Herpes Virus Fusion and Entry: A Story with Many Characters

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
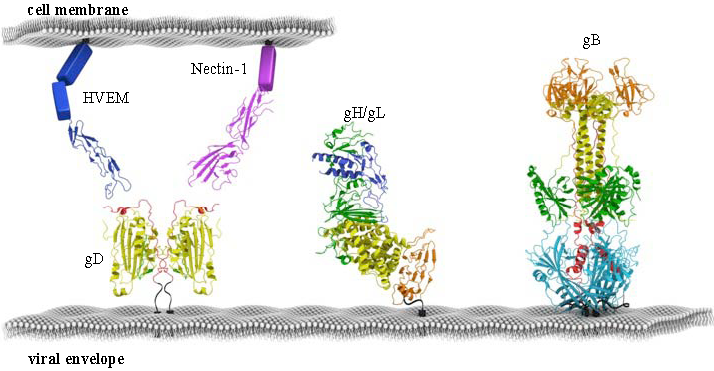
2. The Two Receptors of HSV and Their Roles in Entry and in vivo Tropism
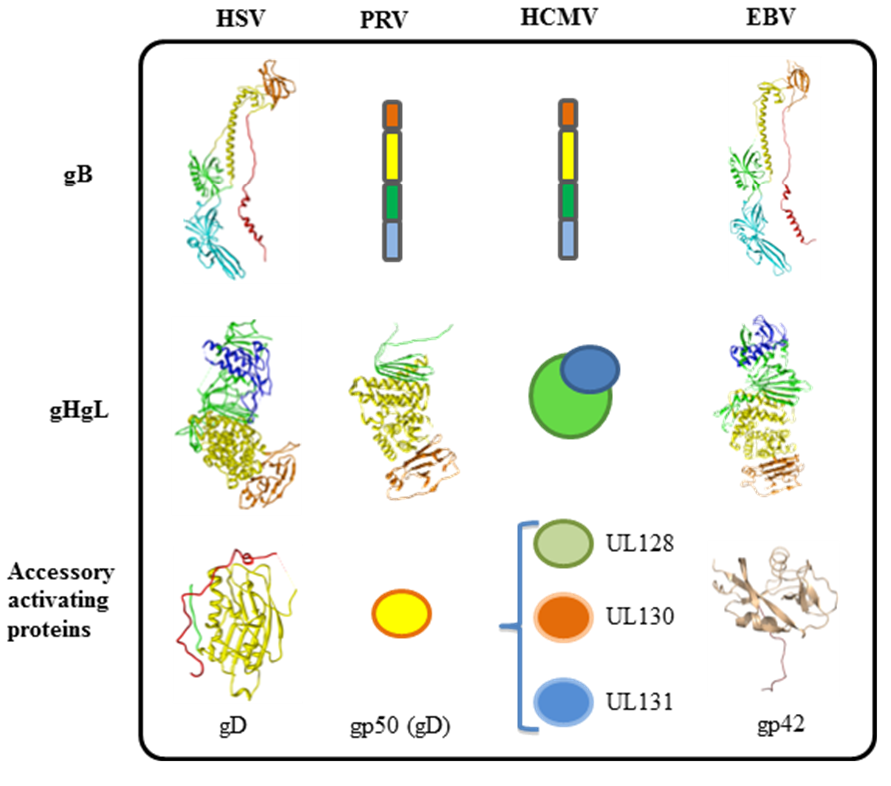
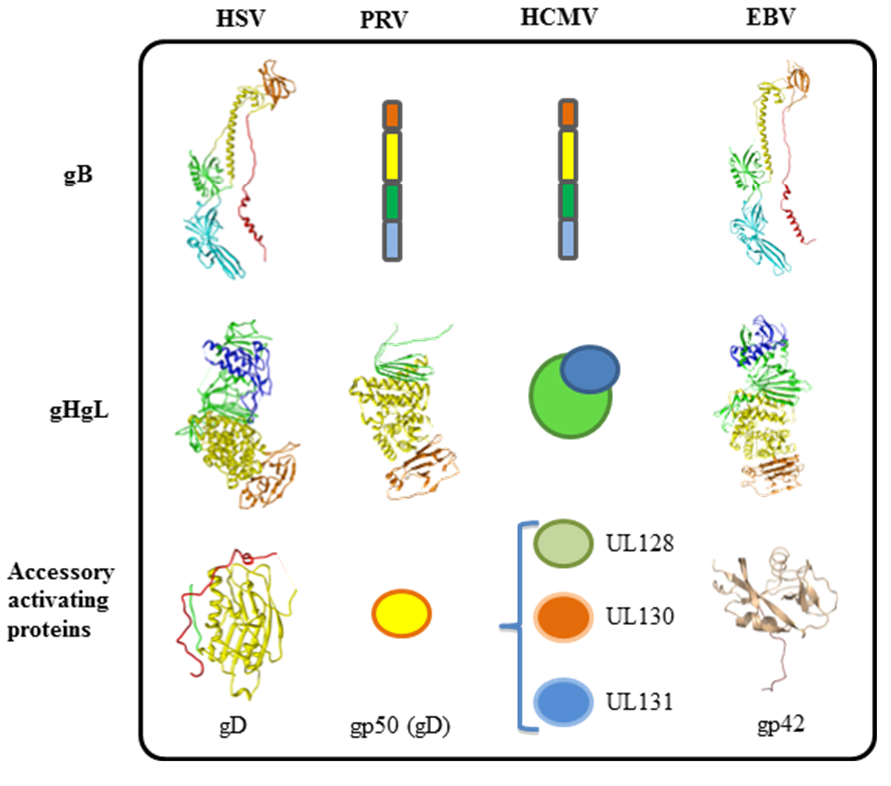
3. Tropism of Herpesviruses is Often Defined by Accessory Receptor Binding Proteins and Receptors
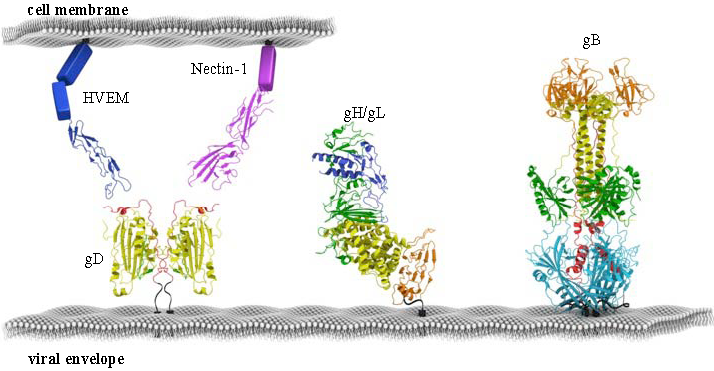
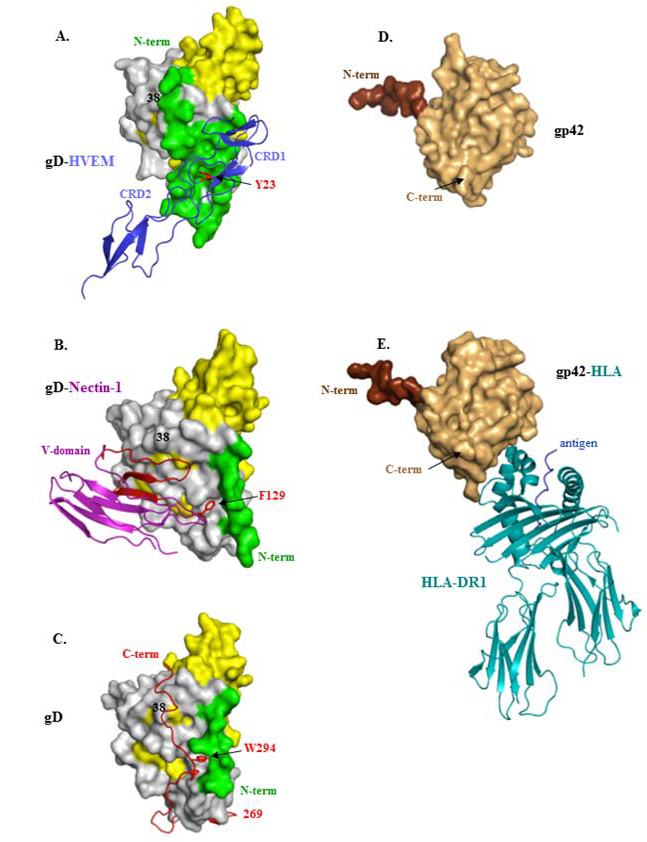
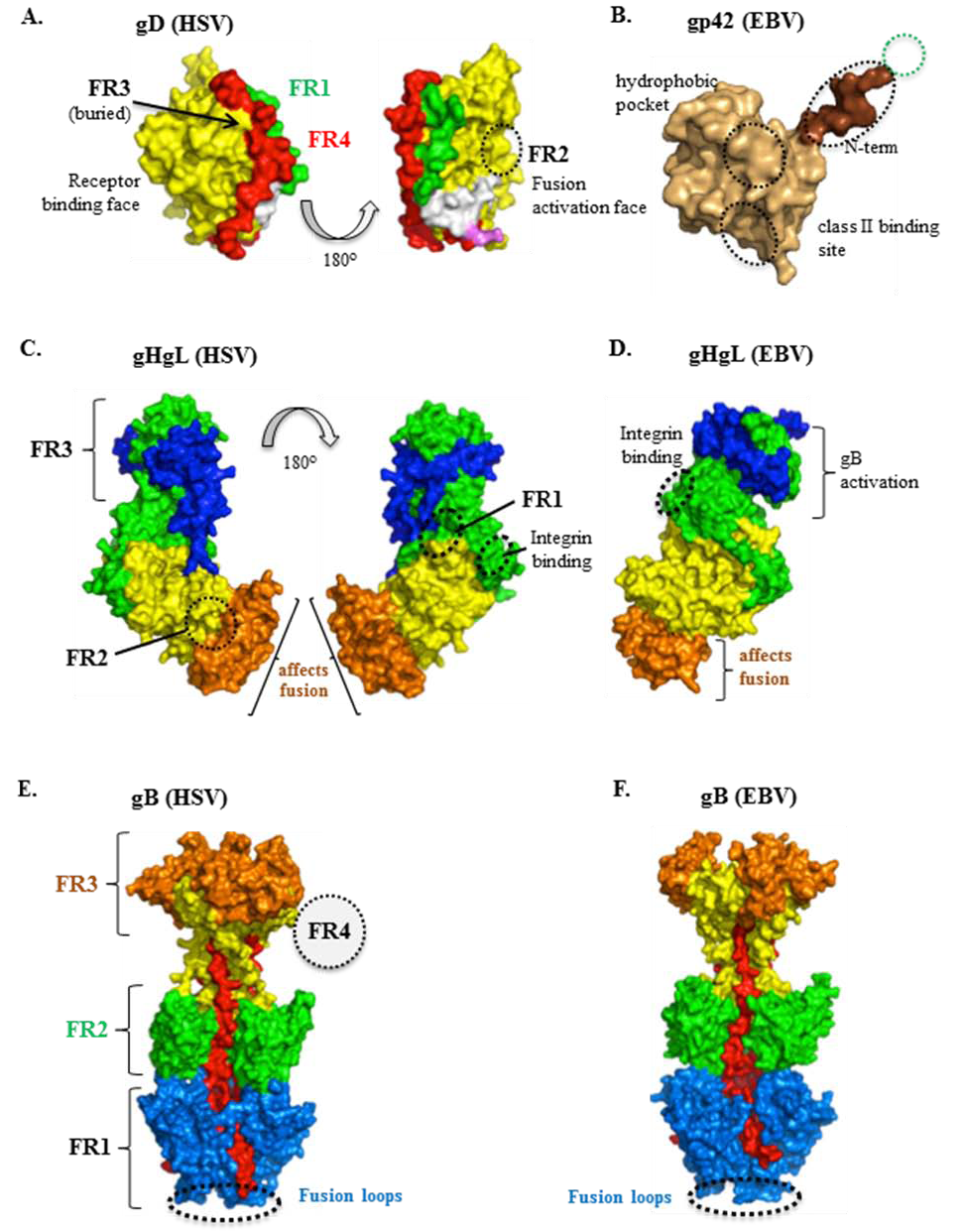
4. Structural Studies of HSV gD and Its Two Protein Receptors
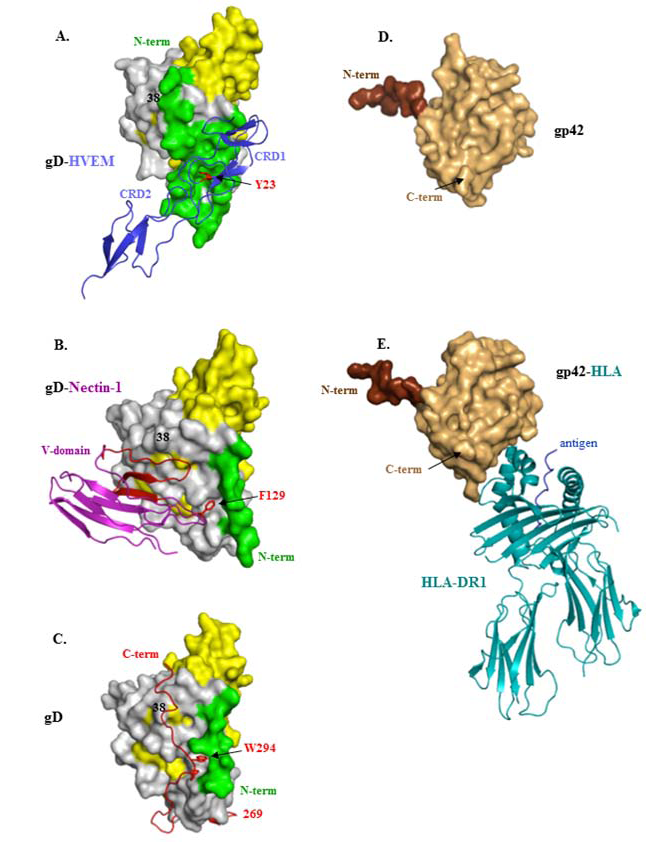
5. Virus Neutralizing Antibodies to gD Target Its Two Separate Functions
6. Structure-Function Analysis of gH/gL

7. The Structure of gB and Its Role in HSV Entry
8. Understanding the Conversion of Pre-Fusion Form of gB to a Post-Fusion State is Hampered by the Lack of a Pre-Fusion Structure
9. Functional Studies of gB with Mutants and Monoclonal Antibodies
10. Interactions Between gB and gH/gL
11. Fusion in trans Reveals Additional Information about gH/gL Function
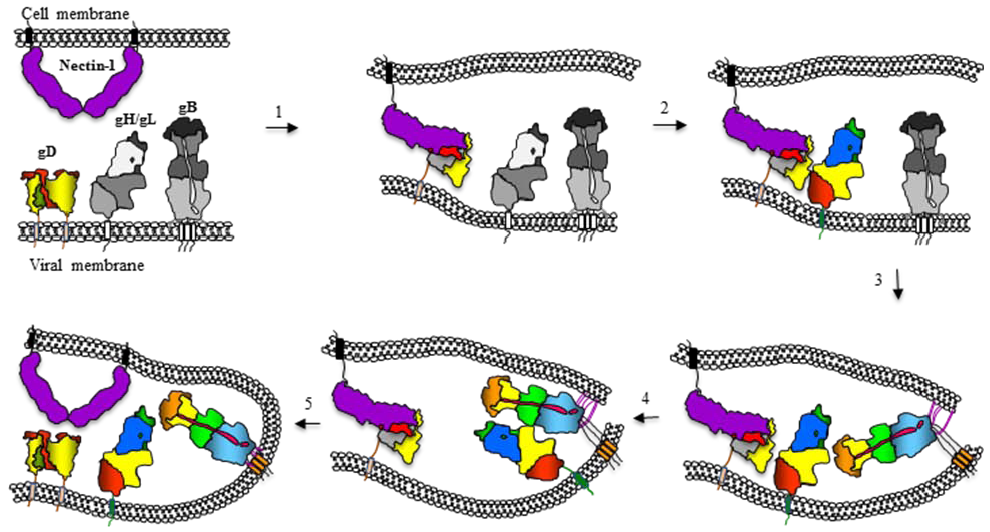
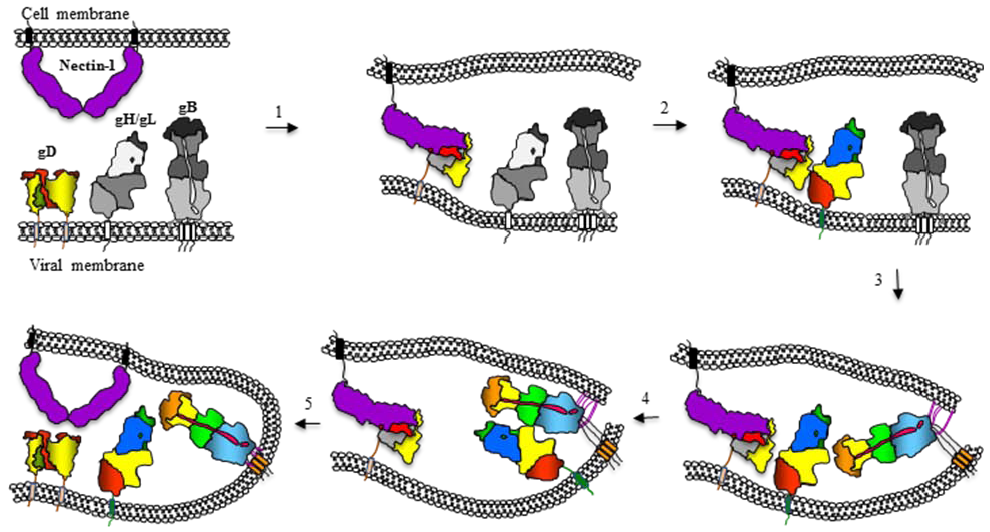
12. Model for Herpesvirus Fusion and How It Relates to Fusion by Other Viruses
Acknowledgments
Conflict of Interest
References
- Campadelli-Fiume, G.; Menotti, L. Entry of Alphaherpesviruses into the Cell. In Source Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007; Chapter 7. [Google Scholar]
- Campadelli-Fiume, G.; Menotti, L.; Avitabile, E.; Gianni, T. Viral and cellular contributions to herpes simplex virus entry into the cell. Curr. Opin. Virol. 2012, 2, 28–36. [Google Scholar]
- Navaratnarajah, C.K.; Miest, T.S.; Carfi, A.; Cattaneo, R. Targeted entry of enveloped viruses: Measles and herpes simplex virus I. Curr. Opin. Virol. 2012, 2, 43–49. [Google Scholar]
- Connolly, S.A.; Jackson, J.O.; Jardetzky, T.S.; Longnecker, R. Fusing structure and function: A structural view of the herpesvirus entry machinery. Nat. Rev. Microbiol. 2011, 9, 369–381. [Google Scholar]
- Hutt-Fletcher, L.M.; Chesnokova, L.S. Integrins as triggers of Epstein-Barr virus fusion and epithelial cell infection. Virulence 2010, 1, 395–398. [Google Scholar]
- Akhtar, J.; Shukla, D. Viral entry mechanisms: Cellular and viral mediators of herpes simplex virus entry. FEBS J. 2009, 276, 7228–7236. [Google Scholar]
- Browne, H.M. The role of glycoprotein H in herpesvirus membrane fusion. Protein Pept. Lett. 2009, 16, 760–765. [Google Scholar]
- Sinzger, C.; Digel, M.; Jahn, G. Cytomegalovirus cell tropism. Curr. Top. Microbiol. Immunol. 2008, 325, 63–83. [Google Scholar]
- Mori, Y. Recent topics related to human herpesvirus 6 cell tropism. Cell. Microbiol. 2009, 11, 1001–1006. [Google Scholar]
- Sinzger, C. Entry route of HCMV into endothelial cells. J. Clin. Virol. 2008, 41, 174–179. [Google Scholar]
- Ryckman, B.J.; Jarvis, M.A.; Drummond, D.D.; Nelson, J.A.; Johnson, D.C. Human cytomegalovirus entry into epithelial and endothelial cells depends on genes UL128 to UL150 and occurs by endocytosis and low-pH fusion. J. Virol. 2006, 80, 710–722. [Google Scholar]
- Spear, P.G.; Eisenberg, R.J.; Cohen, G.H. Three classes of cell surface receptors for alphaherpesvirus entry. Virology 2000, 275, 1–8. [Google Scholar]
- Li, Q.; Spriggs, M.K.; Kovats, S.; Turk, S.M.; Comeau, M.R.; Nepom, B.; Hutt-Fletcher, L.M. Epstein-Barr virus uses HLA class II as a cofactor for infection of B lymphocytes. J. Virol. 1997, 71, 4657–4662. [Google Scholar]
- Wang, X.; Hutt-Fletcher, L.M. Epstein-Barr virus lacking glycoprotein gp42 can bind to B cells but is not able to infect. J. Virol. 1998, 72, 158–163. [Google Scholar]
- Chesnokova, L.S.; Nishimura, S.L.; Hutt-Fletcher, L.M. Fusion of epithelial cells by Epstein-Barr virus proteins is triggered by binding of viral glycoproteins gHgL to integrins alphavbeta6 or alphavbeta8. Proc. Natl. Acad. Sci. USA 2009, 106, 20464–20469. [Google Scholar]
- Chen, J.; Rowe, C.L.; Jardetzky, T.S.; Longnecker, R. The KGD motif of Epstein-Barr virus gH/gL is bifunctional, orchestrating infection of B cells and epithelial cells. MBio 2012, 3. [Google Scholar] [CrossRef]
- Leoni, V.; Gianni, T.; Salvioli, S.; Campadelli-Fiume, G. Herpes simplex virus gH/gL and gB bind TLR2 and soluble gH/gL is sufficient to activate NF-kappaB. J. Virol. 2012. [Google Scholar] [CrossRef]
- Juckem, L.K.; Boehme, K.W.; Feire, A.L.; Compton, T. Differential initiation of innate immune responses induced by human cytomegalovirus entry into fibroblast cells. J. Immunol. 2008, 180, 4965–4977. [Google Scholar]
- Boehme, K.W.; Singh, J.; Perry, S.T.; Compton, T. Human cytomegalovirus elicits a coordinated cellular antiviral response via envelope glycoprotein B. J. Virol. 2004, 78, 1202–1211. [Google Scholar]
- Simmen, K.A.; Singh, J.; Luukkonen, B.G.; Lopper, M.; Bittner, A.; Miller, N.E.; Jackson, M.R.; Compton, T.; Fruh, K. Global modulation of cellular transcription by human cytomegalovirus is initiated by viral glycoprotein B. Proc. Natl. Acad. Sci. USA 2001, 98, 7140–7145. [Google Scholar]
- Krummenacher, C.; Baribaud, F.; Ponce de Leon, M.; Baribaud, I.; Whitbeck, J.C.; Xu, R.; Cohen, G.H.; Eisenberg, R.J. Comparative usage of herpesvirus entry mediator A and nectin-1 by laboratory strains and clinical isolates of herpes simplex virus. Virology 2004, 322, 286–299. [Google Scholar]
- Compaan, D.M.; Gonzalez, L.C.; Tom, I.; Loyet, K.M.; Eaton, D.; Hymowitz, S.G. Attenuating lymphocyte activity: The crystal structure of the BTLA-HVEM complex. J. Biol. Chem. 2005, 280, 39553–39561. [Google Scholar]
- Stiles, K.M.; Whitbeck, J.C.; Lou, H.; Cohen, G.H.; Eisenberg, R.J.; Krummenacher, C. Herpes simplex virus glycoprotein D interferes with binding of herpesvirus entry mediator to its ligands through downregulation and direct competition. J. Virol. 2010, 84, 11646–11660. [Google Scholar]
