The Role of the CoREST/REST Repressor Complex in Herpes Simplex Virus 1 Productive Infection and in Latency

{kind=link}
{kind=link}
Abstract
:1. Background
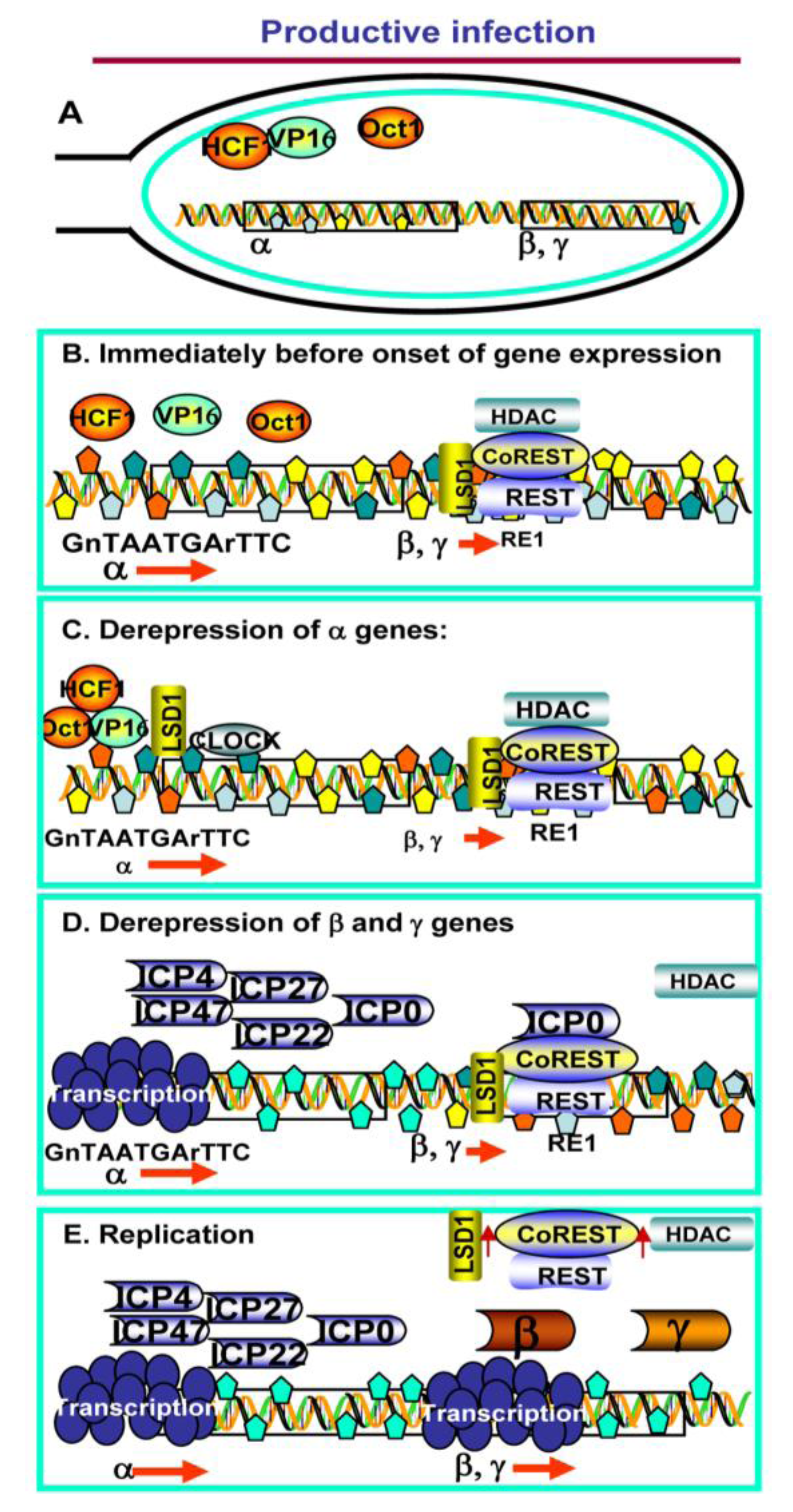
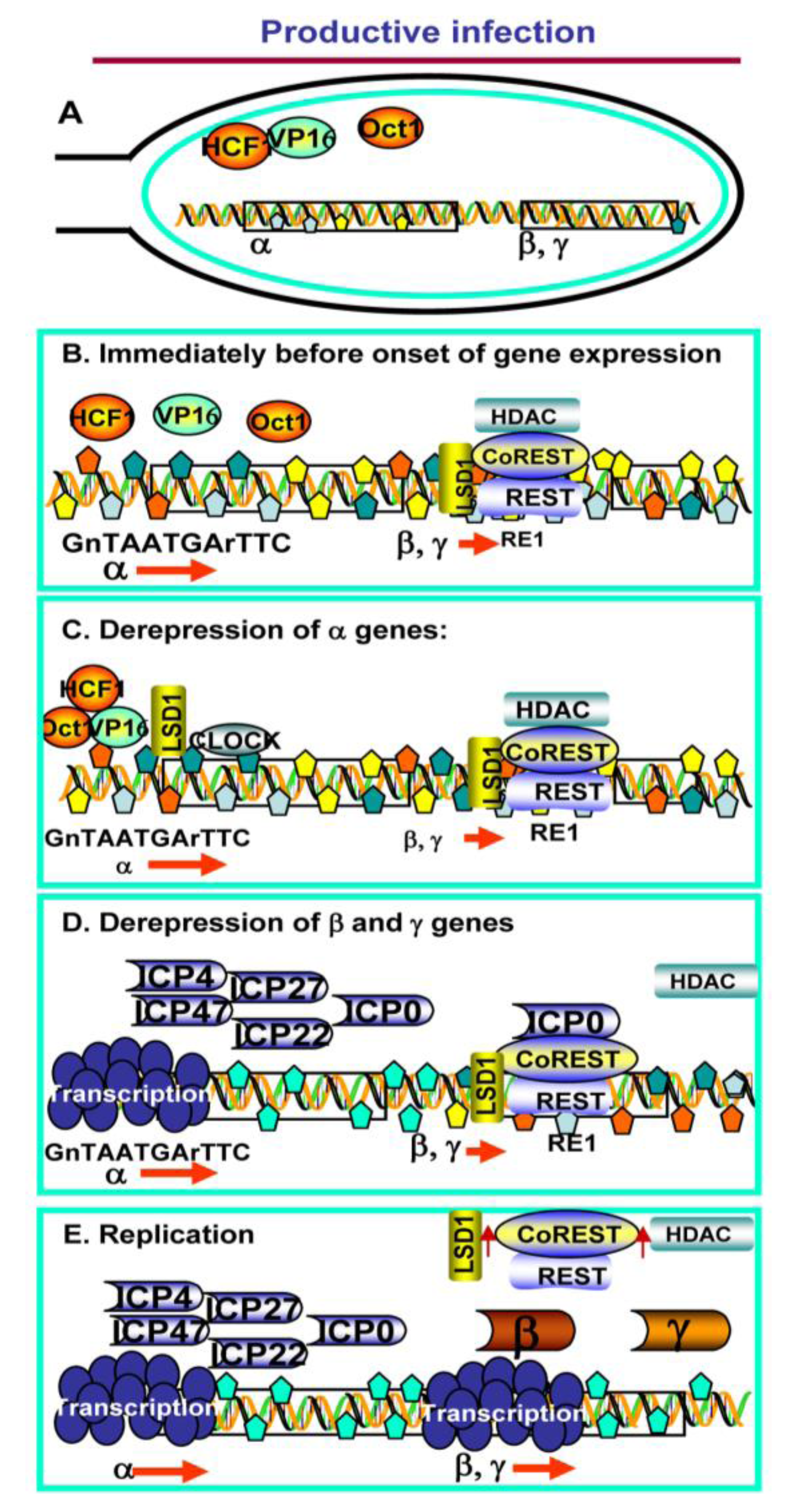
2. Productive Infection
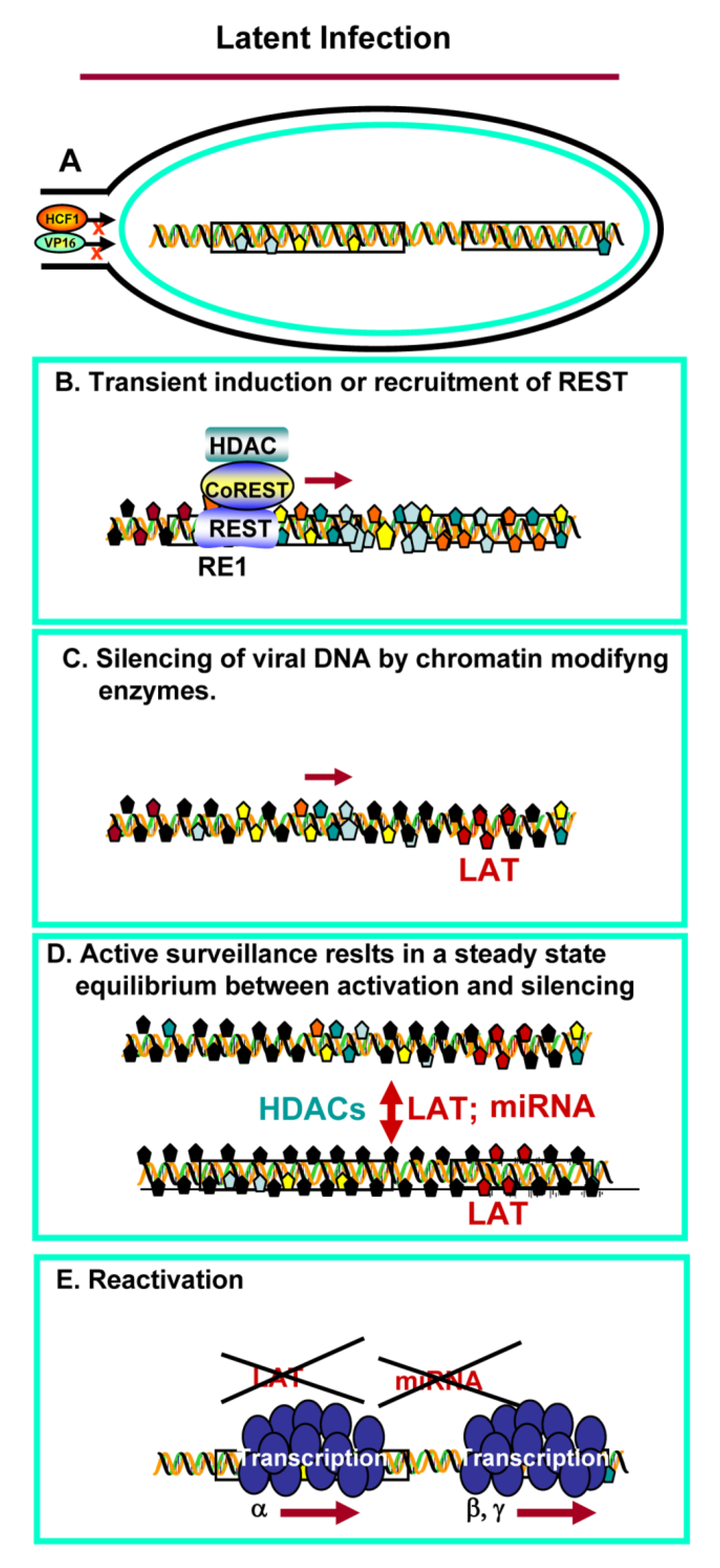
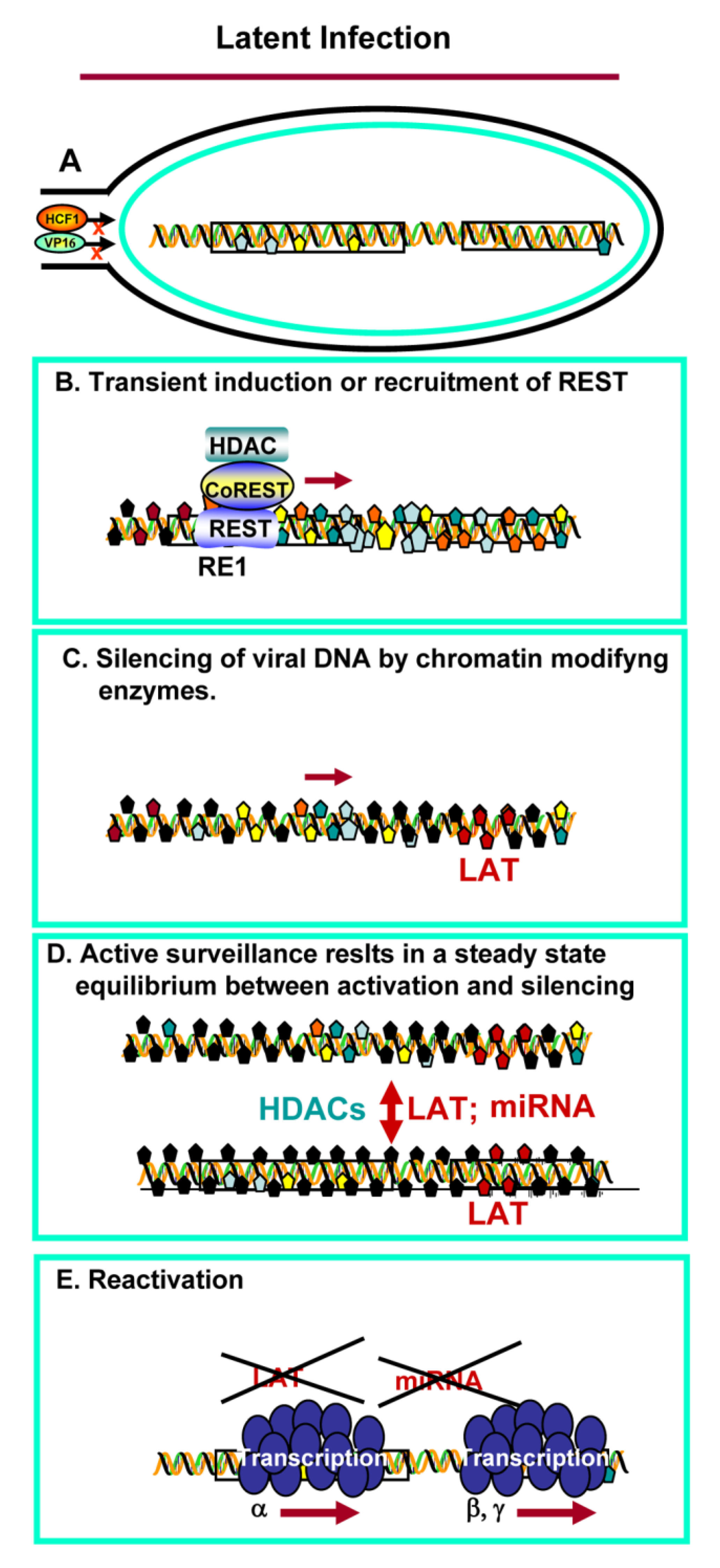
3. Establishment of Latency and Reactivation
4. Reactivation from Latency
5. Conclusions: The Role of the CoREST/REST Repressor Complex in the HSV Lifestyle
- (i)
- All of the data available to date indicate that no viral function is required to establish latent infections [2,42]. Silencing of viral DNA in neurons appears to be a neuronal function, most likely a defense mechanism to block transmission of viruses from the periphery to the central nervous system. Viewed from this prospective, HSV took advantage of the innate neuron defenses to enable itself to be silenced and remain as a reservoir in its human host.
- (ii)
- One response to stress in neurons is activation of the CoREST/REST complex. This has been reported in some degenerative diseases of the CNS [23,24,25,26]. It is conceivable that transient expression of REST following entry of the virus into the CNS, is sufficient to initiate the epigenetic modifications essential to silence viral DNA, but not irreversibly damage the neuron harboring the virus in the silent state.
- (iii)
- In cultured cells productively infected with HSV-1, the CoREST/REST repressor complex is readily overcome by displacement of HDACs from the repressor complex by ICP0, or in high multiplicity infections [8]. The studies on the HSV-1 mutant carrying dnREST [38] suggest that the interaction with the CoREST/REST repressor complex serves to maintain equilibrium between excessive replication—that would irreversibly injure the host and prevent transmission—and minimal replication—which is insufficient to secure establishment of latent virus, and ultimately frequent transmission from infected to uninfected individuals.
