HIV-1 Latency: An Update of Molecular Mechanisms and Therapeutic Strategies

{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
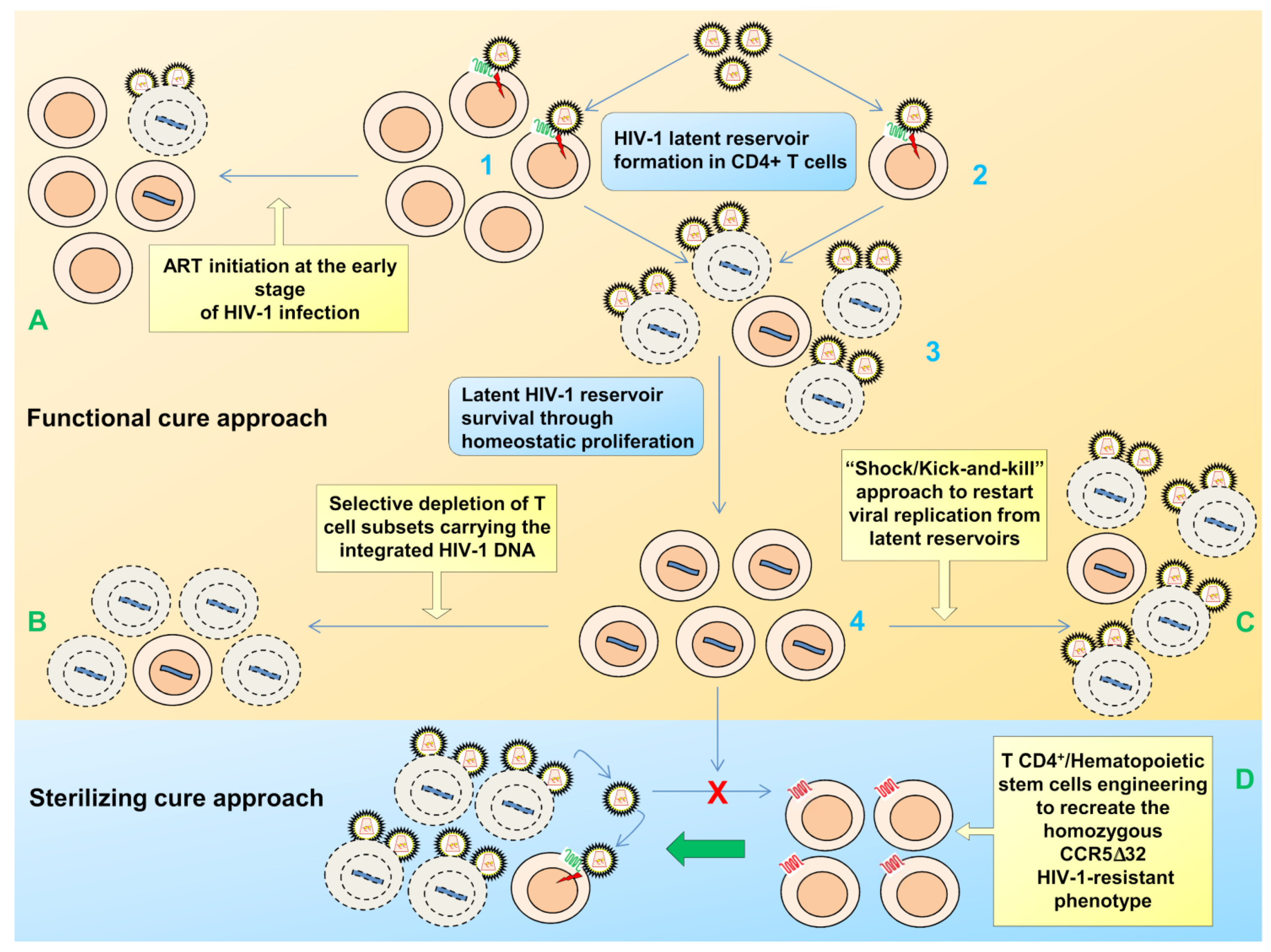
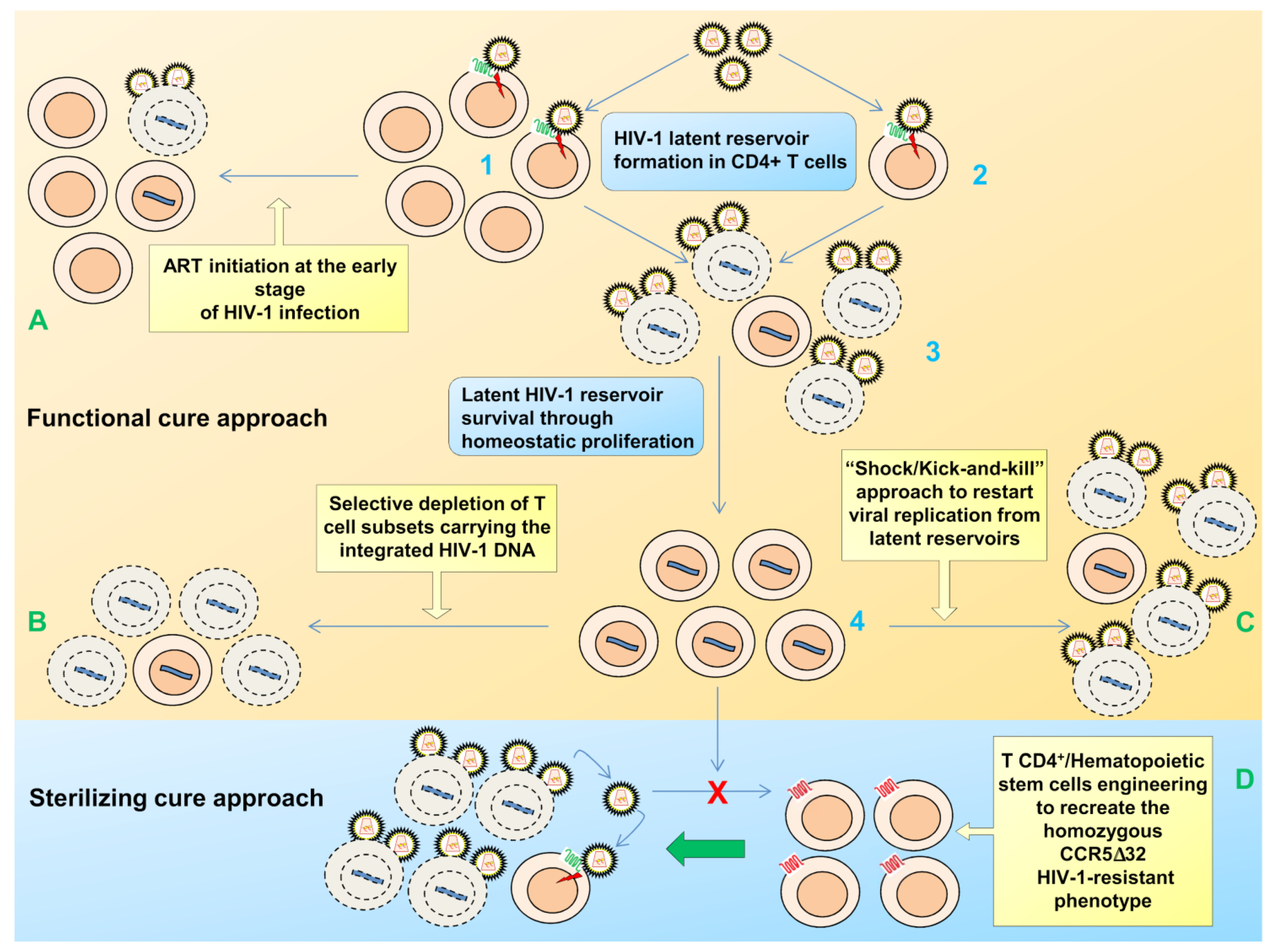
2. Viral Persistence and Reservoirs
3. Mechanisms of Post Integration HIV-1 Latency
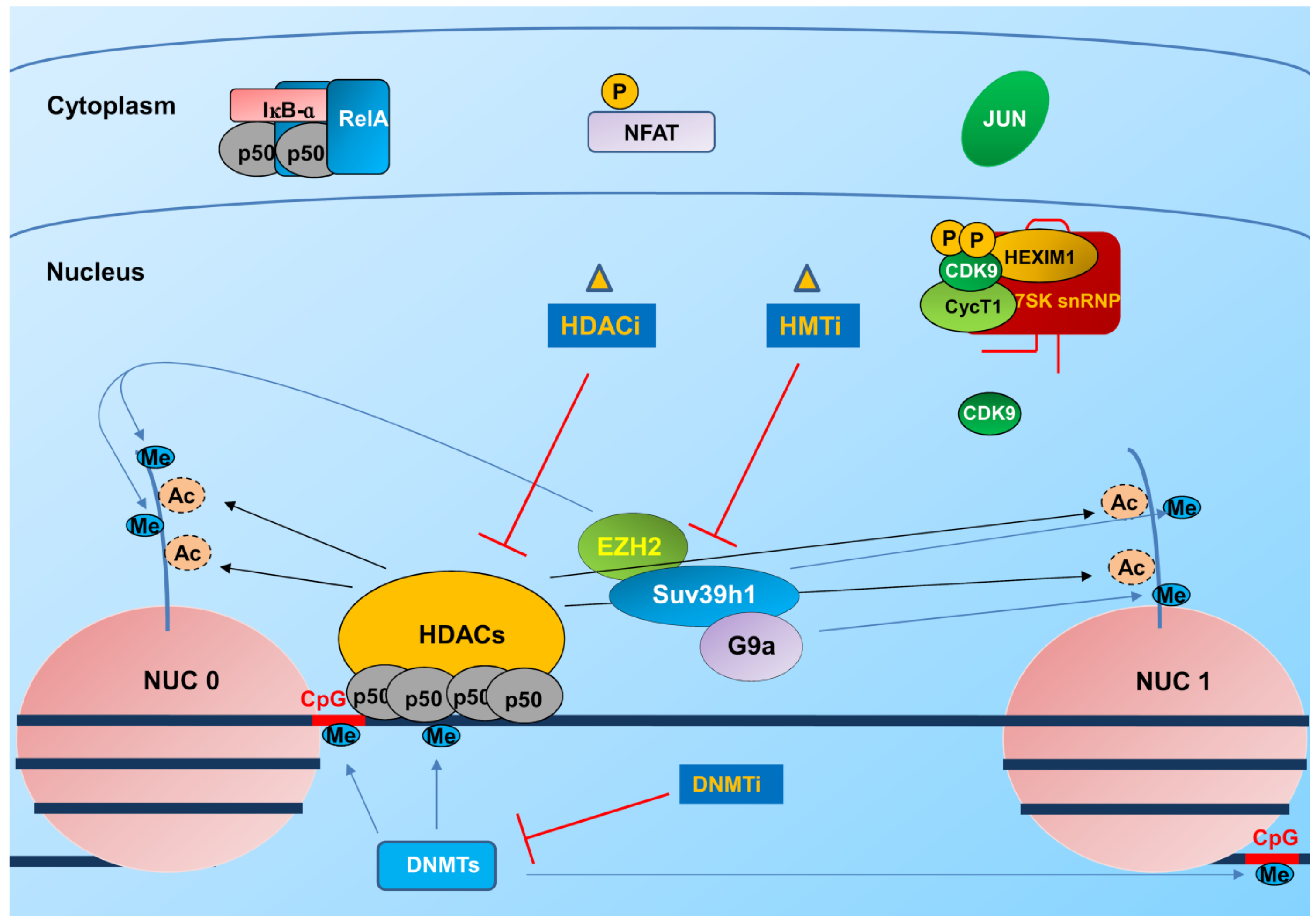
3.1. Integration and Chromatin Organization at the Viral Promoter
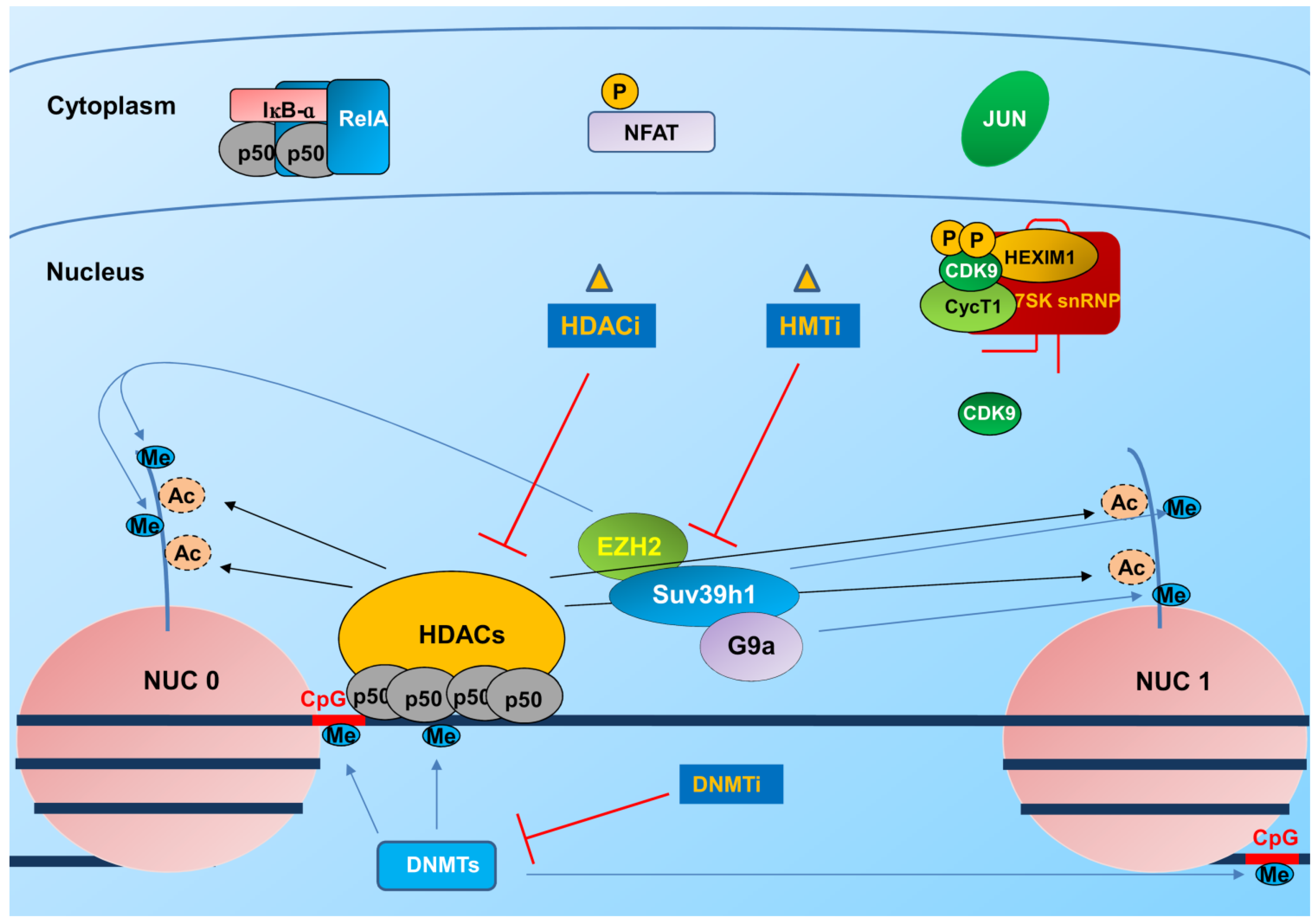
3.2. Transcription and Elongation Factors Relevant for HIV-1 Gene Expression
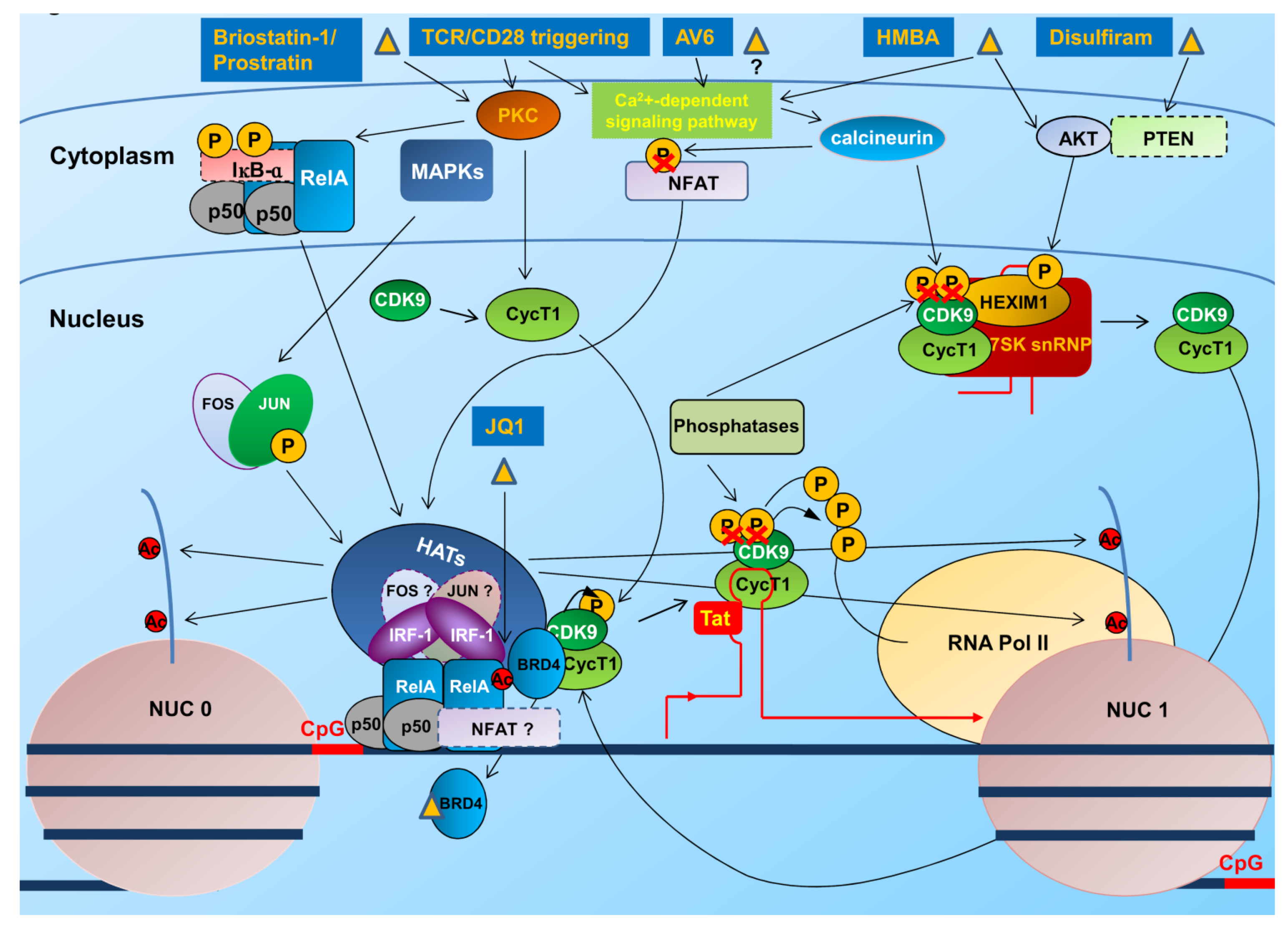
4. Therapeutic Approaches to Overcome Latency
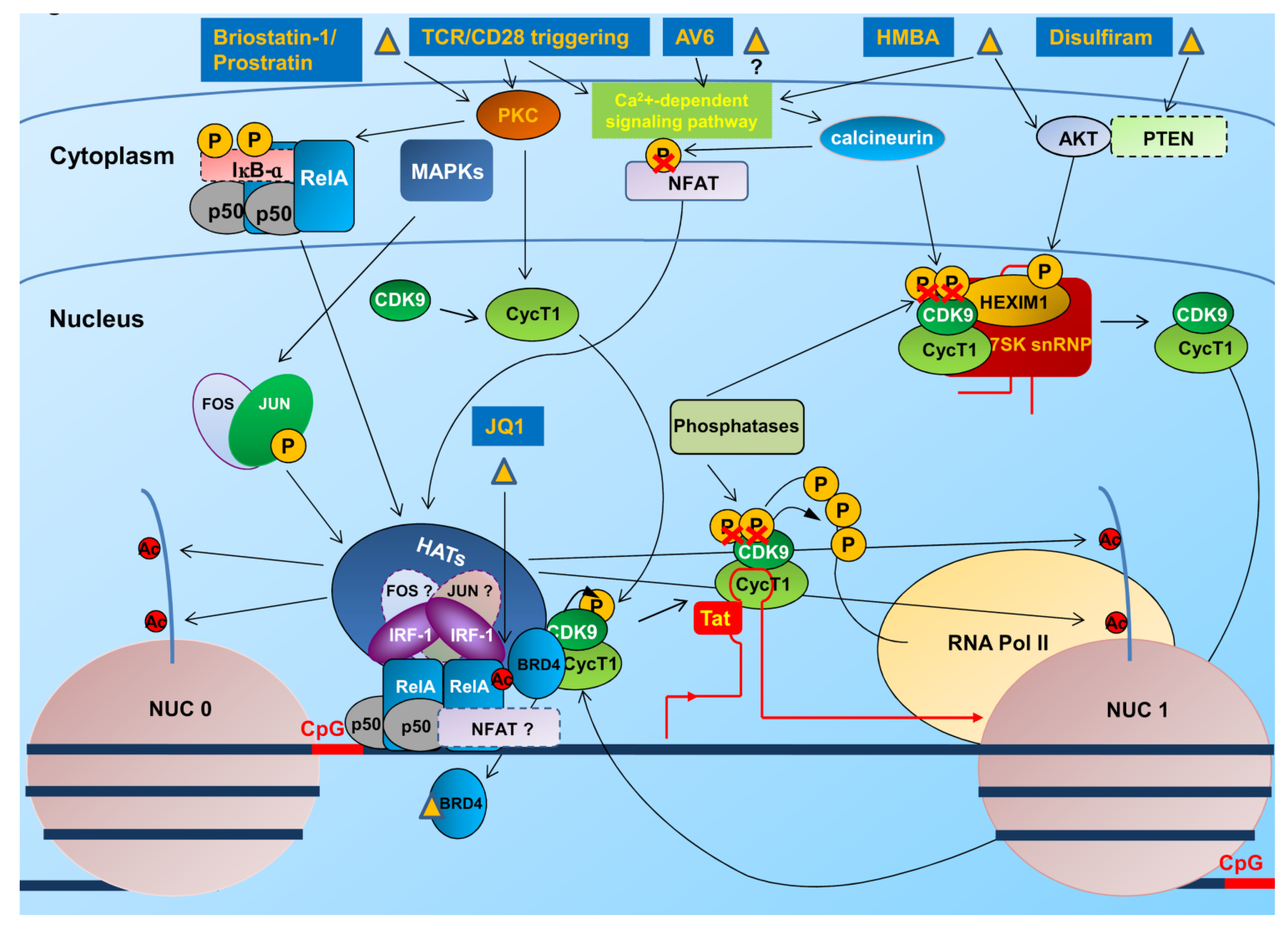
4.1. Interventions to Revert the Latent State
4.2. Immune-Based Therapies
4.3. Gene Therapy Approaches
5. Conclusions and Challenges
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Barre-Sinoussi, F.; Chermann, J.C.; Rey, F.; Nugeyre, M.T.; Chamaret, S.; Gruest, J.; Dauguet, C.; Axler-Blin, C.; Vézinet-Brun, F.; Rouzioux, C.; et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). Science 1983, 220, 868–871. [Google Scholar]
- Gallo, R.C.; Salahuddin, S.Z.; Popovic, M.; Shearer, G.M.; Kaplan, M.; Haynes, B.F.; Palker, T.J.; Redfield, R.; Oleske, J.; Safai, B.; et al. Frequent detection and isolation of cytopathic retroviruses (HTLV-III) from patients with AIDS and at risk for AIDS. Science 1984, 224, 500–503. [Google Scholar]
- Popovic, M.; Sarngadharan, M.G.; Read, E.; Gallo, R.C. Detection, isolation, and continuous production of cytopathic retroviruses (HTLV-III) from patients with AIDS and pre-AIDS. Science 1984, 224, 497–500. [Google Scholar]
- Sarngadharan, M.G.; Popovic, M.; Bruch, L.; Schupbach, J.; Gallo, R.C. Antibodies reactive with human T-lymphotropic retroviruses (HTLV-III) in the serum of patients with AIDS. Science 1984, 224, 506–508. [Google Scholar]
- Global report. Unaids report on the global aids epidemic 2012. Available online: http://www.unaids.org/en/media/unaids/contentassets/documents/epidemiology/2012/gr2012/20121120_UNAIDS_Global_Report_2012_with_annexes_en.pdf (accessed on 1 December 2013).
- Katlama, C.; Deeks, S.G.; Autran, B.; Martinez-Picado, J.; van Lunzen, J.; Rouzioux, C.; Miller, M.; Vella, S.; Schmitz, J.E.; Ahlers, J.; et al. Barriers to a cure for HIV: New ways to target and eradicate HIV-1 reservoirs. Lancet 2013, 381, 2109–2117. [Google Scholar] [CrossRef]
- Este, J.A.; Cihlar, T. Current status and challenges of antiretroviral research and therapy. Antivir. Res. 2010, 85, 25–33. [Google Scholar] [CrossRef]
- Hecht, R.; Stover, J.; Bollinger, L.; Muhib, F.; Case, K.; de Ferranti, D. Financing of HIV/AIDS programme scale-up in low-income and middle-income countries, 2009–31. Lancet 2010, 376, 1254–1260. [Google Scholar] [CrossRef]
- Hill, A.M.; Cho, M.; Mrus, J.M. The costs of full suppression of plasma HIV RNA in highly antiretroviral-experienced patients. AIDS Rev. 2011, 13, 41–48. [Google Scholar]
- Gulick, R.M.; Mellors, J.W.; Havlir, D.; Eron, J.J.; Gonzalez, C.; McMahon, D.; Richman, D.D.; Valentine, F.T.; Jonas, L.; Meibohm, A.; et al. Treatment with indinavir, zidovudine, and lamivudine in adults with human immunodeficiency virus infection and prior antiretroviral therapy. N. Engl. J. Med. 1997, 337, 734–739. [Google Scholar] [CrossRef]
- Hammer, S.M.; Squires, K.E.; Hughes, M.D.; Grimes, J.M.; Demeter, L.M.; Currier, J.S.; Eron, J.J., Jr.; Feinberg, J.E.; Balfour, H.H., Jr.; Deyton, L.R.; et al. A controlled trial of two nucleoside analogues plus indinavir in persons with human immunodeficiency virus infection and CD4 cell counts of 200 per cubic millimeter or less. AIDS Clinical Trials Group 320 Study Team. N. Engl. J. Med. 1997, 337, 725–733. [Google Scholar] [CrossRef]
- Perelson, A.S.; Essunger, P.; Cao, Y.; Vesanen, M.; Hurley, A.; Saksela, K.; Markowitz, M.; Ho, D.D. Decay characteristics of HIV-1-infected compartments during combination therapy. Nature 1997, 387, 188–191. [Google Scholar] [CrossRef]
- Brenchley, J.M.; Price, D.A.; Schacker, T.W.; Asher, T.E.; Silvestri, G.; Rao, S.; Kazzaz, Z.; Bornstein, E.; Lambotte, O.; Altmann, D.; et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat. Med. 2006, 12, 1365–1371. [Google Scholar]
- Hunt, P.W.; Martin, J.N.; Sinclair, E.; Bredt, B.; Hagos, E.; Lampiris, H.; Deeks, S.G. T cell activation is associated with lower CD4+ T cell gains in human immunodeficiency virus-infected patients with sustained viral suppression during antiretroviral therapy. J. Infect. Dis. 2003, 187, 1534–1543. [Google Scholar] [CrossRef]
- Zeng, M.; Southern, P.J.; Reilly, C.S.; Beilman, G.J.; Chipman, J.G.; Schacker, T.W.; Haase, A.T. Lymphoid tissue damage in HIV-1 infection depletes naive T cells and limits T cell reconstitution after antiretroviral therapy. PLoS Pathog. 2012, 8, e1002437. [Google Scholar] [CrossRef]
- Chun, T.W.; Fauci, A.S. HIV reservoirs: Pathogenesis and obstacles to viral eradication and cure. AIDS 2012, 26, 1261–1268. [Google Scholar] [CrossRef]
- Davey, R.T., Jr.; Bhat, N.; Yoder, C.; Chun, T.W.; Metcalf, J.A.; Dewar, R.; Natarajan, V.; Lempicki, R.A.; Adelsberger, J.W.; Miller, K.D.; et al. HIV-1 and T cell dynamics after interruption of highly active antiretroviral therapy (HAART) in patients with a history of sustained viral suppression. Proc. Natl. Acad. Sci. USA 1999, 96, 15109–15114. [Google Scholar] [CrossRef]
- Garcia, F.; Plana, M.; Vidal, C.; Cruceta, A.; O’Brien, W.A.; Pantaleo, G.; Pumarola, T.; Gallart, T.; Miró, J.M.; Gatell, J.M. Dynamics of viral load rebound and immunological changes after stopping effective antiretroviral therapy. AIDS 1999, 13, F79–F86. [Google Scholar] [CrossRef]
- Mata, R.C.; Viciana, P.; de Alarcon, A.; Lopez-Cortes, L.F.; Gomez-Vera, J.; Trastoy, M.; Cisneros, J.M. Discontinuation of antiretroviral therapy in patients with chronic HIV infection: Clinical, virologic, and immunologic consequences. AIDS Patient Care STDS 2005, 19, 550–562. [Google Scholar] [CrossRef]
- Eisele, E.; Siliciano, R.F. Redefining the viral reservoirs that prevent HIV-1 eradication. Immunity 2012, 37, 377–388. [Google Scholar] [CrossRef]
- Siliciano, J.D.; Kajdas, J.; Finzi, D.; Quinn, T.C.; Chadwick, K.; Margolick, J.B.; Kovacs, C.; Gange, S.J.; Siliciano, R.F. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 2003, 9, 727–728. [Google Scholar] [CrossRef]
- Dinoso, J.B.; Kim, S.Y.; Wiegand, A.M.; Palmer, S.E.; Gange, S.J.; Cranmer, L.; O’Shea, A.; Callender, M.; Spivak, A.; Brennan, T.; et al. Treatment intensification does not reduce residual HIV-1 viremia in patients on highly active antiretroviral therapy. Proc. Natl. Acad. Sci. USA 2009, 106, 9403–9408. [Google Scholar] [CrossRef]
- Deeks, S.G.; Autran, B.; Berkhout, B.; Benkirane, M.; Cairns, S.; Chomont, N.; Chun, T.W.; Churchill, M.; di Mascio, M.; Katlama, C.; et al. Towards an HIV cure: A global scientific strategy. Nat. Rev. Immunol. 2012, 12, 607–614. [Google Scholar] [CrossRef]
- Trono, D.; van Lint, C.; Rouzioux, C.; Verdin, E.; Barre-Sinoussi, F.; Chun, T.W.; Chomont, N. HIV persistence and the prospect of long-term drug-free remissions for HIV-infected individuals. Science 2010, 329, 174–180. [Google Scholar] [CrossRef]
- Allers, K.; Hutter, G.; Hofmann, J.; Loddenkemper, C.; Rieger, K.; Thiel, E.; Schneider, T. Evidence for the cure of HIV infection by CCR5Delta32/Delta32 stem cell transplantation. Blood 2011, 117, 2791–2799. [Google Scholar] [CrossRef]
- Hutter, G.; Nowak, D.; Mossner, M.; Ganepola, S.; Mussig, A.; Allers, K.; Schneider, T.; Hofmann, J.; Kücherer, C.; Blau, O.; et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N. Engl. J. Med. 2009, 360, 692–698. [Google Scholar] [CrossRef]
- Yukl, S.A.; Boritz, E.; Busch, M.; Bentsen, C.; Chun, T.W.; Douek, D.; Eisele, E.; Haase, A.; Ho, Y.C.; Hütter, G.; et al. Challenges in detecting HIV persistence during potentially curative interventions: A study of the Berlin patient. PLoS Pathog. 2013, 9, e1003347. [Google Scholar] [CrossRef]
- Saez-Cirion, A.; Bacchus, C.; Hocqueloux, L.; Avettand-Fenoel, V.; Girault, I.; Lecuroux, C.; Potard, V.; Versmisse, P.; Melard, A.; Prazuck, T.; et al. Post-treatment HIV-1 controllers with a long-term virological remission after the interruption of early initiated antiretroviral therapy ANRS VISCONTI Study. PLoS Pathog. 2013, 9, e1003211. [Google Scholar] [CrossRef]
- Hocqueloux, L.; Avettand-Fenoel, V.; Jacquot, S.; Prazuck, T.; Legac, E.; Melard, A.; Niang, M.; Mille, C.; le Moal, G.; Viard, J.P.; et al. Long-term antiretroviral therapy initiated during primary HIV-1 infection is key to achieving both low HIV reservoirs and normal T cell counts. J. Antimicrob. Chemother. 2013, 68, 1169–1178. [Google Scholar] [CrossRef]
- Hocqueloux, L.; Prazuck, T.; Avettand-Fenoel, V.; Lafeuillade, A.; Cardon, B.; Viard, J.P.; Rouzioux, C. Long-term immunovirologic control following antiretroviral therapy interruption in patients treated at the time of primary HIV-1 infection. AIDS 2010, 24, 1598–1601. [Google Scholar] [CrossRef]