- Mizoguchi, A.; Nakanishi, H.; Kimura, K.; Matsubara, K.; Ozaki-Kuroda, K.; Katata, T.; Honda, T.; Kiyohara, Y.; Heo, K.; Higashi, M.; et al. Nectin: An adhesion molecule involved in formation of synapses. J. Cell Biol. 2002, 156, 555–565. [Google Scholar] [CrossRef]
- Galen, B.; Cheshenko, N.; Tuyama, A.; Ramratnam, B.; Herold, B.C. Access to nectin favors herpes simplex virus infection at the apical surface of polarized human epithelial cells. J. Virol. 2006, 80, 12209–12218. [Google Scholar]
- Richart, S.M.; Simpson, S.A.; Krummenacher, C.; Whitbeck, J.C.; Pizer, L.I.; Cohen, G.H.; Eisenberg, R.J.; Wilcox, C.L. Entry of herpes simplex virus type 1 into primary sensory neurons in vitro is mediated by Nectin-1/HveC. J. Virol. 2003, 77, 3307–3311. [Google Scholar]
- Simpson, S.A.; Manchak, M.D.; Hager, E.J.; Krummenacher, C.; Whitbeck, J.C.; Levin, M.J.; Freed, C.R.; Wilcox, C.L.; Cohen, G.H.; Eisenberg, R.J.; et al. Nectin-1/HveC Mediates herpes simplex virus type 1 entry into primary human sensory neurons and fibroblasts. J. Neurovirol. 2005, 11, 208–218. [Google Scholar] [CrossRef]
- Taylor, J.M.; Lin, E.; Susmarski, N.; Yoon, M.; Zago, A.; Ware, C.F.; Pfeffer, K.; Miyoshi, J.; Takai, Y.; Spear, P.G. Alternative entry receptors for herpes simplex virus and their roles in disease. Cell Host Microbe 2007, 2, 19–28. [Google Scholar]
- Kopp, S.J.; Banisadr, G.; Glajch, K.; Maurer, U.E.; Grunewald, K.; Miller, R.J.; Osten, P.; Spear, P.G. Infection of neurons and encephalitis after intracranial inoculation of herpes simplex virus requires the entry receptor nectin-1. Proc. Natl. Acad. Sci. USA 2009, 106, 17916–17920. [Google Scholar]
- Karaba, A.H.; Kopp, S.J.; Longnecker, R. Herpesvirus entry mediator and nectin-1 mediate herpes simplex virus 1 infection of the murine cornea. J. Virol. 2011, 85, 10041–10047. [Google Scholar]
- Geraghty, R.J.; Krummenacher, C.; Cohen, G.H.; Eisenberg, R.J.; Spear, P.G. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 1998, 280, 1618–1620. [Google Scholar]
- Milne, R.S.; Connolly, S.A.; Krummenacher, C.; Eisenberg, R.J.; Cohen, G.H. Porcine HveC, a member of the highly conserved HveC/nectin 1 family, is a functional alphaherpesvirus receptor. Virology 2001, 281, 315–328. [Google Scholar]
- Fan, Q.; Amen, M.; Harden, M.; Severini, A.; Griffiths, A.; Longnecker, R. Herpes B virus utilizes human nectin-1 but not HVEM or PILRalpha for cell-cell fusion and virus entry. J. Virol. 2012, 86, 4468–4476. [Google Scholar]
- Varthakavi, V.; Minocha, H.C. Identification of a 56 kDa putative bovine herpesvirus 1 cellular receptor by anti-idiotype antibodies. J. Gen. Virol. 1996, 77, 1875–1882. [Google Scholar]
- Cole, N.L.; Grose, C. Membrane fusion mediated by herpesvirus glycoproteins: The paradigm of varicella-zoster virus. Rev. Med. Virol. 2003, 13, 207–222. [Google Scholar]
- Li, Q.; Ali, M.A.; Wang, K.; Sayre, D.; Hamel, F.G.; Fischer, E.R.; Bennett, R.G.; Cohen, J.I. Insulin degrading enzyme induces a conformational change in varicella-zoster virus gE, and enhances virus infectivity and stability. PLoS One 2010, 5, e11327. [Google Scholar]
- Li, Q.; Ali, M.A.; Cohen, J.I. Insulin degrading enzyme is a cellular receptor mediating varicella-zoster virus infection and cell-to-cell spread. Cell 2006, 127, 305–316. [Google Scholar]
- Zerboni, L.; Berarducci, B.; Rajamani, J.; Jones, C.D.; Zehnder, J.L.; Arvin, A. Varicella-zoster virus glycoprotein E is a critical determinant of virulence in the SCID mouse-human model of neuropathogenesis. J. Virol. 2011, 85, 98–111. [Google Scholar]
- Cha, T.A.; Tom, E.; Kemble, G.W.; Duke, G.M.; Mocarski, E.S.; Spaete, R.R. Human cytomegalovirus clinical isolates carry at least 19 genes not found in laboratory strains. J. Virol. 1996, 70, 78–83. [Google Scholar]
- Ryckman, B.J.; Rainish, B.L.; Chase, M.C.; Borton, J.A.; Nelson, J.A.; Jarvis, M.A.; Johnson, D.C. Characterization of the human cytomegalovirus gH/gL/UL128–131 complex that mediates entry into epithelial and endothelial cells. J. Virol. 2008, 82, 60–70. [Google Scholar]
- Ryckman, B.J.; Chase, M.C.; Johnson, D.C. HCMV gH/gL/UL128–131 interferes with virus entry into epithelial cells: Evidence for cell type-specific receptors. Proc. Natl. Acad. Sci. USA 2008, 105, 14118–14123. [Google Scholar]
- Ryckman, B.J.; Chase, M.C.; Johnson, D.C. Human cytomegalovirus TR strain glycoprotein O acts as a chaperone promoting gH/gL incorporation into virions but is not present in virions. J. Virol. 2010, 84, 2597–2609. [Google Scholar]
- Wille, P.T.; Knoche, A.J.; Nelson, J.A.; Jarvis, M.A.; Johnson, D.C. A human cytomegalovirus gO-null mutant fails to incorporate gH/gL into the virion envelope and is unable to enter fibroblasts and epithelial and endothelial cells. J. Virol. 2010, 84, 2585–2596. [Google Scholar]
- Vanarsdall, A.L.; Ryckman, B.J.; Chase, M.C.; Johnson, D.C. Human cytomegalovirus glycoproteins gB and gH/gL mediate epithelial cell-cell fusion when expressed either in cis or in trans. J. Virol. 2008, 82, 11837–11850. [Google Scholar]
- York, I.A.; Roop, C.; Andrews, D.W.; Riddell, S.R.; Graham, F.L.; Johnson, D.C. A cytosolic herpes simplex virus protein inhibits antigen presentation to CD8+ T lymphocytes. Cell 1994, 77, 525–535. [Google Scholar]
- Jugovic, P.; Hill, A.M.; Tomazin, R.; Ploegh, H.; Johnson, D.C. Inhibition of major histocompatibility complex class I antigen presentation in pig and primate cells by herpes simplex virus type 1 and 2 ICP47. J. Virol. 1998, 72, 5076–5084. [Google Scholar]
- Tomazin, R.; van Schoot, N.E.; Goldsmith, K.; Jugovic, P.; Sempe, P.; Fruh, K.; Johnson, D.C. Herpes simplex virus type 2 ICP47 inhibits human TAP but not mouse TAP. J. Virol. 1998, 72, 2560–2563. [Google Scholar]
- Vanarsdall, A.L.; Chase, M.C.; Johnson, D.C. Human cytomegalovirus glycoprotein gO complexes with gH/gL, promoting interference with viral entry into human fibroblasts but not entry into epithelial cells. J. Virol. 2011, 85, 11638–11645. [Google Scholar]
