HCMV Infection of Human Trophoblast Progenitor Cells of the Placenta Is Neutralized by a Human Monoclonal Antibody to Glycoprotein B and Not by Antibodies to the Pentamer Complex

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
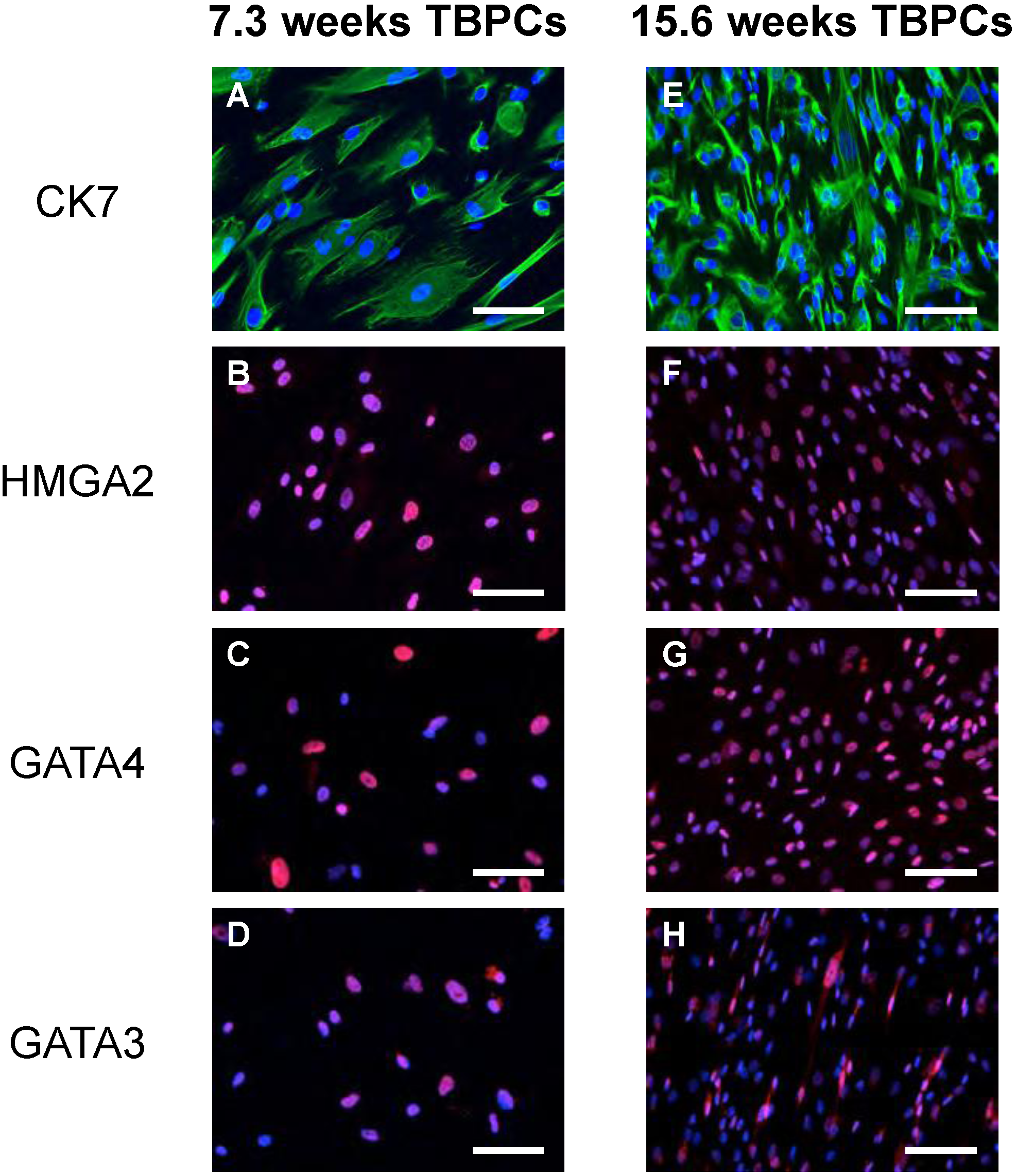
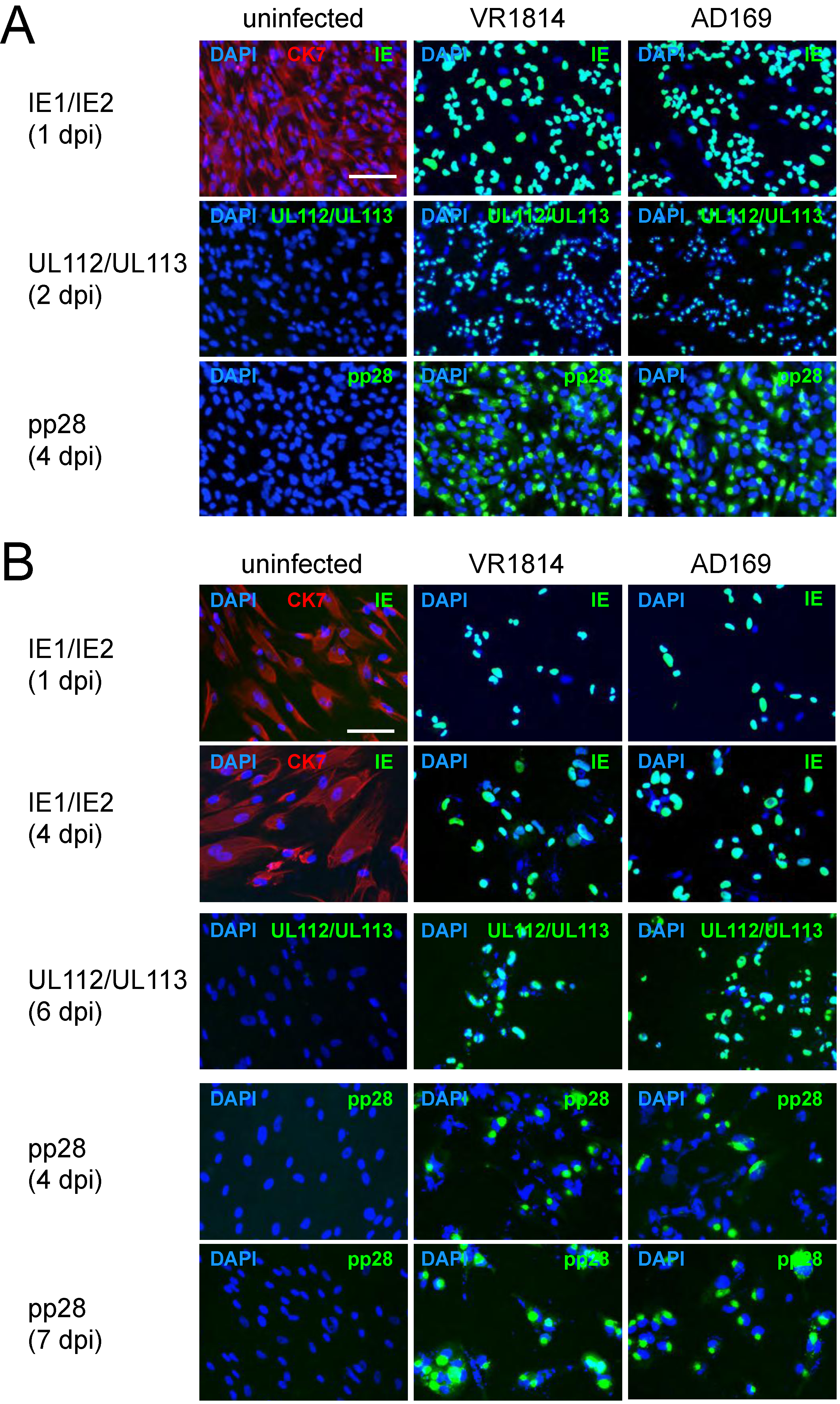
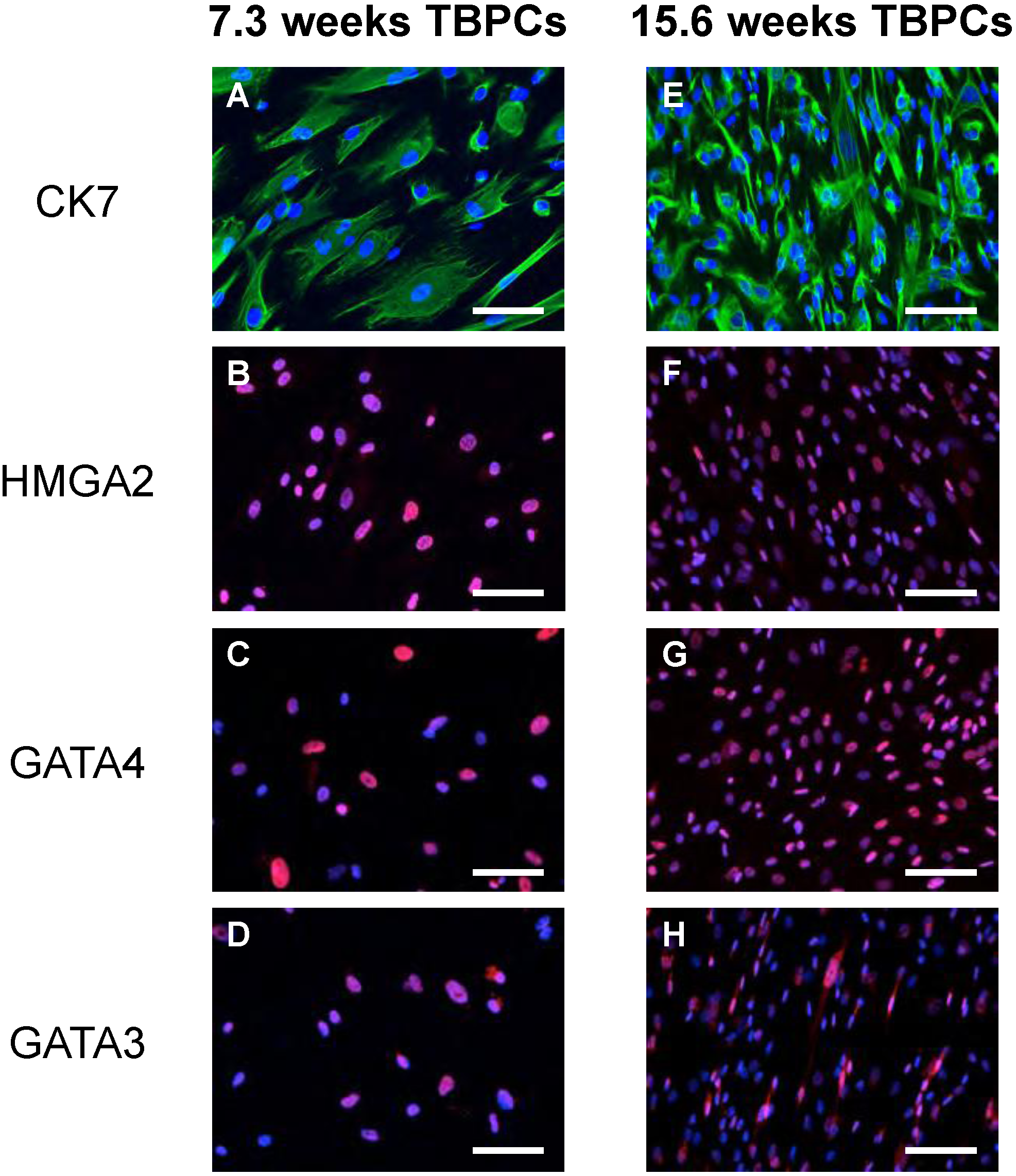
2.1. TBPCs from First and Second Trimesters Are Permissive for Pathogenic and Attenuated HCMV Strains
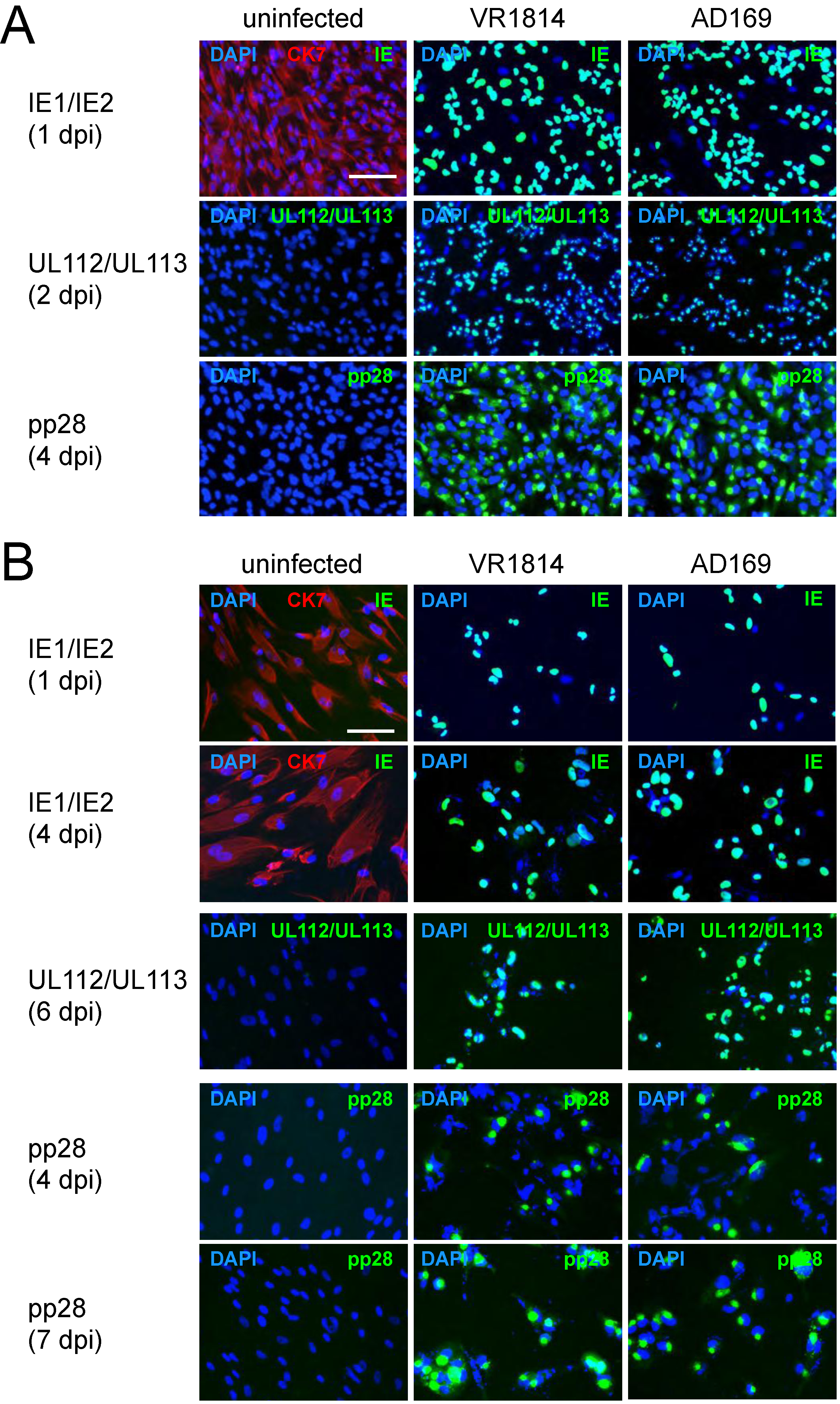
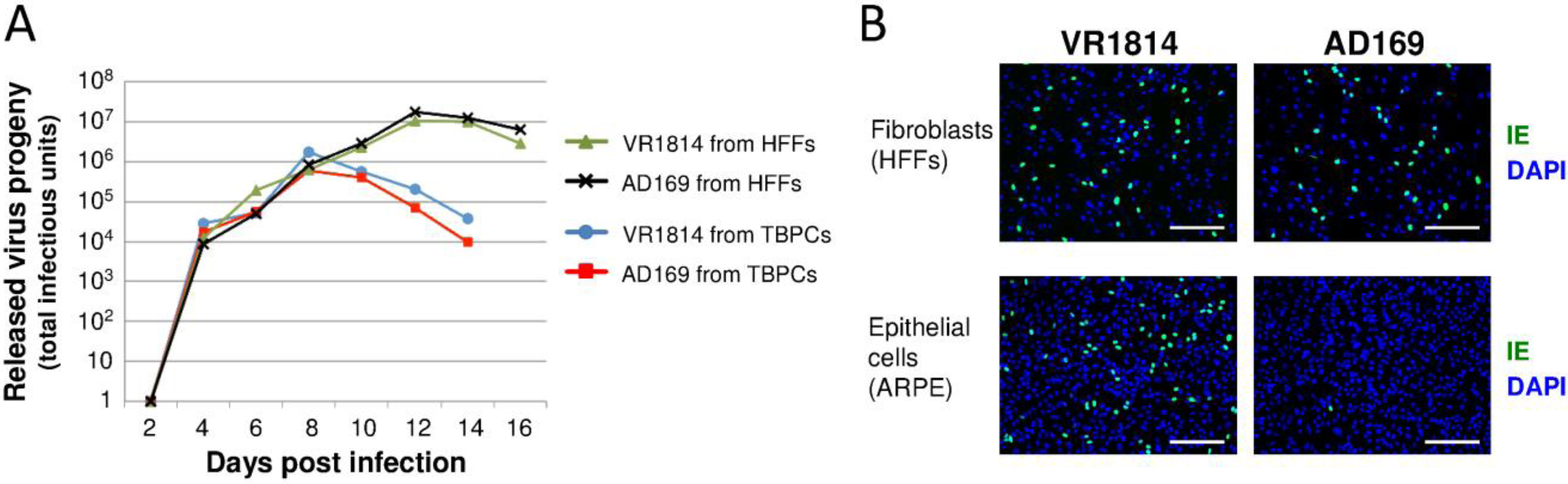
2.2. HCMV-Infected TBPCs Produce Infectious Progeny Virions
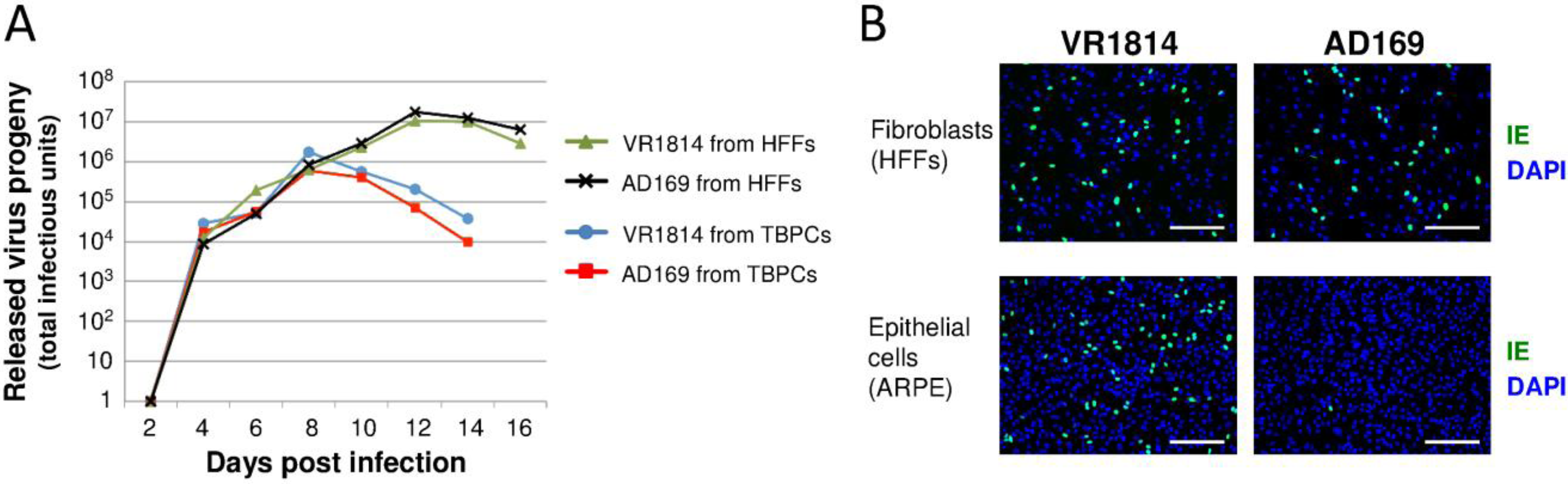
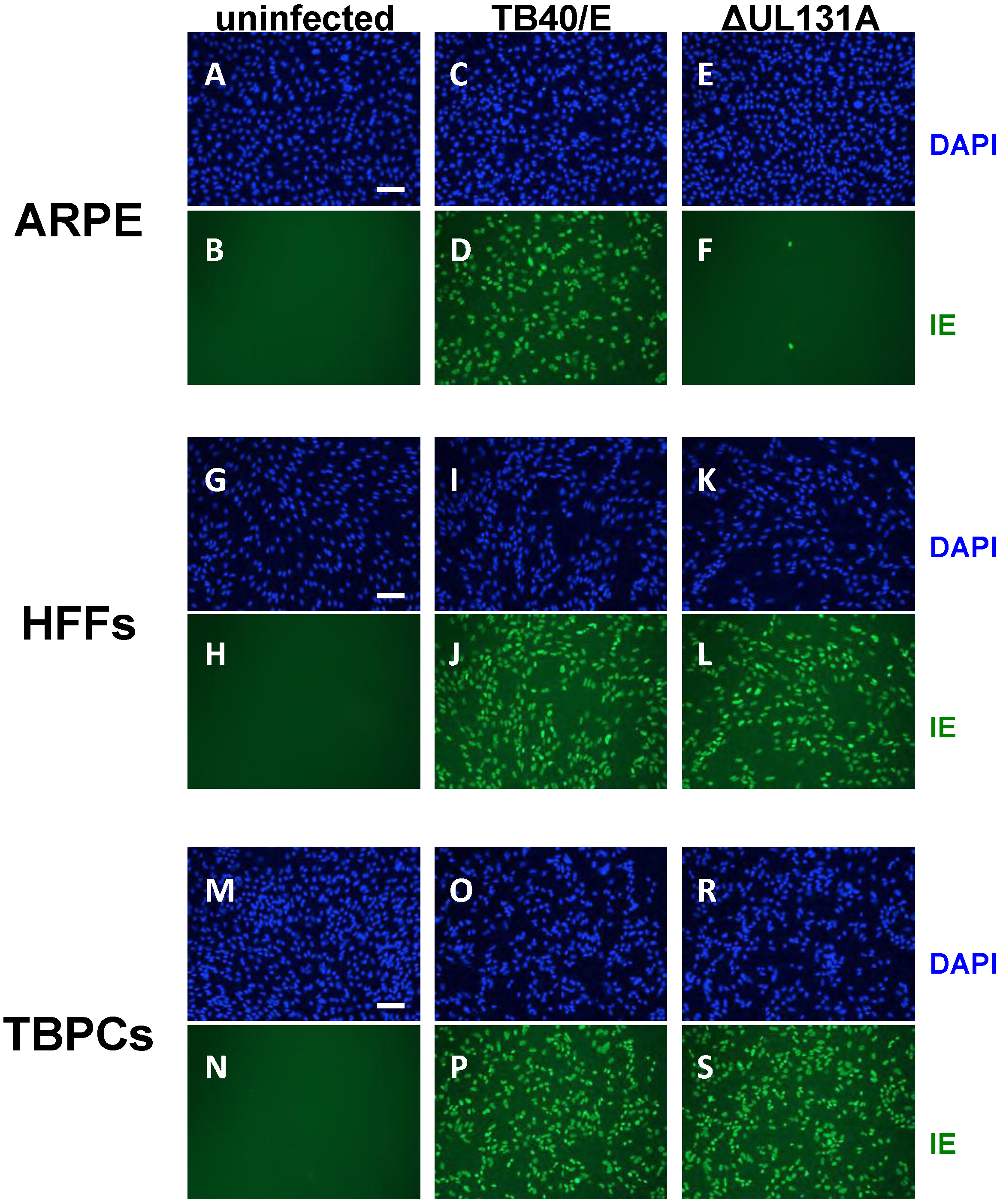
2.3. Viral Entry into TBPCs Is Independent of the Pentamer Complex
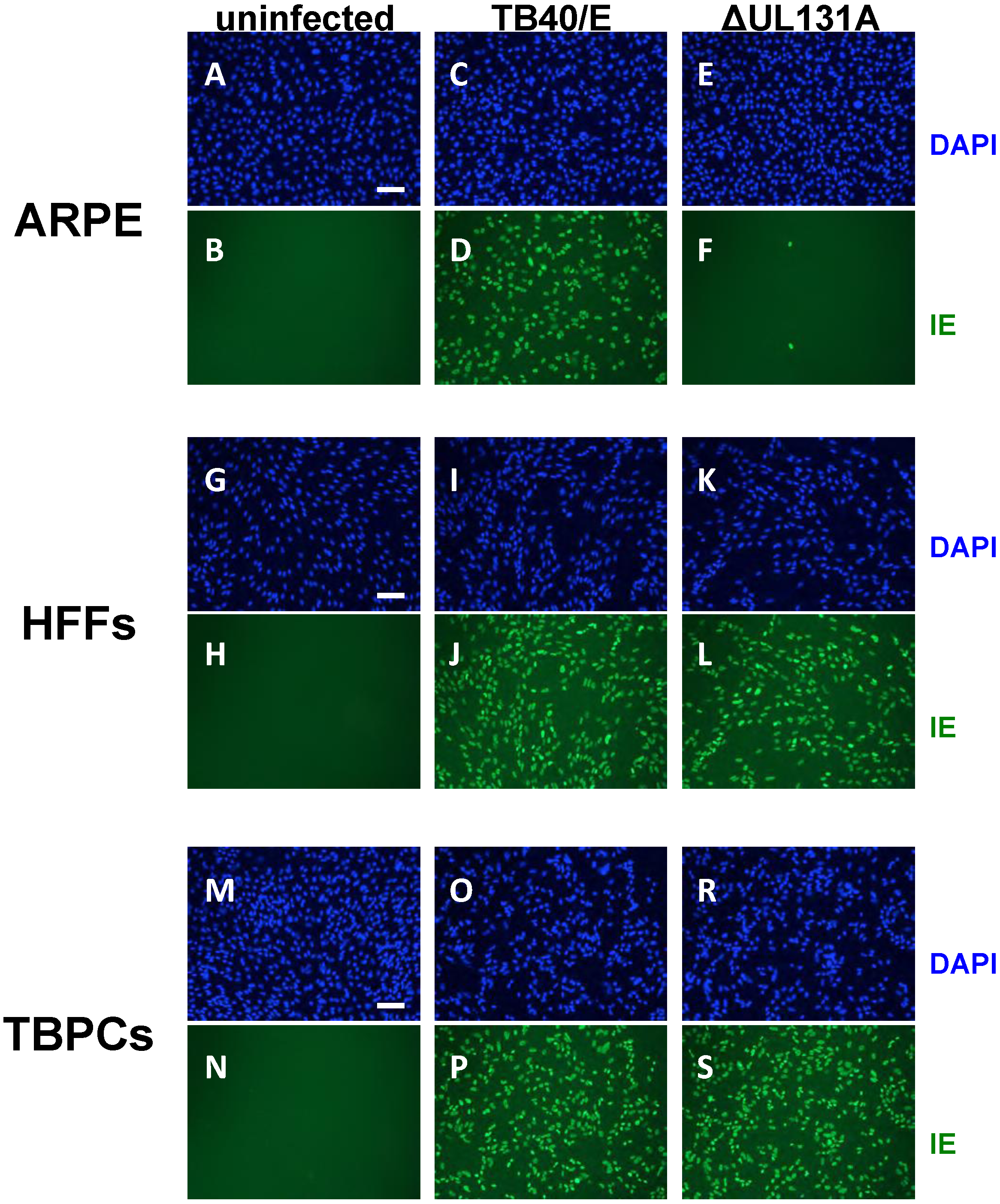
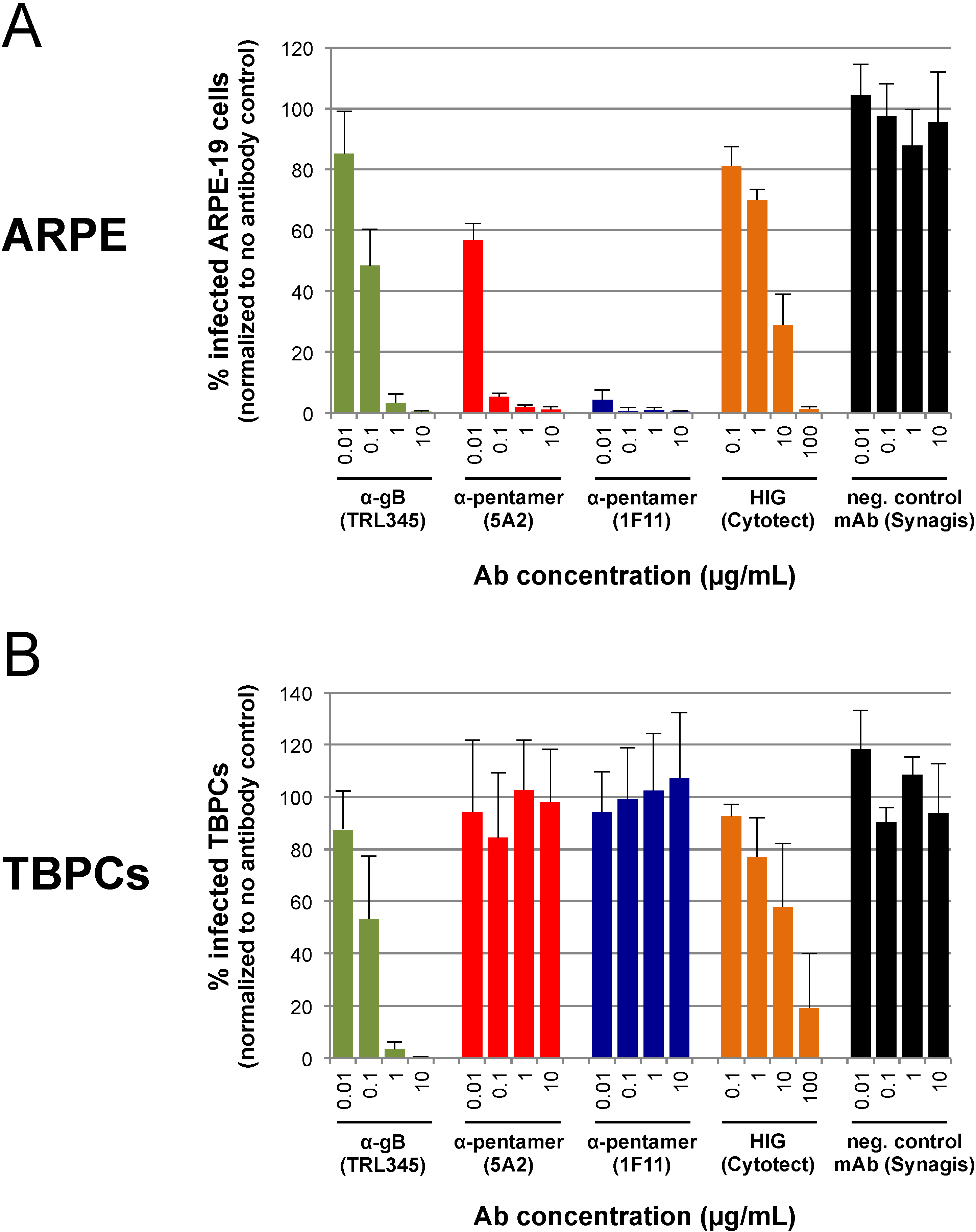
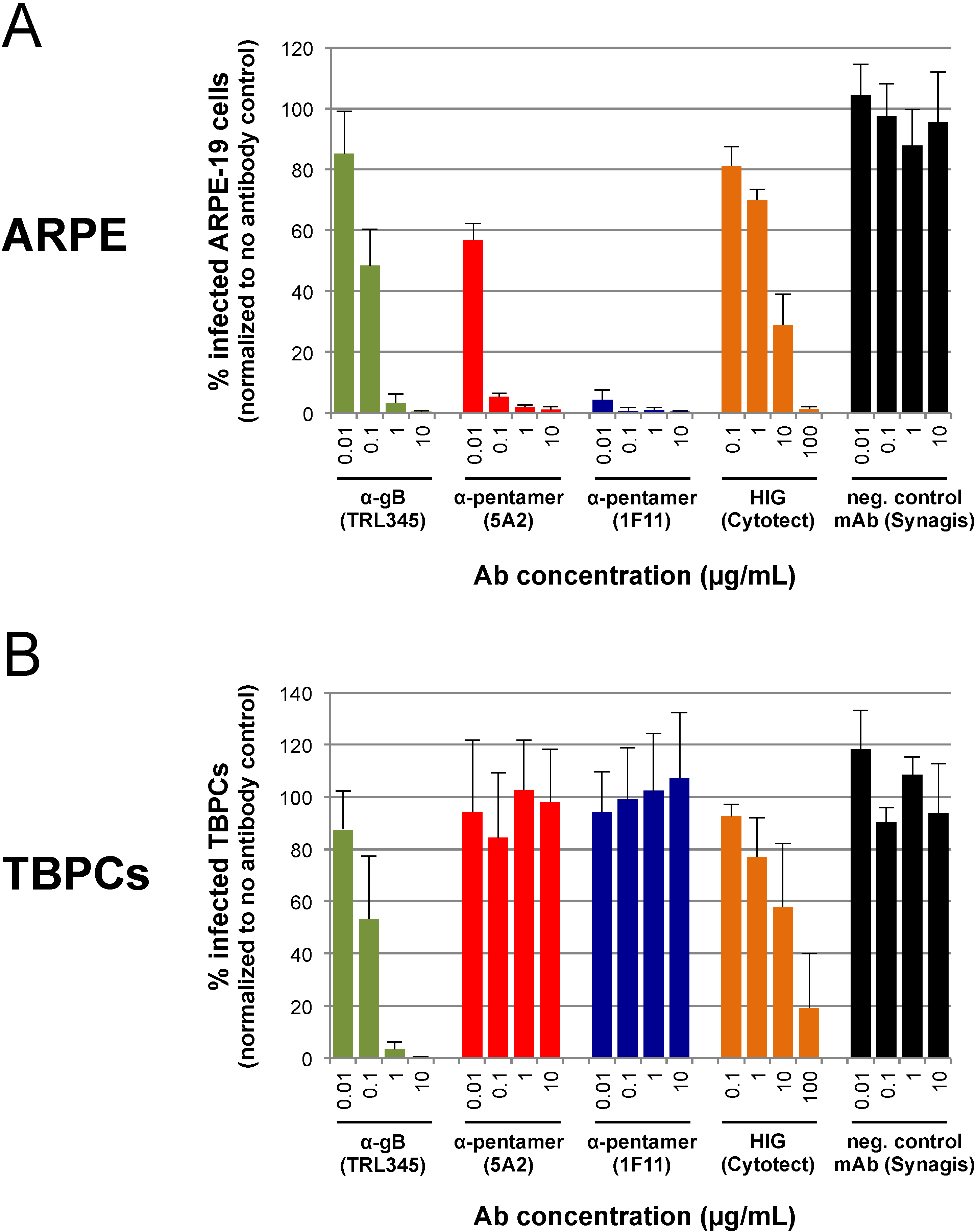
2.4. TBPC Infection Is Blocked by mAbs to gB But Not mAbs to the Pentamer Complex
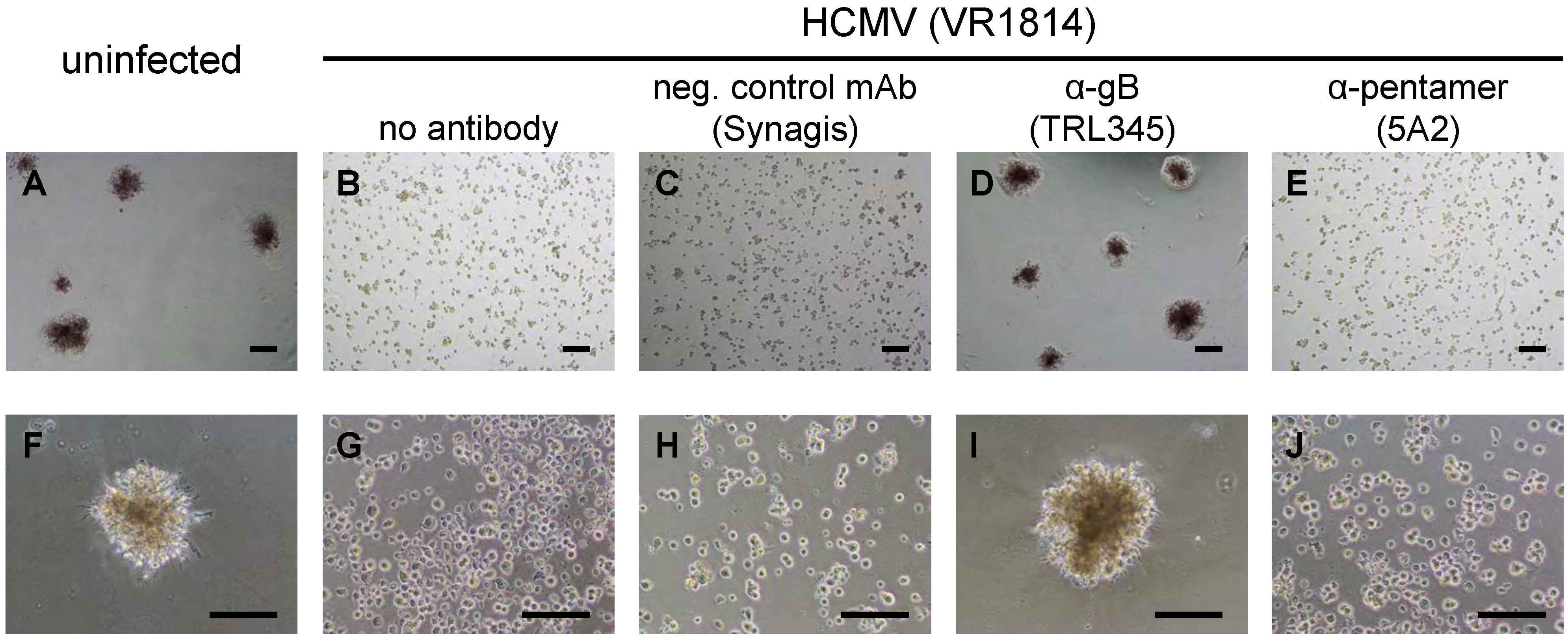
2.5. Neutralization of VR1814 by Anti-gB mAb TRL345 Restores TBPC Differentiation
2.6. Discussion

3. Experimental Section
3.1. Cells
3.2. Viruses and Infections
3.3. Immunofluorescence
3.4. Detection and Quantification of Viral Progeny
3.5. Virus Neutralization Assay
3.6. Trophosphere Formation
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Britt, W.J. Congenital cytomegalovirus infection. In Sexually Transmitted Diseases and Adverse Outcomes of Pregnancy; Hitchcock, P.J., MacKay, H.T., Wasserheit, J.N., Eds.; ASM Press: Washington, DC, USA, 1999; pp. 269–281. [Google Scholar]
- Demmler, G.J. Congenital cytomegalovirus infection and disease. Adv. Pediatr. Infect. Dis. 1996, 11, 135–162. [Google Scholar]
- Rivera, L.B.; Boppana, S.B.; Fowler, K.B.; Britt, W.J.; Stagno, S.; Pass, R.F. Predictors of hearing loss in children with symptomatic congenital cytomegalovirus infection. Pediatrics 2002, 110, 762–767. [Google Scholar] [CrossRef]
- Pereira, L.; Petitt, M.; Fong, A.; Tsuge, M.; Tabata, T.; Fang-Hoover, J.; Maidji, E.; Zydek, M.; Zhou, Y.; Inoue, N.; et al. Intrauterine growth restriction caused by underlying congenital cytomegalovirus infection. J. Infect. Dis. 2014. [Google Scholar] [CrossRef]
- Fowler, K.B.; Stagno, S.; Pass, R.F. Maternal immunity and prevention of congenital cytomegalovirus infection. JAMA 2003, 289, 1008–1011. [Google Scholar]
- Yamamoto, A.Y.; Mussi-Pinhata, M.M.; Isaac Mde, L.; Amaral, F.R.; Carvalheiro, C.G.; Aragon, D.C.; Manfredi, A.K.; Boppana, S.B.; Britt, W.J. Congenital cytomegalovirus infection as a cause of sensorineural hearing loss in a highly immune population. Pediatr. Infect. Dis. J. 2011, 30, 1043–1046. [Google Scholar] [CrossRef]