- Strain, M.C.; Little, S.J.; Daar, E.S.; Havlir, D.V.; Gunthard, H.F.; Lam, R.Y.; Daly, O.A.; Nguyen, J.; Ignacio, C.C.; Spina, C.A.; et al. Effect of treatment, during primary infection, on establishment and clearance of cellular reservoirs of HIV-1. J. Infect. Dis. 2005, 191, 1410–1418. [Google Scholar] [CrossRef]
- Lodi, S.; Meyer, L.; Kelleher, A.D.; Rosinska, M.; Ghosn, J.; Sannes, M.; Porter, K. Immunovirologic control 24 months after interruption of antiretroviral therapy initiated close to HIV seroconversion. Arch. Intern. Med. 2012, 172, 1252–1255. [Google Scholar] [CrossRef]
- Goujard, C.; Girault, I.; Rouzioux, C.; Lecuroux, C.; Deveau, C.; Chaix, M.L.; Jacomet, C.; Talamali, A.; Delfraissy, J.F.; Venet, A.; et al. HIV-1 control after transient antiretroviral treatment initiated in primary infection: Role of patient characteristics and effect of therapy. Antivir. Ther. 2012, 17, 1001–1009. [Google Scholar] [CrossRef]
- Persaud, D.; Gay, H.; Ziemniak, C.; Chen, Y.H.; Piatak, M., Jr.; Chun, T.W.; Strain, M.; Richman, D.; Luzuriaga, K. Absence of detectable HIV-1 viremia after treatment cessation in an infant. N. Engl. J. Med. 2013, 369, 1828–1835. [Google Scholar] [CrossRef]
- Lewis, M.G.; DaFonseca, S.; Chomont, N.; Palamara, A.T.; Tardugno, M.; Mai, A.; Collins, M.; Wagner, W.L.; Yalley-Ogunro, J.; Greenhouse, J.; et al. Gold drug auranofin restricts the viral reservoir in the monkey AIDS model and induces containment of viral load following ART suspension. AIDS 2011, 25, 1347–1356. [Google Scholar] [CrossRef]
- Shytaj, I.L.; Chirullo, B.; Wagner, W.; Ferrari, M.G.; Sgarbanti, R.; Corte, A.D.; LaBranche, C.; Lopalco, L.; Palamara, A.T.; Montefiori, D.; et al. Investigational treatment suspension and enhanced cell-mediated immunity at rebound followed by drug-free remission of simian AIDS. Retrovirology 2013, 10. [Google Scholar] [CrossRef]
- Shytaj, I.L.; Norelli, S.; Chirullo, B.; Corte, A.D.; Collins, M.; Yalley-Ogunro, J.; Greenhouse, J.; Iraci, N.; Acosta, E.P.; Barreca, M.L.; et al. A highly intensified ART regimen induces long-term viral suppression and restriction of the viral reservoir in a simian AIDS model. PLoS Pathog. 2012, 8, e1002774. [Google Scholar] [CrossRef]
- Barton, K.M.; Burch, B.D.; Soriano-Sarabia, N.; Margolis, D.M. Prospects for treatment of latent HIV. Clin. Pharmacol. Ther. 2013, 93, 46–56. [Google Scholar] [CrossRef]
- Hamer, D.H. Can HIV be cured? Mechanisms of HIV persistence and strategies to combat it. Curr. HIV Res. 2004, 2, 99–111. [Google Scholar] [CrossRef]
- Savarino, A.; Mai, A.; Norelli, S.; El Daker, S.; Valente, S.; Rotili, D.; Altucci, L.; Palamara, A.T.; Garaci, E. “Shock and kill” effects of class I-selective histone deacetylase inhibitors in combination with the glutathione synthesis inhibitor buthionine sulfoximine in cell line models for HIV-1 quiescence. Retrovirology 2009, 6. [Google Scholar] [CrossRef]
- Dornadula, G.; Zhang, H.; VanUitert, B.; Stern, J.; Livornese, L., Jr.; Ingerman, M.J.; Witek, J.; Kedanis, R.J.; Natkin, J.; DeSimone, J.; et al. Residual HIV-1 RNA in blood plasma of patients taking suppressive highly active antiretroviral therapy. JAMA 1999, 282, 1627–1632. [Google Scholar] [CrossRef]
- Fischer, M.; Gunthard, H.F.; Opravil, M.; Joos, B.; Huber, W.; Bisset, L.R.; Ott, P.; Böni, J.; Weber, R.; Cone, R.W. Residual HIV-RNA levels persist for up to 2.5 years in peripheral blood mononuclear cells of patients on potent antiretroviral therapy. AIDS Res. Hum. Retrovir. 2000, 16, 1135–1140. [Google Scholar] [CrossRef] [Green Version]
- Palmer, S.; Maldarelli, F.; Wiegand, A.; Bernstein, B.; Hanna, G.J.; Brun, S.C.; Kempf, D.J.; Mellors, J.W.; Coffin, J.M.; King, M.S. Low-level viremia persists for at least 7 years in patients on suppressive antiretroviral therapy. Proc. Natl. Acad. Sci. USA 2008, 105, 3879–3884. [Google Scholar] [CrossRef]
- Wong, J.K.; Hezareh, M.; Gunthard, H.F.; Havlir, D.V.; Ignacio, C.C.; Spina, C.A.; Richman, D.D. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science 1997, 278, 1291–1295. [Google Scholar] [CrossRef]
- Natarajan, V.; Bosche, M.; Metcalf, J.A.; Ward, D.J.; Lane, H.C.; Kovacs, J.A. HIV-1 replication in patients with undetectable plasma virus receiving HAART. Highly active antiretroviral therapy. Lancet 1999, 353, 119–120. [Google Scholar]
- Ramratnam, B.; Mittler, J.E.; Zhang, L.; Boden, D.; Hurley, A.; Fang, F.; Macken, C.A.; Perelson, A.S.; Markowitz, M.; Ho, D.D. The decay of the latent reservoir of replication-competent HIV-1 is inversely correlated with the extent of residual viral replication during prolonged anti-retroviral therapy. Nat. Med. 2000, 6, 82–85. [Google Scholar] [CrossRef]
- Gras, G.; Kaul, M. Molecular mechanisms of neuroinvasion by monocytes-macrophages in HIV-1 infection. Retrovirology 2010, 7. [Google Scholar] [CrossRef]
- Chun, T.W.; Nickle, D.C.; Justement, J.S.; Meyers, J.H.; Roby, G.; Hallahan, C.W.; Kottilil, S.; Moir, S.; Mican, J.M.; Mullins, J.I.; et al. Persistence of HIV in gut-associated lymphoid tissue despite long-term antiretroviral therapy. J. Infect. Dis. 2008, 197, 714–720. [Google Scholar] [CrossRef]
- Schrager, L.K.; D’Souza, M.P. Cellular and anatomical reservoirs of HIV-1 in patients receiving potent antiretroviral combination therapy. JAMA 1998, 280, 67–71. [Google Scholar] [CrossRef]
- Chun, T.W.; Davey, R.T., Jr.; Ostrowski, M.; Shawn Justement, J.; Engel, D.; Mullins, J.I.; Fauci, A.S. Relationship between pre-existing viral reservoirs and the re-emergence of plasma viremia after discontinuation of highly active anti-retroviral therapy. Nat. Med. 2000, 6, 757–761. [Google Scholar] [CrossRef]
- Carter, C.C.; McNamara, L.A.; Onafuwa-Nuga, A.; Shackleton, M.; Riddell, J.T.; Bixby, D.; Savona, M.R.; Morrison, S.J.; Collins, K.L. HIV-1 utilizes the CXCR4 chemokine receptor to infect multipotent hematopoietic stem and progenitor cells. Cell Host Microbe 2011, 9, 223–234. [Google Scholar] [CrossRef]
- Durand, C.M.; Ghiaur, G.; Siliciano, J.D.; Rabi, S.A.; Eisele, E.E.; Salgado, M.; Shan, L.; Lai, J.F.; Zhang, H.; Margolick, J.; et al. HIV-1 DNA is detected in bone marrow populations containing CD4+ T cells but is not found in purified CD34+ hematopoietic progenitor cells in most patients on antiretroviral therapy. J. Infect. Dis. 2012, 205, 1014–1018. [Google Scholar] [CrossRef]
- Josefsson, L.; Eriksson, S.; Sinclair, E.; Ho, T.; Killian, M.; Epling, L.; Shao, W.; Lewis, B.; Bacchetti, P.; Loeb, L.; et al. Hematopoietic precursor cells isolated from patients on long-term suppressive HIV therapy did not contain HIV-1 DNA. J. Infect. Dis. 2012, 206, 28–34. [Google Scholar] [CrossRef]
- Crowe, S.; Zhu, T.; Muller, W.A. The contribution of monocyte infection and trafficking to viral persistence, and maintenance of the viral reservoir in HIV infection. J. Leukoc. Biol. 2003, 74, 635–641. [Google Scholar] [CrossRef]
- Shen, L.; Siliciano, R.F. Viral reservoirs, residual viremia, and the potential of highly active antiretroviral therapy to eradicate HIV infection. J. Allergy Clin. Immunol. 2008, 122, 22–28. [Google Scholar] [CrossRef]
- Le Douce, V.; Herbein, G.; Rohr, O.; Schwartz, C. Molecular mechanisms of HIV-1 persistence in the monocyte-macrophage lineage. Retrovirology 2010, 7. [Google Scholar] [CrossRef] [Green Version]
- Lambotte, O.; Taoufik, Y.; de Goer, M.G.; Wallon, C.; Goujard, C.; Delfraissy, J.F. Detection of infectious HIV in circulating monocytes from patients on prolonged highly active antiretroviral therapy. J. Acquir. Immune Defic. Syndr. 2000, 23, 114–119. [Google Scholar] [CrossRef]
- Zhu, T.; Muthui, D.; Holte, S.; Nickle, D.; Feng, F.; Brodie, S.; Hwangbo, Y.; Mullins, J.I.; Corey, L. Evidence for human immunodeficiency virus type 1 replication in vivo in CD14+ monocytes and its potential role as a source of virus in patients on highly active antiretroviral therapy. J. Virol. 2002, 76, 707–716. [Google Scholar] [CrossRef]
- Orenstein, J.M.; Fox, C.; Wahl, S.M. Macrophages as a source of HIV during opportunistic infections. Science 1997, 276, 1857–1861. [Google Scholar] [CrossRef]
- Redel, L.; le Douce, V.; Cherrier, T.; Marban, C.; Janossy, A.; Aunis, D.; van Lint, C.; Rohr, O.; Schwartz, C. HIV-1 regulation of latency in the monocyte-macrophage lineage and in CD4+ T lymphocytes. J. Leukoc. Biol. 2010, 87, 575–588. [Google Scholar] [CrossRef]
- Marsden, M.D.; Avancena, P.; Kitchen, C.M.; Hubbard, T.; Zack, J.A. Single mutations in HIV integrase confer high-level resistance to raltegravir in primary human macrophages. Antimicrob. Agents Chemother. 2011, 55, 3696–3702. [Google Scholar] [CrossRef]
- Churchill, M.; Nath, A. Where does HIV hide? A focus on the central nervous system. Curr. Opin. HIV AIDS 2013, 8, 165–169. [Google Scholar] [CrossRef]
