Modulation of Homology-Directed Repair in T98G Glioblastoma Cells Due to Interactions between Wildtype p53, Rad51 and HCMV IE1-72

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
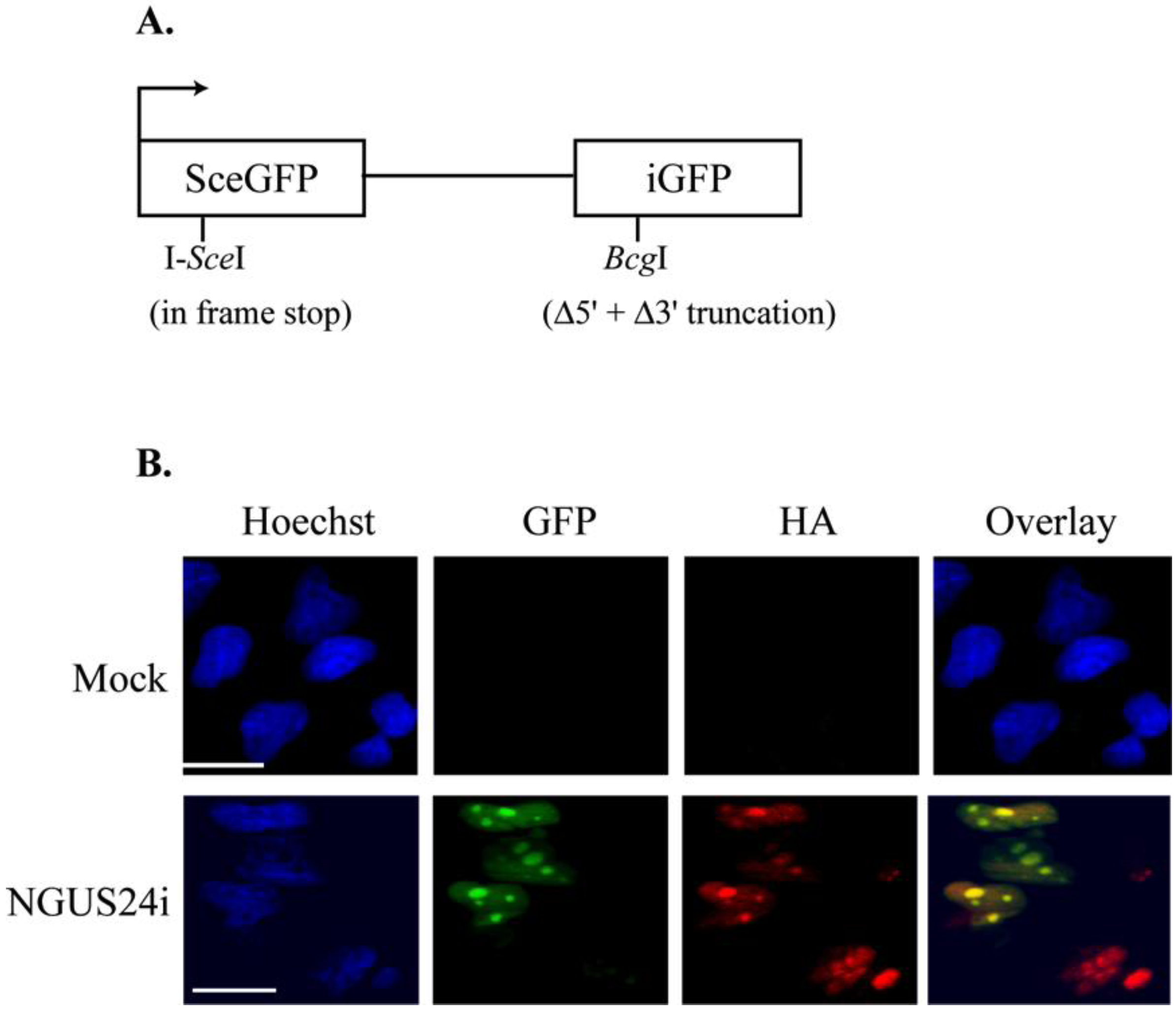
2.1. Introduction of the pDRGFP Substrate into T98G Cells to Assess HDR
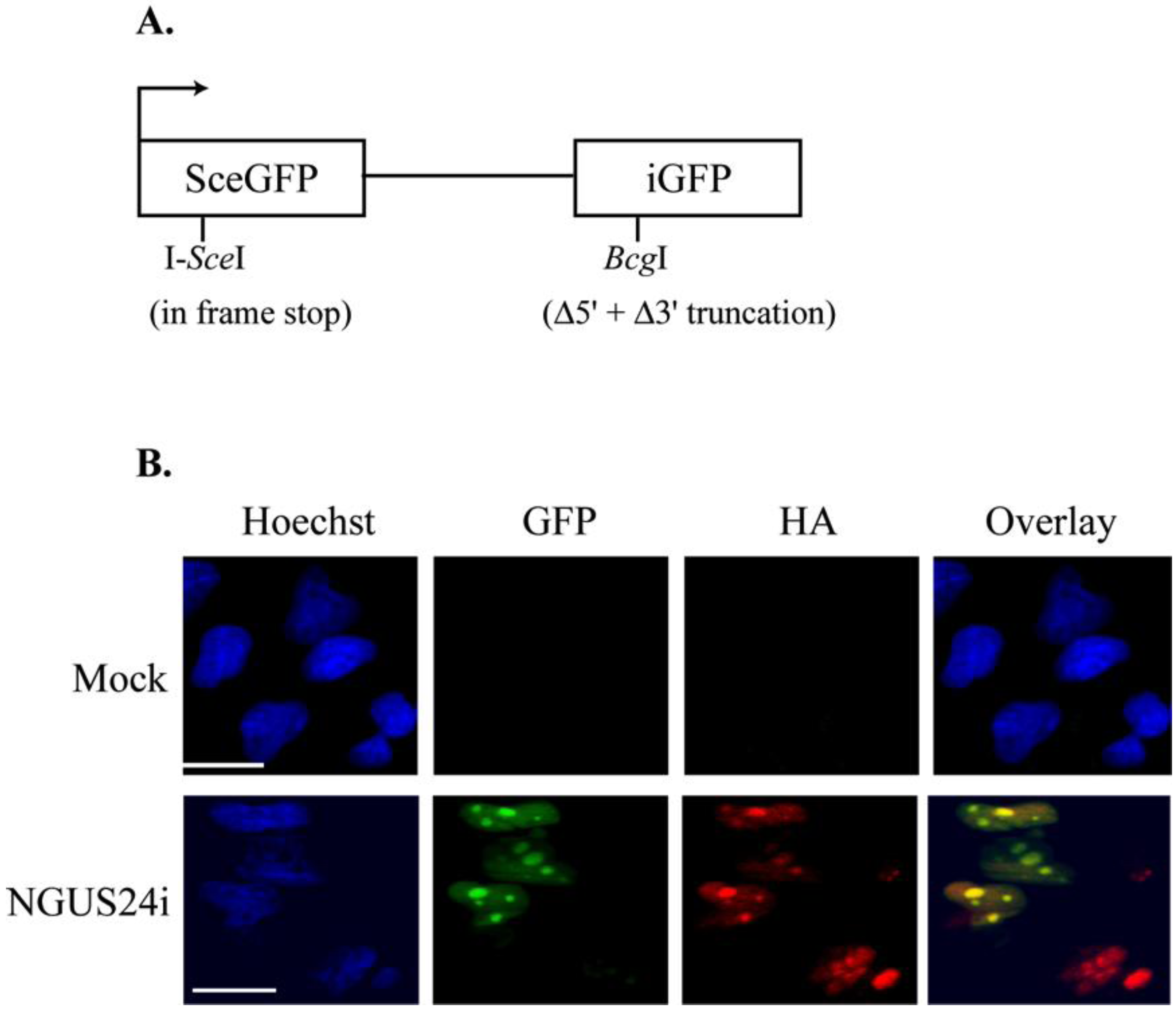
2.2. HDR at I-SceI Induced DSBs Was Downregulated in HCMV-Infected T98G Cells
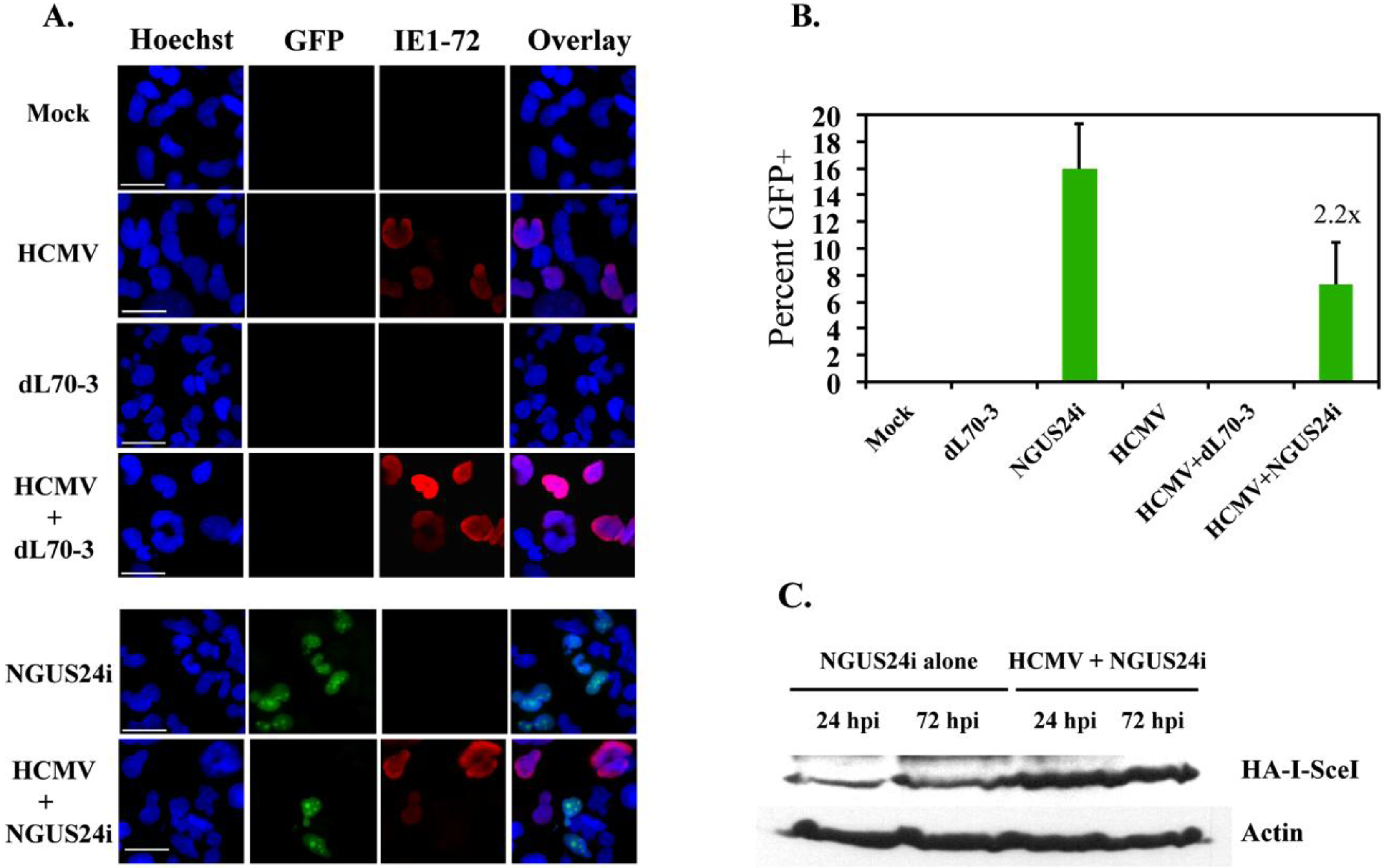
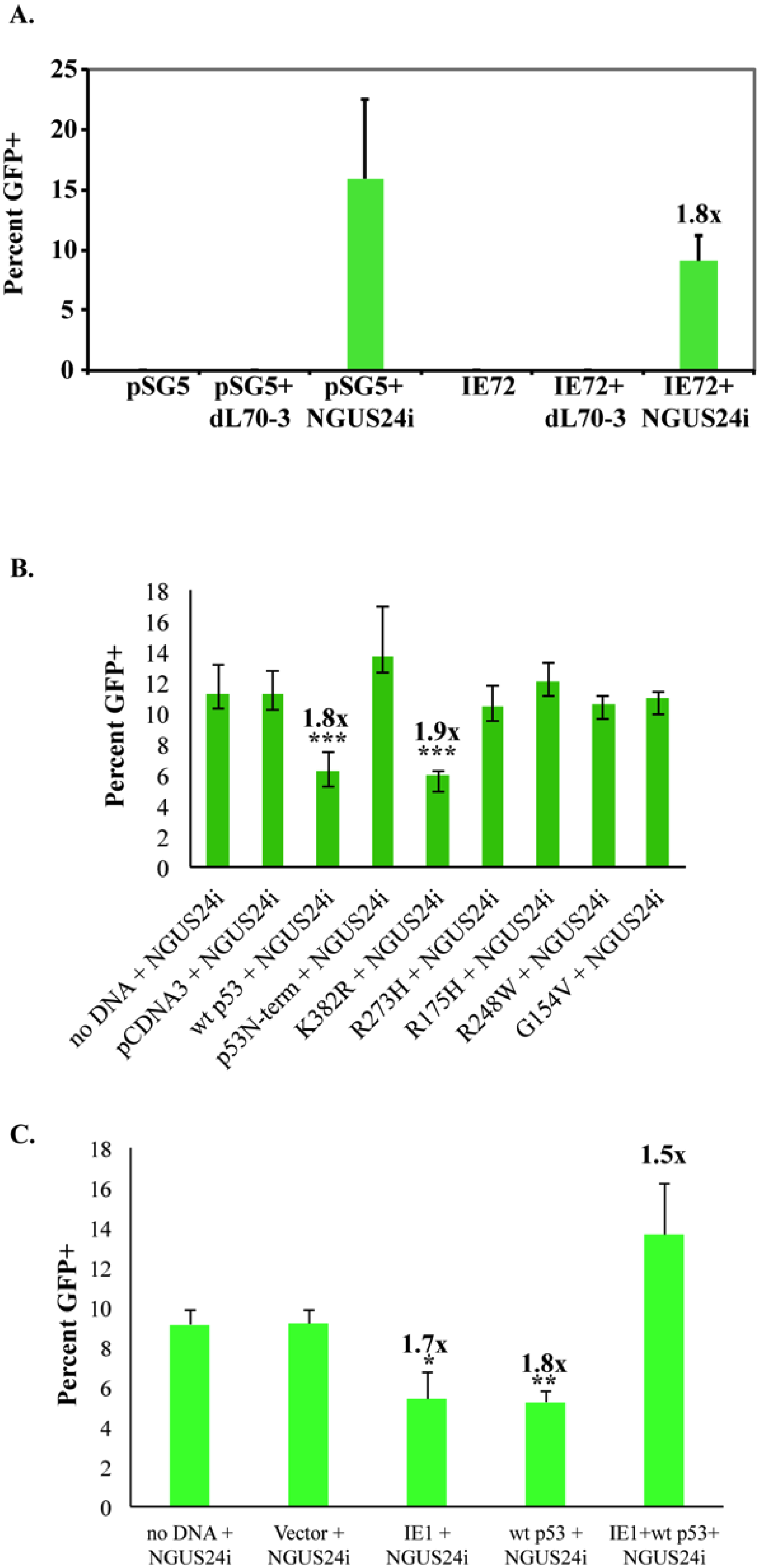
2.3. Transient Expression of IE72 Alone Was Sufficient to Downregulate HDR in T98Gs
2.4. Expression of wt p53 in T98Gs Caused the Same Decrease in HDR as IE72

2.5. Co-Expression of IE72 and wt p53 Negated Their Individual Effects on HDR
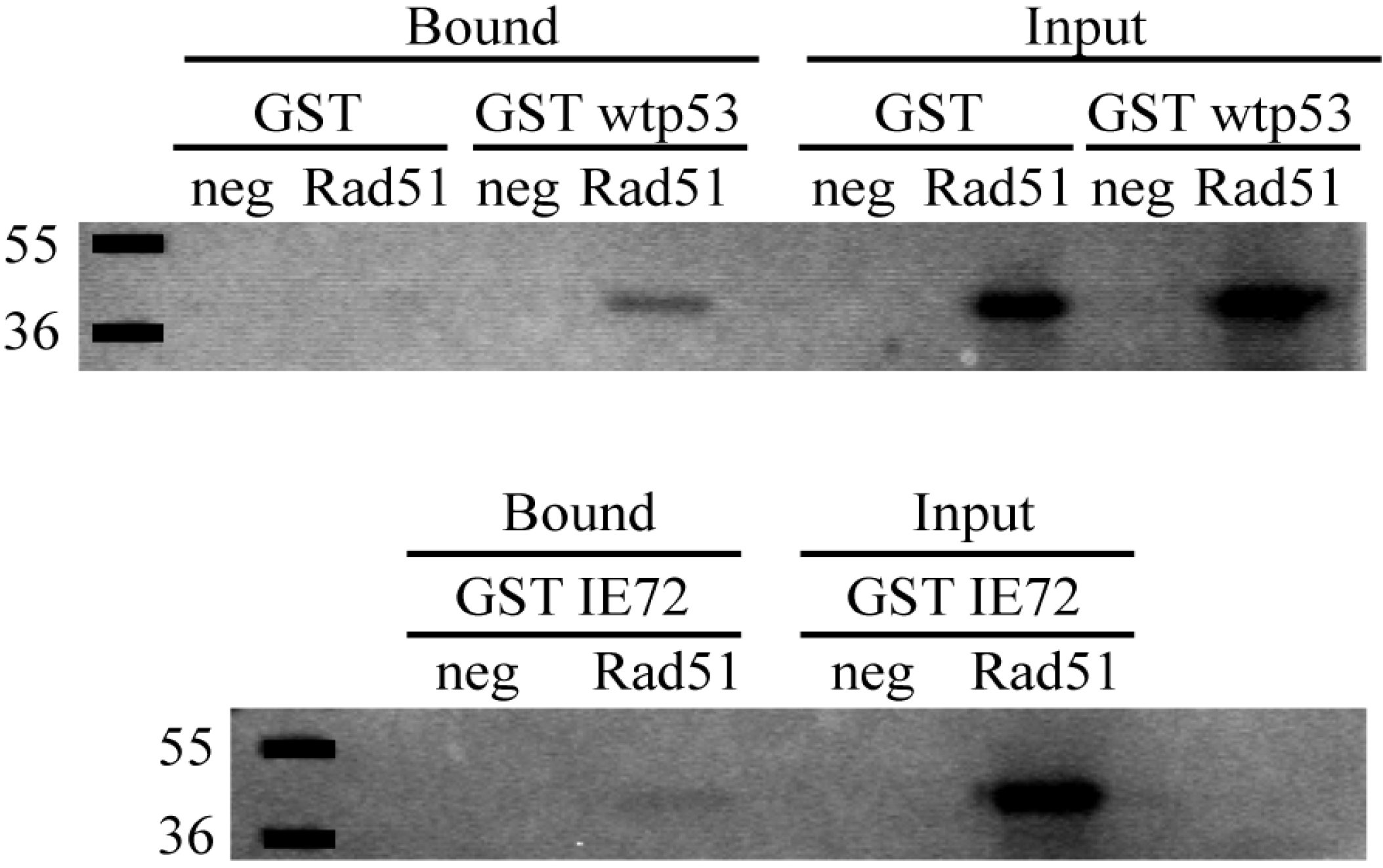
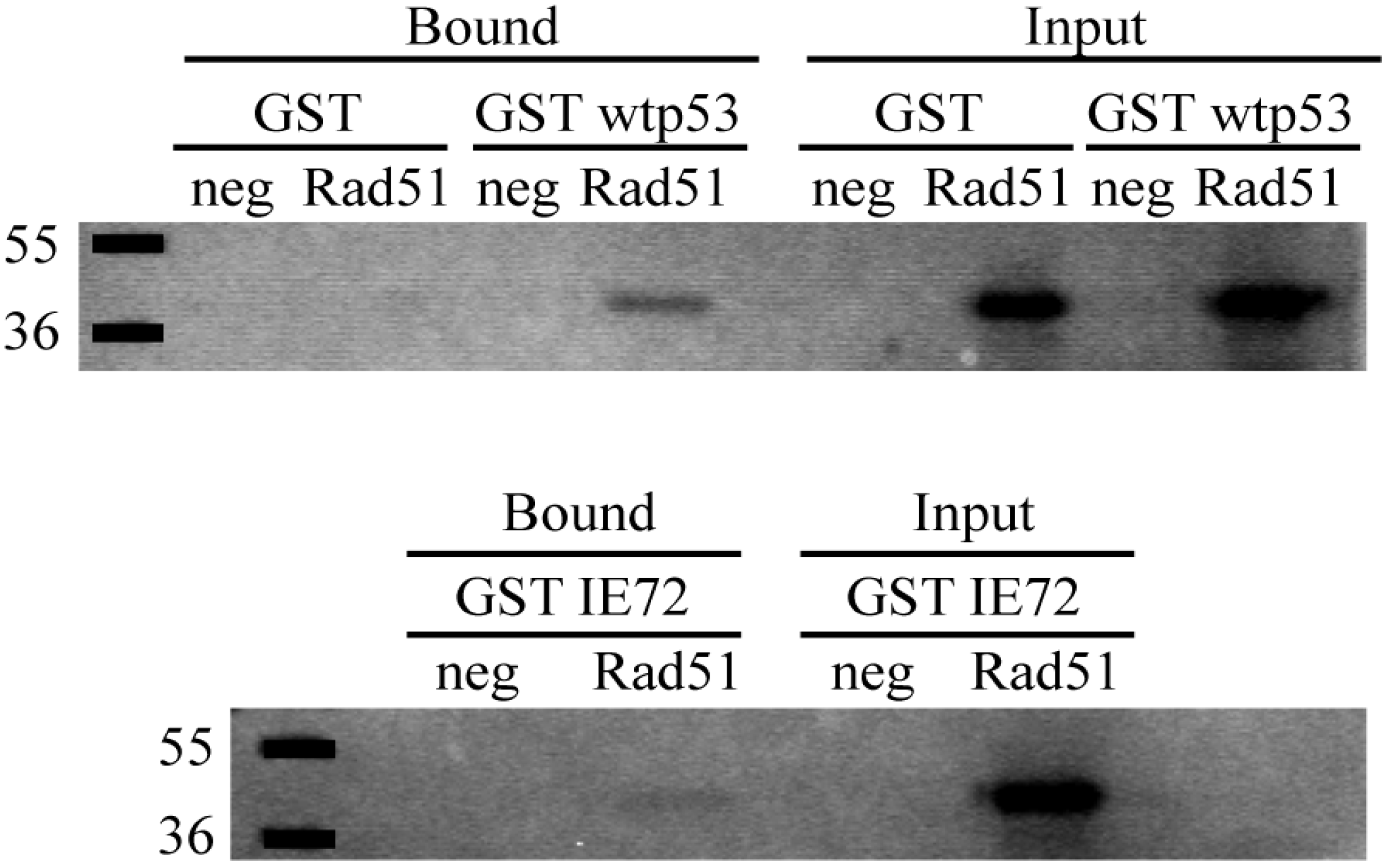
2.6. In Vitro Binding Assays Found Both wt p53 and IE72 Bound Rad51
3. Experimental Section
3.1. Cells and Virus Growth
3.2. Transfections
3.3. Nucleofections
3.4. Molecular Cloning
3.5. DSB Repair Assay
3.6. Virus Infection
3.7. Antibodies (Ab)
3.8. Immunofluorescence (IF)
3.9. Immunoblotting
3.10. GST Binding Studies
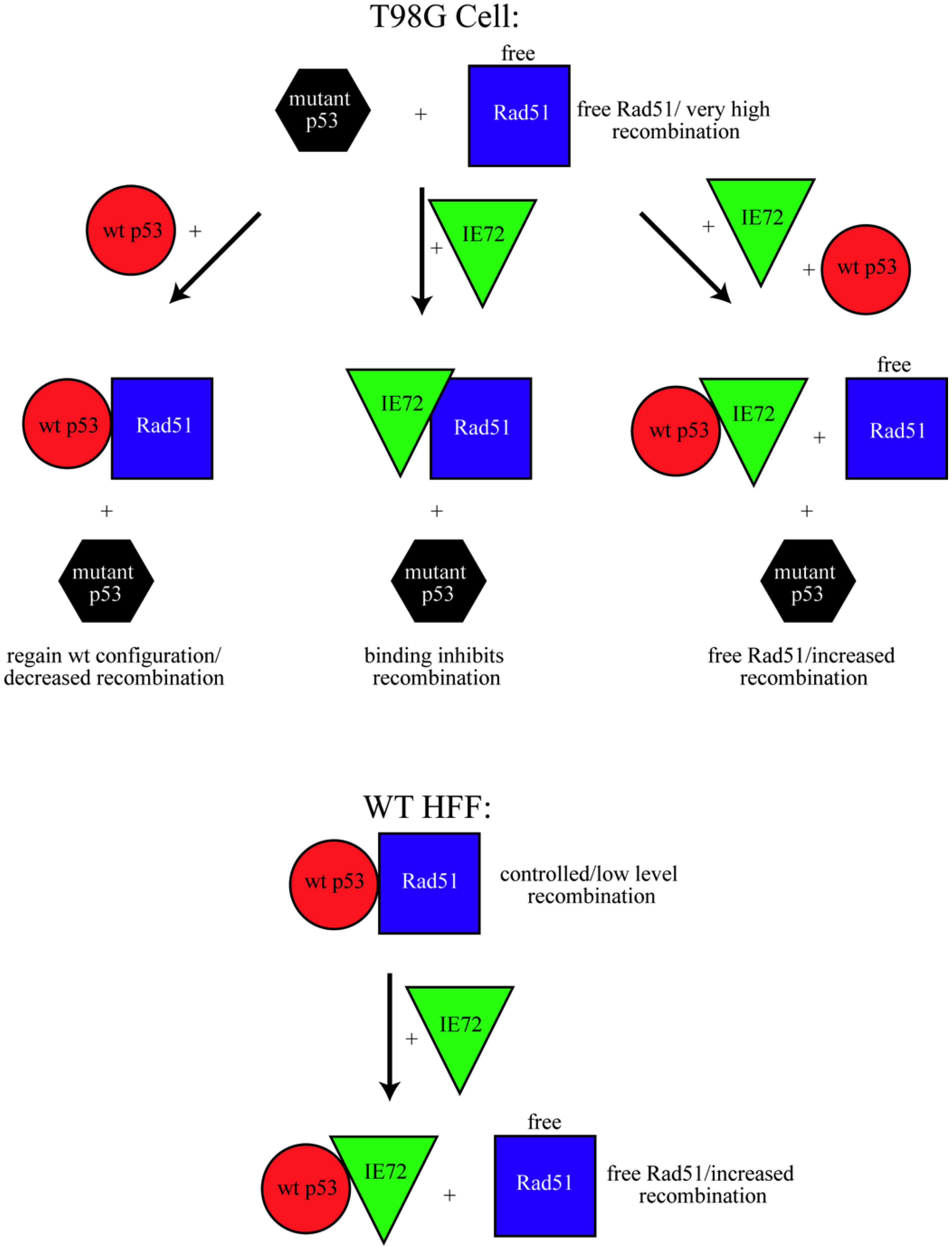
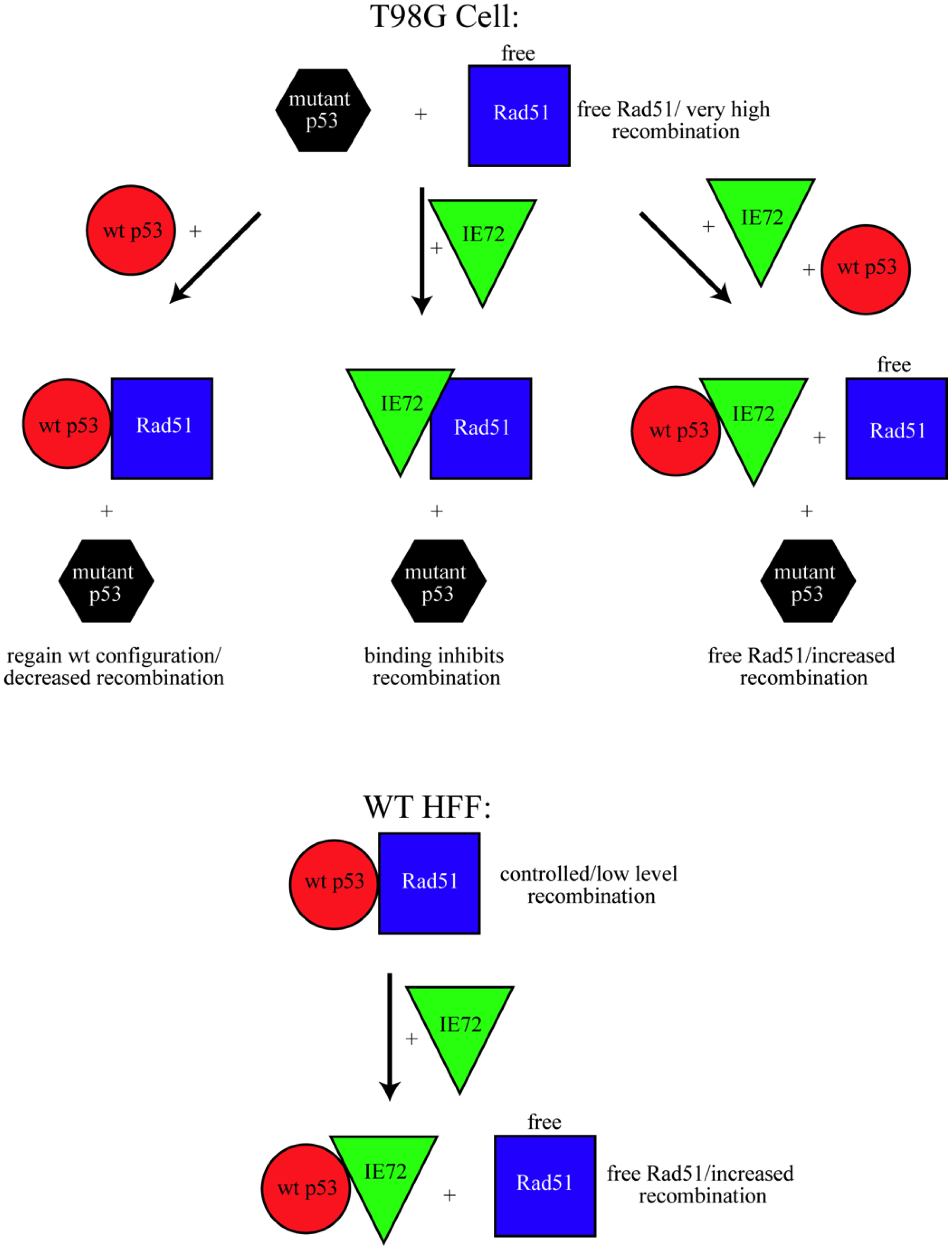
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Boppana, S.B.; Fowler, K.B.; Britt, W.J.; Stagno, S.; Pass, R.F. Symptomatic congenital cytomegalovirus infection in infants born to mothers with preexisting immunity to cytomegalovirus. Pediatrics 1999, 104, 55–60. [Google Scholar] [CrossRef]
- Cinque, P.; Marenzi, R.; Ceresa, D. Cytomegalovirus infections of the nervous system. Intervirology 1997, 40, 85–97. [Google Scholar] [CrossRef]
- Zanghellini, F.; Boppana, S.B.; Emery, V.C.; Griffiths, P.D.; Pass, R.F. Asymptomatic primary cytomegalovirus infection: Virologic and immunologic features. J. Infect. Dis. 1999, 180, 702–707. [Google Scholar] [CrossRef]
- Britt, W.; Alford, C. Cytomegalovirus. In Fields Virology; Lippincott-Raven Publishers: Philadelphia, PA, USA, 1996; pp. 2493–2523. [Google Scholar]