- Ross, S.A.; Fowler, K.B.; Ashrith, G.; Stagno, S.; Britt, W.J.; Pass, R.F.; Boppana, S.B. Hearing loss in children with congenital cytomegalovirus infection born to mothers with preexisting immunity. J. Pediatr. 2006, 148, 332–336. [Google Scholar] [CrossRef]
- Mercorelli, B.; Sinigalia, E.; Loregian, A.; Palu, G. Human cytomegalovirus DNA replication: Antiviral targets and drugs. Rev. Med. Virol. 2008, 18, 177–210. [Google Scholar] [CrossRef]
- Benoist, G.; Leruez-Ville, M.; Magny, J.F.; Jacquemard, F.; Salomon, L.J.; Ville, Y. Management of pregnancies with confirmed cytomegalovirus fetal infection. Fetal. Diagn. Ther. 2013, 33, 203–214. [Google Scholar] [CrossRef]
- Nigro, G.; Adler, S.P.; Parruti, G.; Anceschi, M.M.; Coclite, E.; Pezone, I.; Di Renzo, G.C. Immunoglobulin therapy of fetal cytomegalovirus infection occurring in the first half of pregnancy—A case-control study of the outcome in children. J. Infect. Dis. 2012, 205, 215–227. [Google Scholar] [CrossRef]
- Buxmann, H.; Stackelberg, O.M.; Schlosser, R.L.; Enders, G.; Gonser, M.; Meyer-Wittkopf, M.; Hamprecht, K.; Enders, M. Use of cytomegalovirus hyperimmunoglobulin for prevention of congenital cytomegalovirus disease: A retrospective analysis. J. Perinat. Med. 2012, 40, 439–446. [Google Scholar]
- Nigro, G.; Adler, S.P.; La Torre, R.; Best, A.M. Passive immunization during pregnancy for congenital cytomegalovirus infection. New Engl. J. Med. 2005, 353, 1350–1362. [Google Scholar] [CrossRef]
- Nigro, G.; Torre, R.L.; Pentimalli, H.; Taverna, P.; Lituania, M.; de Tejada, B.M.; Adler, S.P. Regression of fetal cerebral abnormalities by primary cytomegalovirus infection following hyperimmunoglobulin therapy. Prenat. Diagn. 2008, 28, 512–517. [Google Scholar] [CrossRef]
- Maidji, E.; Nigro, G.; Tabata, T.; McDonagh, S.; Nozawa, N.; Shiboski, S.; Muci, S.; Anceschi, M.M.; Aziz, N.; Adler, S.P.; Pereira, L. Antibody treatment promotes compensation for human cytomegalovirus-induced pathogenesis and a hypoxia-like condition in placentas with congenital infection. Am. J. Pathol. 2010, 177, 1298–1310. [Google Scholar] [CrossRef]
- Adler, S.P.; Nigro, G. Findings and conclusions from cmv hyperimmune globulin treatment trials. J. Clin. Virol. 2009, 46, S54–S57. [Google Scholar] [CrossRef]
- Nightingale, S.L. From the food and drug administration. JAMA 1990, 264, 2863. [Google Scholar] [CrossRef]
- Snydman, D.R.; Werner, B.G.; Heinze-Lacey, B.; Berardi, V.P.; Tilney, N.L.; Kirkman, R.L.; Milford, E.L.; Cho, S.I.; Bush, H.L., Jr.; Levey, A.S.; et al. Use of cytomegalovirus immune globulin to prevent cytomegalovirus disease in renal-transplant recipients. New Engl. J. Med. 1987, 317, 1049–1054. [Google Scholar] [CrossRef]