- Bentz, G.L.; Yurochko, A.D. Human CMV infection of endothelial cells induces an angiogenic response through viral binding to EGF receptor and beta1 and beta3 integrins. Proc. Natl. Acad. Sci. USA 2008, 105, 5531–5536. [Google Scholar]
- Tang, H.; Hayashi, M.; Maeki, T.; Yamanishi, K.; Mori, Y. Human herpesvirus 6 glycoprotein complex formation is required for folding and trafficking of the gH/gL/gQ1/gQ2 complex and its cellular receptor binding. J. Virol. 2011, 85, 11121–11130. [Google Scholar]
- Tang, H.; Kawabata, A.; Yoshida, M.; Oyaizu, H.; Maeki, T.; Yamanishi, K.; Mori, Y. Human herpesvirus 6 encoded glycoprotein Q1 gene is essential for virus growth. Virology 2010, 407, 360–367. [Google Scholar]
- Turk, S.M.; Hutt-Fletcher, L.M. The Epstein-Barr virus-associated protein p105 is not encoded by the Epstein-Barr virus genome. Virology 1994, 200, 313–318. [Google Scholar]
- Turk, S.M.; Jiang, R.; Chesnokova, L.S.; Hutt-Fletcher, L.M. Antibodies to gp350/220 enhance the ability of Epstein-Barr virus to infect epithelial cells. J. Virol. 2006, 80, 9628–9633. [Google Scholar]
- Chesnokova, L.S.; Hutt-Fletcher, L.M. Fusion of Epstein-Barr virus with epithelial cells can be triggered by alphavbeta5 in addition to alphavbeta6 and alphavbeta8, and integrin binding triggers a conformational change in glycoproteins gHgL. J. Virol. 2011, 85, 13214–13223. [Google Scholar]
- Borza, C.M.; Morgan, A.J.; Turk, S.M.; Hutt-Fletcher, L.M. Use of gHgL for attachment of Epstein-Barr virus to epithelial cells compromises infection. J. Virol. 2004, 78, 5007–5014. [Google Scholar]
- Borza, C.M.; Hutt-Fletcher, L.M. Alternate replication in B cells and epithelial cells switches tropism of Epstein-Barr virus. Nat. Med. 2002, 8, 594–599. [Google Scholar]
- Mullen, M.M.; Haan, K.M.; Longnecker, R.; Jardetzky, T.S. Structure of the Epstein-Barr virus gp42 protein bound to the MHC class II receptor HLA-DR1. Mol. Cell 2002, 9, 375–385. [Google Scholar]
- Kirschner, A.N.; Sorem, J.; Longnecker, R.; Jardetzky, T.S. Structure of Epstein-Barr virus glycoprotein 42 suggests a mechanism for triggering receptor-activated virus entry. Structure 2009, 17, 223–233. [Google Scholar]
- Sorem, J.; Jardetzky, T.S.; Longnecker, R. Cleavage and secretion of Epstein-Barr virus glycoprotein 42 promote membrane fusion with B lymphocytes. J. Virol. 2009, 83, 6664–6672. [Google Scholar]
- Kirschner, A.N.; Omerovic, J.; Popov, B.; Longnecker, R.; Jardetzky, T.S. Soluble Epstein-Barr virus glycoproteins gH, gL, and gp42 form a 1:1:1 stable complex that acts like soluble gp42 in B-cell fusion but not in epithelial cell fusion. J. Virol. 2006, 80, 9444–9454. [Google Scholar]
- Shaw, P.L.; Kirschner, A.N.; Jardetzky, T.S.; Longnecker, R. Characteristics of Epstein-Barr virus envelope protein gp42. Virus Genes 2010, 40, 307–319. [Google Scholar]
- Carfi, A.; Willis, S.H.; Whitbeck, J.C.; Krummenacher, C.; Cohen, G.H.; Eisenberg, R.J.; Wiley, D.C. Herpes simplex virus glycoprotein D bound to the human receptor HveA. Mol. Cell 2001, 8, 169–179. [Google Scholar]
- Krummenacher, C.; Supekar, V.M.; Whitbeck, J.C.; Lazear, E.; Connolly, S.A.; Eisenberg, R.J.; Cohen, G.H.; Wiley, D.C.; Carfi, A. Structure of unliganded HSV gD reveals a mechanism for receptor-mediated activation of virus entry. EMBO J. 2005, 24, 4144–4153. [Google Scholar]
- Di Giovine, P.; Settembre, E.C.; Bhargava, A.K.; Luftig, M.A.; Lou, H.; Cohen, G.H.; Eisenberg, R.J.; Krummenacher, C.; Carfi, A. Structure of herpes simplex virus glycoprotein D bound to the human receptor nectin-1. PLoS Pathog. 2011, 7, e1002277. [Google Scholar]
- Zhang, N.; Yan, J.; Lu, G.; Guo, Z.; Fan, Z.; Wang, J.; Shi, Y.; Qi, J.; Gao, G.F. Binding of herpes simplex virus glycoprotein D to nectin-1 exploits host cell adhesion. Nat. Commun. 2011, 2, 577. [Google Scholar]
- Rux, A.H.; Willis, S.H.; Nicola, A.V.; Hou, W.; Peng, C.; Lou, H.; Cohen, G.H.; Eisenberg, R.J. Functional region IV of glycoprotein D from herpes simplex virus modulates glycoprotein binding to the herpesvirus entry mediator. J. Virol. 1998, 72, 7091–7098. [Google Scholar]
- Willis, S.H.; Rux, A.H.; Peng, C.; Whitbeck, J.C.; Nicola, A.V.; Lou, H.; Hou, W.; Salvador, L.; Eisenberg, R.J.; Cohen, G.H. Examination of the kinetics of herpes simplex virus glycoprotein D binding to the herpesvirus entry mediator, using surface plasmon resonance. J. Virol. 1998, 72, 5937–5947. [Google Scholar]
- Krummenacher, C.; Rux, A.H.; Whitbeck, J.C.; Ponce-de-Leon, M.; Lou, H.; Baribaud, I.; Hou, W.; Zou, C.; Geraghty, R.J.; Spear, P.G.; et al. The first immunoglobulin-like domain of HveC is sufficient to bind herpes simplex virus gD with full affinity, while the third domain is involved in oligomerization of HveC. J. Virol. 1999, 73, 8127–8137. [Google Scholar]
- Connolly, S.A.; Landsburg, D.J.; Carfi, A.; Wiley, D.C.; Eisenberg, R.J.; Cohen, G.H. Structure-based analysis of the herpes simplex virus glycoprotein D binding site present on herpesvirus entry mediator HveA (HVEM). J. Virol. 2002, 76, 10894–10904. [Google Scholar]
- Connolly, S.A.; Landsburg, D.J.; Carfi, A.; Wiley, D.C.; Cohen, G.H.; Eisenberg, R.J. Structure-based mutagenesis of herpes simplex virus glycoprotein D defines three critical regions at the gD-HveA/HVEM binding interface. J. Virol. 2003, 77, 8127–8140. [Google Scholar] [CrossRef]
- Yoon, M.; Zago, A.; Shukla, D.; Spear, P.G. Mutations in the N termini of herpes simplex virus type 1 and 2 gDs alter functional interactions with the entry/fusion receptors HVEM, nectin-2, and 3-O-sulfated heparan sulfate but not with nectin-1. J. Virol. 2003, 77, 9221–9231. [Google Scholar]