- Narasipura, S.D.; Kim, S.; Al-Harthi, L. Epigenetic regulation of HIV-1 latency in astrocytes. J. Virol. 2014, 88, 3031–3038. [Google Scholar] [CrossRef]
- Blankson, J.N.; Persaud, D.; Siliciano, R.F. The challenge of viral reservoirs in HIV-1 infection. Annu. Rev. Med. 2002, 53, 557–593. [Google Scholar] [CrossRef]
- Buzon, M.J.; Massanella, M.; Llibre, J.M.; Esteve, A.; Dahl, V.; Puertas, M.C.; Gatell, J.M.; Domingo, P.; Paredes, R.; Sharkey, M.; et al. HIV-1 replication and immune dynamics are affected by raltegravir intensification of HAART-suppressed subjects. Nat. Med. 2010, 16, 460–465. [Google Scholar] [CrossRef]
- Joos, B.; Fischer, M.; Kuster, H.; Pillai, S.K.; Wong, J.K.; Boni, J.; Hirschel, B.; Weber, R.; Trkola, A.; Günthard, H.F.; et al. HIV rebounds from latently infected cells, rather than from continuing low-level replication. Proc. Natl. Acad. Sci. USA 2008, 105, 16725–16730. [Google Scholar] [CrossRef]
- Ruff, C.T.; Ray, S.C.; Kwon, P.; Zinn, R.; Pendleton, A.; Hutton, N.; Ashworth, R.; Gange, S.; Quinn, T.C.; Siliciano, R.F.; et al. Persistence of wild-type virus and lack of temporal structure in the latent reservoir for human immunodeficiency virus type 1 in pediatric patients with extensive antiretroviral exposure. J. Virol. 2002, 76, 9481–9492. [Google Scholar] [CrossRef]
- Bailey, J.R.; Sedaghat, A.R.; Kieffer, T.; Brennan, T.; Lee, P.K.; Wind-Rotolo, M.; Haggerty, C.M.; Kamireddi, A.R.; Liu, Y.; Lee, J.; et al. Residual human immunodeficiency virus type 1 viremia in some patients on antiretroviral therapy is dominated by a small number of invariant clones rarely found in circulating CD4+ T cells. J. Virol. 2006, 80, 6441–6457. [Google Scholar] [CrossRef]
- Brennan, T.P.; Woods, J.O.; Sedaghat, A.R.; Siliciano, J.D.; Siliciano, R.F.; Wilke, C.O. Analysis of human immunodeficiency virus type 1 viremia and provirus in resting CD4+ T cells reveals a novel source of residual viremia in patients on antiretroviral therapy. J. Virol. 2009, 83, 8470–8481. [Google Scholar] [CrossRef]
- Chun, T.W.; Finzi, D.; Margolick, J.; Chadwick, K.; Schwartz, D.; Siliciano, R.F. In vivo fate of HIV-1-infected T cells: Quantitative analysis of the transition to stable latency. Nat. Med. 1995, 1, 1284–1290. [Google Scholar] [CrossRef]
- Finzi, D.; Hermankova, M.; Pierson, T.; Carruth, L.M.; Buck, C.; Chaisson, R.E.; Quinn, T.C.; Chadwick, K.; Margolick, J.; Brookmeyer, R.; et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 1997, 278, 1295–1300. [Google Scholar] [CrossRef]
- Chun, T.W.; Stuyver, L.; Mizell, S.B.; Ehler, L.A.; Mican, J.A.; Baseler, M.; Lloyd, A.L.; Nowak, M.A.; Fauci, A.S. Presence of an inducible HIV-1 latent reservoir during highly active antiretroviral therapy. Proc. Natl. Acad. Sci. USA 1997, 94, 13193–13197. [Google Scholar] [CrossRef]
- Finzi, D.; Blankson, J.; Siliciano, J.D.; Margolick, J.B.; Chadwick, K.; Pierson, T.; Smith, K.; Lisziewicz, J.; Lori, F.; Flexner, C.; et al. Latent infection of CD4+ T cells provides a mechanism for lifelong persistence of HIV-1, even in patients on effective combination therapy. Nat. Med. 1999, 5, 512–517. [Google Scholar] [CrossRef]
- Swiggard, W.J.; Baytop, C.; Yu, J.J.; Dai, J.; Li, C.; Schretzenmair, R.; Theodosopoulos, T.; O’Doherty, U. Human immunodeficiency virus type 1 can establish latent infection in resting CD4+ T cells in the absence of activating stimuli. J. Virol. 2005, 79, 14179–14188. [Google Scholar] [CrossRef]
- Siliciano, R.F.; Greene, W.C. HIV latency. Cold Spring Harb. Perspect. Med. 2011, 1, a007096. [Google Scholar]
- Rong, L.; Perelson, A.S. Asymmetric division of activated latently infected cells may explain the decay kinetics of the HIV-1 latent reservoir and intermittent viral blips. Math. Biosci. 2009, 217, 77–87. [Google Scholar] [CrossRef]
- Chomont, N.; El-Far, M.; Ancuta, P.; Trautmann, L.; Procopio, F.A.; Yassine-Diab, B.; Boucher, G.; Boulassel, M.R.; Ghattas, G.; Brenchley, J.M.; et al. HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 2009, 15, 893–900. [Google Scholar] [CrossRef]
- Bosque, A.; Famiglietti, M.; Weyrich, A.S.; Goulston, C.; Planelles, V. Homeostatic proliferation fails to efficiently reactivate HIV-1 latently infected central memory CD4+ T cells. PLoS Pathog. 2011, 7, e1002288. [Google Scholar] [CrossRef]
- Gandhi, R.T.; Zheng, L.; Bosch, R.J.; Chan, E.S.; Margolis, D.M.; Read, S.; Kallungal, B.; Palmer, S.; Medvik, K.; Lederman, M.M.; et al. The effect of raltegravir intensification on low-level residual viremia in HIV-infected patients on antiretroviral therapy: A randomized controlled trial. PLoS Med. 2010, 7. [Google Scholar] [CrossRef] [Green Version]
- Ho, Y.C.; Shan, L.; Hosmane, N.N.; Wang, J.; Laskey, S.B.; Rosenbloom, D.I.; Lai, J.; Blankson, J.N.; Siliciano, J.D.; Siliciano, R.F.; et al. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 Cure. Cell 2013, 155, 540–551. [Google Scholar] [CrossRef]
- Laird, G.M.; Eisele, E.E.; Rabi, S.A.; Lai, J.; Chioma, S.; Blankson, J.N.; Siliciano, J.D.; Siliciano, R.F. Rapid quantification of the latent reservoir for HIV-1 using a viral outgrowth assay. PLoS Pathog. 2013, 9, e1003398. [Google Scholar] [CrossRef]
- Donahue, D.A.; Wainberg, M.A. Cellular and molecular mechanisms involved in the establishment of HIV-1 latency. Retrovirology 2013, 10. [Google Scholar] [CrossRef]
- Han, Y.; Lassen, K.; Monie, D.; Sedaghat, A.R.; Shimoji, S.; Liu, X.; Pierson, T.C.; Margolick, J.B.; Siliciano, R.F.; Siliciano, J.D. Resting CD4+ T cells from human immunodeficiency virus type 1 (HIV-1)-infected individuals carry integrated HIV-1 genomes within actively transcribed host genes. J. Virol. 2004, 78, 6122–6133. [Google Scholar] [CrossRef]
- Schroder, A.R.; Shinn, P.; Chen, H.; Berry, C.; Ecker, J.R.; Bushman, F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell 2002, 110, 521–529. [Google Scholar] [CrossRef]
- Shan, L.; Yang, H.C.; Rabi, S.A.; Bravo, H.C.; Shroff, N.S.; Irizarry, R.A.; Zhang, H.; Margolick, J.B.; Siliciano, J.D.; Siliciano, R.F.; et al. Influence of host gene transcription level and orientation on HIV-1 latency in a primary-cell model. J. Virol. 2011, 85, 5384–5393. [Google Scholar] [CrossRef]
- Lenasi, T.; Contreras, X.; Peterlin, B.M. Transcriptional interference antagonizes proviral gene expression to promote HIV latency. Cell Host Microbe 2008, 4, 123–133. [Google Scholar] [CrossRef]
- Greger, I.H.; Demarchi, F.; Giacca, M.; Proudfoot, N.J. Transcriptional interference perturbs the binding of Sp1 to the HIV-1 promoter. Nucleic Acids Res. 1998, 26, 1294–1301. [Google Scholar] [CrossRef]
- Han, Y.; Lin, Y.B.; An, W.; Xu, J.; Yang, H.C.; O’Connell, K.; Dordai, D.; Boeke, J.D.; Siliciano, J.D.; Siliciano, R.F.; et al. Orientation-dependent regulation of integrated HIV-1 expression by host gene transcriptional readthrough. Cell Host Microbe 2008, 4, 134–146. [Google Scholar] [CrossRef]
- Van Lint, C. Role of chromatin in HIV-1 transcriptional regulation. Adv. Pharmacol. 2000, 48, 121–160. [Google Scholar] [CrossRef]
- Jordan, A.; Defechereux, P.; Verdin, E. The site of HIV-1 integration in the human genome determines basal transcriptional activity and response to Tat transactivation. EMBO J. 2001, 20, 1726–1738. [Google Scholar] [CrossRef]
- Pearson, R.; Kim, Y.K.; Hokello, J.; Lassen, K.; Friedman, J.; Tyagi, M.; Karn, J. Epigenetic silencing of human immunodeficiency virus (HIV) transcription by formation of restrictive chromatin structures at the viral long terminal repeat drives the progressive entry of HIV into latency. J. Virol. 2008, 82, 12291–12303. [Google Scholar] [CrossRef]
- Van Lint, C.; Emiliani, S.; Ott, M.; Verdin, E. Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. EMBO J. 1996, 15, 1112–1120. [Google Scholar]
- Blazkova, J.; Trejbalova, K.; Gondois-Rey, F.; Halfon, P.; Philibert, P.; Guiguen, A.; Verdin, E.; Olive, D.; Van Lint, C.; Hejnar, J.; et al. CpG methylation controls reactivation of HIV from latency. PLoS Pathog. 2009, 5, e1000554. [Google Scholar] [CrossRef]
- Lusic, M.; Marcello, A.; Cereseto, A.; Giacca, M. Regulation of HIV-1 gene expression by histone acetylation and factor recruitment at the LTR promoter. EMBO J. 2003, 22, 6550–6561. [Google Scholar] [CrossRef]
- Quivy, V.; van Lint, C. Diversity of acetylation targets and roles in transcriptional regulation: The human immunodeficiency virus type 1 promoter as a model system. Biochem. Pharmacol. 2002, 64, 925–934. [Google Scholar] [CrossRef]
- Verdin, E.; Paras, P., Jr.; van Lint, C. Chromatin disruption in the promoter of human immunodeficiency virus type 1 during transcriptional activation. EMBO J. 1993, 12, 3249–3259. [Google Scholar]
- Keedy, K.S.; Archin, N.M.; Gates, A.T.; Espeseth, A.; Hazuda, D.J.; Margolis, D.M. A limited group of class I histone deacetylases acts to repress human immunodeficiency virus type 1 expression. J. Virol. 2009, 83, 4749–4756. [Google Scholar] [CrossRef]