- Ballow, M. Mechanisms of action of intravenous immune serum globulin therapy. Pediatr. Infect. Dis. J. 1994, 13, 806–811. [Google Scholar] [CrossRef]
- Macagno, A.; Bernasconi, N.L.; Vanzetta, F.; Dander, E.; Sarasini, A.; Revello, M.G.; Gerna, G.; Sallusto, F.; Lanzavecchia, A. Isolation of human monoclonal antibodies that potently neutralize human cytomegalovirus infection by targeting different epitopes on the gh/gl/ul128–131a complex. J. Virol. 2010, 84, 1005–1013. [Google Scholar] [CrossRef]
- Collarini, E.J.; Lee, F.E.; Foord, O.; Park, M.; Sperinde, G.; Wu, H.; Harriman, W.D.; Carroll, S.F.; Ellsworth, S.L.; Anderson, L.J.; et al. Potent high-affinity antibodies for treatment and prophylaxis of respiratory syncytial virus derived from b cells of infected patients. J. Immunol. 2009, 183, 6338–6345. [Google Scholar] [CrossRef]
- Harriman, W.D.; Collarini, E.J.; Sperinde, G.V.; Strandh, M.; Fatholahi, M.M.; Dutta, A.; Lee, Y.; Mettler, S.E.; Keyt, B.A.; Ellsworth, S.L.; et al. Antibody discovery via multiplexed single cell characterization. J. Immunol. Methods 2009, 341, 135–145. [Google Scholar] [CrossRef]
- Wrammert, J.; Smith, K.; Miller, J.; Langley, W.A.; Kokko, K.; Larsen, C.; Zheng, N.Y.; Mays, I.; Garman, L.; Helms, C.; et al. Rapid cloning of high-affinity human monoclonal antibodies against influenza virus. Nature 2008, 453, 667–671. [Google Scholar] [CrossRef]
- McCutcheon, K.M.; Gray, J.; Chen, N.Y.; Liu, K.; Park, M.; Ellsworth, S.; Tripp, R.A.; Mark Tompkins, S.; Johnson, S.K.; et al. Multiplexed screening of natural humoral immunity identifies antibodies at fine specificity for complex and dynamic viral targets. MAbs 2014, 6, 460–473. [Google Scholar]
- Damsky, C.H.; Fisher, S.J. Trophoblast pseudo-vasculogenesis: Faking it with endothelial adhesion receptors. Curr. Opin. Cell Biol. 1998, 10, 660–666. [Google Scholar] [CrossRef]
- Zhou, Y.; Fisher, S.J.; Janatpour, M.; Genbacev, O.; Dejana, E.; Wheelock, M.; Damsky, C.H. Human cytotrophoblasts adopt a vascular phenotype as they differentiate. A strategy for successful endovascular invasion? J. Clin. Invest. 1997, 99, 2139–2151. [Google Scholar] [CrossRef]
- Hemberger, M.; Udayashankar, R.; Tesar, P.; Moore, H.; Burton, G.J. Elf5-enforced transcriptional networks define an epigenetically regulated trophoblast stem cell compartment in the human placenta. Hum. Mol. Genet. 2010, 19, 2456–2467. [Google Scholar] [CrossRef]
