Proteogenomic Identification of a Novel Protein-Encoding Gene in Bovine Herpesvirus 1 That Is Expressed during Productive Infection

 ,
,  ,
,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Virus
2.2. Infected Whole Cell Sample Preparation
2.3. Liquid Chromatography-Tandem Mass Spectrometry
2.4. Proteogenomic Mapping
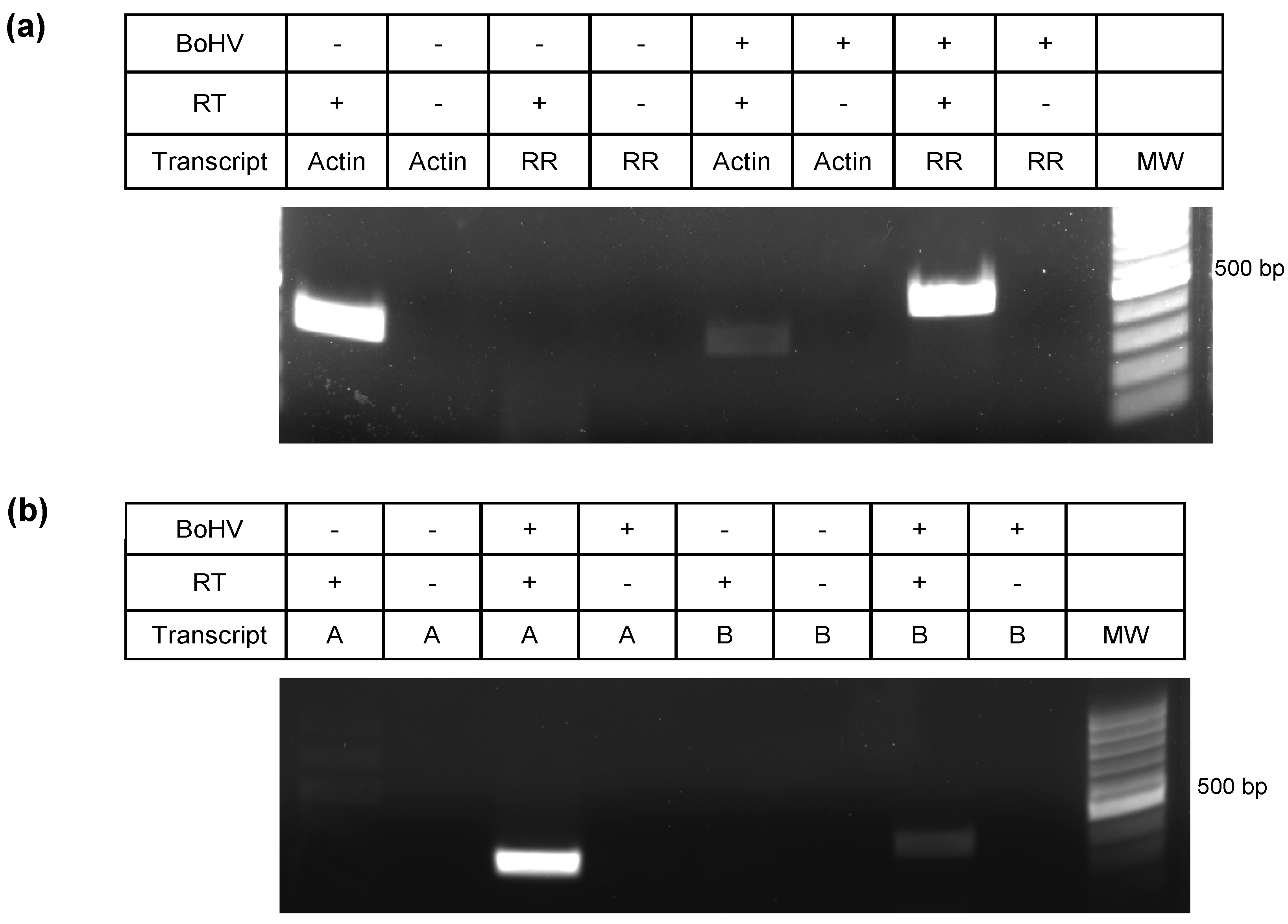