- Coull, J.J.; Romerio, F.; Sun, J.M.; Volker, J.L.; Galvin, K.M.; Davie, J.R.; Shi, Y.; Hansen, U.; Margolis, D.M. The human factors YY1 and LSF repress the human immunodeficiency virus type 1 long terminal repeat via recruitment of histone deacetylase 1. J. Virol. 2000, 74, 6790–6799. [Google Scholar] [CrossRef]
- He, G.; Margolis, D.M. Counterregulation of chromatin deacetylation and histone deacetylase occupancy at the integrated promoter of human immunodeficiency virus type 1 (HIV-1) by the HIV-1 repressor YY1 and HIV-1 activator Tat. Mol. Cell Biol. 2002, 22, 2965–2973. [Google Scholar] [CrossRef]
- Bernhard, W.; Barreto, K.; Raithatha, S.; Sadowski, I. An upstream YY1 binding site on the HIV-1 LTR contributes to latent infection. PLoS One 2013, 8, e77052. [Google Scholar]
- Marban, C.; Suzanne, S.; Dequiedt, F.; de Walque, S.; Redel, L.; van Lint, C.; Aunis, D.; Rohr, O. Recruitment of chromatin-modifying enzymes by CTIP2 promotes HIV-1 transcriptional silencing. EMBO J. 2007, 26, 412–423. [Google Scholar] [CrossRef]
- Tyagi, M.; Karn, J. CBF-1 promotes transcriptional silencing during the establishment of HIV-1 latency. EMBO J. 2007, 26, 4985–4995. [Google Scholar] [CrossRef]
- Williams, S.A.; Chen, L.F.; Kwon, H.; Ruiz-Jarabo, C.M.; Verdin, E.; Greene, W.C. NF-kappaB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional initiation. EMBO J. 2006, 25, 139–149. [Google Scholar] [CrossRef]
- Jiang, G.; Espeseth, A.; Hazuda, D.J.; Margolis, D.M. c-Myc and Sp1 contribute to proviral latency by recruiting histone deacetylase 1 to the human immunodeficiency virus type 1 promoter. J. Virol. 2007, 81, 10914–10923. [Google Scholar] [CrossRef]
- Wen, Y.D.; Cress, W.D.; Roy, A.L.; Seto, E. Histone deacetylase 3 binds to and regulates the multifunctional transcription factor TFII-I. J. Biol. Chem. 2003, 278, 1841–1847. [Google Scholar] [CrossRef]
- Marsili, G.; Remoli, A.L.; Sgarbanti, M.; Perrotti, E.; Fragale, A.; Battistini, A. HIV-1, interferon and the interferon regulatory factor system: An interplay between induction, antiviral responses and viral evasion. Cytokine Growth Factor Rev. 2012, 23, 255–270. [Google Scholar] [CrossRef]
- Fragale, A.; Stellacci, E.; Ilari, R.; Remoli, A.L.; Lanciotti, A.; Perrotti, E.; Shytaj, I.; Orsatti, R.; Lawrence, H.R.; Lawrence, N.J.; et al. Critical role of IRF-8 in negative regulation of TLR3 expression by Src homology 2 domain-containing protein tyrosine phosphatase-2 activity in human myeloid dendritic cells. J. Immunol. 2011, 186, 1951–1962. [Google Scholar] [CrossRef]
- Sgarbanti, M.; Borsetti, A.; Moscufo, N.; Bellocchi, M.C.; Ridolfi, B.; Nappi, F.; Marsili, G.; Marziali, G.; Coccia, E.M.; Ensoli, B.; et al. Modulation of human immunodeficiency virus 1 replication by interferon regulatory factors. J. Exp. Med. 2002, 195, 1359–1370. [Google Scholar] [CrossRef]
- Sgarbanti, M.; Remoli, A.L.; Marsili, G.; Ridolfi, B.; Borsetti, A.; Perrotti, E.; Orsatti, R.; Ilari, R.; Sernicola, L.; Stellacci, E.; et al. IRF-1 is required for full NF-kappaB transcriptional activity at the human immunodeficiency virus type 1 long terminal repeat enhancer. J. Virol. 2008, 82, 3632–3641. [Google Scholar] [CrossRef]
- Munie110. Munier, S.; Delcroix-Genete, D.; Carthagena, L.; Gumez, A.; Hazan, U. Characterization of two candidate genes, NCoA3 and IRF8, potentially involved in the control of HIV-1 latency. Retrovirology 2005, 2. [Google Scholar] [CrossRef] [Green Version]
- Du Chene, I.; Basyuk, E.; Lin, Y.L.; Triboulet, R.; Knezevich, A.; Chable-Bessia, C.; Mettling, C.; Baillat, V.; Reynes, J.; Corbeau, P.; et al. Suv39H1 and HP1gamma are responsible for chromatin-mediated HIV-1 transcriptional silencing and post-integration latency. EMBO J. 2007, 26, 424–435. [Google Scholar] [CrossRef]
- Imai, K.; Togami, H.; Okamoto, T. Involvement of histone H3 lysine 9 (H3K9) methyltransferase G9a in the maintenance of HIV-1 latency and its reactivation by BIX01294. J. Biol. Chem. 2010, 285, 16538–16545. [Google Scholar] [CrossRef]
- Friedman, J.; Cho, W.K.; Chu, C.K.; Keedy, K.S.; Archin, N.M.; Margolis, D.M.; Karn, J. Epigenetic silencing of HIV-1 by the histone H3 lysine 27 methyltransferase enhancer of Zeste 2. J. Virol. 2011, 85, 9078–9089. [Google Scholar] [CrossRef]
- Kauder, S.E.; Bosque, A.; Lindqvist, A.; Planelles, V.; Verdin, E. Epigenetic regulation of HIV-1 latency by cytosine methylation. PLoS Pathog. 2009, 5, e1000495. [Google Scholar] [CrossRef]
- Blazkova, J.; Murray, D.; Justement, J.S.; Funk, E.K.; Nelson, A.K.; Moir, S.; Chun, T.W.; Fauci, A.S. Paucity of HIV DNA methylation in latently infected, resting CD4+ T cells from infected individuals receiving antiretroviral therapy. J. Virol. 2012, 86, 5390–5392. [Google Scholar] [CrossRef]
- Stevenson, M. Molecular mechanisms for the regulation of HIV replication, persistence and latency. AIDS 1997, 11, S25–S33. [Google Scholar]
- Zhong, H.; May, M.J.; Jimi, E.; Ghosh, S. The phosphorylation status of nuclear NF-kappa B determines its association with CBP/p300 or HDAC-1. Mol. Cell 2002, 9, 625–636. [Google Scholar] [CrossRef]
- Kinoshita, S.; Chen, B.K.; Kaneshima, H.; Nolan, G.P. Host control of HIV-1 parasitism in T cells by the nuclear factor of activated T cells. Cell 1998, 95, 595–604. [Google Scholar] [CrossRef]
- Cron, R.Q.; Bartz, S.R.; Clausell, A.; Bort, S.J.; Klebanoff, S.J.; Lewis, D.B. NFAT1 enhances HIV-1 gene expression in primary human CD4 T cells. Clin. Immunol. 2000, 94, 179–191. [Google Scholar] [CrossRef]
- Ruff, V.A.; Leach, K.L. Direct demonstration of NFATp dephosphorylation and nuclear localization in activated HT-2 cells using a specific NFATp polyclonal antibody. J. Biol. Chem. 1995, 270, 22602–22607. [Google Scholar] [CrossRef]
- Garcia-Rodriguez, C.; Rao, A. Nuclear factor of activated T cells (NFAT)-dependent transactivation regulated by the coactivators p300/CREB-binding protein (CBP). J. Exp. Med. 1998, 187, 2031–2036. [Google Scholar] [CrossRef]
- Kinoshita, S.; Su, L.; Amano, M.; Timmerman, L.A.; Kaneshima, H.; Nolan, G.P. The T cell activation factor NF-ATc positively regulates HIV-1 replication and gene expression in T cells. Immunity 1997, 6, 235–244. [Google Scholar] [CrossRef]
- Remoli, A.L.; Marsili, G.; Battistini, A.; Sgarbanti, M. The development of immune-modulating compounds to disrupt HIV latency. Cytokine Growth Factor Rev. 2012, 23, 159–172. [Google Scholar] [CrossRef]
- Yang, X.; Chen, Y.; Gabuzda, D. ERK MAP kinase links cytokine signals to activation of latent HIV-1 infection by stimulating a cooperative interaction of AP-1 and NF-κB. J. Biol. Chem. 1999, 274, 27981–27988. [Google Scholar] [CrossRef]
- Karn, J. The molecular biology of HIV latency: Breaking and restoring the Tat-dependent transcriptional circuit. Curr. Opin. HIV AIDS 2011, 6, 4–11. [Google Scholar] [CrossRef]
- Chen, L.; Fischle, W.; Verdin, E.; Greene, W.C. Duration of nuclear NF-κB action regulated by reversible acetylation. Science 2001, 293, 1653–1657. [Google Scholar] [CrossRef]
- Chen, L.F.; Mu, Y.; Greene, W.C. Acetylation of RelA at discrete sites regulates distinct nuclear functions of NF-κB. EMBO J. 2002, 21, 6539–6548. [Google Scholar] [CrossRef]
- Wei, P.; Garber, M.E.; Fang, S.M.; Fischer, W.H.; Jones, K.A. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell 1998, 92, 451–462. [Google Scholar] [CrossRef]
- Peterlin, B.M.; Price, D.H. Controlling the elongation phase of transcription with P-TEFb. Mol. Cell 2006, 23, 297–305. [Google Scholar] [CrossRef]
- Taube, R.; Peterlin, M. Lost in transcription: Molecular mechanisms that control HIV latency. Viruses 2013, 5, 902–927. [Google Scholar] [CrossRef]
- Ping, Y.H.; Rana, T.M. Tat-associated kinase (P-TEFb): A component of transcription preinitiation and elongation complexes. J. Biol. Chem. 1999, 274, 7399–7404. [Google Scholar] [CrossRef]
- Fujinaga, K.; Irwin, D.; Huang, Y.; Taube, R.; Kurosu, T.; Peterlin, B.M. Dynamics of human immunodeficiency virus transcription: P-TEFb phosphorylates RD and dissociates negative effectors from the transactivation response element. Mol. Cell Biol. 2004, 24, 787–795. [Google Scholar] [CrossRef]
- Nguyen, V.T.; Kiss, T.; Michels, A.A.; Bensaude, O. 7SK small nuclear RNA binds to and inhibits the activity of CDK9/cyclin T complexes. Nature 2001, 414, 322–325. [Google Scholar] [CrossRef]
- Yang, Z.; Zhu, Q.; Luo, K.; Zhou, Q. The 7SK small nuclear RNA inhibits the CDK9/cyclin T1 kinase to control transcription. Nature 2001, 414, 317–322. [Google Scholar] [CrossRef]
- Barboric, M.; Kohoutek, J.; Price, J.P.; Blazek, D.; Price, D.H.; Peterlin, B.M. Interplay between 7SK snRNA and oppositely charged regions in HEXIM1 direct the inhibition of P-TEFb. EMBO J. 2005, 24, 4291–4303. [Google Scholar] [CrossRef]
- Michels, A.A.; Fraldi, A.; Li, Q.; Adamson, T.E.; Bonnet, F.; Nguyen, V.T.; Sedore, S.C.; Price, J.P.; Price, D.H.; Lania, L.; et al. Binding of the 7SK snRNA turns the HEXIM1 protein into a P-TEFb (CDK9/cyclin T) inhibitor. EMBO J. 2004, 23, 2608–2619. [Google Scholar] [CrossRef]