Conflict of Interest
Acknowledgements
References and Notes
- Roizman, B.; Zhou, G.; Du, T. Checkpoints in productive and latent infections with herpes simplex virus 1: Conceptualization of the issues. J. Neurovirol. 2011, 17, 512–517. [Google Scholar] [CrossRef]
- Roizman, B.; Knipe, D.M.; Whitley, R.J. Herpes Simplex Viruses. In Fields Virology, 6th; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2013. [Google Scholar]
- Gu, H.; Liang, Y.; Mandel, G.; Roizman, B. Components of the REST/CoREST/histone deacetylase repressor complex are disrupted, modified, and translocated in HSV-1-infected cells. Proc. Natl. Acad. Sci. USA 2005, 102, 7571–7576. [Google Scholar]
- Gu, H.; Roizman, B. Herpes simplex virus-infected cell protein 0 blocks the silencing of viral DNA by dissociating histone deacetylases from the CoREST/REST complex. Proc. Natl. Acad. Sci. USA 2007, 104, 17134–17139. [Google Scholar] [CrossRef]
- Gu, H.; Roizman, B. Engagement of the lysine-specific demethylase/HDAC1/CoREST/REST complex by herpes simplex virus 1. J. Virol. 2009, 83, 4376–4385. [Google Scholar] [CrossRef]
- Cai, W.; Schaffer, P.A. Herpes simplex virus type 1 ICP0 regulates expression of immediate-early, early, and late genes in productively infected cells. J. Virol. 1992, 66, 2904–2915. [Google Scholar]
- Chen, J.; Panagiotidis, C.; Silverstein, S. Multimerization of ICP0, a herpes simplex virus immediate-early protein. J. Virol. 1992, 66, 5598–5602. [Google Scholar]
- Gu, H.; Roizman, B. The two functions of herpes simplex virus 1 ICP0, inhibition of silencing by the CoREST/REST/HDAC complex and degradation of PML, are executed in tandem. J. Virol. 2009, 83, 181–187. [Google Scholar] [CrossRef]
- Andres, M.E.; Burger, C.; Peral-Rubio, M.J.; Battaglioli, E.; Anderson, M.E.; Grimes, J.; Dallman, J.; Ballas, N.; Mandel, G. CoREST: A functional corepressor required for regulation of neural-specific gene expression. Proc. Natl. Acad. Sci. USA 1999, 96, 9873–9878. [Google Scholar] [CrossRef]
- Chong, J.A.; Tapia-Ramirez, J.; Kim, S.; Toledo-Aral, J.J.; Zheng, Y.; Boutros, M.C.; Altshuller, Y.M.; Frohman, M.A.; Kraner, S.D.; Mandel, G. REST: A mammalian silencer protein that restricts sodium channel gene expression to neurons. Cell 1995, 80, 949–957. [Google Scholar] [CrossRef]
- Schoenherr, C.J.; Anderson, D.J. The neuron-restrictive silencer factor (NRSF): A coordinate repressor of multiple neuron-specific genes. Science 1995, 267, 1360–1363. [Google Scholar] [CrossRef]
- Humphrey, G.W.; Wang, Y.; Russanova, V.R.; Hirai, T.; Qin, J.; Nakatani, Y.; Howard, B.H. Stable histone deacetylase complexes distinguished by the presence of SANT domain proteins CoREST/kiaa0071 and Mta-L1. J. Biol. Chem. 2001, 276, 6817–6824. [Google Scholar]
- You, A.; Tong, J.K.; Grozinger, C.M.; Schreiber, S.L. CoREST is an integral component of the CoREST- human histone deacetylase complex. Proc. Natl. Acad. Sci. USA 2001, 98, 1454–1458. [Google Scholar]
- Ballas, N.; Mandel, G. The many faces of REST oversee epigenetic programming of neuronal genes. Curr. Opin. Neurobiol. 2005, 15, 500–506. [Google Scholar] [CrossRef]
- Gopalakrishnan, V. REST and the RESTless: In stem cells and beyond. Future Neurol. 2009, 4, 317–329. [Google Scholar] [CrossRef]
- Metzger, E.; Wissmann, M.; Yin, N.; Muller, J.M.; Schneider, R.; Peters, A.H.; Schule, R. LSD1 demethylates repressive histone marks to promote androgen-receptor-dependent transcription. Nature 2005, 437, 436–439. [Google Scholar]