- Dziurzynski, K.; Wei, J.; Qiao, W.; Hatiboglu, M.A.; Kong, L.Y.; Wu, A.; Wang, Y.; Cahill, D.; Levine, N.; Prabhu, S.; et al. Glioma-associated cytomegalovirus mediates subversion of the monocyte lineage to a tumor propagating phenotype. Clin. Canc. Res. 2011, 17, 4642–4649. [Google Scholar] [CrossRef]
- Michaelis, M.; Baumgarten, P.; Mittelbronn, M.; Driever, P.H.; Doerr, H.W.; Cinatl, J., Jr. Oncomodulation by human cytomegalovirus: Novel clinical findings open new roads. Med. Microbiol. Immunol. 2011, 200, 1–5. [Google Scholar] [CrossRef]
- Fortunato, E.A.; Dell'Aquila, M.L.; Spector, D.H. Specific chromosome 1 breaks induced by human cytomegalovirus. Proc. Natl. Acad. Sci. USA 2000, 97, 853–858. [Google Scholar] [CrossRef]
- Nystad, M.; Fagerheim, T.; Brox, V.; Fortunato, E.A.; Nilssen, O. Human cytomegalovirus (HCMV) and hearing impairment: Infection of fibroblast cells with HCMV induces chromosome breaks at 1q23.3, between loci DFNA7 and DFNA49—Both involved in dominantly inherited, sensorineural, hearing impairment. Mutat. Res. 2008, 637, 56–65. [Google Scholar] [CrossRef]
- Toesca, A. Central and peripheral myelin in the rat cochlear and vestibular nerves. Neurosci. Lett. 1996, 221, 21–24. [Google Scholar] [CrossRef]
- Chapman, J.R.; Taylor, M.R.; Boulton, S.J. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell 2012, 47, 497–510. [Google Scholar] [CrossRef]
- Rothkamm, K.; Kruger, I.; Thompson, L.H.; Lobrich, M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol. Cell Biol. 2003, 23, 5706–5715. [Google Scholar] [CrossRef]
- Kulkarni, A.S.; Fortunato, E.A. Stimulation of homology-directed repair at I-SceI-induced DNA breaks during the permissive life cycle of human cytomegalovirus. J. Virol. 2011, 85, 6049–6054. [Google Scholar] [CrossRef]
- Pierce, A.J.; Johnson, R.D.; Thompson, L.H.; Jasin, M. Xrcc3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 1999, 13, 2633–2638. [Google Scholar] [CrossRef]
- Wiese, C.; Pierce, A.J.; Gauny, S.S.; Jasin, M.; Kronenberg, A. Gene conversion is strongly induced in human cells by double-strand breaks and is modulated by the expression of bcl-x(l). Canc. Res. 2002, 62, 1279–1283. [Google Scholar]