- Budhiraja, S.; Famiglietti, M.; Bosque, A.; Planelles, V.; Rice, A.P. Cyclin T1 and CDK9 T-loop phosphorylation are downregulated during establishment of HIV-1 latency in primary resting memory CD4+ T cells. J. Virol. 2013, 87, 1211–1220. [Google Scholar] [CrossRef]
- Barboric, M.; Yik, J.H.; Czudnochowski, N.; Yang, Z.; Chen, R.; Contreras, X.; Geyer, M.; Peterlin, B.M.; Zhou, Q. Tat competes with HEXIM1 to increase the active pool of P-TEFb for HIV-1 transcription. Nucleic Acids Res. 2007, 35, 2003–2012. [Google Scholar] [CrossRef]
- Gringhuis, S.I.; van der Vlist, M.; van den Berg, L.M.; den Dunnen, J.; Litjens, M.; Geijtenbeek, T.B. HIV-1 exploits innate signaling by TLR8 and DC-SIGN for productive infection of dendritic cells. Nat. Immunol. 2010, 11, 419–426. [Google Scholar] [CrossRef]
- Masumi, A.; Wang, I.M.; Lefebvre, B.; Yang, X.J.; Nakatani, Y.; Ozato, K. The histone acetylase PCAF is a phorbol-ester-inducible coactivator of the IRF family that confers enhanced interferon responsiveness. Mol. Cell Biol. 1999, 19, 1810–1820. [Google Scholar]
- Sgarbanti, M.; Marsili, G.; Remoli, A.L.; Stellacci, E.; Mai, A.; Rotili, D.; Perrotti, E.; Acchioni, C.; Orsatti, R.; Iraci, N.; et al. IkappaB kinase {varepsilon} targets interferon regulatory factor 1 in activated T lymphocytes. Mol. Cell Biol. 2014, 34, 1054–1065. [Google Scholar] [CrossRef]
- Brasier, A.R.; Tian, B.; Jamaluddin, M.; Kalita, M.K.; Garofalo, R.P.; Lu, M. RelA Ser276 phosphorylation-coupled Lys310 acetylation controls transcriptional elongation of inflammatory cytokines in respiratory syncytial virus infection. J. Virol. 2011, 85, 11752–11769. [Google Scholar] [CrossRef]
- Duverger, A.; Wolschendorf, F.; Zhang, M.; Wagner, F.; Hatcher, B.; Jones, J.; Cron, R.Q.; van der Sluis, R.M.; Jeeninga, R.E.; Berkhout, B.; et al. An AP-1 binding site in the enhancer/core element of the HIV-1 promoter controls the ability of HIV-1 to establish latent infection. J. Virol. 2013, 87, 2264–2277. [Google Scholar] [CrossRef]
- Duverger, A.; Wolschendorf, F.; Anderson, J.C.; Wagner, F.; Bosque, A.; Shishido, T.; Jones, J.; Planelles, V.; Willey, C.; Cron, R.Q.; et al. Kinase control of Latent HIV-1 Infection: PIM-1 kinase as a major contributor to HIV-1 reactivation. J. Virol. 2014, 88, 364–376. [Google Scholar] [CrossRef]
- Lassen, K.G.; Ramyar, K.X.; Bailey, J.R.; Zhou, Y.; Siliciano, R.F. Nuclear retention of multiply spliced HIV-1 RNA in resting CD4+ T cells. PLoS Pathog. 2006, 2, e68. [Google Scholar] [CrossRef]
- Corbeau, P. Interfering RNA and HIV: Reciprocal interferences. PLoS Pathog. 2008, 4, e1000162. [Google Scholar] [CrossRef]
- Chiang, K.; Rice, A.P. MicroRNA-mediated restriction of HIV-1 in resting CD4+ T cells and monocytes. Viruses 2012, 4, 1390–1409. [Google Scholar] [CrossRef]
- Huang, J.; Wang, F.; Argyris, E.; Chen, K.; Liang, Z.; Tian, H.; Huang, W.; Squires, K.; Verlinghieri, G.; Zhang, H. Cellular microRNAs contribute to HIV-1 latency in resting primary CD4+ T lymphocytes. Nat. Med. 2007, 13, 1241–1247. [Google Scholar] [CrossRef]
- Chable-Bessia, C.; Meziane, O.; Latreille, D.; Triboulet, R.; Zamborlini, A.; Wagschal, A.; Jacquet, J.M.; Reynes, J.; Levy, Y.; Saib, A.; et al. Suppression of HIV-1 replication by microRNA effectors. Retrovirology 2009, 6, 26. [Google Scholar] [CrossRef]
- Klase, Z.; Kale, P.; Winograd, R.; Gupta, M.V.; Heydarian, M.; Berro, R.; McCaffrey, T.; Kashanchi, F. HIV-1 TAR element is processed by Dicer to yield a viral micro-RNA involved in chromatin remodeling of the viral LTR. BMC Mol. Biol. 2007, 8. [Google Scholar] [CrossRef]
- Wagschal, A.; Rousset, E.; Basavarajaiah, P.; Contreras, X.; Harwig, A.; Laurent-Chabalier, S.; Nakamura, M.; Chen, X.; Zhang, K.; Meziane, O.; et al. Microprocessor, Setx, Xrn2, and Rrp6 co-operate to induce premature termination of transcription by RNAPII. Cell 2012, 150, 1147–1157. [Google Scholar] [CrossRef]
- Kim, H.J.; Bae, S.C. Histone deacetylase inhibitors: Molecular mechanisms of action and clinical trials as anti-cancer drugs. Am. J. Transl. Res. 2011, 3, 166–179. [Google Scholar]
- Ververis, K.; Hiong, A.; Karagiannis, T.C.; Licciardi, P.V. Histone deacetylase inhibitors (HDACIs): Multitargeted anticancer agents. Biologics 2013, 7, 47–60. [Google Scholar]
- Archin, N.M.; Liberty, A.L.; Kashuba, A.D.; Choudhary, S.K.; Kuruc, J.D.; Crooks, A.M.; Parker, D.C.; Anderson, E.M.; Kearney, M.F.; Strain, M.C.; et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 2012, 487, 482–485. [Google Scholar] [CrossRef]
- Lehrman, G.; Hogue, I.B.; Palmer, S.; Jennings, C.; Spina, C.A.; Wiegand, A.; Landay, A.L.; Coombs, R.W.; Richman, D.D.; Mellors, J.W.; et al. Depletion of latent HIV-1 infection in vivo: A proof-of-concept study. Lancet 2005, 366, 549–555. [Google Scholar] [CrossRef]
- Routy, J.P.; Tremblay, C.L.; Angel, J.B.; Trottier, B.; Rouleau, D.; Baril, J.G.; Harris, M.; Trottier, S.; Singer, J.; Chomont, N.; et al. Valproic acid in association with highly active antiretroviral therapy for reducing systemic HIV-1 reservoirs: Results from a multicentre randomized clinical study. HIV Med. 2012, 13, 291–296. [Google Scholar] [CrossRef]
- Archin, N.M.; Espeseth, A.; Parker, D.; Cheema, M.; Hazuda, D.; Margolis, D.M. Expression of latent HIV induced by the potent HDAC inhibitor suberoylanilide hydroxamic acid. AIDS Res. Hum. Retrovir. 2009, 25, 207–212. [Google Scholar] [CrossRef]
- Contreras, X.; Schweneker, M.; Chen, C.S.; McCune, J.M.; Deeks, S.G.; Martin, J.; Peterlin, B.M. Suberoylanilide hydroxamic acid reactivates HIV from latently infected cells. J. Biol. Chem. 2009, 284, 6782–6789. [Google Scholar] [CrossRef]
- Margolis, D.M. Histone deacetylase inhibitors and HIV latency. Curr. Opin. HIV AIDS 2011, 6, 25–29. [Google Scholar] [CrossRef]
- Matalon, S.; Palmer, B.E.; Nold, M.F.; Furlan, A.; Kassu, A.; Fossati, G.; Mascagni, P.; Dinarello, CA. The histone deacetylase inhibitor ITF2357 decreases surface CXCR4 and CCR5 expression on CD4+ T-cells and monocytes and is superior to valproic acid for latent HIV-1 expression in vitro. J. Acquir. Immune Defic. Syndr. 2010, 54, 1–9. [Google Scholar]
- Rasmussen, T.A.; Schmeltz Sogaard, O.; Brinkmann, C.; Wightman, F.; Lewin, S.R.; Melchjorsen, J.; Dinarello, C.; Østergaard, L.; Tolstrup, M. Comparison of HDAC inhibitors in clinical development: Effect on HIV production in latently infected cells and T-cell activation. Hum. Vaccines Immunother. 2013, 9, 993–1001. [Google Scholar] [CrossRef]
- Yin, H.; Zhang, Y.; Zhou, X.; Zhu, H. Histonedeacetylase inhibitor Oxamflatin increase HIV-1 transcription by inducing histone modification in latently infected cells. Mol. Biol. Rep. 2011, 38, 5071–5078. [Google Scholar] [CrossRef]
- Victoriano, A.F.; Imai, K.; Togami, H.; Ueno, T.; Asamitsu, K.; Suzuki, T.; Miyata, N.; Ochiai, K.; Okamoto, T. Novel histone deacetylase inhibitor NCH-51 activates latent HIV-1 gene expression. FEBS Lett. 2011, 585, 1103–1111. [Google Scholar] [CrossRef]
- Furumai, R.; Matsuyama, A.; Kobashi, N.; Lee, K.H.; Nishiyama, M.; Nakajima, H.; Tanaka, A.; Komatsu, Y.; Nishino, N.; Yoshida, M.; et al. FK228 (depsipeptide) as a natural prodrug that inhibits class I histone deacetylases. Canc. Res. 2002, 62, 4916–4921. [Google Scholar]
- Huber, K.; Doyon, G.; Plaks, J.; Fyne, E.; Mellors, J.W.; Sluis-Cremer, N. Inhibitors of histone deacetylases: Correlation between isoform specificity and reactivation of HIV type 1 (HIV-1) from latently infected cells. J. Biol. Chem. 2011, 286, 22211–22218. [Google Scholar] [CrossRef]
- Glozak, M.A.; Sengupta, N.; Zhang, X.; Seto, E. Acetylation and deacetylation of non-histone proteins. Gene 2005, 363, 15–23. [Google Scholar] [CrossRef]
- Reuse, S.; Calao, M.; Kabeya, K.; Guiguen, A.; Gatot, J.S.; Quivy, V.; Vanhulle, C.; Lamine, A.; Vaira, D.; Demonte, D.; et al. Synergistic activation of HIV-1 expression by deacetylase inhibitors and prostratin: Implications for treatment of latent infection. PLoS One 2009, 4, e6093. [Google Scholar] [CrossRef]
- Marks, P.A.; Richon, V.M.; Breslow, R.; Rifkind, R.A. Histone deacetylase inhibitors as new cancer drugs. Curr. Opin. Oncol. 2001, 13, 477–483. [Google Scholar] [CrossRef]
- Crazzolara, R.; Johrer, K.; Johnstone, R.W.; Greil, R.; Kofler, R.; Meister, B.; Bernhard, D. Histone deacetylase inhibitors potently repress CXCR4 chemokine receptor expression and function in acute lymphoblastic leukaemia. Br. J. Haematol. 2002, 119, 965–969. [Google Scholar] [CrossRef]
- Shirakawa, K.; Chavez, L.; Hakre, S.; Calvanese, V.; Verdin, E. Reactivation of latent HIV by histone deacetylase inhibitors. Trends Microbiol. 2013, 21, 277–285. [Google Scholar] [CrossRef]
- McManamy, M.E.; Hakre, S.; Verdin, E.M.; Margolis, D.M. Therapy for latent HIV-1 infection: The role of histone deacetylase inhibitors. Antivir. Chem. Chemother. 2014, 23, 145–149. [Google Scholar]
