, M.A.; Deyrieux, A.; Wilson, V.G. Proteins of the PIAS family enhance the sumoylation of the papillomavirus E1 protein. Virology 2005, 331, 190–203. [Google Scholar] [CrossRef] [PubMed]
- Fradet-Turcotte, A.; Brault, K.; Titolo, S.; Howley, P.M.; Archambault, J. Characterization of papillomavirus E1 helicase mutants defective for interaction with the SUMO-conjugating enzyme Ubc9. Virology 2009, 395, 190–201. [Google Scholar] [CrossRef] [PubMed]
- Rangasamy, D.; Woytek, K.; Khan, S.A.; Wilson, V.G. SUMO-1 modification of bovine papillomavirus E1 protein is required for intranuclear accumulation. J. Biol. Chem. 2000, 275, 37999–38004. [Google Scholar] [CrossRef] [PubMed]
- Rosas-Acosta, G.; Wilson, V.G. Identification of a nuclear export signal sequence for bovine papillomavirus E1 protein. Virology 2008, 373, 149–162. [Google Scholar] [CrossRef] [PubMed]
- Gali, H.; Juhasz, S.; Morocz, M.; Hajdu, I.; Fatyol, K.; Szukacsov, V.; Burkovics, P.; Haracska, L. Role of SUMO modification of human PCNA at stalled replication fork. Nucleic Acids Res. 2012, 40, 6049–6059. [Google Scholar] [CrossRef] [PubMed]
- Penrose, K.J.; McBride, A.A. Proteasome-mediated degradation of the papillomavirus E2-TA protein is regulated by phosphorylation and can modulate viral genome copy number. J. Virol. 2000, 74, 6031–6038. [Google Scholar] [CrossRef] [PubMed]
- Blachon, S.; Bellanger, S.; Demeret, C.; Thierry, G. Nucleo-cytoplasmic shuttling of high risk human Papillomavirus E2 proteins induces apoptosis. J. Biol. Chem. 2005, 280, 36088–36098. [Google Scholar] [CrossRef] [PubMed]
- Gagnon, D.; Joubert, S.; Senechal, H.; Fradet-Turcotte, A.; Torre, S.; Archambault, J. Proteasomal degradation of the papillomavirus E2 protein is inhibited by overexpression of bromodomain-containing protein 4. J. Virol. 2009, 83, 4127–4139. [Google Scholar] [CrossRef] [PubMed]
- Bellanger, S.; Tan, C.L.; Nei, W.; He, P.P.; Thierry, F. The human papillomavirus type 18 E2 protein is a cell cycle-dependent target of the SCFSkp2 ubiquitin ligase. J. Virol. 2010, 84, 437–444. [Google Scholar] [CrossRef] [PubMed]
- Gammoh, N.; Gardiol, D.; Massimi, P.; Banks, L. The Mdm2 ubiquitin ligase enhances transcriptional activity of human papillomavirus E2. J. Virol. 2009, 83, 1538–1543. [Google Scholar] [CrossRef] [PubMed]
- Bellanger, S.; Blachon, S.; Mechali, F.; Bonne-Andrea, C.; Thierry, F. High-risk but not low-risk HPV E2 proteins bind to the APC activators Cdh1 and Cdc20 and cause genomic instability. Cell Cycle 2005, 4, 1608–1615. [Google Scholar] [CrossRef] [PubMed]
- Maitland, N.J.; Conway, S.; Wilkinson, N.S.; Ramsdale, J.; Morris, J.R.; Sanders, C.M.; Burns, J.E.; Stern, P.L.; Wells, M. Expression patterns of the human papillomavirus type 16 transcription factor E2 in low- and high-grade cervical intraepithelial neoplasia. J. Pathol. 1998, 186, 275–280. [Google Scholar] [CrossRef] [PubMed]
- Zheng, G.; Schweiger, M.R.; Martinez-Noel, G.; Zheng, L.; Smith, J.A.; Harper, J.W.; Howley, P.M. Brd4 regulation of papillomavirus protein E2 stability. J. Virol. 2009, 83, 8683–8692. [Google Scholar] [CrossRef] [PubMed]
- Ilves, I.; Maemets, K.; Silla, T.; Janikson, K.; Ustav, M. Brd4 is involved in multiple processes of the bovine papillomavirus type 1 life cycle. J. Virol. 2006, 80, 3660–3665. [Google Scholar] [CrossRef] [PubMed]
- Lee, A.Y.; Chiang, C.M. Chromatin adaptor Brd4 modulates E2 transcription activity and protein stability. J. Biol. Chem. 2009, 284, 2778–2786. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, Q.; Diaz, J.; You, J. Brd4-mediated nuclear retention of the papillomavirus E2 protein contributes to its stabilization in host cells. Viruses 2014, 6, 319–335. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.Y.; Naidu, S.R.; Sverdrup, F.; Androphy, E.J. Tax1BP1 interacts with papillomavirus E2 and regulates E2-dependent transcription and stability. J. Virol. 2009, 83, 2274–2284. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.W.; Tsao, Y.P.; Lin, C.Y.; Chen, S.L. NRIP, a novel calmodulin binding protein, activates calcineurin to dephosphorylate human papillomavirus E2 protein. J. Virol. 2011, 85, 6750–6763. [Google Scholar] [CrossRef] [PubMed]
