Potential Cellular Functions of Epstein-Barr Nuclear Antigen 1 (EBNA1) of Epstein-Barr Virus


Abstract
:1. Introduction
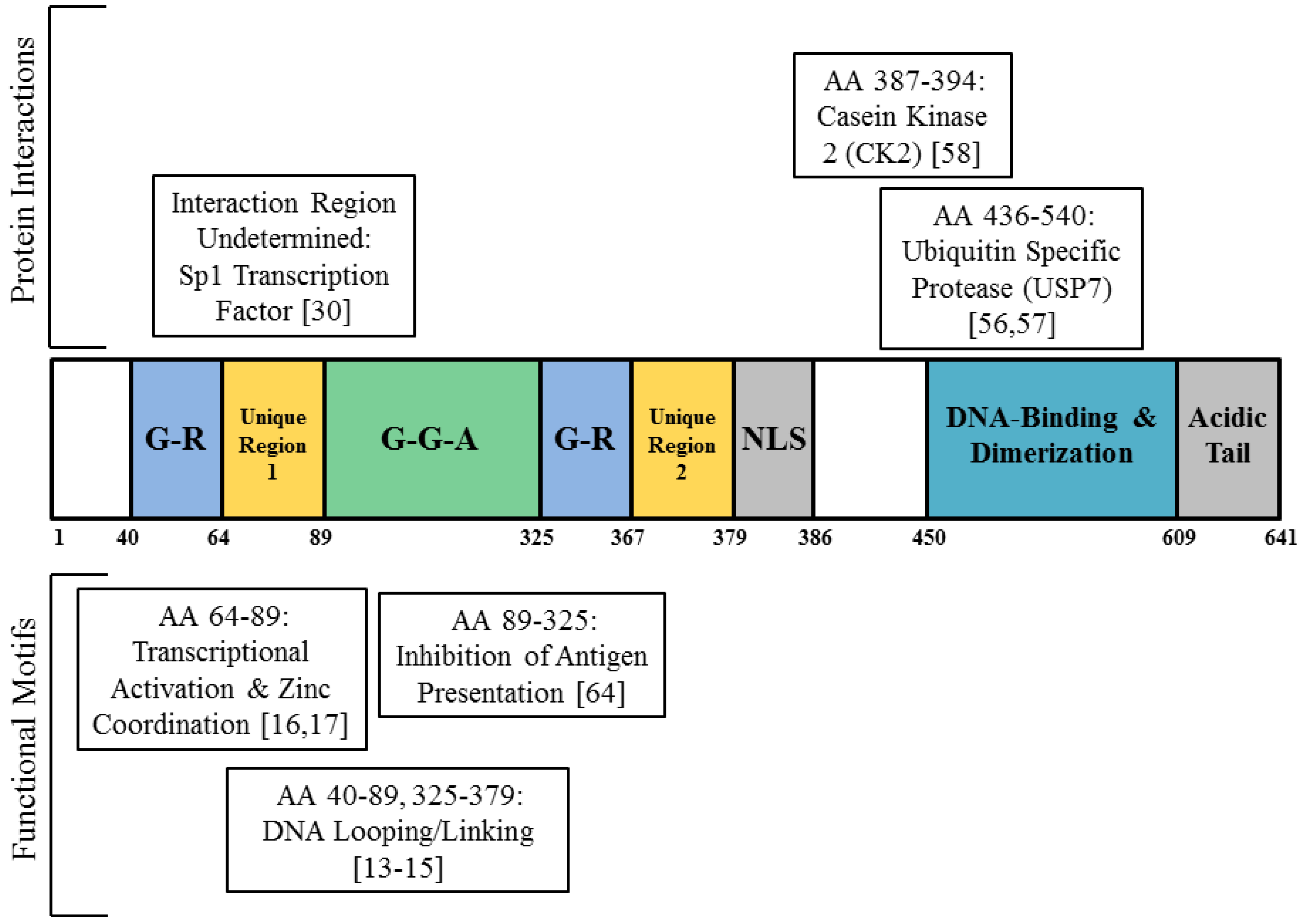
2. EBNA1 Binds the Cellular Genome

{kind=link}
| Gene | Binding sitePosition P-value | Distance fromTranscription Start | Reference |
|---|---|---|---|
| Nox 2 | 4.79E-05 | 16 kbp 5' | [32] |
| HDAC3 | - | None within 20 kbp | |
| MAP3K1 | 4.13E-05 | 13.5 kbp 5' | |
| PBX2 | - | None within 20 kbp | [24] |
| MYO1C-1* | 2.93E-06 | 6.9 kbp 3' | |
| MYO1C-2* | - | 4.9 kbp 5' | |
| MYO1C-3* | - | 5.9 kbp 3' | |
| c-Jun | - | None within 20 kbp | [25,29] |
| ATF2 | - | None within 20 kbp | |
| STAT 1 | 9.37E-06 | 11 kbp 3' | [33] |
| CCL18 | - | None within 20 kbp | |
| CCL3 | - | None within 20 kbp | [23] |
| CCL4 | - | None within 20 kbp | |
| TIAL1 | 9.10E-06 | None within 20 kbp |
3. EBNA1 as a Potential Regulator of Signaling Pathways
4. EBNA1 as an Oncogene
5. EBNA1 Inhibits its Own Antigen Presentation
6. Conclusions
Acknowledgments
Conflict of Interest
References
- Young, L.S.; Murray, P.G. Epstein-barr virus and oncogenesis: From latent genes to tumours. Oncogene 2003, 22, 5108–5121. [Google Scholar] [CrossRef]
- Ambinder, R.F.; Shah, W.A.; Rawlins, D.R.; Hayward, G.S.; Hayward, S.D. Definition of the sequence requirements for binding of the ebna-1 protein to its palindromic target sites in epstein-barr virus DNA. J. Virol. 1990, 64, 2369–2379. [Google Scholar]
- Frappier, L.; O'Donnell, M. Overproduction, purification, and characterization of ebna1, the origin binding protein of epstein-barr virus. J. Biol. Chem. 1991, 266, 7819–7826. [Google Scholar]
- Schepers, A.; Ritzi, M.; Bousset, K.; Kremmer, E.; Yates, J.L.; Harwood, J.; Diffley, J.F.; Hammerschmidt, W. Human origin recognition complex binds to the region of the latent origin of DNA replication of epstein-barr virus. EMBO J. 2001, 20, 4588–4602. [Google Scholar] [CrossRef]
- Sears, J.; Kolman, J.; Wahl, G.M.; Aiyar, A. Metaphase chromosome tethering is necessary for the DNA synthesis and maintenance of orip plasmids but is insufficient for transcription activation by epstein-barr nuclear antigen 1. J. Virol. 2003, 77, 11767–11780. [Google Scholar] [CrossRef]
- Nanbo, A.; Sugden, A.; Sugden, B. The coupling of synthesis and partitioning of ebv's plasmid replicon is revealed in live cells. EMBO J. 2007, 26, 4252–4262. [Google Scholar] [CrossRef]
- Sugden, B.; Warren, N. A promoter of epstein-barr virus that can function during latent infection can be transactivated by ebna-1, a viral protein required for viral DNA replication during latent infection. J. Virol. 1989, 63, 2644–2649. [Google Scholar]
- Altmann, M.; Pich, D.; Ruiss, R.; Wang, J.; Sugden, B.; Hammerschmidt, W. Transcriptional activation by ebv nuclear antigen 1 is essential for the expression of ebv's transforming genes. P. Natl. Acad. Sci. USA 2006, 103, 14188–14193. [Google Scholar]