- Cocchi, F.; Menotti, L.; Mirandola, P.; Lopez, M.; Campadelli-Fiume, G. The ectodomain of a novel member of the immunoglobulin subfamily related to the poliovirus receptor has the attributes of a bona fide receptor for herpes simplex virus types 1 and 2 in human cells. J. Virol. 1998, 72, 9992–10002. [Google Scholar]
- Krummenacher, C.; Baribaud, I.; Ponce de Leon, M.; Whitbeck, J.C.; Lou, H.; Cohen, G.H.; Eisenberg, R.J. Localization of a binding site for herpes simplex virus glycoprotein D on herpesvirus entry mediator C by using antireceptor monoclonal antibodies. J. Virol. 2000, 74, 10863–10872. [Google Scholar]
- Struyf, F.; Plate, A.E.; Spear, P.G. Deletion of the second immunoglobulin-like domain of nectin-1 alters its intracellular processing and localization and ability to mediate entry of herpes simplex virus. J. Virol. 2005, 79, 3841–3845. [Google Scholar]
- Whitbeck, J.C.; Muggeridge, M.I.; Rux, A.; Hou, W.; Krummenacher, C.; Lou, H.; van Geelen, A.; Eisenberg, R.J.; Cohen, G.H. The major neutralizing antigenic site on herpes simplex virus glycoprotein D overlaps a receptor-binding domain. J. Virol. 1999, 73, 9879–9890. [Google Scholar]
- Manoj, S.; Jogger, C.R.; Myscofski, D.; Yoon, M.; Spear, P.G. Mutations in herpes simplex virus glycoprotein D that prevent cell entry via nectins and alter cell tropism. Proc. Natl. Acad. Sci. USA 2004, 101, 12414–12421. [Google Scholar]
- Connolly, S.A.; Landsburg, D.J.; Carfi, A.; Whitbeck, J.C.; Zuo, Y.; Wiley, D.C.; Cohen, G.H.; Eisenberg, R.J. Potential nectin-1 binding site on herpes simplex virus glycoprotein d. J. Virol. 2005, 79, 1282–1295. [Google Scholar]
- Spear, P.G.; Manoj, S.; Yoon, M.; Jogger, C.R.; Zago, A.; Myscofski, D. Different receptors binding to distinct interfaces on herpes simplex virus gD can trigger events leading to cell fusion and viral entry. Virology 2006, 344, 17–24. [Google Scholar]
- Uchida, H.; Shah, W.A.; Ozuer, A.; Frampton, A.R., Jr.; Goins, W.F.; Grandi, P.; Cohen, J.B.; Glorioso, J.C. Generation of herpesvirus entry mediator (HVEM)-restricted herpes simplex virus type 1 mutant viruses: Resistance of HVEM-expressing cells and identification of mutations that rescue nectin-1 recognition. J. Virol. 2009, 83, 2951–2961. [Google Scholar]
- Zago, A.; Spear, P.G. Differences in the N termini of herpes simplex virus type 1 and 2 gDs that influence functional interactions with the human entry receptor Nectin-2 and an entry receptor expressed in Chinese hamster ovary cells. J. Virol. 2003, 77, 9695–9699. [Google Scholar]
- Jogger, C.R.; Montgomery, R.I.; Spear, P.G. Effects of linker-insertion mutations in herpes simplex virus 1 gD on glycoprotein-induced fusion with cells expressing HVEM or nectin-1. Virology 2004, 318, 318–326. [Google Scholar]
- Zago, A.; Jogger, C.R.; Spear, P.G. Use of herpes simplex virus and pseudorabies virus chimeric glycoprotein D molecules to identify regions critical for membrane fusion. Proc. Natl. Acad. Sci. USA 2004, 101, 17498–17503. [Google Scholar]
- Narita, H.; Yamamoto, Y.; Suzuki, M.; Miyazaki, N.; Yoshida, A.; Kawai, K.; Iwasaki, K.; Nakagawa, A.; Takai, Y.; Sakisaka, T. Crystal Structure of the cis-Dimer of Nectin-1: Implications for the architecture of cell-cell junctions. J. Biol. Chem. 2011, 286, 12659–12669. [Google Scholar]
- Cocchi, F.; Fusco, D.; Menotti, L.; Gianni, T.; Eisenberg, R.J.; Cohen, G.H.; Campadelli-Fiume, G. The soluble ectodomain of herpes simplex virus gD contains a membrane-proximal pro-fusion domain and suffices to mediate virus entry. Proc. Natl. Acad. Sci. USA 2004, 101, 7445–7450. [Google Scholar]
- Heldwein, E.E.; Krummenacher, C. Entry of herpesviruses into mammalian cells. Cell. Mol. Life Sci. 2008, 65, 1653–1668. [Google Scholar]
- Lazear, E.; Carfi, A.; Whitbeck, J.C.; Cairns, T.M.; Krummenacher, C.; Cohen, G.H.; Eisenberg, R.J. Engineered disulfide bonds in herpes simplex virus type 1 gD separate receptor binding from fusion initiation and viral entry. J. Virol. 2008, 82, 700–709. [Google Scholar]
- Struyf, F.; Martinez, W.M.; Spear, P.G. Mutations in the N-terminal domains of nectin-1 and nectin-2 reveal differences in requirements for entry of various alphaherpesviruses and for nectin-nectin interactions. J. Virol. 2002, 76, 12940–12950. [Google Scholar]
- Bogan, A.A.; Thorn, K.S. Anatomy of hot spots in protein interfaces. J. Mol. Biol. 1998, 280, 1–9. [Google Scholar]
- Minson, A.C.; Hodgman, T.C.; Digard, P.; Hancock, D.C.; Bell, S.E.; Buckmaster, E.A. An analysis of the biological properties of monoclonal antibodies against glycoprotein D of herpes simplex virus and identification of amino acid substitutions that confer resistance to neutralization. J. Gen. Virol. 1986, 67, 1001–1013. [Google Scholar]
- Chiang, H.Y.; Cohen, G.H.; Eisenberg, R.J. Identification of functional regions of herpes simplex virus glycoprotein gD by using linker-insertion mutagenesis. J. Virol. 1994, 68, 2529–2543. [Google Scholar]
- Muggeridge, M.I.; Wu, T.T.; Johnson, D.C.; Glorioso, J.C.; Eisenberg, R.J.; Cohen, G.H. Antigenic and functional analysis of a neutralization site of HSV-1 glycoprotein D. Virology 1990, 174, 375–387. [Google Scholar]
- Dix, R.D.; Pereira, L.; Baringer, J.R. Use of monoclonal antibody directed against herpes simplex virus glycoproteins to protect mice against acute virus-induced neurological disease. Infect. Immun. 1981, 34, 192–199. [Google Scholar]
- Krummenacher, C.; Nicola, A.V.; Whitbeck, J.C.; Lou, H.; Hou, W.; Lambris, J.D.; Geraghty, R.J.; Spear, P.G.; Cohen, G.H.; Eisenberg, R.J. Herpes simplex virus glycoprotein D can bind to poliovirus receptor-related protein 1 or herpesvirus entry mediator, two structurally unrelated mediators of virus entry. J. Virol. 1998, 72, 7064–7074. [Google Scholar]
- Nicola, A.V.; Ponce de Leon, M.; Xu, R.; Hou, W.; Whitbeck, J.C.; Krummenacher, C.; Montgomery, R.I.; Spear, P.G.; Eisenberg, R.J.; Cohen, G.H. Monoclonal antibodies to distinct sites on herpes simplex virus (HSV) glycoprotein D block HSV binding to HVEM. J. Virol. 1998, 72, 3595–3601. [Google Scholar]