- Bett, A.J.; Haddara, W.; Prevec, L.; Graham, F.L. An efficient and flexible system for construction of adenovirus vectors with insertions or deletions in early regions 1 and 3. Proc. Natl. Acad. Sci. USA 1994, 91, 8802–8806. [Google Scholar] [CrossRef]
- Anglana, M.; Bacchetti, S. Construction of a recombinant adenovirus for efficient delivery of the I-SceI yeast endonuclease to human cells and its application in the in vivo cleavage of chromosomes to expose new potential telomeres. Nucleic Acids Res. 1999, 27, 4276–4281. [Google Scholar] [CrossRef]
- Luo, M.H.; Fortunato, E.A. Long-term infection and shedding of human cytomegalovirus in T98G glioblastoma cells. J. Virol. 2007, 81, 10424–10436. [Google Scholar] [CrossRef]
- Greaves, R.F.; Mocarski, E.S. Defective growth correlates with reduced accumulation of a viral DNA replication protein after low-multiplicity infection by a human cytomegalovirus IE1 mutant. J. Virol. 1998, 72, 366–379. [Google Scholar]
- Castillo, J.P.; Kowalik, T.F. Human cytomegalovirus immediate early proteins and cell growth control. Gene 2002, 290, 19–34. [Google Scholar] [CrossRef]
- Short, S.C.; Bourne, S.; Martindale, C.; Woodcock, M.; Jackson, S.P. DNA damage responses at low radiation doses. Rad. Res. 2005, 164, 292–302. [Google Scholar] [CrossRef]
- Bertrand, P.; Saintigny, Y.; Lopez, B.S. P53's double life: Transactivation-independent repression of homologous recombination. Trends Gen. 2004, 20, 235–243. [Google Scholar] [CrossRef]
- Rosenke, K.; Samuel, M.A.; McDowell, E.T.; Toerne, M.A.; Fortunato, E.A. An intact sequence-specific DNA-binding domain is required for human cytomegalovirus-mediated sequestration of p53 and may promote in vivo binding to the viral genome during infection. Virology 2006, 348, 19–34. [Google Scholar] [CrossRef]
- Arias-Lopez, C.; Lazaro-Trueba, I.; Kerr, P.; Lord, C.J.; Dexter, T.; Iravani, M.; Ashworth, A.; Silva, A. P53 modulates homologous recombination by transcriptional regulation of the Rad51 gene. EMBO Rep. 2006, 7, 219–224. [Google Scholar] [CrossRef] [Green Version]
- Buchhop, S.; Gibson, M.K.; Wang, X.W.; Wagner, P.; Sturzbecher, H.W.; Harris, C.C. Interaction of p53 with the human Rad51 protein. Nucleic Acids Res. 1997, 25, 3868–3874. [Google Scholar] [CrossRef]