- Gahn, T.A.; Sugden, B. An ebna-1-dependent enhancer acts from a distance of 10 kilobase pairs to increase expression of the epstein-barr virus lmp gene. J. Virol. 1995, 69, 2633–2636. [Google Scholar]
- Reisman, D.; Sugden, B. Trans activation of an epstein-barr viral transcriptional enhancer by the epstein-barr viral nuclear antigen 1. Mol. Cell. Biol. 1986, 6, 3838–3846. [Google Scholar]
- Sample, J.; Henson, E.B.; Sample, C. The epstein-barr virus nuclear protein 1 promoter active in type i latency is autoregulated. J. Virol. 1992, 66, 4654–4661. [Google Scholar]
- Sung, N.S.; Wilson, J.; Davenport, M.; Sista, N.D.; Pagano, J.S. Reciprocal regulation of the epstein-barr virus bamhi-f promoter by ebna-1 and an e2f transcription factor. Mol. Cell. Biol. 1994, 14, 7144–7152. [Google Scholar]
- Su, W.; Middleton, T.; Sugden, B.; Echols, H. DNA looping between the origin of replication of epstein-barr virus and its enhancer site: Stabilization of an origin complex with epstein-barr nuclear antigen 1. P. Natl. Acad. Sci. USA 1991, 88, 10870–10874. [Google Scholar] [CrossRef]
- Frappier, L.; O'Donnell, M. Epstein-barr nuclear antigen 1 mediates a DNA loop within the latent replication origin of epstein-barr virus. P. Natl. Acad. Sci. USA 1991, 88, 10875–10879. [Google Scholar] [CrossRef]
- Mackey, D.; Middleton, T.; Sugden, B. Multiple regions within ebna1 can link dnas. J. Virol. 1995, 69, 6199–6208. [Google Scholar]
- Mackey, D.; Sugden, B. The linking regions of ebna1 are essential for its support of replication and transcription. Mol. Cell. Biol. 1999, 19, 3349–3359. [Google Scholar]
- Aras, S.; Singh, G.; Johnston, K.; Foster, T.; Aiyar, A. Zinc coordination is required for and regulates transcription activation by epstein-barr nuclear antigen 1. PLoS Pathogens 2009, 5, e1000469. [Google Scholar] [CrossRef]
- Horner, D.; Lewis, M.; Farrell, P.J. Novel hypotheses for the roles of ebna-1 and bhrf1 in ebv-related cancers. Intervirology 1995, 38, 195–205. [Google Scholar]
- Jones, R.J.; Smith, L.J.; Dawson, C.W.; Haigh, T.; Blake, N.W.; Young, L.S. Epstein-barr virus nuclear antigen 1 (ebna1) induced cytotoxicity in epithelial cells is associated with ebna1 degradation and processing. Virology 2003, 313, 663–676. [Google Scholar] [CrossRef]
- Kennedy, G.; Komano, J.; Sugden, B. Epstein-barr virus provides a survival factor to burkitt's lymphomas. P. Natl. Acad. Sci. USA 2003, 100, 14269–14274. [Google Scholar] [CrossRef]
- Mack, A.A.; Sugden, B. Ebv is necessary for proliferation of dually infected primary effusion lymphoma cells. Cancer Res. 2008, 68, 6963–6968. [Google Scholar]
- Dresang, L.R.; Vereide, D.T.; Sugden, B. Identifying sites bound by epstein-barr virus nuclear antigen 1 (ebna1) in the human genome: Defining a position-weighted matrix to predict sites bound by ebna1 in viral genomes. J. Virol. 2009, 83, 2930–2940. [Google Scholar]