- Lazear, E.; Whitbeck, J.C.; Ponce-de-Leon, M.; Cairns, T.M.; Willis, S.H.; Zuo, Y.; Krummenacher, C.; Cohen, G.H.; Eisenberg, R.J. Antibody-induced conformational changes in herpes simplex virus glycoprotein gD reveal new targets for virus neutralization. J. Virol. 2012, 86, 1563–1576. [Google Scholar]
- Atanasiu, D.; Whitbeck, J.C.; Cairns, T.M.; Reilly, B.; Cohen, G.H.; Eisenberg, R.J. Bimolecular complementation reveals that glycoproteins gB and gH/gL of herpes simplex virus interact with each other during cell fusion. Proc. Natl. Acad. Sci. USA 2007, 104, 18718–18723. [Google Scholar]
- Gianni, T.; Amasio, M.; Campadelli-Fiume, G. Herpes simplex virus gD forms distinct complexes with fusion executors gB and gH/gL in part through the C-terminal profusion domain. J. Biol. Chem. 2009, 284, 17370–17382. [Google Scholar]
- Peng, T.; Ponce-de-Leon, M.; Jiang, H.; Dubin, G.; Lubinski, J.M.; Eisenberg, R.J.; Cohen, G.H. The gH-gL complex of herpes simplex virus (HSV) stimulates neutralizing antibody and protects mice against HSV type 1 challenge. J. Virol. 1998, 72, 65–72. [Google Scholar]
- Macagno, A.; Bernasconi, N.L.; Vanzetta, F.; Dander, E.; Sarasini, A.; Revello, M.G.; Gerna, G.; Sallusto, F.; Lanzavecchia, A. Isolation of human monoclonal antibodies that potently neutralize human cytomegalovirus infection by targeting different epitopes on the gH/gL/UL128-131A complex. J. Virol. 2010, 84, 1005–1013. [Google Scholar]
- Buckmaster, E.A.; Gompels, U.; Minson, A. Characterisation and physical mapping of an HSV-1 glycoprotein of approximately 115 X 10(3) molecular weight. Virology 1984, 139, 408–413. [Google Scholar]
- Roberts, S.R.; Ponce de Leon, M.; Cohen, G.H.; Eisenberg, R.J. Analysis of the intracellular maturation of the herpes simplex virus type 1 glycoprotein gH in infected and transfected cells. Virology 1991, 184, 609–624. [Google Scholar]
- Hutchinson, L.; Browne, H.; Wargent, V.; Davis-Poynter, N.; Primorac, S.; Goldsmith, K.; Minson, A.C.; Johnson, D.C. A novel herpes simplex virus glycoprotein, gL, forms a complex with glycoprotein H (gH) and affects normal folding and surface expression of gH. J. Virol. 1992, 66, 2240–2250. [Google Scholar]
- Cairns, T.M.; Shaner, M.S.; Zuo, Y.; Ponce de Leon, M.; Baribaud, I.; Eisenberg, R.J.; Cohen, G.H.; Whitbeck, J.C. Epitope mapping of herpes simplex virus type 2 gH/gL defines distinct antigenic sites, including some associated with biological function. J. Virol. 2006, 80, 2596–2608. [Google Scholar]
- Roop, C.; Hutchinson, L.; Johnson, D.C. A mutant herpes simplex virus type 1 unable to express glycoprotein L cannot enter cells, and its particles lack glycoprotein H. J. Virol. 1993, 67, 2285–2297. [Google Scholar]
- Dubin, G.; Jiang, H. Expression of herpes simplex virus type 1 glycoprotein L (gL) in transfected mammalian cells: Evidence that gL is not independently anchored to cell membranes. J. Virol. 1995, 69, 4564–4568. [Google Scholar]
- Peng, T.; Ponce de Leon, M.; Novotny, M.J.; Jiang, H.; Lambris, J.D.; Dubin, G.; Spear, P.G.; Cohen, G.H.; Eisenberg, R.J. Structural and antigenic analysis of a truncated form of the herpes simplex virus glycoprotein gH-gL complex. J. Virol. 1998, 72, 6092–6103. [Google Scholar]
- Pertel, P.E. Human herpesvirus 8 glycoprotein B (gB), gH, and gL can mediate cell fusion. J. Virol. 2002, 76, 4390–4400. [Google Scholar]
- Kinzler, E.R.; Compton, T. Characterization of human cytomegalovirus glycoprotein-induced cell-cell fusion. J. Virol. 2005, 79, 7827–7837. [Google Scholar]
- Subramanian, R.P.; Geraghty, R.J. Herpes simplex virus type 1 mediates fusion through a hemifusion intermediate by sequential activity of glycoproteins D, H, L, and B. Proc. Natl. Acad. Sci. USA 2007, 104, 2903–2908. [Google Scholar]
- Jackson, J.O.; Longnecker, R. Reevaluating herpes simplex virus hemifusion. J. Virol. 2010, 84, 11814–11821. [Google Scholar]
- Pasieka, T.J.; Maresova, L.; Shiraki, K.; Grose, C. Regulation of varicella-zoster virus-induced cell-to-cell fusion by the endocytosis-competent glycoproteins gH and gE. J. Virol. 2004, 78, 2884–2896. [Google Scholar] [CrossRef]
- Lopper, M.; Compton, T. Coiled-coil domains in glycoproteins B and H are involved in human cytomegalovirus membrane fusion. J. Virol. 2004, 78, 8333–8341. [Google Scholar]
- Gianni, T.; Piccoli, A.; Bertucci, C.; Campadelli-Fiume, G. Heptad repeat 2 in herpes simplex virus 1 gH interacts with heptad repeat 1 and is critical for virus entry and fusion. J. Virol. 2006, 80, 2216–2224. [Google Scholar]
- Galdiero, S.; Falanga, A.; Vitiello, M.; D’Isanto, M.; Collins, C.; Orrei, V.; Browne, H.; Pedone, C.; Galdiero, M. Evidence for a role of the membrane-proximal region of herpes simplex virus type 1 glycoprotein h in membrane fusion and virus inhibition. Chem. Biochem. 2007, 8, 885–895. [Google Scholar]
- Galdiero, S.; Falanga, A.; Vitiello, M.; D’Isanto, M.; Cantisani, M.; Kampanaraki, A.; Benedetti, E.; Browne, H.; Galdiero, M. Peptides containing membrane-interacting motifs inhibit herpes simplex virus type 1 infectivity. Peptides 2008, 29, 1461–1471. [Google Scholar]
- Galdiero, S.; Vitiello, M.; D’Isanto, M.; Falanga, A.; Collins, C.; Raieta, K.; Pedone, C.; Browne, H.; Galdiero, M. Analysis of synthetic peptides from heptad-repeat domains of herpes simplex virus type 1 glycoproteins H and B. J. Gen. Virol. 2006, 87, 1085–1097. [Google Scholar]
- Bender, F.C.; Whitbeck, J.C.; Ponce de Leon, M.; Lou, H.; Eisenberg, R.J.; Cohen, G.H. Specific association of glycoprotein B with lipid rafts during herpes simplex virus entry. J. Virol. 2003, 77, 9542–9552. [Google Scholar]
- Cairns, T.M.; Whitbeck, J.C.; Lou, H.; Heldwein, E.E.; Chowdary, T.K.; Eisenberg, R.J.; Cohen, G.H. Capturing the herpes simplex virus core fusion complex (gB-gH/gL) in an acidic environment. J. Virol. 2011, 85, 6175–6184. [Google Scholar]
