Molecular and Biological Characterization of a New Isolate of Guinea Pig Cytomegalovirus

 ,
,
Abstract
:1. Introduction
2. Results and Discussion
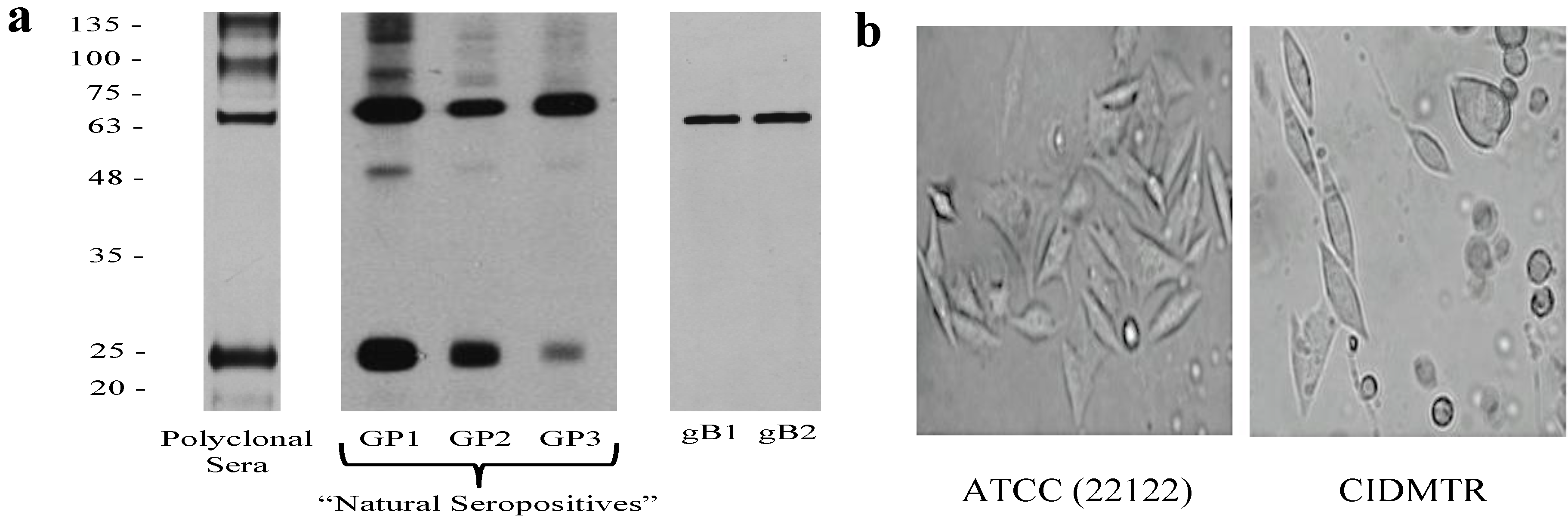
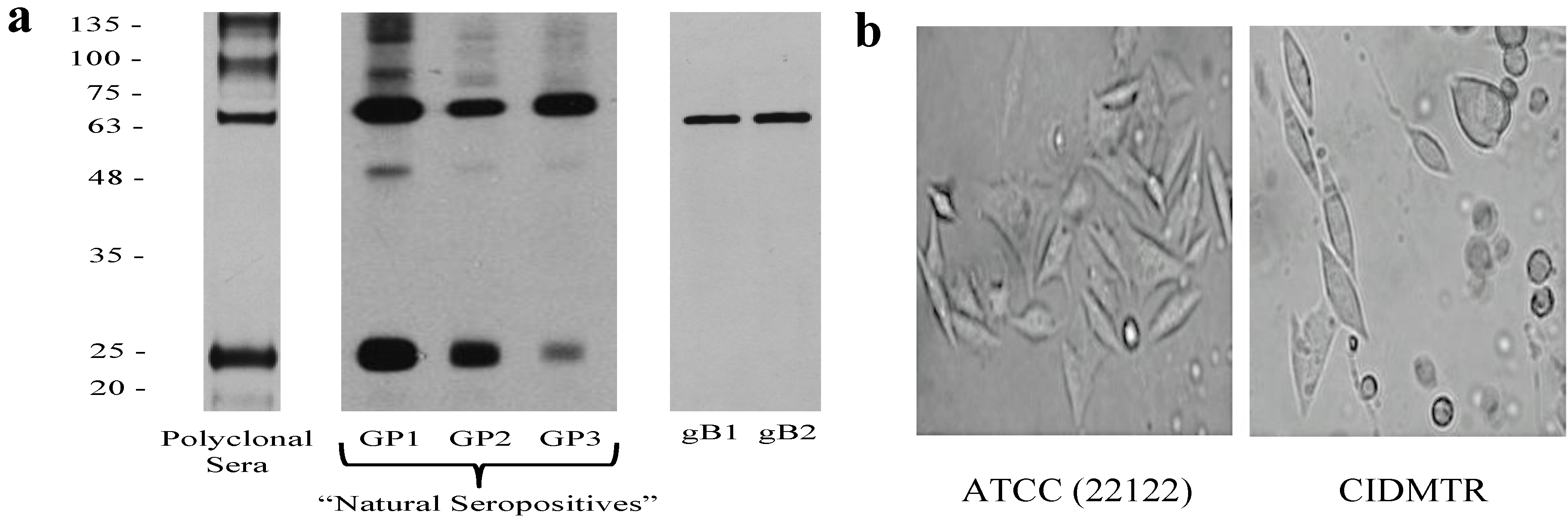
2.1. Isolation of the CIDMTR Strain
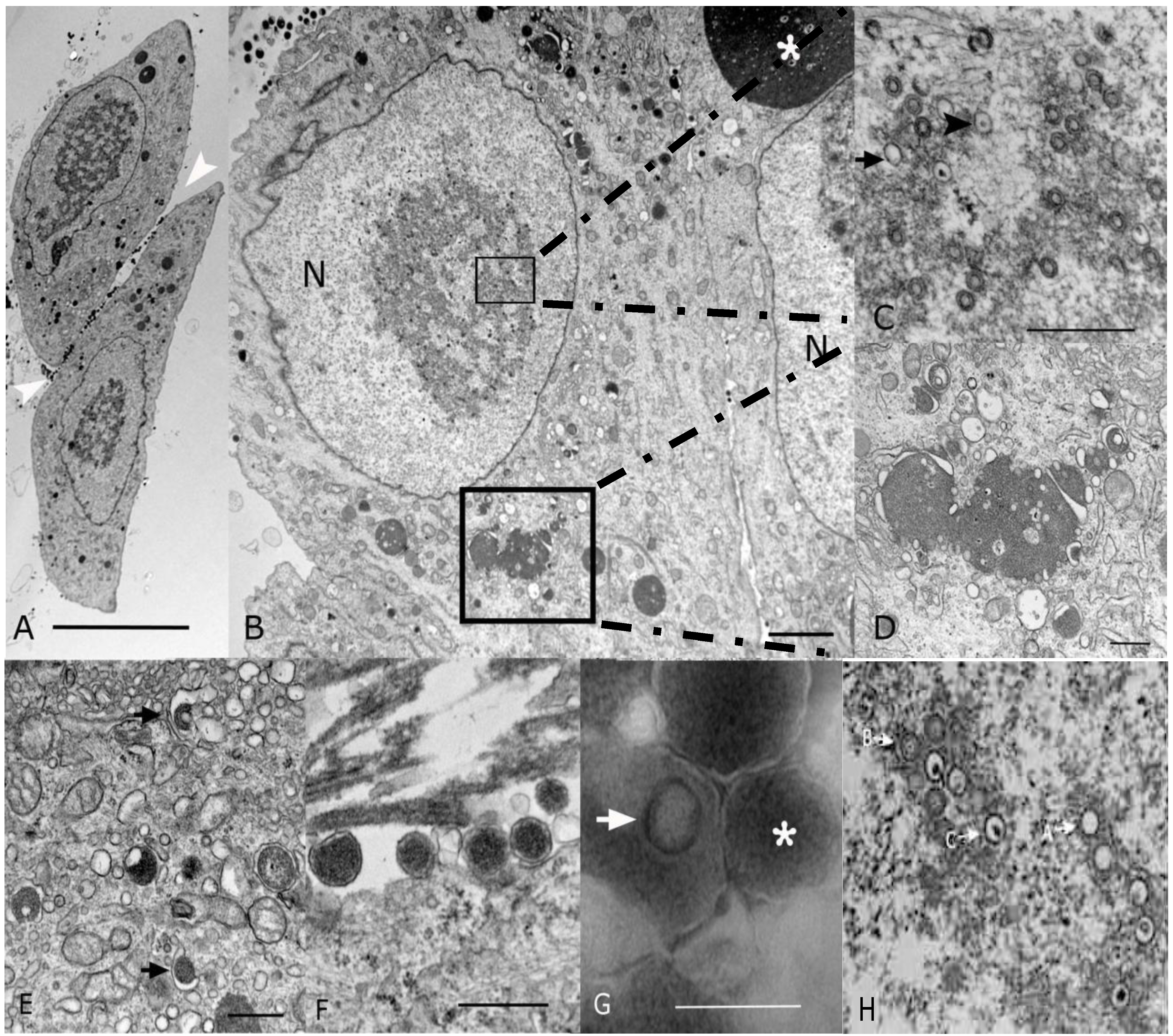
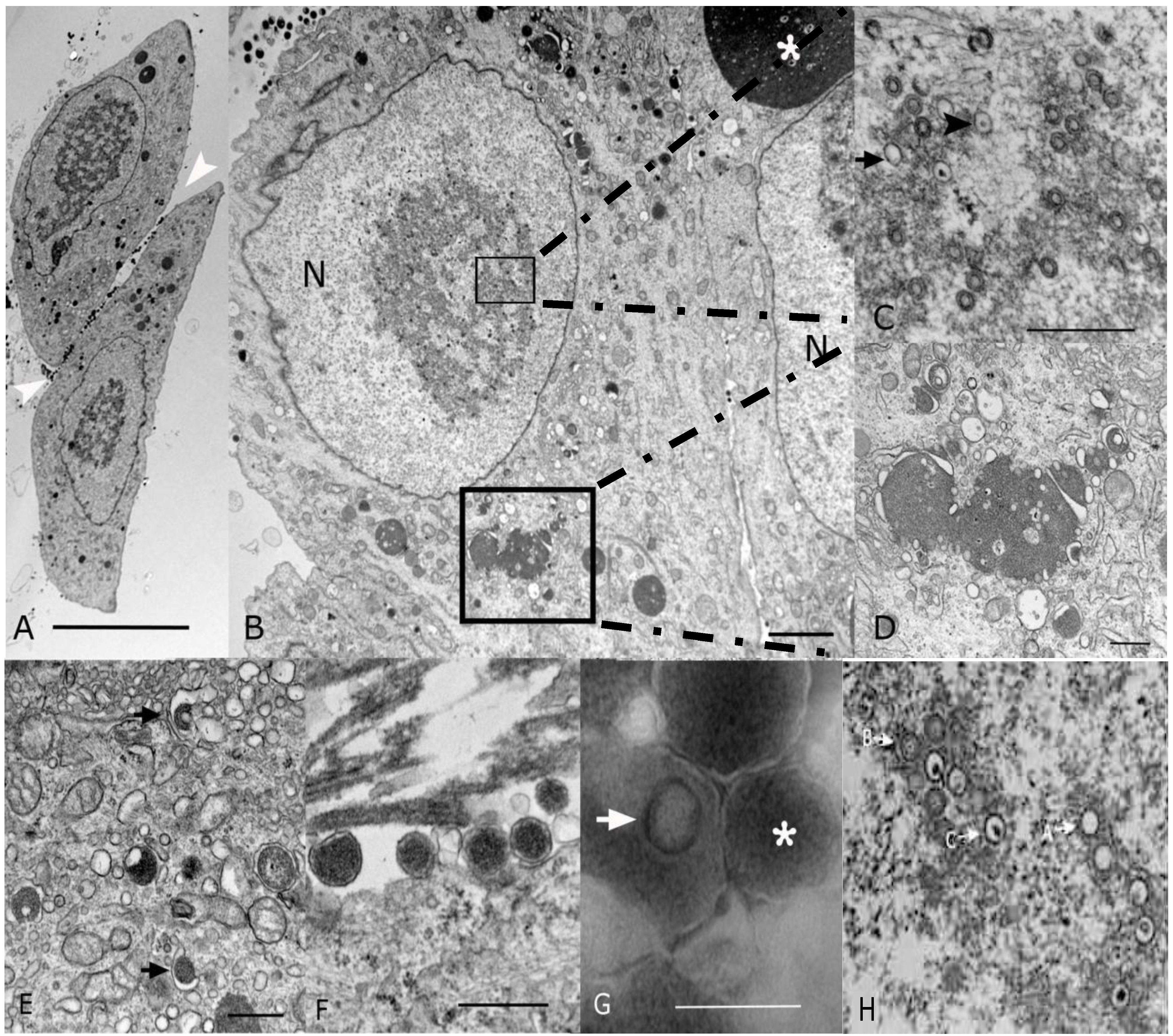
2.2. Morphological Analyses by EM
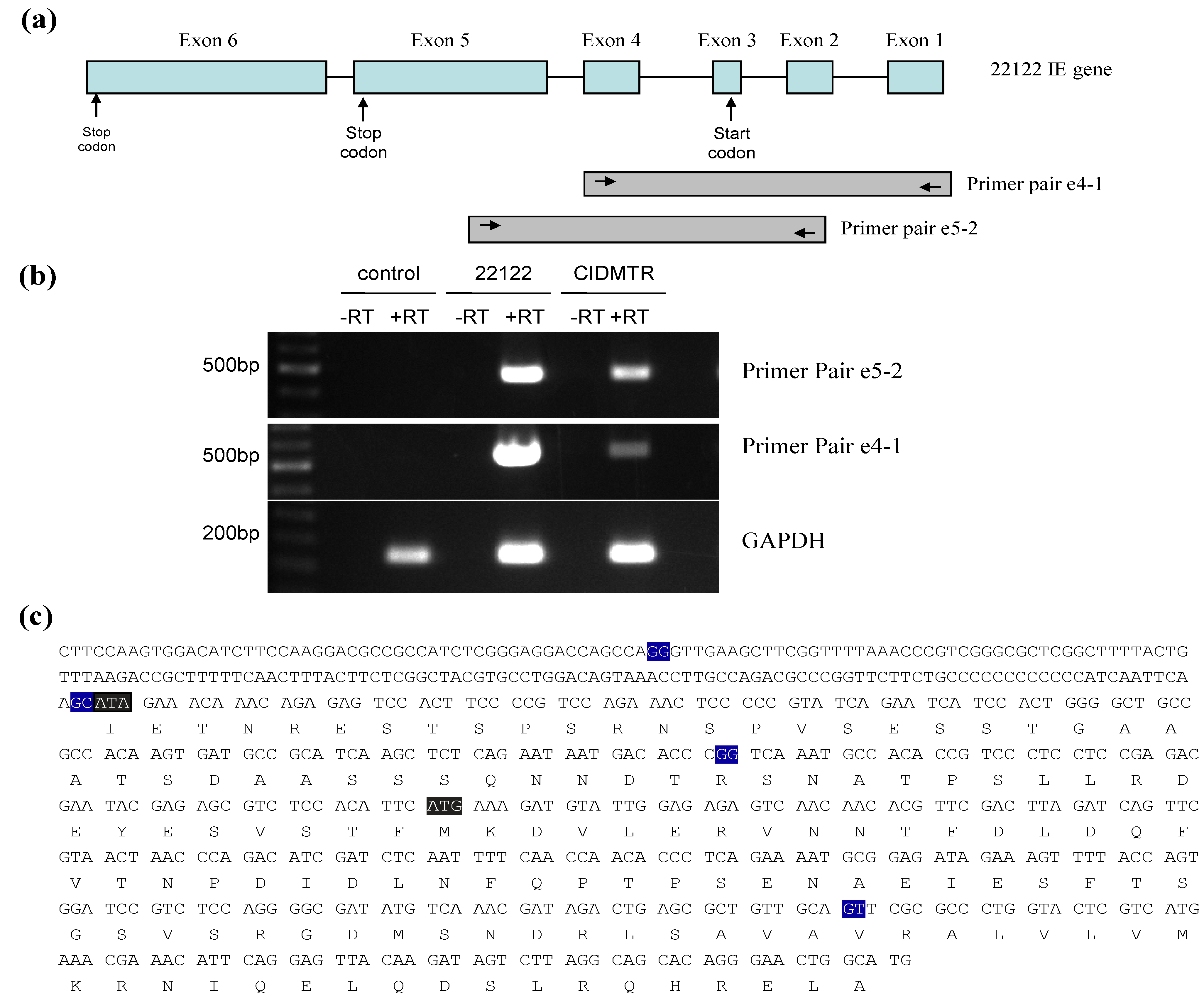
2.3. Sequencing and Sequence Analysis
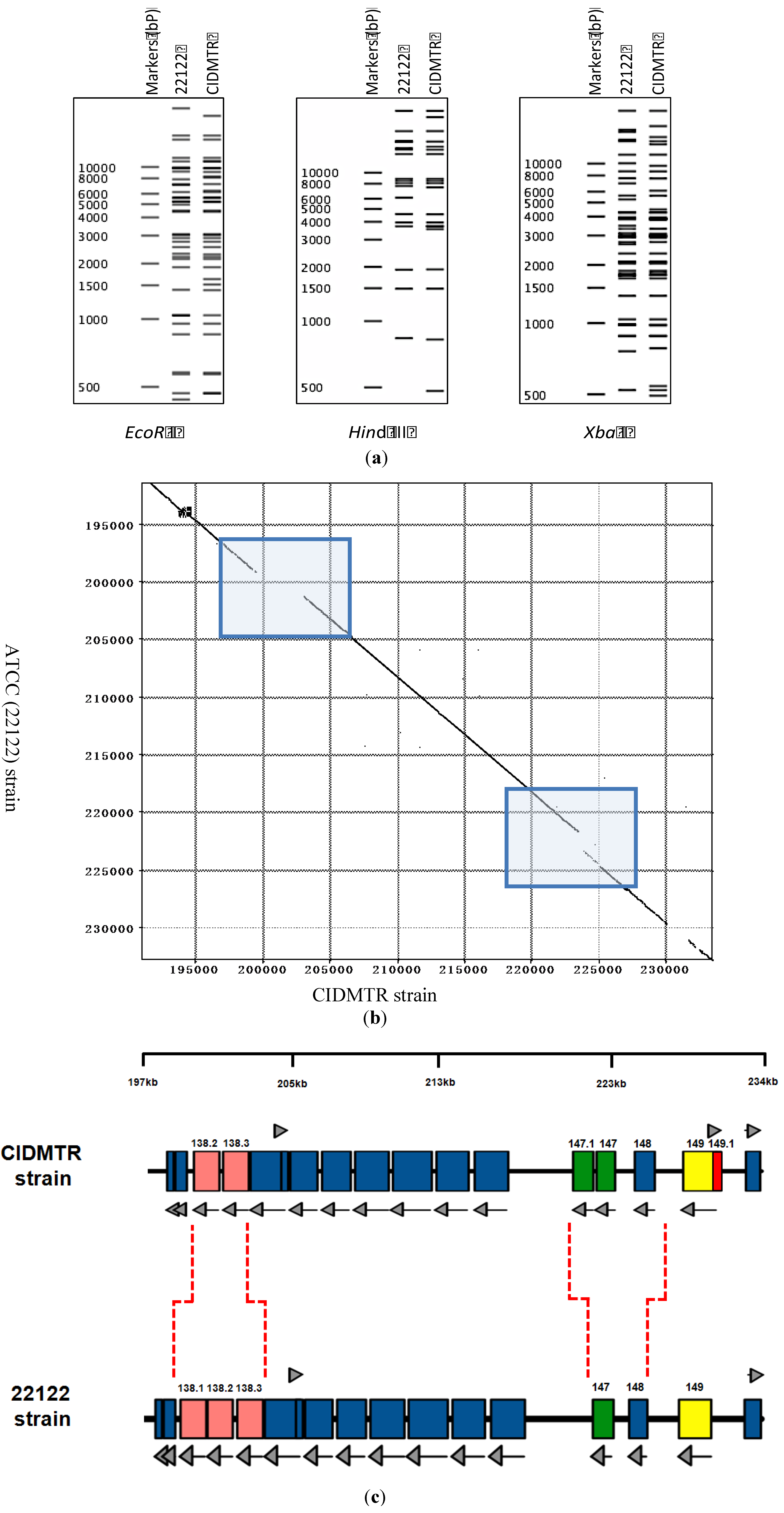

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GPCMV Open Reading Frames (ORFs): CIDMTR Strain | |||||
|---|---|---|---|---|---|
| ORF | Strand | Begin | End | Codons | Notes |
| gp1 | C | 12,464 | 12,769 | 101 | GPCMV MIP 1α; CC chemokine homolog |
| gp2 | 14,840 | 15,685 | 281 | Homology to MCMV M69a | |
| gp3 | C | 17,197 | 19,563 | 788 | Homology to THV T5b; US22 superfamily |
| gp4 | C | 20,829 | 21,152 | 107 | Homology to RCMV r136d |
| gp5 | C | 26,721 | 27,818 | 365 | Homology to MCMV m32a |
| GP23 | C | 33,299 | 34,501 | 400 | UL23 homolog; US22 gene superfamily |
| GP24 | C | 34,739 | 35,956 | 405 | UL24 homolog; US22 superfamily |
| GP25 | 36,542 | 38,194 | 550 | UL25 homolog; tegument protein | |
| GP26 | C | 38,360 | 39,043 | 227 | UL26 homolog |
| GP27 | C | 39,166 | 41,211 | 681 | UL27 homolog |
| GP28 | C | 41,311 | 42,378 | 355 | UL28 homolog; US22 superfamily |
| GP28.1 | C | 42,866 | 44,287 | 473 | UL28 homolog; US22 superfamily |
| GP29 | C | 44,653 | 46,623 | 656 | UL29 homolog; US22 superfamily |
| gp29.1 | C | 47,247 | 47,861 | 204 | US22 superfamily |
| GP30 | C | 49,082 | 50,800 | 572 | UL30 homolog |
| GP31 | 51,094 | 52,572 | 492 | UL31 homolog | |
| GP32 | C | 52,665 | 54,365 | 566 | UL32 homolog |
| GP33 | 54,585 | 55,868 | 427 | UL33 homolog; 7-TMR GPCR superfamily | |
| GP34 | 56,221 | 57,804 | 527 | UL34 homolog | |
| GP35 | 58,008 | 59,666 | 552 | UL35 homolog | |
| GP37 | C | 59,788 | 60,711 | 307 | UL37 homolog |
| GP38 | C | 61,068 | 61,988 | 306 | UL38 homolog |
| gp38.1 | C | 62,705 | 63,262 | 185 | Positional homolog of HCMV UL40 |
| gp38.2 | C | 63,620 | 64,933 | 437 | Positional homolog of HCMV UL41a |
| gp38.3 | C | 65,624 | 66,478 | 284 | Positional homolog of HCMV UL42 |
| gp38.4 | C | 66,997 | 67,362 | 121 | Homology to RCMV r42d |
| GP43 | C | 67,951 | 68,964 | 337 | UL43 homolog |
| GP44 | C | 68,952 | 70,175 | 407 | UL44 homolog |
| GP45 | C | 70,887 | 73,673 | 928 | UL45 homolog |
| GP46 | C | 73,779 | 74,867 | 362 | UL46 homolog |
| GP47 | 74,674 | 77,787 | 1037 | UL47 homolog | |
| GP48 | 77,784 | 84,155 | 2123 | UL48 homolog | |
| GP48.2 | C | 84,238 | 84,468 | 76 | UL48a homolog |
| GP49 | C | 84,479 | 86,119 | 546 | UL49 homolog |