- Rieckmann, T.; Kriegs, M.; Nitsch, L.; Hoffer, K.; Rohaly, G.; Kocher, S.; Petersen, C.; Dikomey, E.; Dornreiter, I.; Dahm-Daphi, J. P53 modulates homologous recombination at I-SceI-induced double-strand breaks through cell-cycle regulation. Oncogene 2013, 32, 968–975. [Google Scholar] [CrossRef]
- Daboussi, F.; Dumay, A.; Delacote, F.; Lopez, B.S. DNA double-strand break repair signalling: The case of Rad51 post-translational regulation. Cell Signal. 2002, 14, 969–975. [Google Scholar] [CrossRef]
- Hwang, E.S.; Zhang, Z.; Cai, H.; Huang, D.Y.; Huong, S.M.; Cha, C.Y.; Huang, E.S. Human cytomegalovirus IE1–72 protein interacts with p53 and inhibits p53-dependent transactivation by a mechanism different from that of IE2–86 protein. J. Virol. 2009, 83, 12388–12398. [Google Scholar] [CrossRef]
- Klucher, K.M.; Sommer, M.; Kadonaga, J.T.; Spector, D.H. In vivo and in vitro analysis of transcriptional activation mediated by the human cytomegalovirus major immediate-early proteins. Mol. Cell Biol. 1993, 13, 1238–1250. [Google Scholar]
- Ashcroft, M.; Kubbutat, M.H.; Vousden, K.H. Regulation of p53 function and stability by phosphorylation. Mol. Cell Biol. 1999, 19, 1751–1758. [Google Scholar]
- Casavant, N.C.; Luo, M.H.; Rosenke, K.; Winegardner, T.; Zurawska, A.; Fortunato, E.A. Potential role for p53 in the permissive life cycle of human cytomegalovirus. J. Virol. 2006, 80, 8390–8401. [Google Scholar] [CrossRef]
- Luo, M.H.; Rosenke, K.; Czornak, K.; Fortunato, E.A. Human cytomegalovirus disrupts both ataxia telangiectasia mutated protein (ATM)- and ATM-Rad3-related kinase-mediated DNA damage responses during lytic infection. J. Virol. 2007, 81, 1934–1950. [Google Scholar] [CrossRef]
- Ayed, A.; Mulder, F.A.; Yi, G.S.; Lu, Y.; Kay, L.E.; Arrowsmith, C.H. Latent and active p53 are identical in conformation. Nat. Struct. Biol. 2001, 8, 756–760. [Google Scholar] [CrossRef]
- Guan, K.L.; Dixon, J.E. Eukaryotic proteins expressed in Escherichia coli: An improved thrombin cleavage and purification procedure of fusion proteins with glutathione s-transferase. Anal. Biochem. 1991, 192, 262–267. [Google Scholar] [CrossRef]