- Genbacev, O.; Donne, M.; Kapidzic, M.; Gormley, M.; Lamb, J.; Gilmore, J.; Larocque, N.; Goldfien, G.; Zdravkovic, T.; McMaster, M.T.; Fisher, S.J. Establishment of human trophoblast progenitor cell lines from the chorion. Stem Cells 2011, 29, 1427–1436. [Google Scholar]
- Genbacev, O.; Lamb, J.D.; Prakobphol, A.; Donne, M.; McMaster, M.T.; Fisher, S.J. Human trophoblast progenitors: Where do they reside? Semin. Reprod. Med. 2013, 31, 56–61. [Google Scholar] [CrossRef]
- Fisher, S.; Genbacev, O.; Maidji, E.; Pereira, L. Human cytomegalovirus infection of placental cytotrophoblasts in vitro and in utero: Implications for transmission and pathogenesis. J. Virol. 2000, 74, 6808–6820. [Google Scholar] [CrossRef]
- Maidji, E.; Genbacev, O.; Chang, H.T.; Pereira, L. Developmental regulation of human cytomegalovirus receptors in cytotrophoblasts correlates with distinct replication sites in the placenta. J. Virol. 2007, 81, 4701–4712. [Google Scholar] [CrossRef]
- Pereira, L.; Maidji, E.; McDonagh, S.; Genbacev, O.; Fisher, S. Human cytomegalovirus transmission from the uterus to the placenta correlates with the presence of pathogenic bacteria and maternal immunity. J. Virol. 2003, 77, 13301–13314. [Google Scholar] [CrossRef]
- Pereira, L.; Maidji, E.; McDonagh, S.; Tabata, T. Insights into viral transmission at the uterine-placental interface. Trends Microbiol. 2005, 13, 164–174. [Google Scholar]
- Tabata, T.; McDonagh, S.; Kawakatsu, H.; Pereira, L. Cytotrophoblasts infected with a pathogenic human cytomegalovirus strain dysregulate cell-matrix and cell-cell adhesion molecules: A quantitative analysis. Placenta 2007, 28, 527–537. [Google Scholar] [CrossRef]
- McDonagh, S.; Maidji, E.; Chang, H.T.; Pereira, L. Patterns of human cytomegalovirus infection in term placentas: A preliminary analysis. J. Clin. Virol. 2006, 35, 210–215. [Google Scholar] [CrossRef]
- Maidji, E.; McDonagh, S.; Genbacev, O.; Tabata, T.; Pereira, L. Maternal antibodies enhance or prevent cytomegalovirus infection in the placenta by neonatal fc receptor-mediated transcytosis. Am. J. Pathol. 2006, 168, 1210–1226. [Google Scholar] [CrossRef]
- Nozawa, N.; Fang-Hoover, J.; Tabata, T.; Maidji, E.; Pereira, L. Cytomegalovirus-specific, high-avidity igg with neutralizing activity in maternal circulation enriched in the fetal bloodstream. J. Clin. Virol. 2009, 46, S58–S63. [Google Scholar]
- Tabata, T.; Petitt, M.; Zydek, M.; Fang-Hoover, J.; Larocque, N.; Tsuge, M.; Gormley, M.; Genbacev, O.; Pereira, L. Human cytomegalovirus infection of trophoblast progenitor cells inhibits early steps of differentiation. To be submitted for publication. 2014. [Google Scholar]