| GP50 | C | 86,088 | 87,158 | 356 | UL50 homolog |
| GP51 | C | 87,281 | 87,580 | 99 | UL51 homolog; terminase subunit |
| GP52 | 87,900 | 89,480 | 526 | UL52 homolog | |
| GP53 | 89,473 | 90,459 | 328 | UL53 homolog | |
| gp53.1 | C | 90,196 | 90,537 | 113 | Homology to RhCMV rh86; YP_068179.1 |
| GP54 | C | 90,551 | 93,904 | 1117 | UL54 homolog; DNA polymerase |
| GP55 | C | 93,947 | 96,652 | 901 | UL55 homolog; glycoprotein B |
| GP56 | C | 96,549 | 98,816 | 755 | UL56 homolog; terminase subunit |
| GP57 | C | 98,967 | 102,650 | 1227 | UL57 homolog |
| gp57.1 | C | 104,411 | 104,926 | 171 | Homolog to RCMV r23.1d |
| GP69 | C | 108,260 | 111,430 | 1056 | UL69 homolog |
| GP70 | C | 112,136 | 115,339 | 1067 | UL70 homolog; helicase-primase |
| GP71 | 115,338 | 116,114 | 258 | UL71 homolog | |
| GP72 | C | 116,277 | 117,350 | 357 | UL72 homolog; dUTPase |
| GP73 | 117,432 | 117,833 | 133 | UL73 homolog; glycoprotein N | |
| GP73.5ex1 | 117,860 | 117,869 | 3 | ||
| GP73.5ex2 | 119,005 | 119,198 | 64 | ||
| GP74 | C | 117,780 | 118,904 | 374 | UL74 homolog; glycoprotein O |
| GP75 | C | 119,349 | 121,529 | 726 | UL75 homolog; glycoprotein H |
| GP76 | 121,694 | 122,533 | 279 | UL76 homolog | |
| GP77 | 122,247 | 124,091 | 614 | UL77 homolog | |
| GP78 | 124,473 | 125,717 | 414 | UL78 homolog; 7-TMR GPCR superfamily | |
| GP79 | C | 125,914 | 126,861 | 315 | UL79 homolog |
| GP80 | 126,722 | 129,031 | 769 | UL80 homolog; CMV protease | |
| GP80.5 | 127,610 | 129,031 | 473 | UL80.5 | |
| GP82 | C | 129,324 | 130,889 | 521 | UL82 homolog; pp71 |
| GP83 | C | 131,109 | 132,809 | 566 | UL83 homolog; pp65 |
| GP84 | C | 133,051 | 134,487 | 478 | UL84 homolog |
| GP85 | C | 134,785 | 135,696 | 303 | UL85 homolog |
| GP86 | C | 135,977 | 140,026 | 1349 | UL86 homolog |
| GP87 | 140,407 | 143,328 | 973 | UL87 homolog | |
| GP88 | 143,231 | 144,499 | 422 | UL88 homolog | |
| GP89ex2 | C | 144,545 | 145,675 | 376 | UL89 homolog; terminase subunit, exon 2 |
| GP91 | 146,102 | 146,365 | 87 | UL91 homolog | |
| GP92 | 146,362 | 146,991 | 209 | UL92 homolog | |
| GP93 | 146,957 | 148,732 | 591 | UL93 homolog | |
| GP94 | 148,644 | 149,681 | 345 | UL94 homolog | |
| GP89ex1 | C | 150,032 | 150,913 | 291 | UL89 homolog; terminase subunit, exon 1 |
| GP95 | 150,967 | 152,235 | 422 | UL95 homolog | |
| GP96 | 152,468 | 152,830 | 120 | UL96 homolog | |
| GP97 | 152,910 | 154,727 | 605 | UL97 homolog; protein kinase | |
| GP98 | 154,747 | 156,531 | 594 | UL98 homolog; alkaline nuclease | |
| GP99 | 156,444 | 156,965 | 173 | UL99 homolog; pp28 | |
| gp99.1 | 157,155 | 157,769 | 204 | Homology to RCMV r4d | |
| GP100 | C | 157,278 | 158,327 | 349 | UL100 homolog; glycoprotein M |
| GP102 | 158,657 | 160,942 | 761 | UL102 homolog | |