- King, L.E.; Dornan, E.S.; Donaldson, M.M.; Morgan, I.M. Human papillomavirus 16 E2 stability and transcriptional activation is enhanced by E1 via a direct protein-protein interaction. Virology 2011, 414, 26–33. [Google Scholar] [CrossRef] [PubMed]
- Davy, C.; McIntosh, P.; Jackson, D.J.; Sorathia, R.; Miell, M.; Wang, Q.; Khan, J.; Soneji, Y.; Doorbar, J. A novel interaction between the human papillomavirus type 16 E2 and E1^E4 proteins leads to stabilization of E2. Virology 2009, 394, 266–275. [Google Scholar] [CrossRef] [PubMed]
- Shembade, N.; Harhaj, N.S.; Liebl, D.J.; Harhaj, E.W. Essential role for TAX1BP1 in the termination of TNF-alpha-, IL-1- and LPS-mediated NF-kappaB and JNK signaling. EMBO J. 2007, 26, 3910–3922. [Google Scholar] [PubMed]
- Johansson, C.; Graham, S.V.; Dornan, E.S.; Morgan, I.M. The human papillomavirus 16 E2 protein is stabilised in S phase. Virology 2009, 394, 194–199. [Google Scholar] [CrossRef] [PubMed]
- Sekhar, V.; McBride, A.A. Phosphorylation regulates binding of the human papillomavirus type 8 E2 protein to host chromosomes. J. Virol. 2012, 86, 10047–10058. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.C.; Deyrieux, A.F.; Wilson, V.G. Papillomaviruses and the host SUMOylation system. Biochem. Soc. Trans. 2007, 35, 1433–1435. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.C.; Roark, A.A.; Bian, X.L.; Wilson, V.G. Modification of papillomavirus E2 proteins by the small ubiquitin-like modifier family members (SUMOs). Virology 2008, 378, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.C.; Bian, X.L.; Heaton, P.H.; Deyrieux, A.F.; Wilson, V.G. Host cell sumoylation level influences papillomavirus E2 protein stability. Virology 2009, 387, 176–183. [Google Scholar] [CrossRef] [PubMed]
- Deyrieux, A.F.; Rosas-Acosta, G.; Ozbun, M.A.; Wilson, V.G. Sumoylation dynamics during keratinocyte differentiation. J. Cell. Sci. 2007, 120, 125–136. [Google Scholar] [CrossRef] [PubMed]
- Maglennon, G.A.; McIntosh, P.; Doorbar, J. Persistence of viral DNA in the epithelial basal layer suggests a model for papillomavirus latency following immune regression. Virology 2011, 414, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Doorbar, J.; Foo, C.; Coleman, N.; Medcalf, L.; Hartley, O.; Prospero, T.; Napthine, S.; Sterling, J.; Winter, G.; Griffin, H. Characterization of events during the late stages of HPV16 infection in vivo using high-affinity synthetic fabs to E4. Virology 1997, 238, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Kennedy, A.; Das, P.; McIntosh, P.B.; Howell, S.A.; Isaacson, E.R.; Hinz, S.A.; Davy, C.; Doorbar, J. Phosphorylation of the human papillomavirus type 16 E1^E4 Protein at T57 by ERK triggers a structural change that enhances keratin binding and protein stability. J. Virol. 2009, 83, 3668–3683. [Google Scholar] [CrossRef] [PubMed]
- Davy, C.E.; Ayub, M.; Jackson, D.J.; Das, P.; McIntosh, P.; Doorbar, J. HPV16 E1^E4 protein is phosphorylated by Cdk2/cyclin A and relocalizes this complex to the cytoplasm. Virology 2006, 349, 230–244. [Google Scholar] [CrossRef] [PubMed]
- Ding, Q.; Li, L.; Whyte, P. Human papillomavirus 18 E1^E4 protein interacts with cyclin A/CDK 2 through an RXL motif. Mol. Cell. Biochem. 2013, 373, 29–40. [Google Scholar] [CrossRef]
- McIntosh, P.B.; Martin, S.R.; Jackson, D.J.; Khan, J.; Isaacson, E.R.; Calder, L.; Raj, K.; Griffin, H.M.; Wang, Q.; Laskey, P.; Eccleston, J.F.; Doorbar, J. Structural analysis reveals an amyloid form of the human papillomavirus type 16 E1^E4 protein and provides a molecular basis for its accumulation. J. Virol. 2008, 82, 8196–8203. [Google Scholar] [CrossRef] [PubMed]