- Snowden, T.; Shim, K.S.; Schmutte, C.; Acharya, S.; Fishel, R. hMSH4-hMSH5 adenosine nucleotide processing and interactions with homologous recombination machinery. J. Biol. Chem. 2008, 283, 145–154. [Google Scholar] [CrossRef]
- Baydoun, H.H.; Pancewicz, J.; Nicot, C. Human T-lymphotropic type 1 virus p30 inhibits homologous recombination and favors unfaithful DNA repair. Blood 2011, 117, 5897–5906. [Google Scholar] [CrossRef]
- Becker, S.A.; Lee, T.H.; Butel, J.S.; Slagle, B.L. Hepatitis B virus X protein interferes with cellular DNA repair. J. Virol. 1998, 72, 266–272. [Google Scholar]
- Chipitsyna, G.; Slonina, D.; Siddiqui, K.; Peruzzi, F.; Skorski, T.; Reiss, K.; Sawaya, B.E.; Khalili, K.; Amini, S. HIV-1 Tat increases cell survival in response to cisplatin by stimulating Rad51 gene expression. Oncogene 2004, 23, 2664–2671. [Google Scholar] [CrossRef]
- Durkin, S.S.; Guo, X.; Fryrear, K.A.; Mihaylova, V.T.; Gupta, S.K.; Belgnaoui, S.M.; Haoudi, A.; Kupfer, G.M.; Semmes, O.J. HTLV-1 Tax oncoprotein subverts the cellular DNA damage response via binding to DNA-dependent protein kinase. J. Biol. Chem. 2008, 283, 36311–36320. [Google Scholar] [CrossRef]
- Groisman, I.J.; Koshy, R.; Henkler, F.; Groopman, J.D.; Alaoui-Jamali, M.A. Downregulation of DNA excision repair by the hepatitis B virus-X protein occurs in p53-proficient and p53-deficient cells. Carcinogenesis 1999, 20, 479–483. [Google Scholar] [CrossRef]
- Gruhne, B.; Sompallae, R.; Masucci, M.G. Three Epstein-Barr virus latency proteins independently promote genomic instability by inducing DNA damage, inhibiting DNA repair and inactivating cell cycle checkpoints. Oncogene 2009, 28, 3997–4008. [Google Scholar] [CrossRef]
- Jia, L.; Wang, X.W.; Harris, C.C. Hepatitis B virus X protein inhibits nucleotide excision repair. Int. J. Canc. 1999, 80, 875–879. [Google Scholar] [CrossRef]
- Liang, X.; Pickering, M.T.; Cho, N.H.; Chang, H.; Volkert, M.R.; Kowalik, T.F.; Jung, J.U. Deregulation of DNA damage signal transduction by herpesvirus latency-associated M2. J. Virol. 2006, 80, 5862–5874. [Google Scholar] [CrossRef]