| GP103 | C | 161,127 | 161,852 | 241 | UL103 homolog |
| GP104 | C | 161,815 | 163,908 | 697 | UL104 homolog; portal |
| GP105 | 163,748 | 166,531 | 927 | UL105 homolog; helicase-primase | |
| GP112ex1 | 176,745 | 177,498 | 315 | UL112 homolog; replication accessory, ex 1 | |
| GP112ex2 | 177,606 | 177,782 | UL112 homolog; replication accessory, ex 2 | ||
| GP112ex3 | 178,115 | 178,131 | UL122 homolog; replication accessory, ex 3 | ||
| GP114 | C | 179,126 | 179,920 | 264 | UL114 homolog; uracil glycosylase |
| GP115 | C | 179,986 | 180,762 | 258 | UL115 homolog; glycoprotein L |
| GP116 | C | 180,755 | 181,654 | 299 | Homology to THV t116b; Fc receptor//Ig |
| GP117 | C | 181,877 | 183,262 | 461 | UL117 homolog |
| gp119.1 | C | 184,418 | 185,167 | 249 | Similar to MCMV in ACE95619.1 |
| GP121.2 | C | 185,160 | 185,834 | 224 | Betaherpesvirus B7D8, accession AFK83957 |
| GP121.4 | C | 186,299 | 187,174 | 291 | UL121 homolog; Tupaia t121.4, NP_116476 |
| GP122ex3 | C | 187,993 | 189,677 | 677 | UL122 homolog; HCMV IE2 |
| GP122ex2 | C | 191,079 | 191,311 | ||
| §GP122ex1 | C | 191,403 | 191,518 | ||
| GP123ex3 | C | 189,907 | 190,985 | 475 | UL123 homolog; HCMV IE1 |
| GP123ex2 | C | 191,079 | 191,311 | ||
| §GP123ex1 | C | 191,403 | 191,518 | ||
| GP128 | 195,400 | 196,455 | 351 | Similar to Bat HSV B126; US22 Family | |
| gp130 | 196,655 | 196,999 | 114 | ||
| GP129ex3 | C | 196,432 | 196,690 | 178 | Homolog of HCMV UL128 |
| GP129ex2 | 196,768 | 196,890 | |||
| GP129ex1 | C | 196,974 | 197,128 | ||
| GP131ex2 | C | 197,133 | 197,469 | 191 | Homolog of HCMV UL130 |
| GP131ex1 | C | 197,550 | 197,788 | ||
| GP133 | C | 197,788 | 198,174 | 128 | Homolog of HCMV UL131 |
| GP134 | C | 198,268 | 198,951 | 227 | |
| gp138.2 | C | 199,367 | 200,875 | 502 | |
| gp138.3 | C | 201,090 | 202,607 | 505 | |
| gp139 | C | 202,706 | 204,793 | 695 | THV T5; US22 superfamily |
| gp140 | 204,522 | 204,929 | 135 | Homology to CCMV UL132 | |
| gp141 | C | 205,053 | 206,660 | 535 | HCMV US23; US22 superfamily |
| gp142 | C | 206,928 | 208,622 | 564 | HCMV US24; US22 superfamily |
| gp143 | C | 208,875 | 210,866 | 663 | THV T5; US22 superfamily |
| gp144 | C | 211,109 | 213,403 | 764 | US26; US22 gene superfamily |
| gp145 | C | 213,677 | 215,575 | 632 | HCMV IRS1/TRS1; US22 superfamily |
| gp146 | C | 215,932 | 217,914 | 660 | HCMV IRS1/TRS1; US22 superfamily |
| gp147.1 | C | 221,739 | 222,935 | 398 | MHC class I homolog |
| gp147 | C | 223,124 | 224,218 | 364 | MHC class I homolog |
| gp148 | C | 225,379 | 226,560 | 393 | MHC class I homolog |
| gp149 | C | 228,236 | 230,209 | 657 | MHC class I homolog |
| gp149.1 | 230,163 | 230,465 | 100 | Unique ORF sequence in CIDMTR | |