- Canaan, A.; Haviv, I.; Urban, A.E.; Schulz, V.P.; Hartman, S.; Zhang, Z.; Palejev, D.; Deisseroth, A.B.; Lacy, J.; Snyder, M.; et al. Ebna1 regulates cellular gene expression by binding cellular promoters. P. Natl. Acad. Sci. USA 2009, 106, 22421–22426. [Google Scholar]
- Lu, F.; Wikramasinghe, P.; Norseen, J.; Tsai, K.; Wang, P.; Showe, L.; Davuluri, R.V.; Lieberman, P.M. Genome-wide analysis of host-chromosome binding sites for epstein-barr virus nuclear antigen 1 (ebna1). J. Virol. 2010, 7, 262. [Google Scholar] [CrossRef]
- d'Herouel, A.F.; Birgersdotter, A.; Werner, M. Fr-like ebna1 binding repeats in the human genome. Virology 2010, 405, 524–529. [Google Scholar] [CrossRef]
- Bashaw, J.M.; Yates, J.L. Replication from orip of epstein-barr virus requires exact spacing of two bound dimers of ebna1 which bend DNA. J. Virol. 2001, 75, 10603–10611. [Google Scholar] [CrossRef]
- Wang, J.; Lindner, S.E.; Leight, E.R.; Sugden, B. Essential elements of a licensed, mammalian plasmid origin of DNA synthesis. Mol. Cell. Biol. 2006, 26, 1124–1134. [Google Scholar] [CrossRef]
- Kennedy, G.; Sugden, B. Ebna-1, a bifunctional transcriptional activator. Mol Cell. Biol. 2003, 23, 6901–6908. [Google Scholar] [CrossRef]
- O'Neil, J.D.; Owen, T.J.; Wood, V.H.; Date, K.L.; Valentine, R.; Chukwuma, M.B.; Arrand, J.R.; Dawson, C.W.; Young, L.S. Epstein-barr virus-encoded ebna1 modulates the ap-1 transcription factor pathway in nasopharyngeal carcinoma cells and enhances angiogenesis in vitro. J. Gen. Virol. 2008, 89, 2833–2842. [Google Scholar] [CrossRef]
- Lu, J.; Murakami, M.; Verma, S.C.; Cai, Q.; Haldar, S.; Kaul, R.; Wasik, M.A.; Middeldorp, J.; Robertson, E.S. Epstein-barr virus nuclear antigen 1 (ebna1) confers resistance to apoptosis in ebv-positive b-lymphoma cells through up-regulation of survivin. Virology 2011, 410, 64–75. [Google Scholar] [CrossRef]
- Bailey, T.L.; Gribskov, M. Combining evidence using p-values: Application to sequence homology searches. Bioinformatics 1998, 14, 48–54. [Google Scholar] [CrossRef]
- Gruhne, B.; Sompallae, R.; Marescotti, D.; Kamranvar, S.A.; Gastaldello, S.; Masucci, M.G. The epstein-barr virus nuclear antigen-1 promotes genomic instability via induction of reactive oxygen species. Proc. Natl. Acad. Sci. USA 2009, 106, 2313–2318. [Google Scholar]
- Wood, V.H.; O'Neil, J.D.; Wei, W.; Stewart, S.E.; Dawson, C.W.; Young, L.S. Epstein-barr virus-encoded ebna1 regulates cellular gene transcription and modulates the stat1 and tgfbeta signaling pathways. Oncogene 2007, 26, 4135–4147. [Google Scholar] [CrossRef]
- Niller, H.H.; Minarovits, J. Similarities between the epstein-barr virus (ebv) nuclear protein ebna1 and the pioneer transcription factor foxa: Is ebna1 a “bookmarking” oncoprotein that alters the host cell epigenotype? Pathogens 2012, 1, 37–51. [Google Scholar] [CrossRef]