- Khan, J.; Davy, C.E.; McIntosh, P.B.; Jackson, D.J.; Hinz, S.; Wang, Q.; Doorbar, J. Role of calpain in the formation of human papillomavirus type 16 E1^E4 amyloid fibers and reorganization of the keratin network. J. Virol. 2011, 85, 9984–9997. [Google Scholar] [CrossRef] [PubMed]
- Grossman, S.R.; Mora, R.; Laimins, L.A. Intracellular localization and DNA-binding properties of human papillomavirus type 18 E6 protein expressed with a baculovirus vector. J. Virol. 1989, 63, 366–374. [Google Scholar] [PubMed]
- Androphy, E.J.; Hubbert, N.L.; Schiller, J.T.; Lowy, D.R. Identification of the HPV-16 E6 protein from transformed mouse cells and human cervical carcinoma cell lines. EMBO J. 1987, 6, 989–992. [Google Scholar]
- Stewart, D.; Kazemi, S.; Li, S.Y.; Massimi, P.; Banks, L.; Koromilas, A.E.; Matlashewski, G. Ubiquitination and proteasome degradation of the E6 proteins of human papillomavirus types 11 and 18. J. Gen. Virol. 2004, 85, 1419–1426. [Google Scholar] [CrossRef] [PubMed]
- Huibregtse, J.M.; Scheffner, M.; Howley, P.M. A cellular protein mediates association of p53 with the E6 oncoprotein of human papillomavirus types 16 or 18. EMBO J. 1991, 10, 4129–4135. [Google Scholar] [PubMed]
- Tomaic, V.; Pim, D.; Banks, L. The stability of the human papillomavirus E6 oncoprotein is E6AP dependent. Virology 2009, 393, 7–10. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Tomaic, V.; Pim, D.; Myers, M.P.; Tommasino, M.; Banks, L. Interactions between E6AP and E6 proteins from alpha and beta HPV types. Virology 2013, 435, 357–362. [Google Scholar] [CrossRef] [PubMed]
- Vos, R.M.; Altreuter, J.; White, E.A.; Howley, P.M. The Ubiquitin-Specific Peptidase USP15 Regulates Human Papillomavirus Type 16 E6 Protein Stability. J. Virol. 2009, 83, 8885–8892. [Google Scholar] [CrossRef] [PubMed]
- Tomaic, V.; Pim, D.; Thomas, M.; Massimi, P.; Myers, M.P.; Banks, L. Regulation of the human papillomavirus type 18 E6/E6AP ubiquitin ligase complex by the HECT domain-containing protein EDD. J. Virol. 2011, 85, 3120–3127. [Google Scholar] [CrossRef] [PubMed]
- Kuhnle, S.; Kogel, U.; Glockzin, S.; Marquardt, A.; Ciechanover, A.; Matentzoglu, K.; Scheffner, M. Physical and functional interaction of the HECT ubiquitin-protein ligases E6AP and HERC2. J. Biol. Chem. 2011, 286, 19410–19416. [Google Scholar] [CrossRef]
- Martinez-Noel, G.; Galligan, J.T.; Sowa, M.E.; Arndt, V.; Overton, T.M.; Harper, J.W.; Howley, P.M. Identification and proteomic analysis of distinct UBE3A/E6AP protein complexes. Mol. Cell. Biol. 2012, 32, 3095–3106. [Google Scholar] [CrossRef] [PubMed]
- Nicolaides, L.; Davy, C.; Raj, K.; Kranjec, C.; Banks, L.; Doorbar, J. Stabilization of HPV16 E6 protein by PDZ proteins, and potential implications for genome maintenance. Virology 2011, 414, 137–145. [Google Scholar] [CrossRef] [PubMed]
- Gardiol, D.; Kuhne, C.; Glaunsinger, B.; Lee, S.S.; Javier, R.; Banks, L. Oncogenic human papillomavirus E6 proteins target the discs large tumour suppressor for proteasome-mediated degradation. Oncogene 1999, 18, 5487–5496. [Google Scholar] [CrossRef] [PubMed]
- Nakagawa, S.; Huibregtse, J.M. Human scribble (Vartul) is targeted for ubiquitin-mediated degradation by the high-risk papillomavirus E6 proteins and the E6AP ubiquitin-protein ligase. Mol. Cell. Biol. 2000, 20, 8244–8253. [Google Scholar] [CrossRef] [PubMed]
- Selvey, L.A.; Dunn, L.A.; Tindle, R.W.; Park, D.S.; Frazer, I.H. Human papillomavirus (HPV) type 18 E7 protein is a short-lived steroid-inducible phosphoprotein in HPV-transformed cell lines. J. Gen. Virol. 1994, 75, 1647–1653. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Sampath, A.; Raychaudhuri, P.; Bagchi, S. Both Rb and E7 are regulated by the ubiquitin proteasome pathway in HPV-containing cervical tumor cells. Oncogene 2001, 20, 4740–4749. [Google Scholar] [CrossRef] [PubMed]