- Prost, S.; Ford, J.M.; Taylor, C.; Doig, J.; Harrison, D.J. Hepatitis B X protein inhibits p53-dependent DNA repair in primary mouse hepatocytes. J. Biol. Chem. 1998, 273, 33327–33332. [Google Scholar]
- Sun, Y.; Huang, Y.C.; Xu, Q.Z.; Wang, H.P.; Bai, B.; Sui, J.L.; Zhou, P.K. HIV-1 Tat depresses DNA-PK(cs) expression and DNA repair, and sensitizes cells to ionizing radiation. Int. J. Radiat. Oncol. Biol. Phys. 2006, 65, 842–850. [Google Scholar] [CrossRef]
- Trojanek, J.; Croul, S.; Ho, T.; Wang, J.Y.; Darbinyan, A.; Nowicki, M.; Valle, L.D.; Skorski, T.; Khalili, K.; Reiss, K. T-antigen of the human polyomavirus JC attenuates faithful DNA repair by forcing nuclear interaction between IRS-1 and Rad51. J. Cell. Physiol. 2006, 206, 35–46. [Google Scholar] [CrossRef]
- Mathonnet, G.; Lachance, S.; Alaoui-Jamali, M.; Drobetsky, E.A. Expression of hepatitis B virus X oncoprotein inhibits transcription-coupled nucleotide excision repair in human cells. Mutat. Res. 2004, 554, 305–318. [Google Scholar] [CrossRef]
- Mohni, K.N.; Mastrocola, A.S.; Bai, P.; Weller, S.K.; Heinen, C.D. DNA mismatch repair proteins are required for efficient herpes simplex virus 1 replication. J. Virol. 2011, 85, 12241–12253. [Google Scholar] [CrossRef]
- O'Dowd, J.M.; Zavala, A.G.; Brown, C.J.; Mori, T.; Fortunato, E.A. HCMV-infected cells maintain efficient nucleotide excision repair of the viral genome while abrogating repair of the host genome. PLoS Pathog. 2012, 8, e1003038. [Google Scholar] [CrossRef]
- Schumacher, A.J.; Mohni, K.N.; Kan, Y.; Hendrickson, E.A.; Stark, J.M.; Weller, S.K. The HSV-1 exonuclease, UL12, stimulates recombination by a single strand annealing mechanis. PLoS Pathog. 2012, 8, e1002862. [Google Scholar] [CrossRef]
- Deng, C.Z.; AbuBakar, S.; Fons, M.P.; Boldogh, I.; Hokanson, J.; Au, W.W.; Albrecht, T. Cytomegalovirus-enhanced induction of chromosome aberrations in human peripheral blood lymphocytes treated with potent genotoxic agents. Environ. Mol. Mutagen. 1992, 19, 304–310. [Google Scholar]