- Bernhard, W.; Barreto, K.; Saunders, A.; Dahabieh, M.S.; Johnson, P.; Sadowski, I. The Suv39H1 methyltransferase inhibitor chaetocin causes induction of integrated HIV-1 without producing a T cell response. FEBS Lett. 2011, 585, 3549–3554. [Google Scholar] [CrossRef]
- Sakane, N.; Kwon, H.S.; Pagans, S.; Kaehlcke, K.; Mizusawa, Y.; Kamada, M.; Lassen, K.G.; Chan, J.; Greene, W.C.; Schnoelzer, M.; et al. Activation of HIV transcription by the viral Tat protein requires a demethylation step mediated bylysine-specific demethylase 1 (LSD1/KDM1). PLoS Pathog. 2011, 7, e1002184. [Google Scholar] [CrossRef]
- Bouchat, S.; Gatot, J.S.; Kabeya, K.; Cardona, C.; Colin, L.; Herbein, G.; De Wit, S.; Clumeck, N.; Lambotte, O.; Rouzioux, C.; et al. Histone methyltransferase inhibitors induce HIV-1 recovery in resting CD4+ T cells from HIV-1-infected HAART-treated patients. AIDS 2012, 26, 1473–1482. [Google Scholar] [CrossRef]
- Sanchez-Duffhues, G.; Vo, M.Q.; Perez, M.; Calzado, M.A.; Moreno, S.; Appendino, G.; Muñoz, E. Activation of latent HIV-1 expression by protein kinase C agonists. A novel therapeutic approach to eradicate HIV-1 reservoirs. Curr. Drug Targets 2011, 12, 348–356. [Google Scholar] [CrossRef]
- Wender, P.A.; Kee, J.M.; Warrington, J.M. Practical synthesis of prostratin, DPP, and their analogs, adjuvant leads against latent HIV. Science 2008, 320, 649–652. [Google Scholar] [CrossRef]
- Mehla, R.; Bivalkar-Mehla, S.; Zhang, R.; Handy, I.; Albrecht, H.; Giri, S.; Nagarkatti, P.; Nagarkatti, M.; Chauhan, A. Bryostatin modulates latent HIV-1 infection via PKC and AMPK signaling but inhibits acute infection in a receptor independent manner. PLoS One 2010, 5, e11160. [Google Scholar] [CrossRef]
- Perez, M.; de Vinuesa, A.G.; Sanchez-Duffhues, G.; Marquez, N.; Bellido, M.L.; Munoz-Fernandez, M.A.; Moreno, S.; Castor, T.P.; Calzado, M.A.; Muñoz, E. Bryostatin-1 synergizes with histone deacetylase inhibitors to reactivate HIV-1 from latency. Curr. HIV Res. 2010, 8, 418–429. [Google Scholar] [CrossRef]
- Bartholomeeusen, K.; Fujinaga, K.; Xiang, Y.; Peterlin, B.M. Histone deacetylase inhibitors (HDACis) that release the positive transcription elongation factor b (P-TEFb) from its inhibitory complex also activate HIV transcription. J. Biol. Chem. 2013, 288, 14400–14407. [Google Scholar] [CrossRef]
- Kulkosky, J.; Culnan, D.M.; Roman, J.; Dornadula, G.; Schnell, M.; Boyd, M.R.; Pomerantz, R.J. Prostratin: Activation of latent HIV-1 expression suggests a potential inductive adjuvant therapy for HAART. Blood 2001, 98, 3006–3015. [Google Scholar] [CrossRef]
- Boto, W.M.; Brown, L.; Chrest, J.; Adler, W.H. Distinct modulatory effects of bryostatin 1 and staurosporine on the biosynthesis and expression of the HIV receptor protein (CD4) by T cells. Cell. Regul. 1991, 2, 95–103. [Google Scholar]
- Biancotto, A.; Grivel, J.C.; Gondois-Rey, F.; Bettendroffer, L.; Vigne, R.; Brown, S.; Margolis, L.B.; Hirsch, I. Dual role of prostratin in inhibition of infection and reactivation of human immunodeficiency virus from latency in primary blood lymphocytes and lymphoid tissue. J. Virol. 2004, 78, 10507–10515. [Google Scholar] [CrossRef]
- Ariza, M.E.; Ramakrishnan, R.; Singh, N.P.; Chauhan, A.; Nagarkatti, P.S.; Nagarkatti, M. Bryostatin-1, a naturally occurring antineoplastic agent, acts as a Toll-like receptor 4 (TLR-4) ligandand induces unique cytokines and chemokines in dendritic cells. J. Biol. Chem. 2011, 286, 24–34. [Google Scholar]
- Cocchi, F.; DeVico, A.L.; Garzino-Demo, A.; Arya, S.K.; Gallo, R.C.; Lusso, P. Identification of RANTES, MIP-1 alpha, and MIP-1 beta as the major HIV-suppressive factors produced by CD8+ T cells. Science 1995, 270, 1811–1815. [Google Scholar]
- DeChristopher, B.A.; Loy, B.A.; Marsden, M.D.; Schrier, A.J.; Zack, J.A.; Wender, P.A. Designed, synthetically accessible bryostatin analogues potently induce activation of latent HIV reservoirs in vitro. Nat. Chem. 2012, 4, 705–710. [Google Scholar] [CrossRef]
- Morgan, R.J., Jr.; Leong, L.; Chow, W.; Gandara, D.; Frankel, P.; Garcia, A.; Lenz, H.J.; Doroshow, J.H. Phase II trial of bryostatin-1 in combination with cisplatin in patients with recurrent or persistent epithelial ovarian cancer: A California cancer consortium study. Investig. New Drugs 2012, 30, 723–728. [Google Scholar]
- Contreras, X.; Barboric, M.; Lenasi, T.; Peterlin, B.M. HMBA releases P-TEFb from HEXIM1 and 7SK snRNA via PI3K/Akt and activates HIV transcription. PLoS Pathog. 2007, 3, 1459–1469. [Google Scholar]
- Choudhary, S.K.; Archin, N.M.; Margolis, D.M. Hexamethylbisacetamide and disruption of human immunodeficiency virus type 1 latency in CD4+ T cells. J. Infect. Dis. 2008, 197, 1162–1170. [Google Scholar] [CrossRef]
- Yang, H.C.; Xing, S.; Shan, L.; O’Connell, K.; Dinoso, J.; Shen, A.; Zhou, Y.; Shrum, C.K.; Han, Y.; Liu, J.O.; et al. Small-molecule screening using a human primary cell model of HIV latency identifies compounds that reverse latency without cellular activation. J. Clin. Investig. 2009, 119, 3473–3486. [Google Scholar]
- Andreeff, M.; Stone, R.; Michaeli, J.; Young, C.W.; Tong, W.P.; Sogoloff, H.; Ervin, T.; Kufe, D.; Rifkind, R.A.; Marks, P.A. Hexamethylene bisacetamide in myelodysplastic syndrome and acute myelogenous leukemia: A phase II clinical trial with a differentiation-inducing agent. Blood 1992, 80, 2604–2609. [Google Scholar]
- Huang, B.; Yang, X.D.; Zhou, M.M.; Ozato, K.; Chen, L.F. Brd4 coactivates transcriptional activation of NF-κB via specific binding to acetylated RelA. Mol. Cell Biol. 2009, 29, 1375–1387. [Google Scholar] [CrossRef]
- Zhou, H.; Xu, M.; Huang, Q.; Gates, A.T.; Zhang, X.D.; Castle, J.C.; Stec, E.; Ferrer, M.; Strulovici, B.; Hazuda, D.J.; et al. Genome-scale RNAi screen for host factors required for HIV replication. Cell Host Microbe 2008, 4, 495–504. [Google Scholar] [CrossRef]
- Brass, A.L.; Dykxhoorn, D.M.; Benita, Y.; Yan, N.; Engelman, A.; Xavier, R.J.; Lieberman, J.; Elledge, S.J. Identification of host proteins required for HIV infection through a functional genomic screen. Science 2008, 319, 921–926. [Google Scholar] [CrossRef]
- Banerjee, C.; Archin, N.; Michaels, D.; Belkina, A.C.; Denis, G.V.; Bradner, J.; Sebastiani, P.; Margolis, D.M.; Montano, M. BET bromodomain inhibition as a novel strategy for reactivation of HIV-1. J. Leukoc. Biol. 2012, 92, 1147–1154. [Google Scholar] [CrossRef]
- Li, Z.; Guo, J.; Wu, Y.; Zhou, Q. The BET bromodomain inhibitor JQ1 activates HIV latency through antagonizing Brd4 inhibition of Tat-transactivation. Nucleic Acids Res. 2013, 41, 277–287. [Google Scholar] [CrossRef]
- Zhu, J.; Gaiha, G.D.; John, S.P.; Pertel, T.; Chin, C.R.; Gao, G.; Qu, H.; Walker, B.D.; Elledge, S.J.; Brass, A.L. Reactivation of latent HIV-1 by inhibition of BRD4. Cell Rep. 2012, 2, 807–816. [Google Scholar] [CrossRef]
- Bartholomeeusen, K.; Xiang, Y.; Fujinaga, K.; Peterlin, B.M. Bromodomain and extra-terminal (BET) bromodomain inhibition activate transcription via transient release of positive transcription elongation factor b (P-TEFb) from 7SK small nuclear ribonucleoprotein. J. Biol. Chem 2012, 287, 36609–36616. [Google Scholar] [CrossRef]
- Bisgrove, D.A.; Mahmoudi, T.; Henklein, P.; Verdin, E. Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proc. Natl. Acad. Sci. USA 2007, 104, 13690–13695. [Google Scholar] [CrossRef]
- Boehm, D.; Calvanese, V.; Dar, R.D.; Xing, S.; Schroeder, S.; Martins, L.; Aull, K.; Li, P.C.; Planelles, V.; Bradner, J.E.; et al. BET bromodomain-targeting compounds reactivate HIV from latency via a Tat-independent mechanism. Cell Cycle 2013, 12, 452–462. [Google Scholar]
- Novis, C.; Archin, N.M.; Buzon, M.J.; Verdin, E.; Round, J.; Lichterfeld, M.; Margolis, D.M.; Planelles, V.; Bosque, A.; et al. Reactivation of latent HIV-1 in central memory CD4+ T cells through TLR-1/2 stimulation. Retrovirology 2013, 10. [Google Scholar] [CrossRef]
- Xing, S.; Bullen, C.K.; Shroff, N.S.; Shan, L.; Yang, H.C.; Manucci, J.L.; Bhat, S.; Zhang, H.; Margolick, J.B.; Quinn, T.C.; et al. Disulfiram reactivates latent HIV-1 in a Bcl-2-transduced primary CD4+ T cell model without inducing global T cell activation. J. Virol. 2011, 85, 6060–6064. [Google Scholar] [CrossRef]
- Doyon, G.; Zerbato, J.; Mellors, J.W.; Sluis-Cremer, N. Disulfiram reactivates latent HIV-1 expression through depletion of the phosphatase and tensin homolog. AIDS 2013, 27, F7–F11. [Google Scholar] [CrossRef]
- Spivak, A.A.A.; Hoh, R.; Bacchetti, P.; Eisele, E.; Buckheit, R., III; Lai, J.; Siliciano, J.; Siliciano, R.; Deeks, S. Safety and feasibility of using disulfiram to enhance HIV transcription among long-term ARV-treated adults: Preliminary results from a pilot study. In Proceedings of the 19th Conference on Retroviruses and Opportunistic Infections, Seattle, WA, USA, 5–8 March 2012.