- Ben-Saadon, R.; Fajerman, I.; Ziv, T.; Hellman, U.; Schwartz, A.L.; Ciechanover, A. The tumor suppressor protein p16(INK4a) and the human papillomavirus oncoprotein-58 E7 are naturally occurring lysine-less proteins that are degraded by the ubiquitin system—Direct evidence for ubiquitination at the N-terminal residue. J. Biol. Chem. 2004, 279, 41414–41421. [Google Scholar] [CrossRef] [PubMed]
- Kamio, M.; Yoshida, T.; Ogata, H.; Douchi, T.; Nagata, Y.; Inoue, M.; Hasegawa, M.; Yonemitsu, Y.; Yoshimura, A. SOC1 inhibits HPV-E7-mediated transformation by inducing degradation of E7 protein. Oncogene 2004, 23, 9449. [Google Scholar] [CrossRef]
- Zhang, J.G.; Farley, A.; Nicholson, S.E.; Willson, T.A.; Zugaro, L.M.; Simpson, R.J.; Moritz, R.L.; Cary, D.; Richardson, R.; Hausmann, G.; et al. The conserved SOCS box motif in suppressors of cytokine signaling binds to elongins B and C and may couple bound proteins to proteasomal degradation. Proc. Natl. Acad. Sci. USA 1999, 96, 2071–2076. [Google Scholar]
- Oh, K.J.; Kalinina, A.; Wang, J.; Nakayama, K.; Nakayama, K.I.; Bagchi, S. The papillomavirus E7 oncoprotein is ubiquitinated by UbcH7 and Cullin 1- and Skp2-containing E3 ligase. J. Virol. 2004, 78, 5338–5346. [Google Scholar] [CrossRef] [PubMed]
- Huh, K.; Zhou, X.; Hayakawa, H.; Cho, J.Y.; Libermann, T.A.; Jin, J.; Harper, J.W.; Munger, K. Human papillomavirus type 16 E7 oncoprotein associates with the cullin 2 ubiquitin ligase complex, which contributes to degradation of the retinoblastoma tumor suppressor. J. Virol. 2007, 81, 9737–9747. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Chang, H.S.; Yu, W.C.Y. USP11 stabilizes HPV-16E7 and further modulates the E7 biological activity. J. Biol. Chem. 2008, 283, 15681–15688. [Google Scholar] [CrossRef] [PubMed]
- Bund, T.; Spoden, G.A.; Koynov, K.; Hellmann, N.; Boukhallouk, F.; Arnold, P.; Hinderberger, D.; Florin, L. An L2 SUMO interacting motif is important for PML localization and infection of human papillomavirus type 16. Cell Microbiol. 2014, 16, 1179–1200. [Google Scholar] [CrossRef] [PubMed]
- Durfee, L.A.; Lyon, N.; Seo, K.; Huibregtse, J.M. The ISG15 conjugation system broadly targets newly synthesized proteins: Implications for the antiviral function of ISG15. Mol. Cell 2010, 38, 722–732. [Google Scholar] [CrossRef]
- Wilson, V.G.; Heaton, P.R. Ubiquitin proteolytic system: Focus on SUMO. Expert Rev. Proteomics 2008, 5, 121–135. [Google Scholar] [CrossRef] [PubMed]
- Porras-Yakushi, T.R.; Hess, S. Recent advances in defining the ubiquitylome. Expert Rev. Proteomics 2014, 11, 477–490. [Google Scholar] [CrossRef] [PubMed]
- Lou, Z.; Wang, S. E3 ubiquitin ligases and human papillomavirus-induced carcinogenesis. J. Int. Med. Res. 2014, 42, 247–260. [Google Scholar] [CrossRef] [PubMed]
- Cote-Martin, A.; Moody, C.; Fradet-Turcotte, A.; D'Abramo, C.M.; Lehoux, M.; Joubert, S.; Poirier, G.G.; Coulombe, B.; Laimins, L.A.; Archambault, J. Human papillomavirus E1 helicase interacts with the WD repeat protein p80 to promote maintenance of the viral genome in keratinocytes. J. Virol. 2008, 82, 1271–1283. [Google Scholar] [CrossRef] [PubMed]
- Lehoux, M.; Fradet-Turcotte, A.; Lussier-Price, M.; Omichinski, J.G.; Archambault, J. Inhibition of human papillomavirus DNA replication by an E1-derived p80/UAF1-binding peptide. J. Virol. 2012, 86, 3486–3500. [Google Scholar] [CrossRef] [PubMed]
- Lehoux, M.; Gagnon, D.; Archambault, J. E1-mediated recruitment of a UAF1-USP deubiquitinase complex facilitates human papillomavirus DNA replication. J. Virol. 2014, 88, 8545–8555. [Google Scholar] [CrossRef] [PubMed]