- Isaacson, M.K.; Compton, T. Human cytomegalovirus glycoprotein b is required for virus entry and cell-to-cell spread but not for virion attachment, assembly, or egress. J. Virol. 2009, 83, 3891–3903. [Google Scholar] [CrossRef]
- Wille, P.T.; Wisner, T.W.; Ryckman, B.; Johnson, D.C. Human cytomegalovirus (hcmv) glycoprotein gb promotes virus entry in trans acting as the viral fusion protein rather than as a receptor-binding protein. mBio 2013, 4, e00332–00313. [Google Scholar]
- Compton, T.; Nowlin, D.M.; Cooper, N.R. Initiation of human cytomegalovirus infection requires initial interaction with cell surface heparan sulfate. Virology 1993, 193, 834–841. [Google Scholar] [CrossRef]
- Carlson, C.; Britt, W.J.; Compton, T. Expression, purification, and characterization of a soluble form of human cytomegalovirus glycoprotein b. Virology 1997, 239, 198–205. [Google Scholar] [CrossRef]
- Wang, X.; Huong, S.M.; Chiu, M.L.; Raab-Traub, N.; Huang, E.S. Epidermal growth factor receptor is a cellular receptor for human cytomegalovirus. Nature 2003, 424, 456–461. [Google Scholar] [CrossRef]
- Soroceanu, L.; Akhavan, A.; Cobbs, C.S. Platelet-derived growth factor-alpha receptor activation is required for human cytomegalovirus infection. Nature 2008, 455, 391–395. [Google Scholar] [CrossRef]
- Feire, A.L.; Roy, R.M.; Manley, K.; Compton, T. The glycoprotein b disintegrin-like domain binds beta 1 integrin to mediate cytomegalovirus entry. J. Virol. 2010, 84, 10026–10037. [Google Scholar] [CrossRef]
- Feire, A.L.; Koss, H.; Compton, T. Cellular integrins function as entry receptors for human cytomegalovirus via a highly conserved disintegrin-like domain. Proc. Natl. Acad. Sci. USA 2004, 101, 15470–15475. [Google Scholar] [CrossRef]
- Vanarsdall, A.L.; Ryckman, B.J.; Chase, M.C.; Johnson, D.C. Human cytomegalovirus glycoproteins gb and gh/gl mediate epithelial cell-cell fusion when expressed either in cis or in trans. J. Virol. 2008, 82, 11837–11850. [Google Scholar] [CrossRef]
- Hahn, G.; Revello, M.G.; Patrone, M.; Percivalle, E.; Campanini, G.; Sarasini, A.; Wagner, M.; Gallina, A.; Milanesi, G.; Koszinowski, U.; et al. Human cytomegalovirus ul131–128 genes are indispensable for virus growth in endothelial cells and virus transfer to leukocytes. J. Virol. 2004, 78, 10023–10033. [Google Scholar] [CrossRef]
- Ryckman, B.J.; Chase, M.C.; Johnson, D.C. Hcmv gh/gl/ul128–131 interferes with virus entry into epithelial cells: Evidence for cell type-specific receptors. Proc. Natl. Acad. Sci. USA 2008, 105, 14118–14123. [Google Scholar] [CrossRef]
- Sinzger, C.; Digel, M.; Jahn, G. Cytomegalovirus cell tropism. Curr. Top. Microbiol. Immunol. 2008, 325, 63–83. [Google Scholar]