- Shi, Y.J.; Matson, C.; Lan, F.; Iwase, S.; Baba, T.; Shi, Y. Regulation of LSD1 histone demethylase activity by its associated factors. Mol. Cell 2005, 19, 857–864. [Google Scholar] [CrossRef]
- Yang, M.; Gocke, C.B.; Luo, X.; Borek, D.; Tomchick, D.R.; Machius, M.; Yu, H. Structural basis for CoREST-dependent demethylation of nucleosomes by the human LSD1 histone demethylase. Mol. Cell 2006, 23, 377–387. [Google Scholar] [CrossRef]
- Ballas, N.; Grunseich, C.; Lu, D.D.; Speh, J.C.; Mandel, G. REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell 2005, 121, 645–657. [Google Scholar] [CrossRef]
- Griffith, E.C.; Cowan, C.W.; Greenberg, M.E. REST acts through multiple deacetylase complexes. Neuron 2001, 31, 339–340. [Google Scholar] [CrossRef]
- Koenigsberger, C.; Chicca, J.J., 2nd; Amoureux, M.C.; Edelman, G.M.; Jones, F.S. Differential regulation by multiple promoters of the gene encoding the neuron-restrictive silencer factor. Proc. Natl. Acad. Sci. USA 2000, 97, 2291–2296. [Google Scholar]
- Shimojo, M.; Hersh, L.B. Regulation of the cholinergic gene locus by the repressor element-1 silencing transcription factor/neuron restrictive silencer factor (REST/NRSF). Life Sci. 2004, 74, 2213–2225. [Google Scholar] [CrossRef]
- Marti, E.; Pantano, L.; Banez-Coronel, M.; Llorens, F.; Minones-Moyano, E.; Porta, S.; Estivill, X. A myriad of miRNA variants in control and Huntington’s disease brain regions detected by massively parallel sequencing. Nucleic Acids Res. 2010, 38, 7219–7235. [Google Scholar] [CrossRef]
- Johnson, R.; Richter, N.; Jauch, R.; Gaughwin, P.M.; Zuccato, C.; Cattaneo, E.; Stanton, L.W. Human accelerated region 1 noncoding RNA is repressed by REST in Huntington’s disease. Physiol. Genomics 2010, 41, 269–274. [Google Scholar] [CrossRef]
- Shimojo, M. Huntingtin regulates RE1-silencing transcription factor/neuron-restrictive silencer factor (REST/NRSF) nuclear trafficking indirectly through a complex with REST/NRSF-interacting LIM domain protein (RILP) and dynactin p150 Glued. J. Biol. Chem. 2008, 283, 34880–34886. [Google Scholar] [CrossRef]
- Zuccato, C.; Belyaev, N.; Conforti, P.; Ooi, L.; Tartari, M.; Papadimou, E.; Cattaneo, E. Widespread disruption of repressor element-1 silencing transcription factor/neuron-restrictive silencer factor occupancy at its target genes in Huntington’s disease. J. Neurosci. 2007, 27, 6972–6983. [Google Scholar]
- Johnson, D.S.; Mortazavi, A.; Myers, R.M.; Wold, B. Genome wide mapping of in vivo protein-DNA interactions. Science 2007, 316, 1497–1502. [Google Scholar] [CrossRef]
- Jothi, R.; Cuddapah, S.; Barski, A.; Cui, K.; Zhao, K. Genome-wide identification of in vivo protein-DNA binding sites from ChIP-Seq data. Nucleic Acids Res. 2008, 36, 5221–5231. [Google Scholar] [CrossRef]
- Otto, S.J.; McCorkle, S.R.; Hover, J.; Conaco, C.; Han, J.J.; Impey, S.; Yochum, G.S.; Dunn, J.J.; Goodman, R.H.; Mandel, G. A new binding motif for the transcriptional repressor REST uncovers large networks devoted to neuronal functions. J. Neurosci. 2007, 27, 6729–6739. [Google Scholar] [CrossRef]
- Tsai, M.C.; Manor, O.; Wan, Y.; Mosammaparast, N.; Wang, J.K.; Lan, F.; Shi, Y.; Segal, E.; Chang, H.Y. Long noncoding RNA as modular scaffolds of histone modification complexes. Science 2010, 329, 689–693. [Google Scholar] [CrossRef]
- Boutell, C.; Sadis, S.; Everett, R.D. Herpes simplex virus type 1 immediate-early protein ICP0 and its isolated RING finger domain act as ubiquitin E3 ligases in vitro. J. Virol. 2002, 76, 841–850. [Google Scholar] [CrossRef]