- Duong, F.H.; Christen, V.; Lin, S.; Heim, M.H. Hepatitis C virus-induced up-regulation of protein phosphatase 2a inhibits histone modification and DNA damage repair. Hepatology 2010, 51, 741–751. [Google Scholar]
- Lilley, C.E.; Chaurushiya, M.S.; Boutell, C.; Landry, S.; Suh, J.; Panier, S.; Everett, R.D.; Stewart, G.S.; Durocher, D.; Weitzman, M.D. A viral E3 ligase targets RNF8 and RNF168 to control histone ubiquitination and DNA damage responses. EMBO J. 2010, 29, 943–955. [Google Scholar] [CrossRef]
- Pal, S.; Polyak, S.J.; Bano, N.; Qiu, W.C.; Carithers, R.L.; Shuhart, M.; Gretch, D.R.; Das, A. Hepatitis C virus induces oxidative stress, DNA damage and modulates the DNA repair enzyme NEIL1. J. Gastroenterol. Hepatol. 2010, 25, 627–634. [Google Scholar] [CrossRef]
- Philpott, S.M.; Buehring, G.C. Defective DNA repair in cells with human T-cell leukemia/bovine leukemia viruses: Role of Tax gene. J. Natl. Canc. Inst. 1999, 91, 933–942. [Google Scholar] [CrossRef]
- Ranneberg-Nilsen, T.; Bjoras, M.; Luna, L.; Slettebakk, R.; Dale, H.A.; Seeberg, E.; Rollag, H. Human cytomegalovirus infection modulates DNA base excision repair in fibroblast cells. Virology 2006, 348, 389–397. [Google Scholar] [CrossRef]
- Bowman, K.K.; Sicard, D.M.; Ford, J.M.; Hanawalt, P.C. Reduced global genomic repair of ultraviolet light-induced cyclobutane pyrimidine dimers in simian virus 40-transformed human cells. Mol. Carcinog. 2000, 29, 17–24. [Google Scholar] [CrossRef]
- Yoon, D.; Wang, Y.; Stapleford, K.; Wiesmuller, L.; Chen, J. P53 inhibits strand exchange and replication fork regression promoted by human Rad51. J. Mol. Biol. 2004, 336, 639–654. [Google Scholar] [CrossRef]
- Restle, A.; Janz, C.; Wiesmuller, L. Differences in the association of p53 phosphorylated on serine 15 and key enzymes of homologous recombination. Oncogene 2005, 24, 4380–4387. [Google Scholar] [CrossRef]
- Mekeel, K.L.; Tang, W.; Kachnic, L.A.; Luo, C.M.; DeFrank, J.S.; Powell, S.N. Inactivation of p53 results in high rates of homologous recombination. Oncogene 1997, 14, 1847–1857. [Google Scholar]