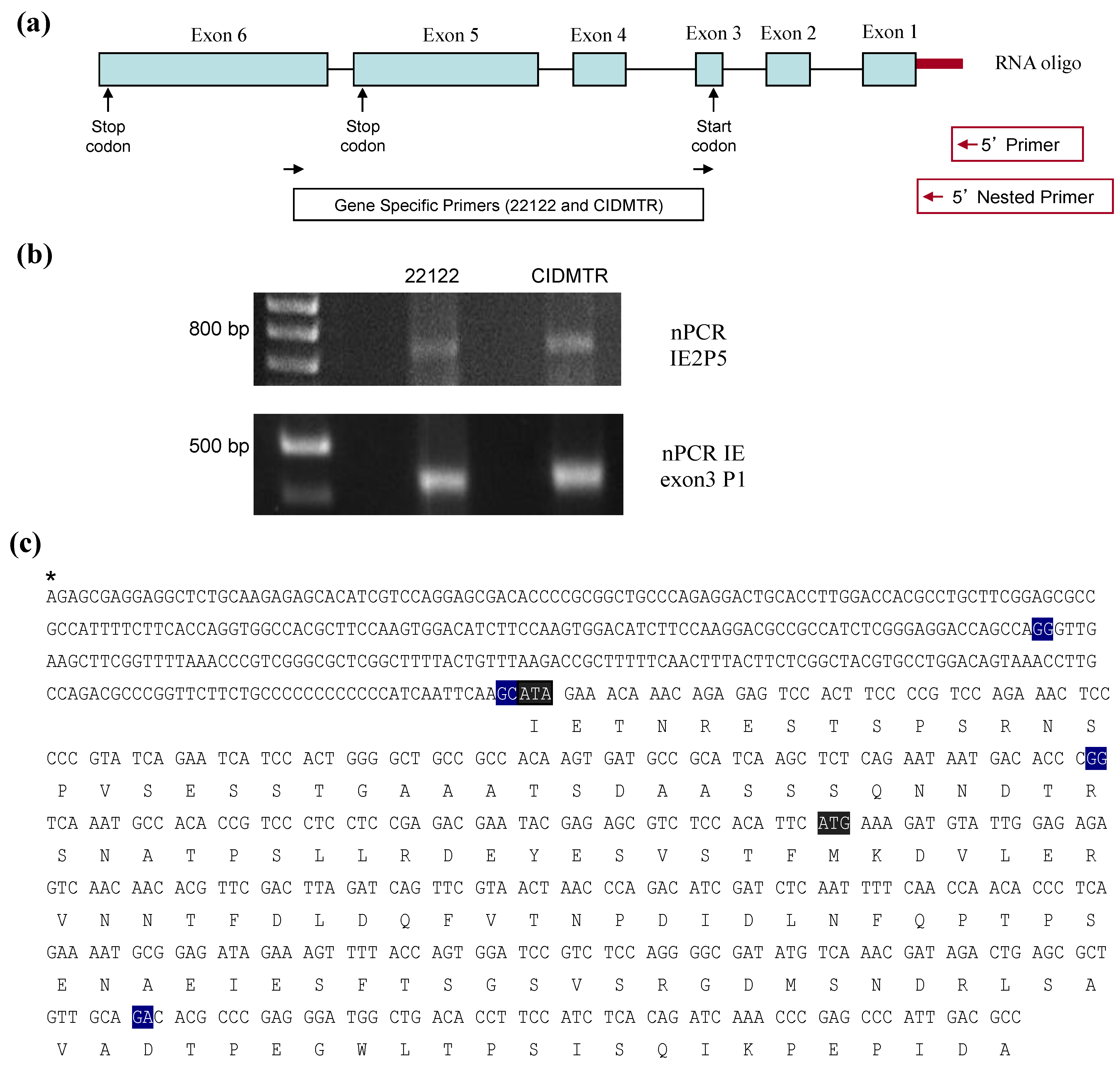
2.4. Sequence Characterization of DNA from Original SG Homogenate in Region of IE1/2 Start Codon

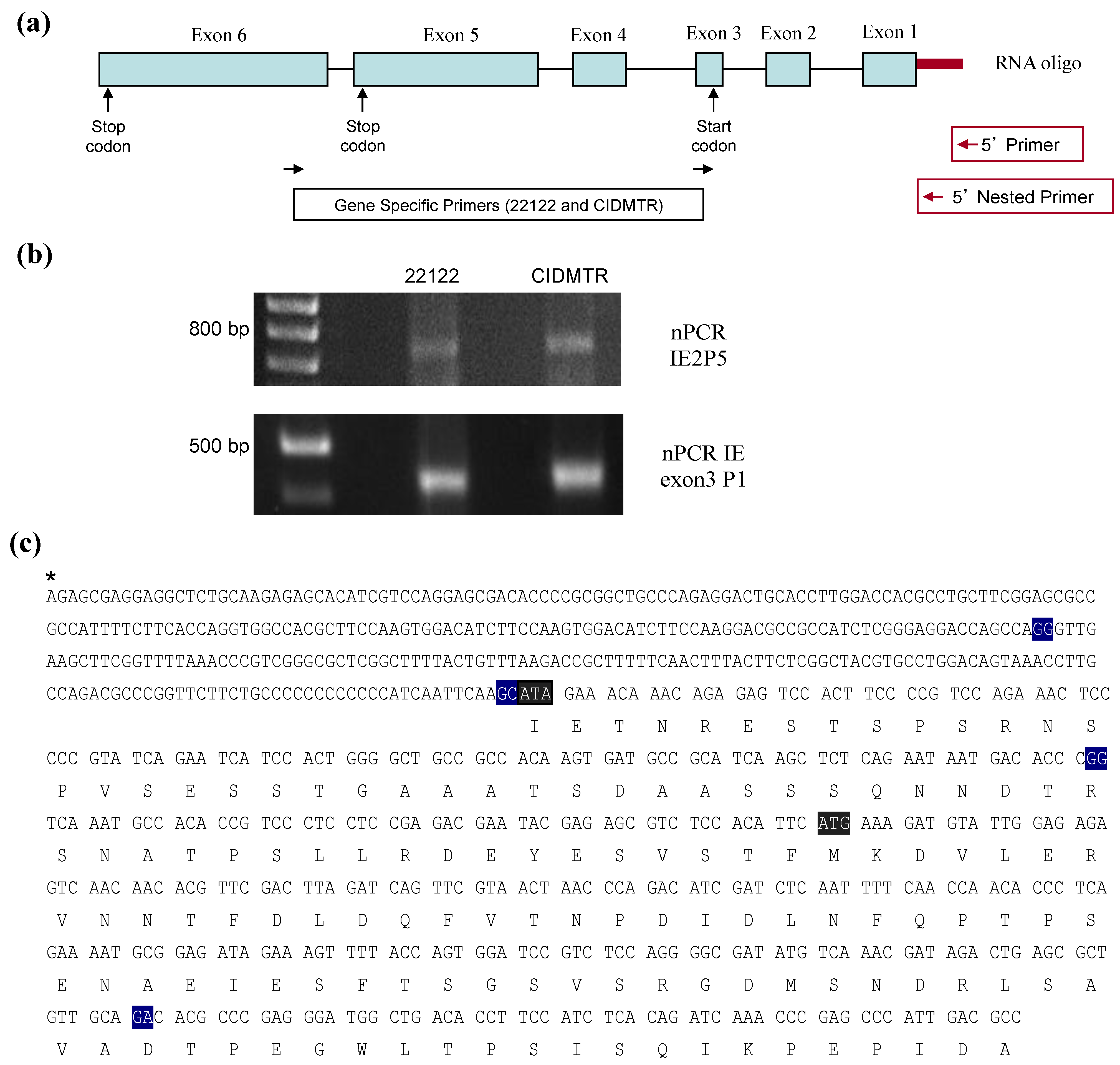
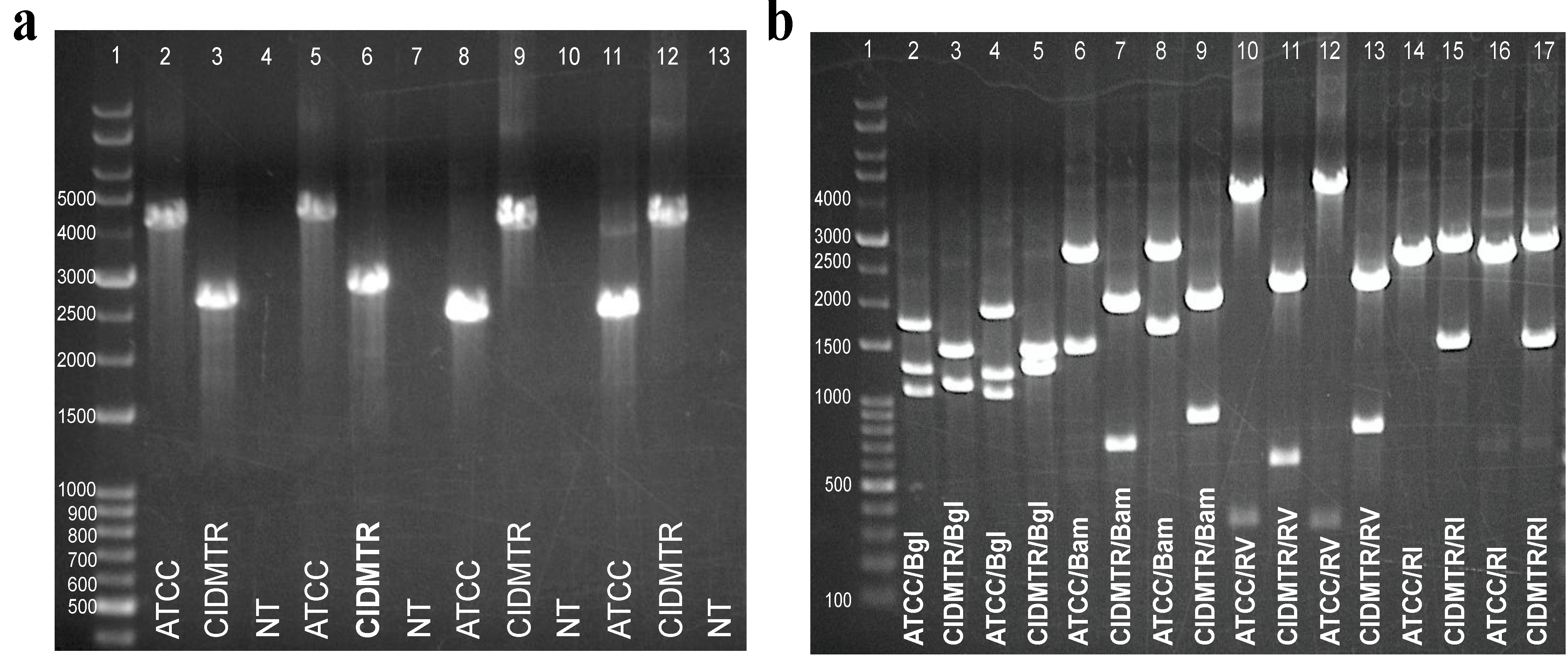
2.5. PCR Confirmation of GPCMV-CIDMTR Genome Structure
| Primer Name | Primer Sequence |
|---|---|
| Mismatch Region 1 F1 | 5'-GTGAGACGTAAGAATAGCTTGC-3' |
| Mismatch Region 1 F2 | 5'-GATCCTTAGACTCTATCACGG-3' |
| Mismatch Region 1 R1 | 5'-GTGTTGTCACAATTGGCACATG-3' |
| Mismatch Region 1 R2 | 5'-ACATGGTCACGACAGAATC-3' |
| Mismatch Region 2 F1 | 5'-GTGGACAGGATCCCCAAATT-3' |
| Mismatch Region 2 F2 | 5'-CCAAATTTCTGTCGTCGGCG-3' |
| Mismatch Region 2 R1 | 5'-TGTTTCCGTGTCTGTCTCCGT-3' |
| Mismatch Region 2 R2 | 5'-GTCTTAGCCCGAGACCTTC-3' |
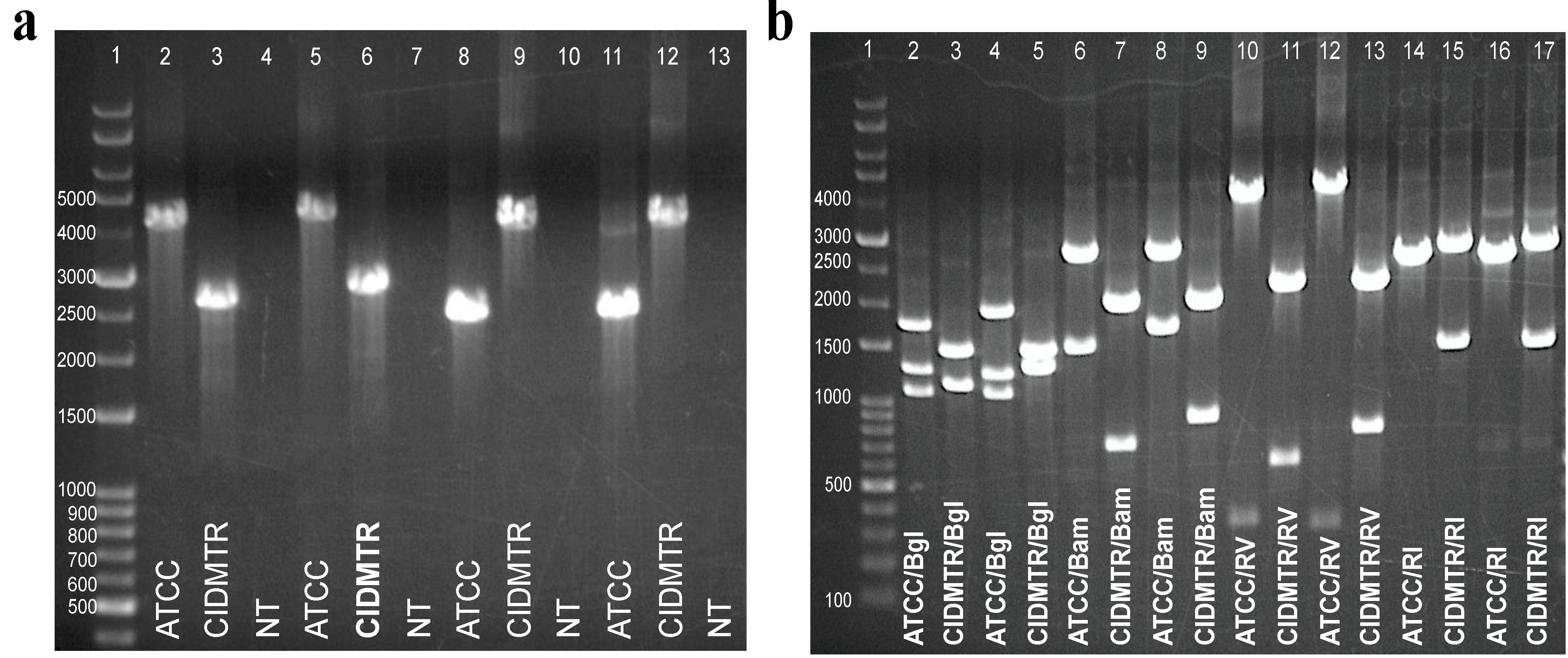
2.6. Infection In Vivo and Development of a Real-Time PCR Assay for Detection of CIDMTR DNA
3. Experimental Section
3.1. Cells, Virus and DNA Preparation
3.2. Transmission Electron Microscopy
3.3. Deep Sequencing and Sequence Analyses
3.4. RT-PCR and RACE Analyses
3.5. GPCMV-CIDMTR Strain PCR Assay
3.6. Animal Challenge Studies
4. Conclusions
Acknowledgments
Author Contributions
Conflict of Interest
EMBL Accession Number
References and Notes
- Sung, H.; Schleiss, M.R. Update on the current status of cytomegalovirus vaccines. Expert Rev. Vaccines 2010, 9, 1303–1314. [Google Scholar] [CrossRef]
- Nigro, G.; Adler, S.P.; La Torre, R.; Best, A.M. Passive immunization during pregnancy for congenital cytomegalovirus infection. N. Engl. J. Med. 2005, 353, 1350–1362. [Google Scholar] [CrossRef]
- Griffiths, P.; Plotkin, S.; Mocarski, E.; Pass, R.; Schleiss, M.; Krause, P.; Bialek, S. Desirability and feasibility of a vaccine against cytomegalovirus. Vaccine 2013, 31, B197–B203. [Google Scholar] [CrossRef]
- Krause, P.R.; Bialek, S.R.; Boppana, S.B.; Griffiths, P.D.; Laughlin, C.A.; Ljungman, P.; Mocarski, E.S.; Pass, R.F.; Read, J.S.; Schleiss, M.R.; et al. Priorities for cmv vaccine development. Vaccine 2013, 32, 4–10. [Google Scholar] [CrossRef]
- Pass, R.F.; Zhang, C.; Evans, A.; Simpson, T.; Andrews, W.; Huang, M.L.; Corey, L.; Hill, J.; Davis, E.; Flanigan, C.; et al. Vaccine prevention of maternal cytomegalovirus infection. N. Engl. J. Med. 2009, 360, 1191–1199. [Google Scholar] [CrossRef]
- Griffiths, P.D.; Stanton, A.; McCarrell, E.; Smith, C.; Osman, M.; Harber, M.; Davenport, A.; Jones, G.; Wheeler, D.C.; O'Beirne, J.; et al. Cytomegalovirus glycoprotein-b vaccine with mf59 adjuvant in transplant recipients: A phase 2 randomised placebo-controlled trial. Lancet 2011, 377, 1256–1263. [Google Scholar] [CrossRef]