- Holowaty, M.N.; Zeghouf, M.; Wu, H.; Tellam, J.; Athanasopoulos, V.; Greenblatt, J.; Frappier, L. Protein profiling with epstein-barr nuclear antigen-1 reveals an interaction with the herpesvirus-associated ubiquitin-specific protease hausp/usp7. J. Biol. Chem. 2003, 278, 29987–29994. [Google Scholar]
- Lin, A.; Wang, S.; Nguyen, T.; Shire, K.; Frappier, L. The ebna1 protein of epstein-barr virus functionally interacts with brd4. J. Virol. 2008, 82, 12009–12019. [Google Scholar]
- Wang, S.; Frappier, L. Nucleosome assembly proteins bind to epstein-barr virus nuclear antigen 1 and. J. Virol. 2009, 83, 11704–11714. [Google Scholar]
- Malik-Soni, N.; Frappier, L. Proteomic profiling of ebna1-host protein interactions in latent and lytic. J. Virol. 2012, 86, 6999–7002. [Google Scholar] [CrossRef]
- Vereide, D.; Seto, E.; Chiu, Y.-F.; Hayes, M.; Tagawa, T.; Grundhoff, A.; Hammerschmidt, W.; Sugden, B. Epstein-barr virus maintains lymphomas via its mirnas. Oncogene 2013, in press. [Google Scholar]
- Vereide, D.T.; Sugden, B. Lymphomas differ in their dependence on epstein-barr virus. Blood 2011, 117, 1977–1985. [Google Scholar] [CrossRef]
- Flavell, J.R.; Baumforth, K.R.; Wood, V.H.; Davies, G.L.; Wei, W.; Reynolds, G.M.; Morgan, S.; Boyce, A.; Kelly, G.L.; Young, L.S.; et al. Down-regulation of the tgf-beta target gene, ptprk, by the epstein-barr virus encoded ebna1 contributes to the growth and survival of hodgkin lymphoma cells. Blood 2008, 111, 292–301. [Google Scholar] [CrossRef]
- Valentine, R.; Dawson, C.W.; Hu, C.; Shah, K.M.; Owen, T.J.; Date, K.L.; Maia, S.P.; Shao, J.; Arrand, J.R.; Young, L.S.; et al. Epstein-barr virus-encoded ebna1 inhibits the canonical nf-kappab pathway in carcinoma cells by inhibiting ikk phosphorylation. Mol. Cancer 2010, 9, 1. [Google Scholar]
- Dirmeier, U.; Hoffmann, R.; Kilger, E.; Schultheiss, U.; Briseno, C.; Gires, O.; Kieser, A.; Eick, D.; Sugden, B.; Hammerschmidt, W. Latent membrane protein 1 of epstein-barr virus coordinately regulates proliferation with control of apoptosis. Oncogene 2005, 24, 1711–1717. [Google Scholar] [CrossRef]
- Wilson, J.B.; Bell, J.L.; Levine, A.J. Expression of epstein-barr virus nuclear antigen-1 induces b cell neoplasia in transgenic mice. EMBO J. 1996, 15, 3117–3126. [Google Scholar]
- Tsimbouri, P.; Drotar, M.E.; Coy, J.L.; Wilson, J.B. Bcl-xl and rag genes are induced and the response to il-2 enhanced in emuebna-1 transgenic mouse lymphocytes. Oncogene 2002, 21, 5182–5187. [Google Scholar] [CrossRef]
- Drotar, M.E.; Silva, S.; Barone, E.; Campbell, D.; Tsimbouri, P.; Jurvansu, J.; Bhatia, P.; Klein, G.; Wilson, J.B. Epstein-barr virus nuclear antigen-1 and myc cooperate in lymphomagenesis. Int. J. Cancer 2003, 106, 388–395. [Google Scholar] [CrossRef]