- Xing, S.; Bhat, S.; Shroff, N.S.; Zhang, H.; Lopez, J.A.; Margolick, J.B.; Liu, J.O.; Siliciano, R.F.; et al. Novel structurally related compounds reactivate latent HIV-1 in a bcl-2-transduced primary CD4+ T cell model without inducing global T cell activation. J. Antimicrob. Chemother. 2012, 67, 398–403. [Google Scholar] [CrossRef]
- Micheva-Viteva, S.; Kobayashi, Y.; Edelstein, L.C.; Pacchia, A.L.; Lee, H.L.; Graci, J.D.; Breslin, J.; Phelan, B.D.; Miller, L.K.; Colacino, J.M.; et al. High-throughput screening uncovers a compound that activates latent HIV-1 and acts cooperatively with a histone deacetylase (HDAC) inhibitor. J. Biol. Chem. 2011, 286, 21083–21091. [Google Scholar] [CrossRef]
- Shan, L.; Deng, K.; Shroff, N.S.; Durand, C.M.; Rabi, S.A.; Yang, H.C.; Zhang, H.; Margolick, J.B.; Blankson, J.N.; Siliciano, R.F.; et al. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity 2012, 36, 491–501. [Google Scholar] [CrossRef]
- Boasso, A.; Shearer, G.M. Chronic innate immune activation as a cause of HIV-1 immunopathogenesis. Clin. Immunol. 2008, 126, 235–242. [Google Scholar] [CrossRef]
- Borrow, P. Innate immunity in acute HIV-1 infection. Curr. Opin. HIV AIDS 2011, 6, 353–363. [Google Scholar] [CrossRef]
- Autran, B.; Descours, B.; Bacchus, C. Immune control of HIV-1 reservoirs. Curr. Opin. HIV AIDS 2013, 8, 204–210. [Google Scholar] [CrossRef]
- Catalfamo, M.; Wilhelm, C.; Tcheung, L.; Proschan, M.; Friesen, T.; Park, J.H.; Adelsberger, J.; Baseler, M.; Maldarelli, F.; Davey, R.; et al. CD4 and CD8 T cell immune activation during chronic HIV infection: Roles of homeostasis, HIV, type I IFN, and IL-7. J. Immunol. 2011, 186, 2106–2116. [Google Scholar] [CrossRef]
- Chun, T.W.; Engel, D.; Mizell, S.B.; Ehler, L.A.; Fauci, A.S. Induction of HIV-1 replication in latently infected CD4+ T cells using a combination of cytokines. J. Exp. Med. 1998, 188, 83–91. [Google Scholar] [CrossRef]
- Chun, T.W.; Engel, D.; Mizell, S.B.; Hallahan, C.W.; Fischette, M.; Park, S.; Davey, R.T., Jr.; Dybul, M.; Kovacs, J.A.; Metcalf, J.A.; et al. Effect of interleukin-2 on the pool of latently infected, resting CD4+ T cells in HIV-1-infected patients receiving highly active anti-retroviral therapy. Nat. Med. 1999, 5, 651–655. [Google Scholar] [CrossRef]
- Vandergeeten, C.; Fromentin, R.; Chomont, N. The role of cytokines in the establishment, persistence and eradication of the HIV reservoir. Cytokine Growth Factor Rev. 2012, 23, 143–149. [Google Scholar] [CrossRef]
- Emery, S.; Capra, W.B.; Cooper, D.A.; Mitsuyasu, R.T.; Kovacs, J.A.; Vig, P.; Smolskis, M.; Saravolatz, L.D.; Lane, H.C; Fyfe, G.A.; et al. Pooled analysis of 3 randomized, controlled trials of interleukin-2 therapy in adult human immunodeficiency virus type 1 disease. J. Infect. Dis. 2000, 182, 428–434. [Google Scholar] [CrossRef]
- Stellbrink, H.J.; van Lunzen, J.; Westby, M.; O’Sullivan, E.; Schneider, C.; Adam, A.; Weitner, L.; Kuhlmann, B.; Hoffmann, C.; Fenske, S.; et al. Effects of interleukin-2 plus highly active antiretroviral therapy on HIV-1 replication and proviral DNA (COSMIC trial). AIDS 2002, 16, 1479–1487. [Google Scholar] [CrossRef]
- Group, I.E.S.; Committee, S.S.; Abrams, D.; Levy, Y.; Losso, M.H.; Babiker, A.; Collins, G.; Cooper, D.A.; Darbyshire, J.; Emery, S.; et al. Interleukin-2 therapy in patients with HIV infection. N. Engl. J. Med. 2009, 361, 1548–1559. [Google Scholar] [CrossRef]
- Prins, J.M.; Jurriaans, S.; van Praag, R.M.; Blaak, H.; van Rij, R.; Schellekens, P.T.; ten Berge, I.J.; Yong, S.L.; Fox, C.H.; Roos, M.T. Immuno-activation with anti-CD3 and recombinant human IL-2 in HIV-1-infected patients on potent antiretroviral therapy. AIDS 1999, 13, 2405–2410. [Google Scholar] [CrossRef]
- Van Praag, R.M.; Prins, J.M.; Roos, M.T.; Schellekens, P.T.; Ten Berge, I.J.; Yong, S.L.; Schuitemaker, H.; Eerenberg, A.J.; Jurriaans, S.; de Wolf, F.; et al. OKT3 and IL-2 treatment for purging of the latent HIV-1 reservoir in vivo results in selective long-lasting CD4+ T cell depletion. J. Clin. Immunol. 2001, 21, 218–226. [Google Scholar] [CrossRef]
- Sereti, I.; Imamichi, H.; Natarajan, V.; Imamichi, T.; Ramchandani, M.S.; Badralmaa, Y.; Berg, S.C.; Metcalf, J.A.; Hahn, B.K.; Shen, J.M.; et al. In vivo expansion of CD4CD45RO-CD25 T cells expressing foxP3 in IL-2-treated HIV-infected patients. J. Clin. Investig. 2005, 115, 1839–1847. [Google Scholar] [CrossRef]
- Wang, F.X.; Xu, Y.; Sullivan, J.; Souder, E.; Argyris, E.G.; Acheampong, E.A.; Fisher, J.; Sierra, M.; Thomson, M.M.; Najera, R.; et al. IL-7 is a potent and proviral strain-specific inducer of latent HIV-1 cellular reservoirs of infected individuals on virally suppressive HAART. J. Clin. Investig. 2005, 115, 128–137. [Google Scholar] [CrossRef]
- Saleh, S.; Wightman, F.; Ramanayake, S.; Alexander, M.; Kumar, N.; Khoury, G.; Pereira, C.; Purcell, D.; Cameron, P.U.; Lewin, S.R. Expression and reactivation of HIV in a chemokine induced model of HIV latency in primary resting CD4+ T cells. Retrovirology 2011, 8. [Google Scholar] [CrossRef]
- Levy, Y.; Lacabaratz, C.; Weiss, L.; Viard, J.P.; Goujard, C.; Lelievre, J.D.; Boue, F.; Molina, J.M.; Rouzioux, C.; Avettand-Fenoel, V.; et al. Enhanced T cell recovery in HIV-1-infected adults through IL-7 treatment. J. Clin. Investig. 2009, 119, 997–1007. [Google Scholar]
- Levy, Y.; Sereti, I.; Tambussi, G.; Routy, J.P.; Lelievre, J.D.; Delfraissy, J.F.; Molina, J.M.; Fischl, M.; Goujard, C.; Rodriguez, B.; et al. Effects of recombinant human interleukin 7 on T-cell recovery and thymic output in HIV-infected patients receiving antiretroviral therapy: Results of a phase I/IIa randomized, placebo-controlled, multicenter study. Clin. Infect. Dis. 2012, 55, 291–300. [Google Scholar] [CrossRef]
- Sereti, I.; Dunham, R.M.; Spritzler, J.; Aga, E.; Proschan, M.A.; Medvik, K.; Battaglia, C.A.; Landay, A.L.; Pahwa, S.; Fischl, M.A.; et al. IL-7 administration drives T cell-cycle entry and expansion in HIV-1 infection. Blood 2009, 113, 6304–6314. [Google Scholar] [CrossRef]
- Imamichi, H.; Degray, G.; Asmuth, D.M.; Fischl, M.A.; Landay, A.L.; Lederman, M.M.; Sereti, I. HIV-1 viruses detected during episodic blips following interleukin-7 administration are similar to the viruses present before and after interleukin-7 therapy. AIDS 2011, 25, 159–164. [Google Scholar] [CrossRef]
- Vandergeeten, C.; Fromentin, R.; DaFonseca, S.; Lawani, M.B.; Sereti, I.; Lederman, M.M.; Ramgopal, M.; Routy, J.P.; Sekaly, R.P.; Chomont, N. Interleukin-7 promotes HIV persistence during antiretroviral therapy. Blood 2013, 121, 4321–4329. [Google Scholar] [CrossRef]
- Croce, M.; Orengo, A.M.; Azzarone, B.; Ferrini, S. Immunotherapeutic applications of IL-15. Immunotherapy 2012, 4, 957–969. [Google Scholar] [CrossRef]
- Vandergeeten, C.; da Fonseca, S.; Sereti, I.; Lederman, M.M.; Sékaly, R.P.; Chomont, N. Differential impact of IL-7 and IL-15 on HIV reservoir persistence. In Proceedings of the 6th IAS Conference on HIV Pathogenesis, treatment and Prevention, Rome, Italy, 17 July 2011.
- Lugli, E.; Mueller, Y.M.; Lewis, M.G.; Villinger, F.; Katsikis, P.D.; Roederer, M. IL-15 delays suppression and fails to promote immune reconstitution in virally suppressed chronically SIV-infected macaques. Blood 2011, 118, 2520–2529. [Google Scholar] [CrossRef]
- Azzoni, L.; Foulkes, A.S.; Papasavvas, E.; Mexas, A.M.; Lynn, K.M.; Mounzer, K.; Tebas, P.; Jacobson, J.M.; Frank, I.; Busch, M.P.; et al. Pegylated Interferon alfa-2a monotherapy results in suppression of HIV type 1 replication and decreased cell-associated HIV DNA integration. J. Infect. Dis. 2013, 207, 213–222. [Google Scholar] [CrossRef]
- Migueles, S.A.; Weeks, K.A.; Nou, E.; Berkley, A.M.; Rood, J.E.; Osborne, C.M.; Hallahan, C.W.; Cogliano-Shutta, N.A.; Metcalf, J.A.; McLaughlin, M.; et al. Defective human immunodeficiency virus-specific CD8+ T-cell polyfunctionality, proliferation, and cytotoxicity are not restored by antiretroviral therapy. J. Virol. 2009, 83, 11876–11889. [Google Scholar] [CrossRef]
- D’Ettorre, G.; Paiardini, M.; Ceccarelli, G.; Silvestri, G.; Vullo, V. HIV-associated immune activation: From bench to bedside. AIDS Res. Hum. Retrovir. 2011, 27, 355–364. [Google Scholar] [CrossRef]
- Miedema, F.; Hazenberg, M.D.; Tesselaar, K.; van Baarle, D.; de Boer, R.J.; Borghans, J.A. Immune activation and collateral damage in AIDS pathogenesis. Front. Immunol. 2013, 4, 298. [Google Scholar]
- Okazaki, T.; Chikuma, S.; Iwai, Y.; Fagarasan, S.; Honjo, T. A rheostat for immune responses: The unique properties of PD-1 and their advantages for clinical application. Nat. Immunol. 2013, 14, 1212–1218. [Google Scholar] [CrossRef] [Green Version]
- Porichis, F.; Kaufmann, D.E. Role of PD-1 in HIV pathogenesis and as target for therapy. Curr. HIV/AIDS Rep. 2012, 9, 81–90. [Google Scholar] [CrossRef]
- Kulpa, D.A.; Lawani, M.; Cooper, A.; Peretz, Y.; Ahlers, J.; Sekaly, R.P. PD-1 coinhibitory signals: The link between pathogenesis and protection. Semin. Immunol. 2013, 25, 219–227. [Google Scholar] [CrossRef]
- Said, E.A.; Dupuy, F.P.; Trautmann, L.; Zhang, Y.; Shi, Y.; El-Far, M.; Hill, B.J.; Noto, A.; Ancuta, P.; Peretz, Y.; et al. Programmeddeath-1-induced interleukin-10 production by monocytes impairs CD4+ T cell activation during HIV infection. Nat. Med. 2010, 16, 452–459. [Google Scholar] [CrossRef]
- Rosignoli, G.; Lim, C.H.; Bower, M.; Gotch, F.; Imami, N. Programmed death (PD)-1 molecule and its ligand PD-L1 distribution among memory CD4 and CD8 T cell subsets in human immunodeficiency virus-1-infected individuals. Clin. Exp. Immunol. 2009, 157, 90–97. [Google Scholar] [CrossRef]
- Day, C.L.; Kaufmann, D.E.; Kiepiela, P.; Brown, J.A.; Moodley, E.S.; Reddy, S.; Mackey, E.W.; Miller, J.D.; Leslie, A.J.; DePierres, C.; et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 2006, 443, 350–354. [Google Scholar] [CrossRef]
- Trautmann, L.; Janbazian, L.; Chomont, N.; Said, E.A.; Gimmig, S.; Bessette, B.; Boulassel, M.R.; Delwart, E.; Sepulveda, H.; Balderas, R.S.; et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat. Med. 2006, 12, 1198–1202. [Google Scholar] [CrossRef]
- Hatano, H.; Jain, V.; Hunt, P.W.; Lee, T.H.; Sinclair, E.; Do, T.D.; Hoh, R.; Martin, J.N.; McCune, J.M.; Hecht, F.; et al. Cell-based measures of viral persistence are associated with immune activation and programmed cell death protein 1 (PD-1)-expressing CD4+ T cells. J. Infect. Dis. 2013, 208, 50–56. [Google Scholar] [CrossRef]
- Whittall, T.; Peters, B.; Rahman, D.; Kingsley, C.I.; Vaughan, R.; Lehner, T. Immunogenic and tolerogenic signatures in human immunodeficiency virus (HIV)-infected controllers compared with progressors and a conversion strategy of virus control. Clin. Exp. Immunol. 2011, 166, 208–217. [Google Scholar] [CrossRef]
- Peretz, Y.; He, Z.; Shi, Y.; Yassine-Diab, B.; Goulet, J.P.; Bordi, R.; Filali-Mouhim, A.; Loubert, J.B.; El-Far, M.; Dupuy, F.P.; et al. CD160 and PD-1co-expression on HIV-specific CD8 T cells defines a subset with advanced dysfunction. PLoS Pathog. 2012, 8, e1002840. [Google Scholar] [CrossRef]
- DaFonseca, S.C.N.; El-Far, M.; Tanel, A.; Fonseca, S.; Procopio, F.; Boulassel, M.; Routy, R.; Sékaly, R. Purging the HIV-1 reservoir through the disruption of the PD-1 pathway. In Proceedings of the 18th Conference on Retroviruses and Opportunistic Infections, Boston, MA, USA, 27 February–2 March 2011.