- Ahlfors, K.; Ivarsson, S.A.; Harris, S. Report on a long-term study of maternal and congenital cytomegalovirus infection in sweden. Review of prospective studies available in the literature. Scand. J. Infect. Dis. 1999, 31, 443–457. [Google Scholar] [CrossRef]
- Ahlfors, K.; Harris, S.; Ivarsson, S.; Svanberg, L. Secondary maternal cytomegalovirus infection causing symptomatic congenital infection. N. Engl. J. Med. 1981, 305, 284. [Google Scholar]
- Boppana, S.B.; Rivera, L.B.; Fowler, K.B.; Mach, M.; Britt, W.J. Intrauterine transmission of cytomegalovirus to infants of women with preconceptional immunity. N. Engl. J. Med. 2001, 344, 1366–1371. [Google Scholar] [CrossRef]
- Ross, S.A.; Arora, N.; Novak, Z.; Fowler, K.B.; Britt, W.J.; Boppana, S.B. Cytomegalovirus reinfections in healthy seroimmune women. J. Infect. Dis. 2010, 201, 386–389. [Google Scholar] [CrossRef]
- Yamamoto, A.Y.; Mussi-Pinhata, M.M.; Boppana, S.B.; Novak, Z.; Wagatsuma, V.M.; Oliveira Pde, F.; Duarte, G.; Britt, W.J. Human cytomegalovirus reinfection is associated with intrauterine transmission in a highly cytomegalovirus-immune maternal population. Am. J. Obstet. Gynecol. 2010, 202, e291–298, 297 e291–298. [Google Scholar]
- Mussi-Pinhata, M.M.; Yamamoto, A.Y.; Moura Brito, R.M.; de Lima Isaac, M.; de Carvalho e Oliveira, P.F.; Boppana, S.; Britt, W.J. Birth prevalence and natural history of congenital cytomegalovirus infection in a highly seroimmune population. Clin. Infect. Dis. 2009, 49, 522–528. [Google Scholar] [CrossRef]
- Ross, S.A.; Fowler, K.B.; Ashrith, G.; Stagno, S.; Britt, W.J.; Pass, R.F.; Boppana, S.B. Hearing loss in children with congenital cytomegalovirus infection born to mothers with preexisting immunity. J. Pediatr. 2006, 148, 332–336. [Google Scholar] [CrossRef]
- Yamamoto, A.Y.; Mussi-Pinhata, M.M.; Isaac, M.D.; Amaral, F.R.; Carvalheiro, C.G.; Aragon, D.C.; Manfredi, A.K.; Boppana, S.B.; Britt, W.J. Congenital cytomegalovirus infection as a cause of sensorineural hearing loss in a highly immune population. J. Pediatr. Infect. Dis. 2011, 30, 1043–1046. [Google Scholar] [CrossRef]
- Novak, Z.; Ross, S.A.; Patro, R.K.; Pati, S.K.; Reddy, M.K.; Purser, M.; Britt, W.J.; Boppana, S.B. Enzyme-linked immunosorbent assay method for detection of cytomegalovirus strain-specific antibody responses. Clin. Vaccine Immunol. 2009, 16, 288–290. [Google Scholar] [CrossRef]
- Yamamoto, A.Y.; Mussi-Pinhata, M.M.; de Deus Wagatsuma, V.M.; Marin, L.J.; Duarte, G.; Figueiredo, L.T. Human cytomegalovirus glycoprotein b genotypes in brazilian mothers and their congenitally infected infants. J. Med. Virol. 2007, 79, 1164–1168. [Google Scholar] [CrossRef]
- Murthy, S.; Hayward, G.S.; Wheelan, S.; Forman, M.S.; Ahn, J.H.; Pass, R.F.; Arav-Boger, R. Detection of a single identical cytomegalovirus (cmv) strain in recently seroconverted young women. PLoS One 2011, 6, e15949. [Google Scholar]