- Kang, M.S.; Lu, H.; Yasui, T.; Sharpe, A.; Warren, H.; Cahir-McFarland, E.; Bronson, R.; Hung, S.C.; Kieff, E. Epstein-barr virus nuclear antigen 1 does not induce lymphoma in transgenic fvb mice. P. Natl. Acad. Sci. USA 2005, 102, 820–825. [Google Scholar]
- Kang, M.S.; Soni, V.; Bronson, R.; Kieff, E. Epstein-barr virus nuclear antigen 1 does not cause lymphoma in c57bl/6j mice. J. Virol. 2008, 82, 4180–4183. [Google Scholar]
- Gruhne, B.; Sompallae, R.; Masucci, M.G. Three epstein-barr virus latency proteins independently promote genomic instability by inducing DNA damage, inhibiting DNA repair and inactivating cell cycle checkpoints. Oncogene 2009, 28, 3997–4008. [Google Scholar] [CrossRef]
- Cao, J.Y.; Mansouri, S.; Frappier, L. Changes in the nasopharyngeal carcinoma nuclear proteome induced by the ebna1 protein of epstein-barr virus reveal potential roles for ebna1 in metastasis and oxidative stress responses. J. Virol. 2012, 86, 382–394. [Google Scholar]
- Kamranvar, S.A.; Masucci, M.G. The epstein-barr virus nuclear antigen-1 promotes telomere dysfunction via induction of oxidative stress. Leukemia 2011, 25, 1017–1025. [Google Scholar] [CrossRef]
- Kamranvar, S.A.; Gruhne, B.; Szeles, A.; Masucci, M.G. Epstein-barr virus promotes genomic instability in burkitt's lymphoma. Oncogene 2007, 26, 5115–5123. [Google Scholar] [CrossRef]
- Lassoued, S.; Ben Ameur, R.; Ayadi, W.; Gargouri, B.; Ben Mansour, R.; Attia, H. Epstein-barr virus induces an oxidative stress during the early stages of infection in b lymphocytes, epithelial, and lymphoblastoid cell lines. Mol. Cell. Biochem. 2008, 313, 179–186. [Google Scholar] [CrossRef]
- Robbiani, D.F.; Bothmer, A.; Callen, E.; Reina-San-Martin, B.; Dorsett, Y.; Difilippantonio, S.; Bolland, D.J.; Chen, H.T.; Corcoran, A.E.; Nussenzweig, A.; et al. Aid is required for the chromosomal breaks in c-myc that lead to c-myc/igh translocations. Cell 2008, 135, 1028–1038. [Google Scholar] [CrossRef]
- Boerma, E.G.; Siebert, R.; Kluin, P.M.; Baudis, M. Translocations involving 8q24 in burkitt lymphoma and other malignant lymphomas: A historical review of cytogenetics in the light of todays knowledge. Leukemia 2009, 23, 225–234. [Google Scholar] [CrossRef] [Green Version]
- Nikitin, P.A.; Yan, C.M.; Forte, E.; Bocedi, A.; Tourigny, J.P.; White, R.E.; Allday, M.J.; Patel, A.; Dave, S.S.; Kim, W.; et al. An atm/chk2-mediated DNA damage-responsive signaling pathway suppresses epstein-barr virus transformation of primary human b cells. Cell Host Microbe 2010, 8, 510–522. [Google Scholar] [CrossRef]
- Lacoste, S.; Wiechec, E.; Dos Santos Silva, A.G.; Guffei, A.; Williams, G.; Lowbeer, M.; Benedek, K.; Henriksson, M.; Klein, G.; Mai, S. Chromosomal rearrangements after ex vivo epstein-barr virus (ebv) infection of human b cells. Oncogene 2010, 29, 503–515. [Google Scholar] [CrossRef]