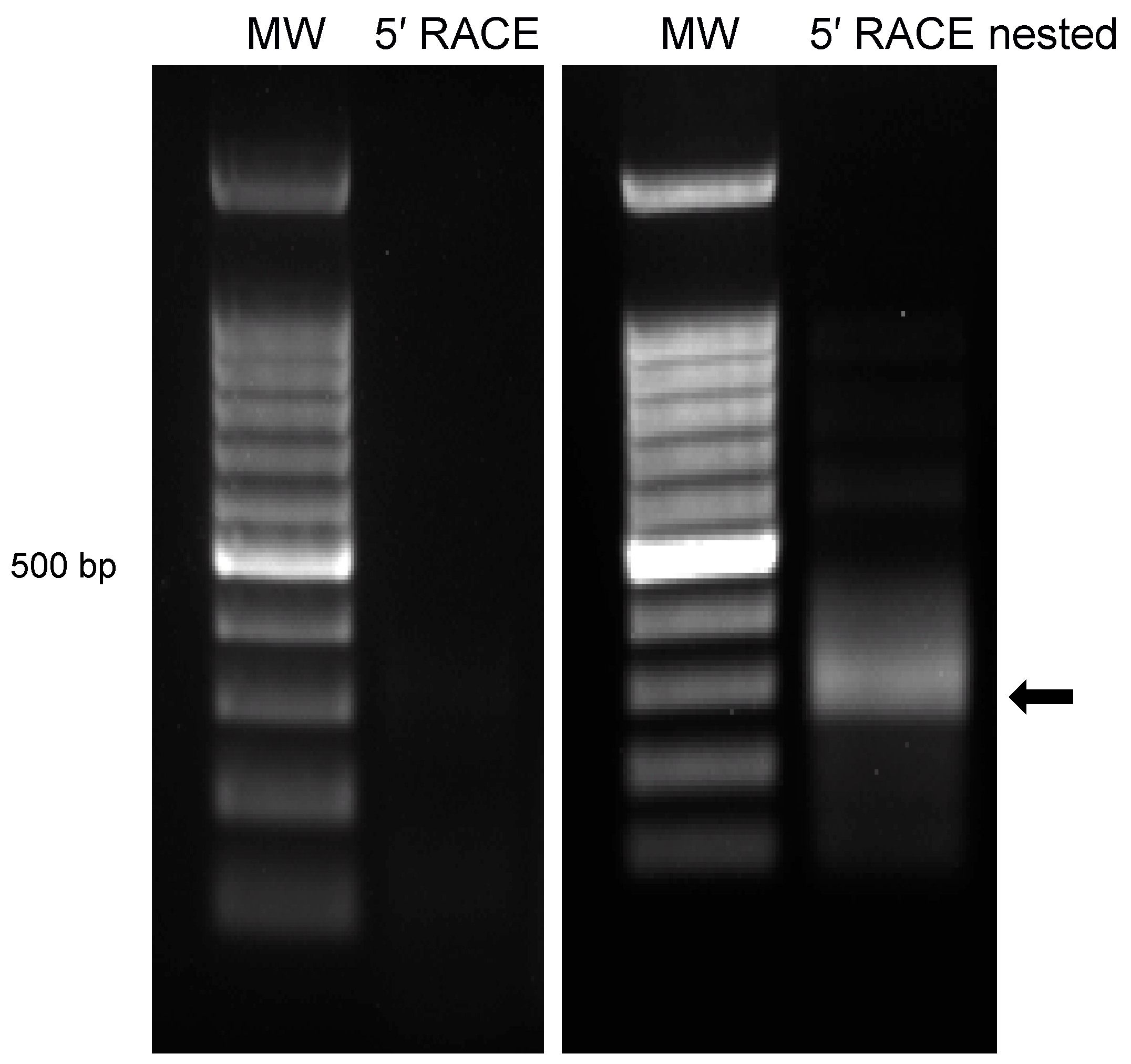
2.5. RNA Extraction, RT-PCR, and RACE
2.6. DNA Gel Electrophoresis, Purification, and Sequencing
2.7. Generation of Peptide Specific