- Chowdary, T.K.; Cairns, T.M.; Atanasiu, D.; Cohen, G.H.; Eisenberg, R.J.; Heldwein, E.E. Crystal structure of the conserved herpesvirus fusion regulator complex gH-gL. Nat. Struct. Mol. Biol. 2010, 17, 882–888. [Google Scholar]
- Backovic, M.; DuBois, R.M.; Cockburn, J.J.; Sharff, A.J.; Vaney, M.C.; Granzow, H.; Klupp, B.G.; Bricogne, G.; Mettenleiter, T.C.; Rey, F.A. Structure of a core fragment of glycoprotein H from pseudorabies virus in complex with antibody. Proc. Natl. Acad. Sci. USA 2010, 107, 22635–22640. [Google Scholar]
- Matsuura, H.; Kirschner, A.N.; Longnecker, R.; Jardetzky, T.S. Crystal structure of the Epstein-Barr virus (EBV) glycoprotein H/glycoprotein L (gH/gL) complex. Proc. Natl. Acad. Sci. USA 2010, 107, 22641–22646. [Google Scholar]
- Grunewald, K.; Desai, P.; Winkler, D.C.; Heymann, J.B.; Belnap, D.M.; Baumeister, W.; Steven, A.C. Three-dimensional structure of herpes simplex virus from cryo-electron tomography. Science 2003, 302, 1396–1398. [Google Scholar]
- Harrison, S.C. Viral membrane fusion. Nat. Struct. Mol. Biol. 2008, 15, 690–698. [Google Scholar]
- Kielian, M.; Rey, F.A. Virus membrane-fusion proteins: More than one way to make a hairpin. Nat. Rev. Microbiol. 2006, 4, 67–76. [Google Scholar]
- Backovic, M.; Jardetzky, T.S. Class III viral membrane fusion proteins. Curr. Opin. Struct. Biol. 2009, 19, 189–196. [Google Scholar]
- Gianni, T.; Fato, R.; Bergamini, C.; Lenaz, G.; Campadelli-Fiume, G. Hydrophobic alpha-helices 1 and 2 of herpes simplex virus gH interact with lipids, and their mimetic peptides enhance virus infection and fusion. J. Virol. 2006, 80, 8190–8198. [Google Scholar]
- Gompels, U.A.; Craxton, M.A.; Honess, R.W. Conservation of glycoprotein H (gH) in herpesviruses: Nucleotide sequence of the gH gene from herpesvirus saimiri. J. Gen. Virol. 1988, 69, 2819–2829. [Google Scholar]
- Galdiero, M.; Whiteley, A.; Bruun, B.; Bell, S.; Minson, T.; Browne, H. Site-Directed and linker Insertion Mutangenesis of Herpes Simplex Virus Type 1 Glycoprotein H. J. Virol. 1997, 71, 2163–2170. [Google Scholar]
- Wu, L.; Borza, C.M.; Hutt-Fletcher, L.M. Mutations of Epstein-Barr virus gH that are differentially able to support fusion with B cells or epithelial cells. J. Virol. 2005, 79, 10923–10930. [Google Scholar]
- Vleck, S.E.; Oliver, S.L.; Brady, J.J.; Blau, H.M.; Rajamani, J.; Sommer, M.H.; Arvin, A.M. Structure-function analysis of varicella-zoster virus glycoprotein H identifies domain-specific roles for fusion and skin tropism. Proc. Natl. Acad. Sci. USA 2011, 108, 18412–18417. [Google Scholar]
- Gompels, U.A.; Carss, A.L.; Saxby, C.; Hancock, D.C.; Forrester, A.; Minson, A.C. Characterization and sequence analyses of antibody-selected antigenic variants of herpes simplex virus show a conformationally complex epitope on glycoprotein H. J. Virol. 1991, 65, 2393–2401. [Google Scholar]
- Omerovic, J.; Lev, L.; Longnecker, R. The amino terminus of Epstein-Barr virus glycoprotein gH is important for fusion with epithelial and B cells. J. Virol. 2005, 79, 12408–12415. [Google Scholar]
- Plate, A.E.; Smajlovic, J.; Jardetzky, T.S.; Longnecker, R. Functional analysis of glycoprotein L (gL) from rhesus lymphocryptovirus in Epstein-Barr virus-mediated cell fusion indicates a direct role of gL in gB-induced membrane fusion. J. Virol. 2009, 83, 7678–7689. [Google Scholar]
- Atanasiu, D.; Whitbeck, J.C.; de Leon, M.P.; Lou, H.; Hannah, B.P.; Cohen, G.H.; Eisenberg, R.J. Bimolecular complementation defines functional regions of Herpes simplex virus gB that are involved with gH/gL as a necessary step leading to cell fusion. J. Virol. 2010, 84, 3825–3834. [Google Scholar]
- Cairns, T.M.; Friedman, L.S.; Lou, H.; Whitbeck, J.C.; Shaner, M.S.; Cohen, G.H.; Eisenberg, R.J. N-terminal mutants of herpes simplex virus type 2 gH are transported without gL but require gL for function. J. Virol. 2007, 81, 5102–5111. [Google Scholar]
- Avitabile, E.; Forghieri, C.; Campadelli-Fiume, G. Complexes between herpes simplex virus glycoproteins gD, gB, and gH detected in cells by complementation of split enhanced green fluorescent protein. J. Virol. 2007, 81, 11532–11537. [Google Scholar]
- Parry, C.; Bell, S.; Minson, T.; Browne, H. Herpes simplex virus type 1 glycoprotein H binds to alphavbeta3 integrins. J. Gen. Virol. 2005, 86, 7–10. [Google Scholar]
- Gianni, T.; Cerretani, A.; Dubois, R.; Salvioli, S.; Blystone, S.S.; Rey, F.; Campadelli-Fiume, G. Herpes simplex virus glycoproteins H/L bind to cells independently of {alpha}V{beta}3 integrin and inhibit virus entry, and their constitutive expression restricts infection. J. Virol. 2010, 84, 4013–4025. [Google Scholar]
- Roche, S.; Albertini, A.A.; Lepault, J.; Bressanelli, S.; Gaudin, Y. Structures of vesicular stomatitis virus glycoprotein: Membrane fusion revisited. Cell. Mol. Life Sci. 2008, 65, 1716–1728. [Google Scholar]
- Diederich, S.; Dietzel, E.; Maisner, A. Nipah virus fusion protein: Influence of cleavage site mutations on the cleavability by cathepsin L, trypsin and furin. Virus Res. 2009, 145, 300–306. [Google Scholar]
- Diederich, S.; Moll, M.; Klenk, H.D.; Maisner, A. The nipah virus fusion protein is cleaved within the endosomal compartment. J. Biol. Chem. 2005, 280, 29899–29903. [Google Scholar]
- Bzik, D.J.; Fox, B.A.; DeLuca, N.A.; Person, S. Nucleotide sequence of a region of the herpes simplex virus type 1 gB glycoprotein gene: Mutations affecting rate of virus entry and cell fusion. Virology 1984, 137, 185–190. [Google Scholar]
- Heldwein, E.E.; Lou, H.; Bender, F.C.; Cohen, G.H.; Eisenberg, R.J.; Harrison, S.C. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 2006, 313, 217–220. [Google Scholar]
- Roche, S.; Bressanelli, S.; Rey, F.A.; Gaudin, Y. Crystal structure of the low-pH form of the vesicular stomatitis virus glycoprotein G. Science 2006, 313, 187–191. [Google Scholar]