- Boichuk, S.; Hu, L.; Hein, J.; Gjoerup, O.V. Multiple DNA damage signaling and repair pathways deregulated by simian virus 40 large T antigen. J. Virol. 2010, 84, 8007–8020. [Google Scholar] [CrossRef]
- Kudoh, A.; Iwahori, S.; Sato, Y.; Nakayama, S.; Isomura, H.; Murata, T.; Tsurumi, T. Homologous recombinational repair factors are recruited and loaded onto the viral DNA genome in Epstein-Barr virus replication compartments. J. Virol. 2009, 83, 6641–6651. [Google Scholar] [CrossRef]
- Cosnefroy, O.; Tocco, A.; Lesbats, P.; Thierry, S.; Calmels, C.; Wiktorowicz, T.; Reigadas, S.; Kwon, Y.; De Cian, A.; Desfarges, S.; et al. Stimulation of the human Rad51 nucleofilament restricts HIV-1 integration in vitro and in infected cells. J. Virol. 2012, 86, 513–526. [Google Scholar] [CrossRef]
- Fortunato, E.A.; Spector, D.H. P53 and RPA are sequestered in viral replication centers in the nuclei of cells infected with human cytomegalovirus. J. Virol. 1998, 72, 2033–2039. [Google Scholar]
- Dziurzynski, K.; Chang, S.M.; Heimberger, A.B.; Kalejta, R.F.; McGregor Dallas, S.R.; Smit, M.; Soroceanu, L.; Cobbs, C.S. HCMV Glioma Symposium. Consensus on the role of human cytomegalovirus in glioblastoma. Neuro Oncol. 2012, 14, 246–255. [Google Scholar] [CrossRef]
- Ranganathan, P.; Clark, P.A.; Kuo, J.S.; Salamat, M.S.; Kalejta, R.F. Significant association of multiple human cytomegalovirus genomic loci with glioblastoma multiforme samples. J. Virol. 2012, 86, 854–864. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kulkarni, A.S.; Fortunato, E.A. Modulation of Homology-Directed Repair in T98G Glioblastoma Cells Due to Interactions between Wildtype p53, Rad51 and HCMV IE1-72. Viruses 2014, 6, 968-985. https://doi.org/10.3390/v6030968
Kulkarni AS, Fortunato EA. Modulation of Homology-Directed Repair in T98G Glioblastoma Cells Due to Interactions between Wildtype p53, Rad51 and HCMV IE1-72. Viruses. 2014; 6(3):968-985. https://doi.org/10.3390/v6030968
Chicago/Turabian StyleKulkarni, Amit S., and Elizabeth A. Fortunato. 2014. "Modulation of Homology-Directed Repair in T98G Glioblastoma Cells Due to Interactions between Wildtype p53, Rad51 and HCMV IE1-72" Viruses 6, no. 3: 968-985. https://doi.org/10.3390/v6030968