- Allday, M.J. How does epstein-barr virus (ebv) complement the activation of myc in the pathogenesis of burkitt's lymphoma? Semin. Cancer Biol. 2009, 19, 366–376. [Google Scholar] [CrossRef]
- Altmann, M.; Hammerschmidt, W. Epstein-barr virus provides a new paradigm: A requirement for the immediate inhibition of apoptosis. PLoS Biol. 2005, 3, e404. [Google Scholar]
- Paschos, K.; Parker, G.A.; Watanatanasup, E.; White, R.E.; Allday, M.J. Bim promoter directly targeted by ebna3c in polycomb-mediated repression by ebv. Nucleic Acids Res. 2012, 40, 7233–7246. [Google Scholar] [CrossRef]
- Maruo, S.; Zhao, B.; Johannsen, E.; Kieff, E.; Zou, J.; Takada, K. Epstein-barr virus nuclear antigens 3c and 3a maintain lymphoblastoid cell growth by repressing p16ink4a and p14arf expression. P. Natl. Acad. Sci. USA 2011, 108, 1919–1924. [Google Scholar]
- Anderton, E.; Yee, J.; Smith, P.; Crook, T.; White, R.E.; Allday, M.J. Two epstein-barr virus (ebv) oncoproteins cooperate to repress expression of the proapoptotic tumour-suppressor bim: Clues to the pathogenesis of burkitt's lymphoma. Oncogene 2008, 27, 421–433. [Google Scholar] [CrossRef]
- Saridakis, V.; Sheng, Y.; Sarkari, F.; Holowaty, M.N.; Shire, K.; Nguyen, T.; Zhang, R.G.; Liao, J.; Lee, W.; Edwards, A.M.; et al. Structure of the p53 binding domain of hausp/usp7 bound to epstein-barr nuclear antigen 1 implications for ebv-mediated immortalization. Mol. Cell 2005, 18, 25–36. [Google Scholar] [CrossRef]
- Sivachandran, N.; Cao, J.Y.; Frappier, L. Epstein-barr virus nuclear antigen 1 hijacks the host kinase ck2 to disrupt pml nuclear bodies. J. Virol. 2010, 84, 11113–11123. [Google Scholar] [CrossRef]
- Sivachandran, N.; Sarkari, F.; Frappier, L. Epstein-barr nuclear antigen 1 contributes to nasopharyngeal carcinoma through disruption of pml nuclear bodies. PLoS Pathog. 2008, 4, e1000170. [Google Scholar] [CrossRef]
- Allday, M.J.; Sinclair, A.; Parker, G.; Crawford, D.H.; Farrell, P.J. Epstein-barr virus efficiently immortalizes human b cells without neutralizing the function of p53. EMBO J. 1995, 14, 1382–1391. [Google Scholar]
- O'Nions, J.; Turner, A.; Craig, R.; Allday, M.J. Epstein-barr virus selectively deregulates DNA damage responses in normal b cells but has no detectable effect on regulation of the tumor suppressor p53. J. Virol. 2006, 80, 12408–12413. [Google Scholar]
- Schmitz, R.; Young, R.M.; Ceribelli, M.; Jhavar, S.; Xiao, W.; Zhang, M.; Wright, G.; Shaffer, A.L.; Hodson, D.J.; Buras, E.; et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature 2012, 490, 116–120. [Google Scholar]
- Lindstrom, M.S.; Wiman, K.G. Role of genetic and epigenetic changes in burkitt lymphoma. Semin. Cancer Biol. 2002, 12, 381–387. [Google Scholar] [CrossRef]
- Levitskaya, J.; Coram, M.; Levitsky, V.; Imreh, S.; Steigerwald-Mullen, P.M.; Klein, G.; Kurilla, M.G.; Masucci, M.G. Inhibition of antigen processing by the internal repeat region of the epstein-barr virus nuclear antigen-1. Nature 1995, 375, 685–688. [Google Scholar]