- Everett, R.D.; Meredith, M.; Orr, A. The ability of herpes simplex virus type 1 immediate-early protein Vmw110 to bind to a ubiquitin-specific protease contributes to its roles in the activation of gene expression and stimulation of virus replication. J. Virol. 1999, 73, 417–426. [Google Scholar]
- Kawaguchi, Y.; Bruni, R.; Roizman, B. Interaction of herpes simplex virus 1 alpha regulatory protein ICP0 with elongation factor 1δ: ICP0 affects translational machinery. J. Virol. 1997, 71, 1019–1024. [Google Scholar]
- Lopez, P.; van Sant, C.; Roizman, B. Requirements for the nuclear-cytoplasmic translocation of infected-cell protein 0 of herpes simplex virus 1. J. Virol. 2001, 75, 3832–3840. [Google Scholar] [CrossRef]
- Yang, M.; Gocke, C.; Luo, X.; Borek, D.; Tomchick, D.; Machius, M.; Otwinowski, Z.; Yu, H. Structural basis for CoREST-dependent demethylation of nucleosomes by the human LSD1 histone demethylase. Mol. Cell 2006, 23, 377–387. [Google Scholar] [CrossRef]
- Lee, M.G.; Wynder, C.; Cooch, N.; Shiekhattar, R. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature 2005, 437, 432–435. [Google Scholar]
- Zhou, G.; Te, D.; Roizman, B. The CoREST/REST repressor is both necessary and inimical for expression of herpes simplex virus genes. mBio 2011, 2, e00313-10. [Google Scholar]
- Du, T.; Zhou, G.; Khan, S.; Gu, H.; Roizman, B. Disruption of HDAC/CoREST/REST repressor by dnREST reduces genome silencing and increases virulence of herpes simplex virus. Proc. Natl. Acad. Sci. USA 2010, 107, 15904–15909. [Google Scholar] [CrossRef]
- Roizman, B. An Inquiry into the Mechanisms of Recurrent Herpes Infection of Man. In Perspectives in Virology; Pollard, M., Ed.; Hocher Medical Division: New York, NY, USA, 1966; Volume IV, pp. 283–304. [Google Scholar]
- Roizman, B.; Sears, A.E. An inquiry into the mechanism of herpes simplex virus latency. Annu. Rev. Microbiol. 1987, 41, 543–571. [Google Scholar] [CrossRef]
- Sainz, B.; Loutsch, J.M.; Marquart, M.E.; Hill, J.M. Stress-associated immunomodulation and herpes simplex virus infections. Med. Hypotheses 2001, 56, 348–356. [Google Scholar] [CrossRef]
- Bloom, D.C.; Giordani, N.V.; Kwiatkowski, D.L. Epigenetic regulation of latent HSV-1 gene expression. Biochim. Biophys. Acta 2010, 1799, 246–256. [Google Scholar] [CrossRef]
- Whitley, R.J. Herpes simplex virus infection. Semin. Pediatr. Infect. Dis. 2002, 13, 6–11. [Google Scholar] [CrossRef]
- Toma, H.S.; Murina, A.T.; Areaux, R.G., Jr.; Neumann, D.M.; Bhattacharjee, P.S.; Foster, T.P.; Kaufman, H.E.; Hill, J.M. Ocular HSV-1 latency, reactivation and recurrent disease. Semin. Ophthalmol. 2008, 23, 249–273. [Google Scholar] [CrossRef]
- Divito, S.; Cherpes, T.L.; Hendricks, R.L. A triple entente: Virus, neurons, and CD8+ T cells maintain HSV-1 latency. Immunol. Res. 2006, 36, 119–126. [Google Scholar] [CrossRef]
- Baringer, J.R. Herpes simplex virus infection of nervous tissue in animals and man. Prog. Med. Virol. 1975, 20, 1–26. [Google Scholar]
- Baringer, J.R.; Swoveland, P. Recovery of herpes-simplex virus from human trigeminal ganglions. N. Engl. J. Med. 1973, 288, 648–650. [Google Scholar] [CrossRef]
- Perng, G.C.; Jones, C. Towards an understanding of the herpes simplex virus type 1 latency reactivation cycle. Interdiscip. Perspect. Infect. Dis. 2010, 2010, 262415. [Google Scholar] [CrossRef]