- Kropff, B.; Burkhardt, C.; Schott, J.; Nentwich, J.; Fisch, T.; Britt, W.; Mach, M. Glycoprotein n of human cytomegalovirus protects the virus from neutralizing antibodies. PLoS Pathog. 2012, 8, e1002999. [Google Scholar] [CrossRef]
- Pati, S.K.; Novak, Z.; Purser, M.; Arora, N.; Mach, M.; Britt, W.J.; Boppana, S.B. Strain-specific neutralizing antibody responses against human cytomegalovirus envelope glycoprotein n. Clin. Vaccine Immunol. 2012, 19, 909–913. [Google Scholar] [CrossRef]
- Burkhardt, C.; Himmelein, S.; Britt, W.; Winkler, T.; Mach, M. Glycoprotein n subtypes of human cytomegalovirus induce a strain-specific antibody response during natural infection. J. Gen. Virol. 2009, 90, 1951–1961. [Google Scholar] [CrossRef]
- Sabbaj, S.; Pass, R.F.; Goepfert, P.A.; Pichon, S. Glycoprotein b vaccine is capable of boosting both antibody and cd4 t-cell responses to cytomegalovirus in chronically infected women. J. Infect. Dis. 2011, 203, 1534–1541. [Google Scholar] [CrossRef]
- Schleiss, M.R. Could therapeutic vaccination of cytomegalovirus-seropositive persons prevent reinfection and congenital virus transmission? J. Infect. Dis. 2011, 203, 1513–1516. [Google Scholar] [CrossRef]
- Kern, E.R. Pivotal role of animal models in the development of new therapies for cytomegalovirus infections. Antivir. Res. 2006, 71, 164–171. [Google Scholar] [CrossRef]
- Schleiss, M.R. Nonprimate models of congenital cytomegalovirus (cmv) infection: Gaining insight into pathogenesis and prevention of disease in newborns. ILAR J. 2006, 47, 65–72. [Google Scholar] [CrossRef]
- Yue, Y.; Wang, Z.; Abel, K.; Li, J.; Strelow, L.; Mandarino, A.; Eberhardt, M.K.; Schmidt, K.A.; Diamond, D.J.; Barry, P.A. Evaluation of recombinant modified vaccinia ankara virus-based rhesus cytomegalovirus vaccines in rhesus macaques. Med. Microbiol. Immunol. 2008, 197, 117–123. [Google Scholar] [CrossRef]
- Bia, F.J.; Miller, S.A.; Davidson, K.H. The guinea pig cytomegalovirus model of congenital human cytomegalovirus infection. Birth. Defects Orig. Artic. Ser. 1984, 20, 233–241. [Google Scholar]
- Hartley, J.W.; Rowe, W.P.; Huebner, R.J. Serial propagation of the guinea pig salivary gland virus in tissue culture. Proc. Soc. Exp. Biol. Med. 1957, 96, 281–285. [Google Scholar] [CrossRef]
- Johnson, K.P.; Connor, W.S. Guinea pig cytomegalovirus: Transplacental transmission. Brief report. Arch. Virol. 1979, 59, 263–267. [Google Scholar] [CrossRef]
- Schleiss, M.R.; Bourne, N.; Stroup, G.; Bravo, F.J.; Jensen, N.J.; Bernstein, D.I. Protection against congenital cytomegalovirus infection and disease in guinea pigs, conferred by a purified recombinant glycoprotein b vaccine. J. Infect. Dis. 2004, 189, 1374–1381. [Google Scholar] [CrossRef]