- Joo, H.Y.; Jones, A.; Yang, C.; Zhai, L.; Smith, A.D.T.; Zhang, Z.; Chandrasekharan, M.B.; Sun, Z.W.; Renfrow, M.B.; Wang, Y.; et al. Regulation of histone H2A and H2B deubiquitination and Xenopus development by USP12 and USP46. J. Biol. Chem. 2011, 286, 7190–7201. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.T.; Nijman, S.M.; Mirchandani, K.D.; Galardy, P.J.; Cohn, M.A.; Haas, W.; Gygi, S.P.; Ploegh, H.L.; Bernards, R.; D'Andrea, A.D. Regulation of monoubiquitinated PCNA by DUB autocleavage. Nat. Cell Biol. 2006, 8, 339–347. [Google Scholar] [PubMed]
- Nijman, S.M.; Huang, T.T.; Dirac, A.M.; Brummelkamp, T.R.; Kerkhoven, R.M.; D'Andrea, A.D.; Bernards, R. The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Mol. Cell 2005, 17, 331–339. [Google Scholar] [CrossRef] [PubMed]
- Geng, F.; Wenzel, S.; Tansey, W.P. Ubiquitin and proteasomes in transcription. Annu. Rev. Biochem. 2012, 81, 177–201. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.; Demeret, C. The HPV E2-host protein-protein interactions: A complex hijacking of the cellular network. Open Virol. J. 2012, 6, 173–189. [Google Scholar] [CrossRef] [PubMed]
- Muller, M.; Jacob, Y.; Jones, L.; Weiss, A.; Brino, L.; Chantier, T.; Lotteau, V.; Favre, M.; Demeret, C. Large scale genotype comparison of human papillomavirus E2-host interaction networks provides new insights for E2 molecular functions. PLoS Pathog. 2012, 8, e1002761. [Google Scholar] [CrossRef]
- Palvimo, J.J. PIAS proteins as regulators of small ubiquitin-related modifier (SUMO) modifications and transcription. Biochem. Soc. Trans. 2007, 35, 1405–1408. [Google Scholar] [CrossRef] [PubMed]
- Bryan, J.T.; Brown, D.R. Association of the human papillomavirus type 11 E1^E4 protein with cornified cell envelopes derived from infected genital epithelium. Virology 2000, 277, 262–269. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, P.B.; Laskey, P.; Sullivan, K.; Davy, C.; Wang, Q.A.; Jackson, D.J.; Griffin, H.M.; Doorbar, J. E1^E4-mediated keratin phosphorylation and ubiquitylation: A mechanism for keratin depletion in HPV16-infected epithelium. J. Cell. Sci. 2010, 123, 2810–2822. [Google Scholar] [CrossRef] [PubMed]
- Venuti, A.; Paolini, F.; Nasir, L.; Corteggio, A.; Roperto, S.; Campo, M.S.; Borzacchiello, G. Papillomavirus E5: The smallest oncoprotein with many functions. Mol. Cancer 2011, 10, 140. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.Y.; Srirangam, A.; Potter, D.A.; Roman, A. HPV16 E5 protein disrupts the c-Cbl-EGFR interaction and EGFR ubiquitination in human foreskin keratinocytes. Oncogene 2005, 24, 2585–2588. [Google Scholar] [CrossRef] [PubMed]
- Belleudi, F.; Leone, L.; Purpura, V.; Cannella, F.; Scrofani, C.; Torrisi, M.R. HPV16 E5 affects the KGFR/FGFR2b-mediated epithelial growth through alteration of the receptor expression, signaling and endocytic traffic. Oncogene 2011, 30, 4963–4976. [Google Scholar] [CrossRef] [PubMed]
- Miura, S.; Kawana, K.; Schust, D.J.; Fujii, T.; Yokoyama, T.; Iwasawa, Y.; Nagamatsu, T.; Adachi, K.; Tomio, A.; Tomio, K.; et al. CD1d, a sentinel molecule bridging innate and adaptive immunity, is downregulated by the human papillomavirus (HPV) E5 protein: A possible mechanism for immune evasion by HPV. J. Virol. 2010, 84, 11614–11623. [Google Scholar] [CrossRef] [PubMed]
- Oh, J.M.; Kim, S.H.; Cho, E.A.; Song, Y.S.; Kim, W.H.; Juhnn, Y.S. Human papillomavirus type 16 E5 protein inhibits hydrogen-peroxide-induced apoptosis by stimulating ubiquitin-proteasome-mediated degradation of Bax in human cervical cancer cells. Carcinogenesis 2010, 31, 402–410. [Google Scholar] [CrossRef] [PubMed]
- Pedroza-Saavedra, A.; Lam, E.W.F.; Esquivel-Guadarrama, F.; Gutierrez-Xicotencatl, L. The human papillomavirus type 16 E5 oncoprotein synergizes with EGF-receptor signaling to enhance cell cycle progression and the down-regulation of p27(Kip1). Virology 2010, 400, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Kabsch, K.; Alonso, A. The human papillomavirus type 16 E5 protein impairs TRAIL- and FasL-mediated apoptosis in HaCaT cells by different mechanisms. J. Virol. 2002, 76, 12162–12172. [Google Scholar] [CrossRef] [PubMed]