- Izumi, K.M.; McKelvey, A.M.; Devi-Rao, G.B.; Wagner, E.K.; Stevens, J.G. Molecular and biological characterization of a type 1 herpes simplex virus (HSV-1) specifically deleted for expression of the latency-associated transcript (LAT). Microb. Pathog. 1989, 7, 121–134. [Google Scholar] [CrossRef]
- Javier, R.T.; Stevens, J.G.; Dissette, V.B.; Wagner, E.K. A herpes simplex virus transcript abundant in latently infected neurons is dispensable for establishment of the latent state. Virology 1988, 166, 254–257. [Google Scholar] [CrossRef]
- Cui, C.; Griffiths, A.; Li, G.; Silva, L.M.; Kramer, M.F.; Gaasterland, T.; Wang, X.J.; Coen, D.M. Prediction and identification of herpes simplex virus 1-encoded microRNAs. J. Virol. 2006, 80, 5499–5508. [Google Scholar] [CrossRef]
- Tang, S.; Bertke, A.S.; Patel, A.; Wang, K.; Cohen, J.I.; Krause, P.R. An acutely and latently expressed herpes simplex virus 2 viral microRNA inhibits expression of ICP34.5, a viral neurovirulence factor. Proc. Natl. Acad. Sci. USA 2008, 105, 10931–10936. [Google Scholar]
- Umbach, J.L.; Kramer, M.F.; Jurak, I.; Karnowski, H.W.; Coen, D.M.; Cullen, B.R. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature 2008, 454, 780–783. [Google Scholar]
- Umbach, J.L.; Nagel, M.; Cohrs, R.; Gilden, D.; Cullen, B.R. Analysis of human alphaherpesvirus microRNA expression in latently infected human trigeminal ganglia. J. Virol. 2009, 83, 10677–10683. [Google Scholar] [CrossRef]
- Knipe, D.M.; Lieberman, P.M.; Jung, J.U.; McBride, A.A.; Morris, K.V.; Ott, M.; Kristie, T.M. Snapshots: Chromatin control of viral infection. Virology 2013, 435, 141–156. [Google Scholar] [CrossRef]
- Knipe, D.M.; Cliffe, A. Chromatin control of herpes simplex virus lytic and latent infection. Nat. Rev. Microbiol. 2008, 6, 211–221. [Google Scholar] [CrossRef]
- Neumann, D.M.; Bhattacharjee, P.S.; Giordani, N.V.; Bloom, D.C.; Hill, J.M. In vivo changes in the patterns of chromatin structure associated with the latent herpes simplex virus type 1 genome in mouse trigeminal ganglia can be detected at early times after butyrate treatment. J. Virol. 2007, 81, 13248–13253. [Google Scholar] [CrossRef]
- Kwiatkowski, D.L.; Thompson, H.W.; Bloom, D.C. The polycomb group protein Bmi1 binds to the herpes simplex virus 1 latent genome and maintains repressive histone marks during latency. J. Virol. 2009, 83, 8173–8181. [Google Scholar] [CrossRef]
- Du, T.; Zhou, G.; Roizman, B. HSV-1 gene expression from reactivated ganglia is disordered and concurrent with suppression of latency-associated transcript and miRNAs. Proc. Natl. Acad. Sci. USA 2011, 108, 18820–18824. [Google Scholar]
- Zhou, G.; Du, T.; Roizman, B. HSV carrying wild-type REST establishes latency but reactivates only if the synthesis of REST is suppressed. Proc. Natl. Acad. Sci. USA 2013, 110, E498–E506. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zhou, G.; Du, T.; Roizman, B. The Role of the CoREST/REST Repressor Complex in Herpes Simplex Virus 1 Productive Infection and in Latency. Viruses 2013, 5, 1208-1218. https://doi.org/10.3390/v5051208
Zhou G, Du T, Roizman B. The Role of the CoREST/REST Repressor Complex in Herpes Simplex Virus 1 Productive Infection and in Latency. Viruses. 2013; 5(5):1208-1218. https://doi.org/10.3390/v5051208
Chicago/Turabian StyleZhou, Guoying, Te Du, and Bernard Roizman. 2013. "The Role of the CoREST/REST Repressor Complex in Herpes Simplex Virus 1 Productive Infection and in Latency" Viruses 5, no. 5: 1208-1218. https://doi.org/10.3390/v5051208