- Bratcher, D.F.; Bourne, N.; Bravo, F.J.; Schleiss, M.R.; Slaoui, M.; Myers, M.G.; Bernstein, D.I. Effect of passive antibody on congenital cytomegalovirus infection in guinea pigs. J. Infect. Dis. 1995, 172, 944–950. [Google Scholar] [CrossRef]
- Britt, W.J.; Harrison, C. Identification of an abundant disulfide-linked complex of glycoproteins in the envelope of guinea pig cytomegalovirus. Virology 1994, 201, 294–302. [Google Scholar] [CrossRef]
- Schleiss, M.R.; Bernstein, D.I.; McVoy, M.A.; Stroup, G.; Bravo, F.; Creasy, B.; McGregor, A.; Henninger, K.; Hallenberger, S. The non-nucleoside antiviral, bay 38–4766, protects against cytomegalovirus (cmv) disease and mortality in immunocompromised guinea pigs. Antivir. Res. 2005, 65, 35–43. [Google Scholar] [CrossRef]
- Fong, C.K.; Bia, F.; Hsiung, G.D. Ultrastructural development and persistence of guinea pig cytomegalovirus in duet cells of guinea pig submaxillary gland. Arch. Virol. 1980, 64, 97–108. [Google Scholar] [CrossRef]
- Fong, C.K.; Bia, F.; Hsiung, G.D.; Madore, P.; Chang, P.W. Ultrastructural development of guinea pig cytomegalovirus in cultured guinea pig embryo cells. J. Gen. Virol. 1979, 42, 127–140. [Google Scholar] [CrossRef]
- Fong, C.K.; Brigati, D. Ultrastructural localization of viral antigen in nuclear inclusions of cytomegalovirus infected guinea pig cells. Arch. Virol. 1982, 74, 125–133. [Google Scholar] [CrossRef]
- Cui, X.; McGregor, A.; Schleiss, M.R.; McVoy, M.A. The impact of genome length on replication and genome stability of the herpesvirus guinea pig cytomegalovirus. Virology 2009, 386, 132–138. [Google Scholar] [CrossRef]
- Smith, L.M.; McWhorter, A.R.; Masters, L.L.; Shellam, G.R.; Redwood, A.J. Laboratory strains of murine cytomegalovirus are genetically similar to but phenotypically distinct from wild strains of virus. J. Virol. 2008, 82, 6689–6696. [Google Scholar] [CrossRef]
- Pignatelli, S.; Dal Monte, P.; Rossini, G.; Chou, S.; Gojobori, T.; Hanada, K.; Guo, J.J.; Rawlinson, W.; Britt, W.; Mach, M.; et al. Human cytomegalovirus glycoprotein n (gpul73-gn) genomic variants: Identification of a novel subgroup, geographical distribution and evidence of positive selective pressure. J. Gen. Virol. 2003, 84, 647–655. [Google Scholar] [CrossRef]
- Pignatelli, S.; Dal Monte, P.; Rossini, G.; Lazzarotto, T.; Gatto, M.R.; Landini, M.P. Intrauterine cytomegalovirus infection and glycoprotein n (gn) genotypes. J. Clin. Virol. 2003, 28, 38–43. [Google Scholar] [CrossRef]
- CLC Workbench software, version 6.0; CLC: Cambridge, MA, USA. Available online: http://www.clcbio.com/products/clc-main-workbench/ (accessed on 23 January 2014).