- Kelley, M.L.; Keiger, K.E.; Lee, C.J.; Huibregtse, J.M. The global transcriptional effects of the human papillomavirus E6 protein in cervical carcinoma cell lines are mediated by the E6AP ubiquitin ligase. J. Virol. 2005, 79, 3737–3747. [Google Scholar] [PubMed]
- Kuhnle, S.; Mothes, B.; Matentzoglu, K.; Scheffner, M. Role of the ubiquitin ligase E6AP/UBE3A in controlling levels of the synaptic protein. Proc. Natl. Acad. Sci. USA 2013, 110, 8888–8893. [Google Scholar] [PubMed]
- Shai, A.; Pitot, H.C.; Lambert, P.F. E6-associated protein is required for human papillomavirus type 16 E6 to cause cervical cancer in mice. Cancer Res. 2010, 70, 5064–5073. [Google Scholar] [PubMed]
- Scheffner, M.; Huibregtse, J.M.; Vierstra, R.D.; Howley, P.M. The HPV-16 E6 and E6-AP complex functions as a ubiquitin-protein ligase in the ubiquitination of p53. Cell 1993, 75, 495–505. [Google Scholar] [CrossRef] [PubMed]
- Scheffner, M.; Werness, B.A.; Huibregtse, J.M.; Levine, A.J.; Howley, P.M. The E6 oncoprotein encoded by human papillomavirus types 16 and 18 promotes the degradation of p53. Cell 1990, 63, 1129–1136. [Google Scholar] [CrossRef] [PubMed]
- Beaudenon, S.; Huibregtse, J.M. HPV E6, E6AP and cervical cancer. BMC Biochem. 2008, 9, S4. [Google Scholar] [PubMed]
- Kiyono, T.; Hiraiwa, A.; Fujita, M.; Hayashi, Y.; Akiyama, T.; Ishibashi, M. Binding of high-risk human papillomavirus E6 oncoproteins to the human homologue of the drosophila discs large tumor suppressor protein. Proc. Natl. Acad. Sci. USA 1997, 94, 11612–11616. [Google Scholar] [CrossRef]
- Glaunsinger, B.A.; Lee, S.S.; Thomas, M.; Banks, L.; Javier, R. Interactions of the PDZ-protein MAGI-1 with adenovirus E4-ORF1 and high-risk papillomavirus E6 oncoproteins. Oncogene 2000, 19, 5270–5280. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.S.; Glaunsinger, B.; Mantovani, F.; Banks, L.; Javier, R.T. Multi-PDZ domain protein MUPP1 is a cellular target for both adenovirus E4-ORF1 and high-risk papillomavirus type 18 E6 oncoproteins. J. Virol. 2000, 74, 9680–9693. [Google Scholar] [CrossRef] [PubMed]
- Lee, H.J.; Zheng, J.J. PDZ domains and their binding partners: Structure, specificity, and modification. Cell Commun. Signal. 2010, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Kuballa, P.; Matentzoglu, K.; Scheffner, M. The role of the ubiquitin ligase E6-AP in human papillomavirus E6-mediated degradation of PDZ domain-containing proteins. J. Biol. Chem. 2007, 282, 65–71. [Google Scholar] [CrossRef] [PubMed]
- Brimer, N.; Lyons, C.; Pol, S.B.V. Association of E6AP (UBE3A) with human papillomavirus type 11 E6 protein. Virology 2007, 358, 303–310. [Google Scholar] [CrossRef] [PubMed]
- Huibregtse, J.M.; Scheffner, M.; Howley, P.M. Localization of the E6-AP regions that direct human papillomavirus E6 binding, association with p53, and ubiquitination of associated proteins. Mol. Cell. Biol. 1993, 13, 4918–4927. [Google Scholar]
- Tong, X.; Boll, W.; Kirchhausen, T.; Howley, P.M. Interaction of the bovine papillomavirus E6 Protein with the clathrin adaptor complex AP-1. J. Virol. 1998, 72, 476–482. [Google Scholar] [PubMed]
- Chen, J.J.; Hong, Y.H.; Rustamzadeh, E.; Baleja, J.D.; Androphy, E.J. Identification of an alpha helical motif sufficient for association with papillomavirus E6. J. Biol. Chem. 1998, 273, 13537–13544. [Google Scholar] [CrossRef] [PubMed]
- Werness, B.A.; Levine, A.J.; Howley, P.M. Association of human papillomavirus types 16 and 18 E6 proteins with p53. Science 1990, 248, 76–79. [Google Scholar] [CrossRef] [PubMed]