- Vanarsdall, A.L.; Johnson, D.C. Human cytomegalovirus entry into cells. Curr. Opin. Virol. 2012, 2, 37–42. [Google Scholar] [CrossRef]
- Wang, D.; Shenk, T. Human cytomegalovirus ul131 open reading frame is required for epithelial cell tropism. J. Virol. 2005, 79, 10330–10338. [Google Scholar] [CrossRef]
- Dolan, A.; Cunningham, C.; Hector, R.D.; Hassan-Walker, A.F.; Lee, L.; Addison, C.; Dargan, D.J.; McGeoch, D.J.; Gatherer, D.; Emery, V.C.; et al. Genetic content of wild-type human cytomegalovirus. J. Gen. Virol. 2004, 85, 1301–1312. [Google Scholar] [CrossRef]
- Scrivano, L.; Sinzger, C.; Nitschko, H.; Koszinowski, U.H.; Adler, B. Hcmv spread and cell tropism are determined by distinct virus populations. PLoS Pathog. 2011, 7, e1001256. [Google Scholar] [CrossRef] [Green Version]
- Adler, B.; Scrivano, L.; Ruzcics, Z.; Rupp, B.; Sinzger, C.; Koszinowski, U. Role of human cytomegalovirus ul131a in cell type-specific virus entry and release. J. Gen. Virol. 2006, 87, 2451–2460. [Google Scholar] [CrossRef]
- Stinski, M.F. Sequence of protein synthesis in cells infected by human cytomegalovirus: Early and late virus-induced polypeptides. J. Virol. 1978, 26, 686–701. [Google Scholar]
- Compton, T.; Nepomuceno, R.R.; Nowlin, D.M. Human cytomegalovirus penetrates host cells by ph-independent fusion at the cell surface. Virology 1992, 191, 387–395. [Google Scholar] [CrossRef]
- Ryckman, B.J.; Jarvis, M.A.; Drummond, D.D.; Nelson, J.A.; Johnson, D.C. Human cytomegalovirus entry into epithelial and endothelial cells depends on genes ul128 to ul150 and occurs by endocytosis and low-ph fusion. J. Virol. 2006, 80, 710–722. [Google Scholar] [CrossRef]
- Sinzger, C. Entry route of hcmv into endothelial cells. J. Clin. Virol. 2008, 41, 174–179. [Google Scholar] [CrossRef]
- Weisblum, Y.; Panet, A.; Zakay-Rones, Z.; Haimov-Kochman, R.; Goldman-Wohl, D.; Ariel, I.; Falk, H.; Natanson-Yaron, S.; Goldberg, M.D.; Gilad, R.; Lurain, N.S.; Greenfield, C.; Yagel, S.; Wolf, D.G. Modeling of human cytomegalovirus maternal-fetal transmission in a novel decidual organ culture. J. Virol. 2011, 85, 13204–13213. [Google Scholar] [CrossRef]
- Bratcher, D.F.; Bourne, N.; Bravo, F.J.; Schleiss, M.R.; Slaoui, M.; Myers, M.G.; Bernstein, D.I. Effect of passive antibody on congenital cytomegalovirus infection in guinea pigs. J. Infect. Dis. 1995, 172, 944–950. [Google Scholar] [CrossRef]
- Schleiss, M.R.; Bourne, N.; Stroup, G.; Bravo, F.J.; Jensen, N.J.; Bernstein, D.I. Protection against congenital cytomegalovirus infection and disease in guinea pigs, conferred by a purified recombinant glycoprotein b vaccine. J. Infect. Dis. 2004, 189, 1374–1381. [Google Scholar] [CrossRef]