- XPlasMap, version 0.99; Available online: http://www.iayork.com/XPlasMap/ (accessed on 16 January 2014).
- Yamada, S.; Nozawa, N.; Katano, H.; Fukui, Y.; Tsuda, M.; Tsutsui, Y.; Kurane, I.; Inoue, N. Characterization of the guinea pig cytomegalovirus genome locus that encodes homologs of human cytomegalovirus major immediate-early genes, ul128, and ul130. Virology 2009, 391, 99–106. [Google Scholar] [CrossRef]
- Nozawa, N.; Yamamoto, Y.; Fukui, Y.; Katano, H.; Tsutsui, Y.; Sato, Y.; Yamada, S.; Inami, Y.; Nakamura, K.; Yokoi, M.; et al. Identification of a 1.6 kb genome locus of guinea pig cytomegalovirus required for efficient viral growth in animals but not in cell culture. Virology 2008, 379, 45–54. [Google Scholar] [CrossRef]
- Yang, D.; Tamburro, K.; Dittmer, D.; Cui, X.; McVoy, M.A.; Hernandez-Alvarado, N.; Schleiss, M.R. Complete genome sequence of pathogenic guinea pig cytomegalovirus from salivary gland homogenates of infected animals. Genome Announc. 2013, 1, e0005413. [Google Scholar]
- Kozak, M. An analysis of 5'-noncoding sequences from 699 vertebrate messenger rnas. Nucleic Acids Res. 1987, 15, 8125–8148. [Google Scholar] [CrossRef]
- iTEM software, Version 9.0; Olympus SIS: Münster, Germany, 2014.
- Simpson, J.T.; Wong, K.; Jackman, S.D.; Schein, J.E.; Jones, S.J.; Birol, I. Abyss: A parallel assembler for short read sequence data. Genome Res. 2009, 19, 1117–1123. [Google Scholar] [CrossRef]
- Kanai, K.; Yamada, S.; Yamamoto, Y.; Fukui, Y.; Kurane, I.; Inoue, N. Re-evaluation of the genome sequence of guinea pig cytomegalovirus. J. Gen. Virol. 2011, 92, 1005–1020. [Google Scholar] [CrossRef]
- Schleiss, M.R.; McGregor, A.; Choi, K.Y.; Date, S.V.; Cui, X.; McVoy, M.A. Analysis of the nucleotide sequence of the guinea pig cytomegalovirus (gpcmv) genome. Virol. J. 2008, 5, 139. [Google Scholar] [CrossRef]
- Milne, I.; Bayer, M.; Cardle, L.; Shaw, P.; Stephen, G.; Wright, F.; Marshall, D. Tablet--next generation sequence assembly visualization. Bioinformatics 2010, 26, 401–402. [Google Scholar] [CrossRef]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The sequence alignment/map format and samtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Waterhouse, A.M.; Procter, J.B.; Martin, D.M.; Clamp, M.; Barton, G.J. Jalview version 2—A multiple sequence alignment editor and analysis workbench. Bioinformatics 2009, 25, 1189–1191. [Google Scholar] [CrossRef]
- SMRT Analysis software, Version 1.4.0; Pacific Biosciences: Menlo Park, CA, USA, 2014. Available online: http://www.pacificbiosciences.com/products/software/secondary-analysis/ (accessed on 16 January 2014).
- LightCycler Data Analysis Software, version 1.5; Roche: Indianapolis, IN, USA, 2014.
- Crumpler, M.M.; Choi, K.Y.; McVoy, M.A.; Schleiss, M.R. A live guinea pig cytomegalovirus vaccine deleted of three putative immune evasion genes is highly attenuated but remains immunogenic in a vaccine/challenge model of congenital cytomegalovirus infection. Vaccine 2009, 27, 4209–4218. [Google Scholar] [CrossRef]
- Hsiung, G.D.; Bia, F.J.; Fong, C.K. Viruses of guinea pigs: Considerations for biomedical research. Microbiol. Rev. 1980, 44, 468–490. [Google Scholar]
- Hummel, M.; Zhang, Z.; Yan, S.; DePlaen, I.; Golia, P.; Varghese, T.; Thomas, G.; Abecassis, M.I. Allogeneic transplantation induces expression of cytomegalovirus immediate-early genes in vivo: A model for reactivation from latency. J. Virol. 2001, 75, 4814–4822. [Google Scholar] [CrossRef]
- Soderberg-Naucler, C.; Fish, K.N.; Nelson, J.A. Reactivation of latent human cytomegalovirus by allogeneic stimulation of blood cells from healthy donors. Cell 1997, 91, 119–126. [Google Scholar] [CrossRef]
- Middelkamp, J.N.; Patrizi, G.; Reed, C.A. Light and electron microscopic studies of the guinea pig cytomegalovirus. J. Ultrastruct. Res. 1967, 18, 85–101. [Google Scholar] [CrossRef]
- Choi, Y.C.; Hsiung, G.D. Cytomegalovirus infection in guinea pigs. II. Transplacental and horizontal transmission. J. Infect. Dis. 1978, 138, 197–202. [Google Scholar] [CrossRef]
- ATCC History of GPCMV. Available online: http://www.atcc.org/products/all/VR-682.aspx#history/ (accessed on 23 January 2014).
- Brondke, H.; Schmitz, B.; Doerfler, W. Nucleotide sequence comparisons between several strains and isolates of human cytomegalovirus reveal alternate start codon usage. Arch. Virol. 2007, 152, 2035–2046. [Google Scholar] [CrossRef]
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Schleiss, M.R.; McAllister, S.; Armién, A.G.; Hernandez-Alvarado, N.; Fernández-Alarcón, C.; Zabeli, J.C.; Ramaraj, T.; Crow, J.A.; McVoy, M.A. Molecular and Biological Characterization of a New Isolate of Guinea Pig Cytomegalovirus. Viruses 2014, 6, 448-475. https://doi.org/10.3390/v6020448
Schleiss MR, McAllister S, Armién AG, Hernandez-Alvarado N, Fernández-Alarcón C, Zabeli JC, Ramaraj T, Crow JA, McVoy MA. Molecular and Biological Characterization of a New Isolate of Guinea Pig Cytomegalovirus. Viruses. 2014; 6(2):448-475. https://doi.org/10.3390/v6020448
Chicago/Turabian StyleSchleiss, Mark R., Shane McAllister, Anibal G. Armién, Nelmary Hernandez-Alvarado, Claudia Fernández-Alarcón, Jason C. Zabeli, Thiruvarangan Ramaraj, John A. Crow, and Michael A. McVoy. 2014. "Molecular and Biological Characterization of a New Isolate of Guinea Pig Cytomegalovirus" Viruses 6, no. 2: 448-475. https://doi.org/10.3390/v6020448