- Kadlec, J.; Loureiro, S.; Abrescia, N.G.; Stuart, D.I.; Jones, I.M. The postfusion structure of baculovirus gp64 supports a unified view of viral fusion machines. Nat. Struct. Mol. Biol. 2008, 15, 1024–1030. [Google Scholar]
- Stannard, L.M.; Fuller, A.O.; Spear, P.G. Herpes simplex virus glycoproteins associated with different morphological entities projecting from the virion envelope. J. Gen. Virol. 1987, 68, 715–725. [Google Scholar]
- Backovic, M.; Jardetzky, T.S.; Longnecker, R. Hydrophobic residues that form putative fusion loops of Epstein-Barr virus glycoprotein B are critical for fusion activity. J. Virol. 2007, 81, 9596–9600. [Google Scholar]
- Hannah, B.P.; Heldwein, E.E.; Bender, F.C.; Cohen, G.H.; Eisenberg, R.J. Mutational evidence of internal fusion loops in herpes simplex virus glycoprotein B. J. Virol. 2007, 81, 4858–4865. [Google Scholar]
- Hannah, B.P.; Cairns, T.M.; Bender, F.C.; Whitbeck, J.C.; Lou, H.; Eisenberg, R.J.; Cohen, G.H. Herpes simplex virus glycoprotein B associates with target membranes via its fusion loops. J. Virol. 2009, 83, 6825–6836. [Google Scholar]
- Wright, C.C.; Wisner, T.W.; Hannah, B.P.; Eisenberg, R.J.; Cohen, G.H.; Johnson, D.C. Fusion between perinuclear virions and the outer nuclear membrane requires the fusogenic activity of herpes simplex virus gB. J. Virol. 2009, 83, 11847–11856. [Google Scholar]
- Roche, S.; Rey, F.A.; Gaudin, Y.; Bressanelli, S. Structure of the prefusion form of the vesicular stomatitis virus glycoprotein G. Science 2007, 315, 843–848. [Google Scholar]
- Dollery, S.J.; Wright, C.C.; Johnson, D.C.; Nicola, A.V. Low-pH-dependent changes in the conformation and oligomeric state of the prefusion form of herpes simplex virus glycoprotein B are separable from fusion activity. J. Virol. 2011, 85, 9964–9973. [Google Scholar]
- Stampfer, S.D.; Lou, H.; Cohen, G.H.; Eisenberg, R.J.; Heldwein, E.E. Structural basis of local, pH-dependent conformational changes in glycoprotein B from herpes simplex virus type 1. J. Virol. 2010, 84, 12924–12933. [Google Scholar]
- Backovic, M.; Longnecker, R.; Jardetzky, T.S. Structure of a trimeric variant of the Epstein-Barr virus glycoprotein B. Proc. Natl. Acad. Sci. USA 2009, 106, 2880–2885. [Google Scholar]
- Sorem, J.; Longnecker, R. Cleavage of Epstein-Barr virus glycoprotein B is required for full function in cell-cell fusion with both epithelial and B cells. J. Gen. Virol. 2009, 90, 591–595. [Google Scholar]
- Kopp, A.; Blewett, E.; Misra, V.; Mettenleiter, T.C. Proteolytic cleavage of bovine herpesvirus 1 (BHV-1) glycoprotein gB is not necessary for its function in BHV-1 or pseudorabies virus. J. Virol. 1994, 68, 1667–1674. [Google Scholar]
- Kousoulas, K.G.; Pellett, P.E.; Pereira, L.; Roizman, B. Mutations affecting conformation or sequence of neutralizing epitopes identified by reactivity of viable plaques segregate from syn and ts domains of HSV-1(F) gB gene. Virology 1984, 135, 379–394. [Google Scholar]
- Cai, W.Z.; Person, S.; Warner, S.C.; Zhou, J.H.; DeLuca, N.A. Linker-insertion nonsense and restriction-site deletion mutations of the gB glycoprotein gene of herpes simplex virus type 1. J. Virol. 1987, 61, 714–721. [Google Scholar]
- Laquerre, S.; Anderson, D.B.; Argnani, R.; Glorioso, J.C. Herpes simplex virus type 1 glycoprotein B requires a cysteine residue at position 633 for folding, processing, and incorporation into mature infectious virus particle. J. Virol. 1998, 72, 4940–4949. [Google Scholar]
- Lin, E.; Spear, P.G. Random linker-insertion mutagenesis to identify functional domains of herpes simplex virus type 1 glycoprotein B. Proc. Natl. Acad. Sci. USA 2007, 104, 13140–13145. [Google Scholar]
- Arii, J.; Wang, J.; Morimoto, T.; Suenaga, T.; Akashi, H.; Arase, H.; Kawaguchi, Y. A single-amino-acid substitution in herpes simplex virus 1 envelope glycoprotein B at a site required for binding to the paired immunoglobulin-like type 2 receptor alpha (PILRalpha) abrogates PILRalpha-dependent viral entry and reduces pathogenesis. J. Virol. 2010, 84, 10773–10783. [Google Scholar]
- Fan, Q.; Lin, E.; Satoh, T.; Arase, H.; Spear, P.G. Differential effects on cell fusion activity of mutations in herpes simplex virus 1 glycoprotein B (gB) dependent on whether a gD receptor or a gB receptor is overexpressed. J. Virol. 2009, 83, 7384–7390. [Google Scholar]
- Backovic, M.; Leser, G.P.; Lamb, R.A.; Longnecker, R.; Jardetzky, T.S. Characterization of EBV gB indicates properties of both class I and class II viral fusion proteins. Virology 2007, 368, 102–113. [Google Scholar]
- Oliver, S.L.; Sommer, M.; Zerboni, L.; Rajamani, J.; Grose, C.; Arvin, A.M. Mutagenesis of varicella-zoster virus glycoprotein B: Putative fusion loop residues are essential for viral replication, and the furin cleavage motif contributes to pathogenesis in skin tissue in vivo. J. Virol. 2009, 83, 7495–7506. [Google Scholar]
- Highlander, S.L.; Cai, W.; Person, S.; Levine, M.; Glorioso, J.C. Monoclonal antibodies define a domain on herpes simplex virus glycoprotein B involved in virus penetration. J. Virol. 1988, 62, 1881–1888. [Google Scholar]
- Kousoulas, K.G.; Huo, B.; Pereira, L. Antibody-resistant mutations in cross-reactive and type-specific epitopes of herpes simplex virus 1 glycoprotein B map in separate domains. Virology 1988, 166, 423–431. [Google Scholar]
- Pereira, L.; Ali, M.; Kousoulas, K.; Huo, B.; Banks, T. Domain structure of herpes simplex virus 1 glycoprotein B: Neutralizing epitopes map in regions of continuous and discontinuous residues. Virology 1989, 172, 11–24. [Google Scholar]
- Bender, F.C.; Samanta, M.; Heldwein, E.E.; de Leon, M.P.; Bilman, E.; Lou, H.; Whitbeck, J.C.; Eisenberg, R.J.; Cohen, G.H. Antigenic and mutational analyses of herpes simplex virus glycoprotein B reveal four functional regions. J. Virol. 2007, 81, 3827–3841. [Google Scholar]