- Velu, V.; Titanji, K.; Zhu, B.; Husain, S.; Pladevega, A.; Lai, L.; Vanderford, T.H.; Chennareddi, L.; Silvestri, G.; Freeman, G.J.; et al. Enhancing SIV-specific immunity in vivo by PD-1 blockade. Nature 2009, 458, 206–210. [Google Scholar] [CrossRef]
- Titanji, K.; Velu, V.; Chennareddi, L.; Vijay-Kumar, M.; Gewirtz, A.T.; Freeman, G.J.; Amara, R.R. Acute depletion of activated memory B cells involves the PD-1 pathway in rapidly progressing SIV-infected macaques. J. Clin. Investig. 2010, 120, 3878–3890. [Google Scholar] [CrossRef]
- Shetty, R.D.; Velu, V.; Titanji, K.; Bosinger, S.E.; Freeman, G.J.; Silvestri, G.; Amara, R.R. PD-1blockade during chronic SIV infection reduces hyperimmune activation and microbial translocation in rhesus macaques. J. Clin. Investig. 2012, 122, 1712–1716. [Google Scholar] [CrossRef]
- Brahmer, J.R.; Tykodi, S.S.; Chow, L.Q.; Hwu, W.J.; Topalian, S.L.; Hwu, P.; Drake, C.G.; Camacho, L.H.; Kauh, J.; Odunsi, K.; et al. Safety and activity of anti-PD-L1 antibody in patients with advanced cancer. N. Engl. J. Med. 2012, 366, 2455–2465. [Google Scholar] [CrossRef]
- Topalian, S.L.; Hodi, F.S.; Brahmer, J.R.; Gettinger, S.N.; Smith, D.C.; McDermott, D.F.; Powderly, J.D.; Carvajal, R.D.; Sosman, J.A.; Atkins, M.B.; et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N. Engl. J. Med. 2012, 366, 2443–2454. [Google Scholar] [CrossRef]
- Zeng, M.; Haase, A.T.; Schacker, T.W. Lymphoid tissue structure and HIV-1 infection: Life or death for T cells. Trends Immunol. 2012, 33, 306–314. [Google Scholar] [CrossRef]
- Schacker, T.W.; Nguyen, P.L.; Beilman, G.J.; Wolinsky, S.; Larson, M.; Reilly, C.; Haase, A.T. Collagen deposition in HIV-1 infected lymphatic tissues and T cell homeostasis. J. Clin. Investig. 2002, 110, 1133–1139. [Google Scholar] [CrossRef]
- Estes, J.; Baker, J.V.; Brenchley, J.M.; Khoruts, A.; Barthold, J.L.; Bantle, A.; Reilly, C.S.; Beilman, G.J.; George, M.E.; Douek, D.C.; et al. Collagen deposition limits immune reconstitution in the gut. J. Infect. Dis. 2008, 198, 456–464. [Google Scholar] [CrossRef]
- Brilla, C.G.; Funck, R.C.; Rupp, H. Lisinopril-mediated regression of myocardial fibrosis in patients with hypertensive heart disease. Circulation 2000, 102, 1388–1393. [Google Scholar] [CrossRef]
- Sharma, K.; Eltayeb, B.O.; McGowan, T.A.; Dunn, S.R.; Alzahabi, B.; Rohde, R.; Ziyadeh, F.N.; Lewis, E.J. Captopril-induced reduction of serum levels of transforming growth factor-beta1 correlates with long-term renoprotection in insulin-dependent diabetic patients. Am. J. Kidney Dis. 1999, 34, 818–823. [Google Scholar] [CrossRef]
- Peters, J.M.; Shah, Y.M.; Gonzalez, F.J. The role of peroxisome proliferator-activated receptors in carcinogenesis and chemoprevention. Nat. Rev. Cancer 2012, 12, 181–195. [Google Scholar]
- Mencarelli, A.; Francisci, D.; Renga, B.; D’Amore, C.; Cipriani, S.; Basile, F.; Schiaroli, E.; Baldelli, F.; Fiorucci, S. Ritonavir-induced lipoatrophy and dyslipidaemia is reversed by the anti-inflammatory drug leflunomide in a PPAR-gamma-dependent manner. Antivir. Ther. 2012, 17, 669–678. [Google Scholar] [CrossRef]
- Descours, B.; Avettand-Fenoel, V.; Blanc, C.; Samri, A.; Melard, A.; Supervie, V.; Theodorou, I.; Carcelain, G.; Rouzioux, C.; Autran, B.; et al. Immune responses driven by protective human leukocyte antigen alleles from long-term nonprogressors are associated with low HIV reservoir in central memory CD4 T cells. Clin. Infect. Dis. 2012, 54, 1495–1503. [Google Scholar] [CrossRef]
- Almeida, J.R.; Price, D.A.; Papagno, L.; Arkoub, Z.A.; Sauce, D.; Bornstein, E.; Asher, T.E.; Samri, A.; Schnuriger, A.; Theodorou, I.; et al. Superior control of HIV-1 replication by CD8+ T cells is reflected by their avidity, polyfunctionality, and clonal turnover. J. Exp. Med. 2007, 204, 2473–2485. [Google Scholar] [CrossRef]
- Berger, C.T.; Frahm, N.; Price, D.A.; Mothe, B.; Ghebremichael, M.; Hartman, K.L.; Henry, L.M.; Brenchley, J.M.; Ruff, L.E.; Venturi, V.; et al. High-functional-avidity cytotoxic T lymphocyte responses to HLA-B-restricted Gag-derived epitopes associated with relative HIV control. J. Virol. 2011, 85, 9334–9345. [Google Scholar] [CrossRef]
- Persaud, D.; Luzuriaga, K.; Ziemniak, C.; Muresan, P.; Greenough, T.; Fenton, T.; Blackford, A.; Ferguson, K.; Neu, N.; Cunningham, C.K. Effect of therapeutic HIV recombinant poxvirus vaccines on the size of the resting CD4+ T-cell latent HIV reservoir. AIDS 2011, 25, 2227–2234. [Google Scholar] [CrossRef]
- Autran, B.; Costagliola, D.; Murphy, R.; Katlama, C. Evaluating therapeutic vaccines in patients infected with HIV. Expert Rev. Vaccines 2004, 3, S169–S177. [Google Scholar] [CrossRef]
- Garcia, F.; Leon, A.; Gatell, J.M.; Plana, M.; Gallart, T. Therapeutic vaccines against HIV infection. Hum. Vaccines Immunother. 2012, 8, 569–581. [Google Scholar] [CrossRef]
- Shete, A.; Thakar, M.; Singh, D.P.; Gangakhedkar, R.; Gaikwad, A.; Pawar, J.; Paranjape, R. Short communication: HIV antigen-specific reactivation of HIV infection from cellular reservoirs: Implications in the settings of therapeutic vaccinations. AIDS Res. Hum. Retrovir. 2012, 28, 835–843. [Google Scholar] [CrossRef]
- Winckelmann, A.A.; Munk-Petersen, L.V.; Rasmussen, T.A.; Melchjorsen, J.; Hjelholt, T.J.; Montefiori, D.; Meraviglia, P.; Capetti, A.; Biasin, M.; Trabattoni, D.; et al. Administration of a Toll-like receptor 9 agonist decreases the proviral reservoir in virologically suppressed HIV-infected patients. PLoS One 2013, 8, e62074. [Google Scholar] [CrossRef]
- Piconi, S.; Parisotto, S.; Rizzardini, G.; Passerini, S.; Terzi, R.; Argenteri, B.; Meraviglia, P.; Capetti, A.; Biasin, M.; Trabattoni, D.; et al. Hydroxychloroquine drastically reduces immune activation in HIV-infected, antiretroviral therapy-treated immunologic nonresponders. Blood 2011, 118, 3263–3272. [Google Scholar] [CrossRef]
- Murray, S.M.; Down, C.M.; Boulware, D.R.; Stauffer, W.M.; Cavert, W.P.; Schacker, T.W.; Cavert, W.P.; Schacker, T.W.; Brenchley, J.M.; Douek, D.C. Reduction of immune activation with chloroquine therapy during chronic HIV infection. J. Virol. 2010, 84, 12082–12086. [Google Scholar] [CrossRef]
- Paton, N.I.; Goodall, R.L.; Dunn, D.T.; Franzen, S.; Collaco-Moraes, Y.; Gazzard, B.G.; Williams, I.G.; Fisher, M.J.; Winston, A.; Fox, J.; et al. Effects of hydroxychloroquine on immune activation and disease progression among HIV-infected patients not receiving antiretroviral therapy: A randomized controlled trial. JAMA 2012, 308, 353–361. [Google Scholar]
- Henrich, T.J.; Hu, Z.; Li, J.Z.; Sciaranghella, G.; Busch, M.P.; Keating, S.M.; Gallien, S.; Lin, N.H.; Giguel, F.F.; Lavoie, L.; et al. Long-term reduction in peripheral blood HIV type 1 reservoirs following reduced-intensity conditioning allogeneic stem cell transplantation. J. Infect. Dis. 2013, 207, 1694–1702. [Google Scholar]
- Buchholz, F.; Hauber, J. Engineered DNA modifying enzymes: Components of a future strategy to cure HIV/AIDS. Antivir. Res. 2013, 97, 211–217. [Google Scholar] [CrossRef]
- Perez, E.E.; Wang, J.; Miller, J.C.; Jouvenot, Y.; Kim, K.A.; Liu, O.; Wang, N.; Lee, G.; Bartsevich, V.V.; Lee, Y.L.; et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat. Biotechnol. 2008, 26, 808–816. [Google Scholar] [CrossRef]
- Mussolino, C.; Morbitzer, R.; Lutge, F.; Dannemann, N.; Lahaye, T.; Cathomen, T. A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic Acids Res. 2011, 39, 9283–9293. [Google Scholar] [CrossRef]
- Schiffer, J.T.; Aubert, M.; Weber, N.D.; Mintzer, E.; Stone, D.; Jerome, K.R. Targeted DNA mutagenesis for the cure of chronic viral infections. J. Virol. 2012, 86, 8920–8936. [Google Scholar] [CrossRef]
- Holt, N.; Wang, J.; Kim, K.; Friedman, G.; Wang, X.; Taupin, V.; Crooks, G.M.; Kohn, D.B.; Gregory, P.D.; Holmes, M.C.; et al. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat. Biotechnol. 2010, 28, 839–847. [Google Scholar] [CrossRef]
- Tebas, P.; Stein, D.; Tang, W.W.; Frank, I.; Wang, S.Q.; Lee, G.; Spratt, S.K.; Surosky, R.T.; Giedlin, M.A.; Nichol, G.; et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N. Engl. J. Med. 2014, 370, 901–910. [Google Scholar] [CrossRef]
- Lalezari, J.M.R.; Wang, S.; Lee, G.; Giedlin, M.; Tang, W.; Spratt, K.; Surosky, R.; Dubois-Stringfellow, N.; Ando, D. A single infusion of zinc finger nuclease CCR5 modified autologous CD4 T cells (SB-728-T) increases CD4 counts and leads to decrease in HIV proviral load in an aviremic HIV-infected subject. In Proceedings of the 19th Conference on Retroviruses and Opportunistic Infections, Seattle, WA, USA, 5–8 March 2012.
- June, C.T.P.; Stein, D.; Mitsuyasu, R.; Lalezari, J.; Wang, S.; Lee, G.; Levine, B.; Tang, W.; Ando, D. Introduction of acquired CCR5 deficiency with zinc finger nuclease-modified autologous CD4 T cells (SB-728-T) correlates with increases in CD4 count and effects on viral load in HIV-infected subjects. In Proceedings of the 19th Conference on Retroviruses and Opportunistic Infections, Seattle, WA, USA, 5–8 March 2012.
- Westby, M.; Lewis, M.; Whitcomb, J.; Youle, M.; Pozniak, A.L.; James, I.T.; Jenkins, T.M.; Perros, M.; van der Ryst, E. Emergence of CXCR4-using human immunodeficiency virus type 1 (HIV-1) variants in a minority ofHIV-1-infected patients following treatment with the CCR5 antagonist maraviroc is from a pretreatment CXCR4-using virus reservoir. J. Virol. 2006, 80, 4909–4920. [Google Scholar] [CrossRef]
- Nedellec, R.; Coetzer, M.; Lederman, M.M.; Offord, R.E.; Hartley, O.; Mosier, D.E. Resistance to the CCR5 inhibitor 5P12-RANTES requires a difficult evolution from CCR5 to CXCR4 coreceptor use. PLoS One 2011, 6, e22020. [Google Scholar]
- Wilen, C.B.; Wang, J.; Tilton, J.C.; Miller, J.C.; Kim, K.A.; Rebar, E.J.; Sherrill-Mix, S.A.; Patro, S.C.; Secreto, A.J.; Jordan, A.P.; et al. EngineeringHIV-resistant human CD4+ T cells with CXCR4-specific zinc-finger nucleases. PLoS Pathog. 2011, 7, e1002020. [Google Scholar] [CrossRef]
- Didigu, C.A.; Wilen, C.B.; Wang, J.; Duong, J.; Secreto, A.J.; Danet-Desnoyers, G.A.; Riley, J.L.; Gregory, P.D.; June, C.H.; Holmes, M.C.; et al. Simultaneous zinc-finger nuclease editing of the HIV coreceptors CCR5 and CXCR4 protects CD4+ T cells from HIV-1 infection. Blood 2014, 123, 61–69. [Google Scholar] [CrossRef]
- Mitsuyasu, R.T.; Merigan, T.C.; Carr, A.; Zack, J.A.; Winters, M.A.; Workman, C.; Bloch, M.; Lalezari, J.; Becker, S.; Thornton, L.; et al. Phase 2 gene therapy trial of an anti-HIV ribozyme in autologous CD34+ cells. Nat. Med. 2009, 15, 285–292. [Google Scholar]
- Di Giusto, D.L.; Krishnan, A.; Li, L.; Li, H.; Li, S.; Rao, A.; Mi, S.; Yam, P.; Stinson, S.; Kalos, M.; et al. RNA-based gene therapy for HIV with lentiviral vector-modified CD34+ cells in patients undergoing transplantation for AIDS-related lymphoma. Sci. Transl. Med. 2010, 2, 36–43. [Google Scholar]
- Montoya-Durango, D.E.; Liu, Y.; Teneng, I.; Kalbfleisch, T.; Lacy, M.E.; Steffen, M.C.; Ramos, K.S. Epigenetic control of mammalian LINE-1 retrotransposon by retinoblastoma proteins. Mutat. Res. 2009, 665, 20–28. [Google Scholar] [CrossRef]
- Maksakova, I.A.; Mager, D.L.; Reiss, D. Keeping active endogenous retroviral-like elements in check: The epigenetic perspective. Cell. Mol. Life Sci. 2008, 65, 3329–3347. [Google Scholar] [CrossRef]
- Gonzalez-Hernandez, M.J.; Swanson, M.D.; Contreras-Galindo, R.; Cookinham, S.; King, S.R.; Noel, R.J., Jr.; Kaplan, M.H.; Markovitz, D.M. Expression of human endogenous retrovirus type K (HML-2) is activated by the Tat protein of HIV-1. J. Virol. 2012, 86, 7790–7805. [Google Scholar] [CrossRef]
- Jern, P.; Coffin, J.M. Effects of retroviruses on host genome function. Annu. Rev. Genet. 2008, 42, 709–732. [Google Scholar] [CrossRef]
- Mitsuyasu, R. Curing HIV: Lessons from cancer therapy. Curr. Opin. HIV AIDS 2013, 8, 224–229. [Google Scholar] [CrossRef]
- Shan, L.; Siliciano, R.F. From reactivation of latent HIV-1 to elimination of the latent reservoir: The presence of multiple barriers to viral eradication. Bioessays 2013, 35, 544–552. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Battistini, A.; Sgarbanti, M. HIV-1 Latency: An Update of Molecular Mechanisms and Therapeutic Strategies. Viruses 2014, 6, 1715-1758. https://doi.org/10.3390/v6041715
Battistini A, Sgarbanti M. HIV-1 Latency: An Update of Molecular Mechanisms and Therapeutic Strategies. Viruses. 2014; 6(4):1715-1758. https://doi.org/10.3390/v6041715
Chicago/Turabian StyleBattistini, Angela, and Marco Sgarbanti. 2014. "HIV-1 Latency: An Update of Molecular Mechanisms and Therapeutic Strategies" Viruses 6, no. 4: 1715-1758. https://doi.org/10.3390/v6041715