- Straschewski, S.; Patrone, M.; Walther, P.; Gallina, A.; Mertens, T.; Frascaroli, G. Protein pul128 of human cytomegalovirus is necessary for monocyte infection and blocking of migration. J. Virol. 2011, 85, 5150–5158. [Google Scholar] [CrossRef]
- Saccoccio, F.M.; Sauer, A.L.; Cui, X.; Armstrong, A.E.; Habib el, S.E.; Johnson, D.C.; Ryckman, B.J.; Klingelhutz, A.J.; Adler, S.P.; McVoy, M.A. Peptides from cytomegalovirus ul130 and ul131 proteins induce high titer antibodies that block viral entry into mucosal epithelial cells. Vaccine 2011, 29, 2705–2711. [Google Scholar] [CrossRef]
- Fouts, A.E.; Chan, P.; Stephan, J.P.; Vandlen, R.; Feierbach, B. Antibodies against the gh/gl/ul128/ul130/ul131 complex comprise the majority of the anti-cytomegalovirus (anti-cmv) neutralizing antibody response in cmv hyperimmune globulin. J. Virol. 2012, 86, 7444–7447. [Google Scholar] [CrossRef]
- Lilleri, D.; Kabanova, A.; Revello, M.G.; Percivalle, E.; Sarasini, A.; Genini, E.; Sallusto, F.; Lanzavecchia, A.; Corti, D.; Gerna, G. Fetal human cytomegalovirus transmission correlates with delayed maternal antibodies to gh/gl/pul128–130–131 complex during primary infection. PLoS One 2013, 8, e59863. [Google Scholar] [CrossRef]
- Ohlin, M.; Sundqvist, V.A.; Mach, M.; Wahren, B.; Borrebaeck, C.A. Fine specificity of the human immune response to the major neutralization epitopes expressed on cytomegalovirus gp58/116 (gb), as determined with human monoclonal antibodies. J. Virol. 1993, 67, 703–710. [Google Scholar]
- Tugizov, S.; Maidji, E.; Xiao, J.; Pereira, L. An acidic cluster in the cytosolic domain of human cytomegalovirus glycoprotein b is a signal for endocytosis from the plasma membrane. J. Virol. 1999, 73, 8677–8688. [Google Scholar]
- Navarro, D.; Paz, P.; Tugizov, S.; Topp, K.; La Vail, J.; Pereira, L. Glycoprotein b of human cytomegalovirus promotes virion penetration into cells, transmission of infection from cell to cell, and fusion of infected cells. Virology 1993, 197, 143–158. [Google Scholar] [CrossRef]
- Grazia Revello, M.; Baldanti, F.; Percivalle, E.; Sarasini, A.; De-Giuli, L.; Genini, E.; Lilleri, D.; Labo, N.; Gerna, G. In vitro selection of human cytomegalovirus variants unable to transfer virus and virus products from infected cells to polymorphonuclear leukocytes and to grow in endothelial cells. J. Gen. Virol. 2001, 82, 1429–1438. [Google Scholar]
- Maidji, E.; Percivalle, E.; Gerna, G.; Fisher, S.; Pereira, L. Transmission of human cytomegalovirus from infected uterine microvascular endothelial cells to differentiating/invasive placental cytotrophoblasts. Virology 2002, 304, 53–69. [Google Scholar] [CrossRef]