- Bender, F.C.; Whitbeck, J.C.; Lou, H.; Cohen, G.H.; Eisenberg, R.J. Herpes simplex virus glycoprotein B binds to cell surfaces independently of heparan sulfate and blocks virus entry. J. Virol. 2005, 79, 11588–11597. [Google Scholar]
- Satoh, T.; Arase, H. HSV-1 infection through inhibitory receptor, PILRalpha. Uirusu 2008, 58, 27–36. [Google Scholar]
- Satoh, T.; Arii, J.; Suenaga, T.; Wang, J.; Kogure, A.; Uehori, J.; Arase, N.; Shiratori, I.; Tanaka, S.; Kawaguchi, Y.; et al. PILRalpha is a herpes simplex virus-1 entry coreceptor that associates with glycoprotein B. Cell 2008, 132, 935–944. [Google Scholar] [CrossRef]
- Fuller, A.O.; Santos, R.E.; Spear, P.G. Neutralizing antibodies specific for glycoprotein H of herpes simplex virus permit viral attachment to cells but prevent penetration. J. Virol. 1989, 63, 3435–3443. [Google Scholar]
- Avitabile, E.; Forghieri, C.; Campadelli-Fiume, G. Cross talking among the glycoproteins involved in herpes simplex virus entry and fusion: The interaction between gB and gH/gL does not necessarily require gD. J. Virol. 2009, 83, 10752–10760. [Google Scholar]
- Hu, C.D.; Chinenov, Y.; Kerppola, T.K. Visualization of interactions among bZIP and Rel family proteins in living cells using bimolecular fluorescence complementation. Mol. Cell 2002, 9, 789–798. [Google Scholar]
- Kerppola, T.K. Design and implementation of bimolecular fluorescence complementation (BiFC) assays for the visualization of protein interactions in living cells. Nat. Protoc. 2006, 1, 1278–1286. [Google Scholar]
- Kerppola, T.K. Bimolecular fluorescence complementation: Visualization of molecular interactions in living cells. Methods Cell Biol. 2008, 85, 431–470. [Google Scholar]
- Ventura, S. Bimolecular fluorescence complementation: Illuminating cellular protein interactions. Curr. Mol. Med. 2010, 11, 582–598. [Google Scholar]
- Hu, C.D.; Kerppola, T.K. Simultaneous visualization of multiple protein interactions in living cells using multicolor fluorescence complementation analysis. Nat. Biotechnol. 2003, 21, 539–545. [Google Scholar]
- Kerppola, T.K. Visualization of molecular interactions using bimolecular fluorescence complementation analysis: Characteristics of protein fragment complementation. Chem. Soc. Rev. 2009, 38, 2876–2886. [Google Scholar]
- Gompels, U.; Minson, A. The properties and sequence of glycoprotein H of herpes simplex virus type 1. Virology 1986, 153, 230–247. [Google Scholar]
- Browne, H.M.; Bruun, B.C.; Minson, A.C. Characterization of herpes simplex virus type 1 recombinants with mutations in the cytoplasmic tail of glycoprotein H. J. Gen. Virol. 1996, 77, 2569–2573. [Google Scholar]
- Davis-Poynter, N.; Bell, S.; Minson, T.; Browne, H. Analysis of the contributions of herpes simplex virus type 1 membrane proteins to the induction of cell-cell fusion. J. Virol. 1994, 68, 7586–7590. [Google Scholar]
- Harman, A.; Browne, H.; Minson, T. The transmembrane domain and cytoplasmic tail of herpes simplex virus type 1 glycoprotein H play a role in membrane fusion. J. Virol. 2002, 76, 10708–10716. [Google Scholar]
- Atanasiu, D.; Saw, W.T.; Cohen, G.H.; Eisenberg, R.J. Cascade of events governing cell-cell fusion induced by herpes simplex virus glycoproteins gD, gH/gL, and gB. J. Virol. 2010, 84, 12292–12299. [Google Scholar]
- Browne, H.; Bruun, B.; Minson, T. Plasma membrane requirements for cell fusion induced by herpes simplex virus type 1 glycoproteins gB, gD, gH and gL. J. Gen. Virol. 2001, 82, 1419–1422. [Google Scholar]
- Pertel, P.E.; Fridberg, A.; Parish, M.L.; Spear, P.G. Cell fusion induced by herpes simplex virus glycoproteins gB, gD, and gH-gL requires a gD receptor but not necessarily heparan sulfate. Virology 2001, 279, 313–324. [Google Scholar]
- Kirschner, A.N.; Lowrey, A.S.; Longnecker, R.; Jardetzky, T.S. Binding-site interactions between Epstein-Barr virus fusion proteins gp42 and gH/gL reveal a peptide that inhibits both epithelial and B-cell membrane fusion. J. Virol. 2007, 81, 9216–9229. [Google Scholar]
- Liu, F.; Marquardt, G.; Kirschner, A.N.; Longnecker, R.; Jardetzky, T.S. Mapping the N-terminal residues of Epstein-Barr virus gp42 that bind gH/gL by using fluorescence polarization and cell-based fusion assays. J. Virol. 2010, 84, 10375–10385. [Google Scholar]
- Heldwein, E.E. Entry of herpesviruses into cells: More than one way to pull the trigger. Structure 2009, 17, 147–149. [Google Scholar]
- Connolly, S.A.; Leser, G.P.; Yin, H.S.; Jardetzky, T.S.; Lamb, R.A. Refolding of a paramyxovirus F protein from prefusion to postfusion conformations observed by liposome binding and electron microscopy. Proc. Natl. Acad. Sci. USA 2006, 103, 17903–17908. [Google Scholar]
- Smith, E.C.; Popa, A.; Chang, A.; Masante, C.; Dutch, R.E. Viral entry mechanisms: The increasing diversity of paramyxovirus entry. FEBS J. 2009, 276, 7217–7227. [Google Scholar]
- Lee, B.; Ataman, Z.A. Modes of paramyxovirus fusion: A Henipavirus perspective. Trends Microbiol. 2011, 19, 389–399. [Google Scholar]
- Dutch, R.E. Entry and fusion of emerging paramyxoviruses. PLoS Pathog. 2010, 6, e1000881. [Google Scholar] [CrossRef]
- Lee, B.; Ataman, Z.A.; Jin, L. Evil versus ‘eph-ective’ use of ephrin-B2. Nat. Struct. Mol. Biol. 2008, 15, 540–542. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Eisenberg, R.J.; Atanasiu, D.; Cairns, T.M.; Gallagher, J.R.; Krummenacher, C.; Cohen, G.H. Herpes Virus Fusion and Entry: A Story with Many Characters. Viruses 2012, 4, 800-832. https://doi.org/10.3390/v4050800
Eisenberg RJ, Atanasiu D, Cairns TM, Gallagher JR, Krummenacher C, Cohen GH. Herpes Virus Fusion and Entry: A Story with Many Characters. Viruses. 2012; 4(5):800-832. https://doi.org/10.3390/v4050800
Chicago/Turabian StyleEisenberg, Roselyn J., Doina Atanasiu, Tina M. Cairns, John R. Gallagher, Claude Krummenacher, and Gary H. Cohen. 2012. "Herpes Virus Fusion and Entry: A Story with Many Characters" Viruses 4, no. 5: 800-832. https://doi.org/10.3390/v4050800