- Leen, A.; Meij, P.; Redchenko, I.; Middeldorp, J.; Bloemena, E.; Rickinson, A.; Blake, N. Differential immunogenicity of epstein-barr virus latent-cycle proteins for human cd4(+) t-helper 1 responses. J. Virol. 2001, 75, 8649–8659. [Google Scholar] [CrossRef]
- Munz, C.; Bickham, K.L.; Subklewe, M.; Tsang, M.L.; Chahroudi, A.; Kurilla, M.G.; Zhang, D.; O'Donnell, M.; Steinman, R.M. Human cd4(+) t lymphocytes consistently respond to the latent epstein-barr virus nuclear antigen ebna1. J. Exp. Med. 2000, 191, 1649–1660. [Google Scholar] [CrossRef]
- Long, H.M.; Haigh, T.A.; Gudgeon, N.H.; Leen, A.M.; Tsang, C.W.; Brooks, J.; Landais, E.; Houssaint, E.; Lee, S.P.; Rickinson, A.B.; et al. Cd4+ t-cell responses to epstein-barr virus (ebv) latent-cycle antigens and the recognition of ebv-transformed lymphoblastoid cell lines. J. Virol. 2005, 79, 4896–4907. [Google Scholar]
- Leung, C.S.; Haigh, T.A.; Mackay, L.K.; Rickinson, A.B.; Taylor, G.S. Nuclear location of an endogenously expressed antigen, ebna1, restricts access to macroautophagy and the range of cd4 epitope display. P. Natl. Acad. Sci. USA 2010, 107, 2165–2170. [Google Scholar]
- Paludan, C.; Schmid, D.; Landthaler, M.; Vockerodt, M.; Kube, D.; Tuschl, T.; Munz, C. Endogenous mhc class ii processing of a viral nuclear antigen after autophagy. Science 2005, 307, 593–596. [Google Scholar] [CrossRef]
- Nimmerjahn, F.; Milosevic, S.; Behrends, U.; Jaffee, E.M.; Pardoll, D.M.; Bornkamm, G.W.; Mautner, J. Major histocompatibility complex class ii-restricted presentation of a cytosolic antigen by autophagy. Eur. J. Immunol. 2003, 33, 1250–1259. [Google Scholar]
- Mautner, J.; Pich, D.; Nimmerjahn, F.; Milosevic, S.; Adhikary, D.; Christoph, H.; Witter, K.; Bornkamm, G.W.; Hammerschmidt, W.; Behrends, U. Epstein-barr virus nuclear antigen 1 evades direct immune recognition by cd4+ t helper cells. Eur. J. Immunol. 2004, 34, 2500–2509. [Google Scholar]
- Paludan, C.; Bickham, K.; Nikiforow, S.; Tsang, M.L.; Goodman, K.; Hanekom, W.A.; Fonteneau, J.F.; Stevanovic, S.; Munz, C. Epstein-barr nuclear antigen 1-specific cd4(+) th1 cells kill burkitt's lymphoma cells. J. Immunol. 2002, 169, 1593–1603. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Smith, D.W.; Sugden, B. Potential Cellular Functions of Epstein-Barr Nuclear Antigen 1 (EBNA1) of Epstein-Barr Virus. Viruses 2013, 5, 226-240. https://doi.org/10.3390/v5010226
Smith DW, Sugden B. Potential Cellular Functions of Epstein-Barr Nuclear Antigen 1 (EBNA1) of Epstein-Barr Virus. Viruses. 2013; 5(1):226-240. https://doi.org/10.3390/v5010226
Chicago/Turabian StyleSmith, Danielle Westhoff, and Bill Sugden. 2013. "Potential Cellular Functions of Epstein-Barr Nuclear Antigen 1 (EBNA1) of Epstein-Barr Virus" Viruses 5, no. 1: 226-240. https://doi.org/10.3390/v5010226