- Tan, M.J.A.; White, E.A.; Sowa, M.E.; Harper, J.W.; Aster, J.C.; Howley, P.M. Cutaneous beta-human papillomavirus E6 proteins bind Mastermind-like coactivators and repress Notch signaling. Proc. Natl. Acad. Sci. USA 2012, 109, E1473–E1480. [Google Scholar] [CrossRef] [PubMed]
- Brimer, N.; Lyons, C.; Wallberg, A.E.; Vande Pol, S.B. Cutaneous papillomavirus E6 oncoproteins associate with MAML1 to repress transactivation and NOTCH signaling. Oncogene 2012, 31, 4639–4646. [Google Scholar] [CrossRef] [PubMed]
- Pim, D.; Bergant, M.; Boon, S.S.; Ganti, K.; Kranjec, C.; Massimi, P.; Subbaiah, V.K.; Thomas, M.; Tomaic, V.; Banks, L. Human papillomaviruses and the specificity of PDZ domain targeting. FEBS J. 2012, 279, 3530–3537. [Google Scholar] [CrossRef] [PubMed]
- Klingelhutz, A.J.; Roman, A. Cellular transformation by human papillomaviruses: Lessons learned by comparing high- and low-risk viruses. Virology 2012, 424, 77–98. [Google Scholar] [CrossRef] [PubMed]
- Mighty, K.K.; Laimins, L.A. The role of human papillomaviruses in oncogenesis. Recent Results Cancer Res. 2014, 193, 135–148. [Google Scholar] [PubMed]
- Howie, H.L.; Katzenellenbogen, R.A.; Galloway, D.A. Papillomavirus E6 proteins. Virology 2009, 384, 324–334. [Google Scholar] [CrossRef] [PubMed]
- Bedard, K.M.; Underbrink, M.P.; Howie, H.L.; Galloway, D.A. The E6 oncoproteins from human betapapillomaviruses differentially activate telomerase through an E6AP-dependent mechanism and prolong the lifespan of primary keratinocytes. J. Virol. 2008, 82, 3894–3902. [Google Scholar] [CrossRef] [PubMed]
- Underbrink, M.P.; Howie, H.L.; Bedard, K.M.; Koop, J.I.; Galloway, D.A. E6 proteins from multiple human betapapillomavirus types degrade Bak and protect keratinocytes from apoptosis after UVB irradiation. J. Virol. 2008, 82, 10408–10417. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Banks, L. Inhibition of Bak-induced apoptosis by HPV-18 E6. Oncogene 1998, 17, 2943–2954. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.; Banks, L. Human papillomavirus (HPV) E6 interactions with Bak are conserved amongst E6 proteins from high and low risk HPV types. J. Gen. Virol. 1999, 80, 1513–1517. [Google Scholar] [PubMed]
- Westphal, D.; Dewson, G.; Czabotar, P.E.; Kluck, R.M. Molecular biology of Bax and Bak activation and action. Biochim. Biophys. Acta 2011, 1813, 521–531. [Google Scholar] [CrossRef] [PubMed]
- Tomaic, V.; Ganti, K.; Pim, D.; Bauer, C.; Blattner, C.; Banks, L. Interaction of HPV E6 oncoproteins with specific proteasomal subunits. Virology 2013, 446, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Bischof, O.; Schwamborn, K.; Martin, N.; Werner, A.; Sustmann, C.; Grosschedl, R.; Dejean, A. The E3 SUMO ligase PIASy is a regulator of cellular senescence and apoptosis. Mol. Cell 2006, 22, 783–794. [Google Scholar] [CrossRef] [PubMed]
- Heaton, P.R.; Deyrieux, A.F.; Bian, X.L.; Wilson, V.G. HPV E6 proteins target Ubc9, the SUMO conjugating enzyme. Virus Res. 2011, 158, 199–208. [Google Scholar] [CrossRef] [PubMed]
- Heaton, P.R.; Santos, A.; Rosas-Acosta, G.; Wilson, V.G. Analysis of global sumoylation changes occurring during keratinocyte differentiation. PLoS One 2012, 7, e30165. [Google Scholar] [CrossRef] [PubMed]
- Chand, V.; John, R.; Jaiswal, N.; Johar, S.S.; Nag, A. High-risk HPV16E6 stimulates hADA3 degradation by enhancing its SUMOylation. Carcinogenesis 2014, 35, 1830–1839. [Google Scholar]
- Berezutskaya, E.; Yu, B.; Morozov, A.; Raychaudhuri, P.; Bagchi, S. Differential regulation of the pocket domains of the retinoblastoma family proteins by the HPV16 E7 oncoprotein. Cell Growth Differen. 1997, 8, 1277–1286. [Google Scholar]
- Boyer, S.N.; Wazer, D.E.; Band, V. E7 protein of human papilloma virus-16 induces degradation of retinoblastoma protein through the ubiquitin-proteasome pathway. Cancer Res. 1996, 56, 4620–4624. [Google Scholar] [PubMed]