- Damsky, C.H.; Fitzgerald, M.L.; Fisher, S.J. Distribution patterns of extracellular matrix components and adhesion receptors are intricately modulated during first trimester cytotrophoblast differentiation along the invasive pathway, in vivo. J. Clin. Invest. 1992, 89, 210–222. [Google Scholar] [CrossRef]
- Pereira, L.; Hoffman, M.; Gallo, D.; Cremer, N. Monoclonal antibodies to human cytomegalovirus: Three surface membrane proteins with unique immunological and electrophoretic properties specify cross-reactive determinants. Infect. Immun. 1982, 36, 924–932. [Google Scholar]
- Iwayama, S.; Yamamoto, T.; Furuya, T.; Kobayashi, R.; Ikuta, K.; Hirai, K. Intracellular localization and DNA-binding activity of a class of viral early phosphoproteins in human fibroblasts infected with human cytomegalovirus (towne strain). J. Gen. Virol. 1994, 75, 3309–3318. [Google Scholar] [CrossRef]
- Nis-elements, 4.0, Laboratory Imaging, Ltd.: Prague, Czech Republic, 2011.
- Schneider, C.A.; Rasband, W.S.; Eliceiri, K.W. Nih image to imagej: 25 years of image analysis. Nat. Methods 2012, 9, 671–675. [Google Scholar] [CrossRef]
- Fiji (fiji is just imagej), version 1.0; University of Wisconsin at Madison: Madison, WI, USA, 2011.
- Null, D.; Bimle, C.; Weisman, L.; Johnson, K.; Steichen, J.; Gratton, T.; Singh, S.; Wang, E.; Asztalos, E.; Loeffler, A.M.; et al. Palivizumab, a humanized respiratory syncytial virus monoclonal antibody, reduces hospitalization from respiratory syncytial virus infection in high-risk infants. Pediatrics 1998, 102, 531–537. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zydek, M.; Petitt, M.; Fang-Hoover, J.; Adler, B.; Kauvar, L.M.; Pereira, L.; Tabata, T. HCMV Infection of Human Trophoblast Progenitor Cells of the Placenta Is Neutralized by a Human Monoclonal Antibody to Glycoprotein B and Not by Antibodies to the Pentamer Complex. Viruses 2014, 6, 1346-1364. https://doi.org/10.3390/v6031346
Zydek M, Petitt M, Fang-Hoover J, Adler B, Kauvar LM, Pereira L, Tabata T. HCMV Infection of Human Trophoblast Progenitor Cells of the Placenta Is Neutralized by a Human Monoclonal Antibody to Glycoprotein B and Not by Antibodies to the Pentamer Complex. Viruses. 2014; 6(3):1346-1364. https://doi.org/10.3390/v6031346
Chicago/Turabian StyleZydek, Martin, Matthew Petitt, June Fang-Hoover, Barbara Adler, Lawrence M. Kauvar, Lenore Pereira, and Takako Tabata. 2014. "HCMV Infection of Human Trophoblast Progenitor Cells of the Placenta Is Neutralized by a Human Monoclonal Antibody to Glycoprotein B and Not by Antibodies to the Pentamer Complex" Viruses 6, no. 3: 1346-1364. https://doi.org/10.3390/v6031346