- Demers, G.W.; Foster, S.A.; Halbert, C.L.; Galloway, D.A. Growth arrest by induction of p53 in DNA damaged keratinocytes is bypassed by human papillomavirus 16 E7. Proc. Natl. Acad. Sci. USA 1994, 91, 4382–4386. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.L.; Munger, K. Analysis of the p53-mediated G1 growth arrest pathway in cells expressing the human papillomavirus type 16 E7 oncoprotein. J. Virol. 1997, 71, 2905–2912. [Google Scholar] [PubMed]
- Oh, K.J.; Kalinina, A.; Bagchi, S. Destabilization of Rb by human papillomavirus E7 is cell cycle dependent: E2–25K is involved in the proteolysis. Virology 2010, 396, 118–124. [Google Scholar] [CrossRef] [PubMed]
- Classon, M.; Dyson, N. p107 and p130: Versatile proteins with interesting pockets. Exp. Cell. Res. 2001, 264, 135–147. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, S.L.; Stremlau, M.; He, X.; Basile, J.R.; Munger, K. Degradation of the retinoblastoma tumor suppressor by the human papillomavirus type 16 E7 oncoprotein is important for functional inactivation and is separable from proteasomal degradation of E7. J. Virol. 2001, 75, 7583–7591. [Google Scholar] [PubMed]
- Helt, A.M.; Galloway, D.A. Destabilization of the retinoblastoma tumor suppressor by human papillomavirus type 16 E7 is not sufficient to overcome cell cycle arrest in human keratinocytes. J. Virol. 2001, 75, 6737–6747. [Google Scholar] [CrossRef] [PubMed]
- Zhang, B.Y.; Chen, W.; Roman, A. The E7 proteins of low- and high-risk human papillomaviruses share the ability to target the pRB family member p130 for degradation. Proc. Natl. Acad. Sci. USA 2006, 103, 437–442. [Google Scholar] [CrossRef] [PubMed]
- Westphal, K.; Akgul, B.; Storey, A.; Nindl, I. Cutaneous human papillomavirus E7 type-specific effects on differentiation and proliferation of organotypic skin cultures. Cell. Oncol. 2009, 31, 213–226. [Google Scholar] [PubMed]
- Spardy, N.; Covella, K.; Cha, E.; Hoskins, E.E.; Wells, S.I.; Duensing, A.; Duensing, S. Human papillomavirus 16 E7 oncoprotein attenuates DNA damage checkpoint control by increasing the proteolytic turnover of claspin. Cancer Res. 2009, 69, 7022–7029. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Munger, K. Human papillomavirus type 16 E7 oncoprotein inhibits the anaphase promoting complex/cyclosome activity by dysregulating EMI1 expression in mitosis. Virology 2013, 446, 251–259. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.Y.; Munger, K. Human papillomavirus type 16 E7 oncoprotein engages but does not abrogate the mitotic spindle assembly checkpoint. Virology 2012, 432, 120–126. [Google Scholar] [CrossRef] [PubMed]
- Mo, M.; Shahar, S.; Fleming, S.B.; Mercer, A.A. How viruses affect the cell cycle through manipulation of the APC/C. Trends Microbiol. 2012, 20, 440–448. [Google Scholar] [CrossRef] [PubMed]
- Berezutskaya, E.; Bagchi, S. The human papillomavirus E7 oncoprotein functionally interacts with the S4 subunit of the 26S proteasome. J. Biol. Chem. 1997, 272, 30135–30140. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.A.; Shim, J.H.; Kho, C.W.; Park, S.G.; Park, B.C.; Kim, J.W.; Lim, J.S.; Choe, Y.K.; Paik, S.G.; Yoon, D.Y. Protein profiling and identification of modulators regulated by the E7 oncogene in the C33A cell line by proteomics and genomics. Proteomics 2004, 4, 839–848. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
MDPI and ACS Style
Wilson, V.G. The Role of Ubiquitin and Ubiquitin-Like Modification Systems in Papillomavirus Biology. Viruses 2014, 6, 3584-3611. https://doi.org/10.3390/v6093584
AMA Style
Wilson VG. The Role of Ubiquitin and Ubiquitin-Like Modification Systems in Papillomavirus Biology. Viruses. 2014; 6(9):3584-3611. https://doi.org/10.3390/v6093584
Chicago/Turabian StyleWilson, Van G. 2014. "The Role of Ubiquitin and Ubiquitin-Like Modification Systems in Papillomavirus Biology" Viruses 6, no. 9: 3584-3611. https://doi.org/10.3390/v6093584