Antiserum Directed against ORF-A
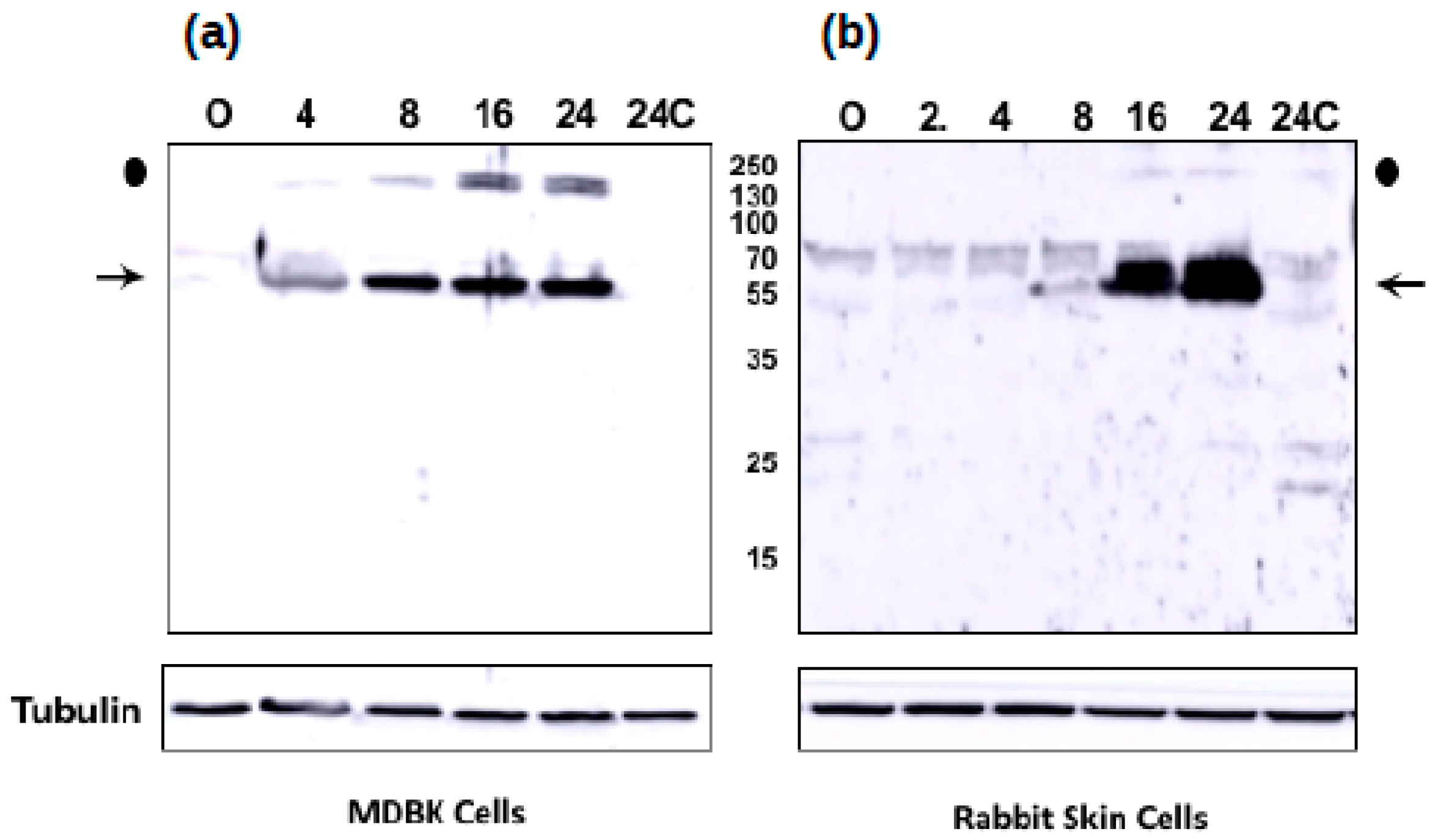
2.8. Western Blot Analysis
3. Results
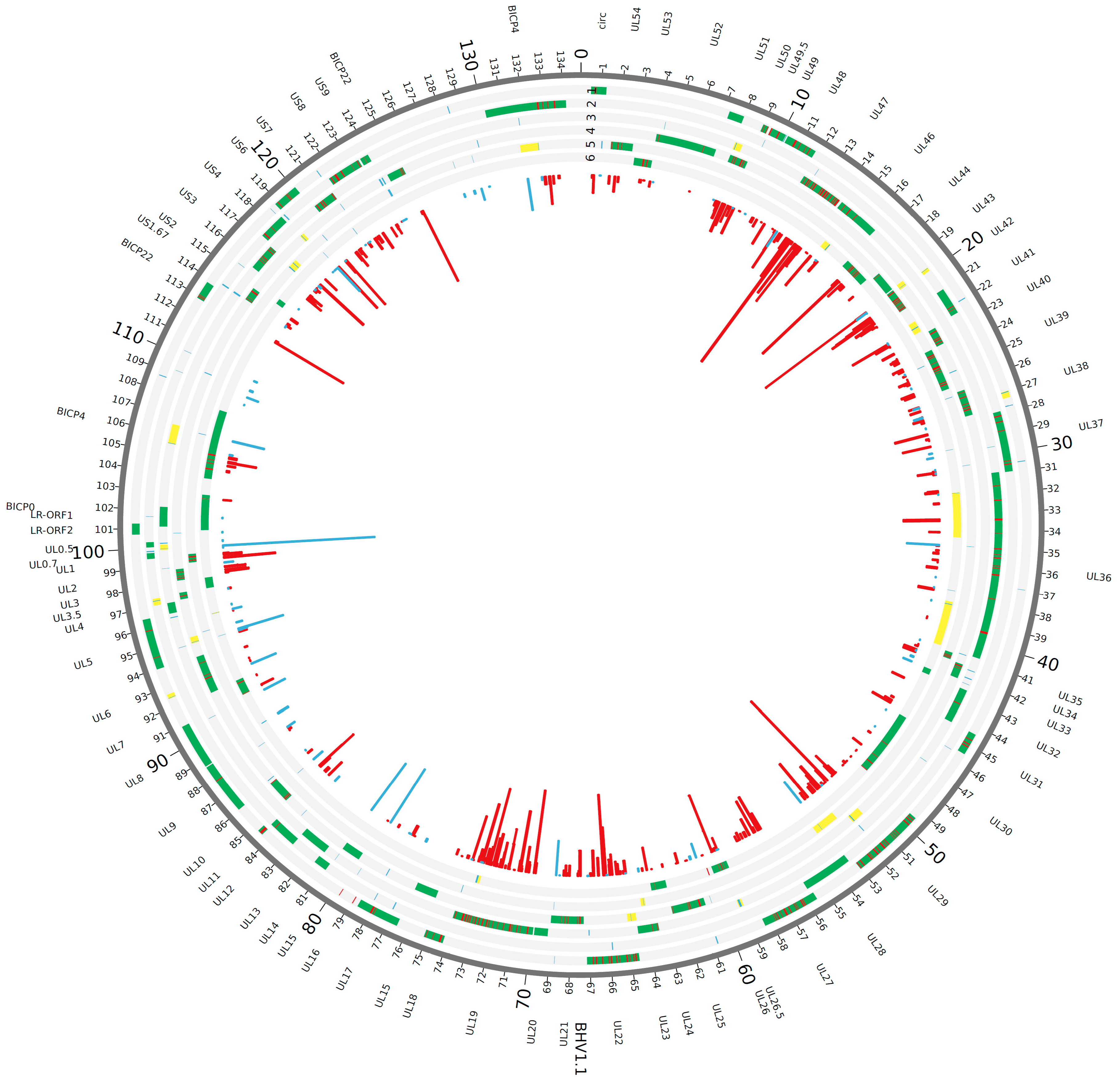
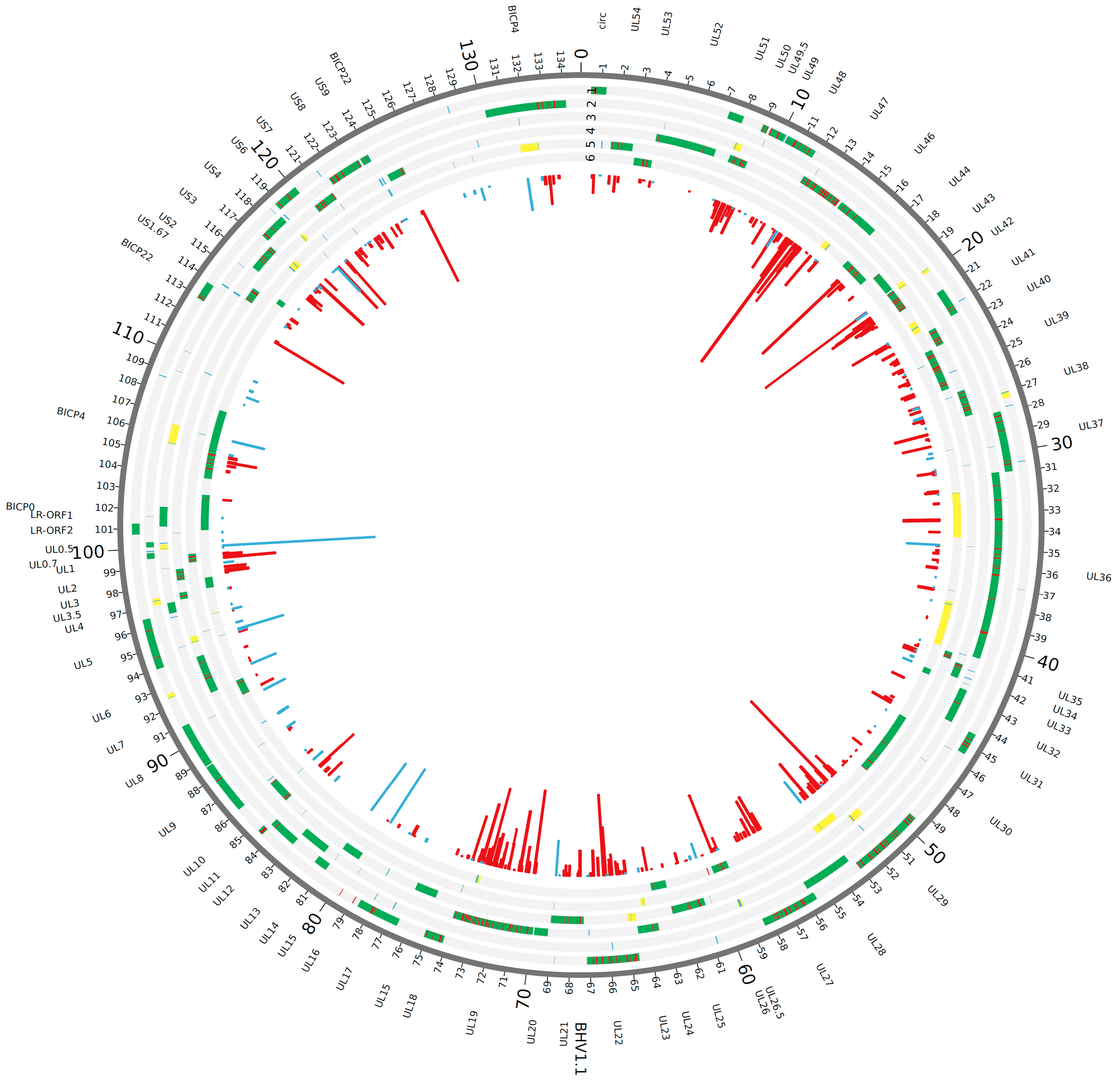
3.1. Proteogenomic Analysis of Infected Cells Identified Potential Novel Open Reading Frames
3.2. Validation of Intergenic Peptides with Surrounding Viable ORFs
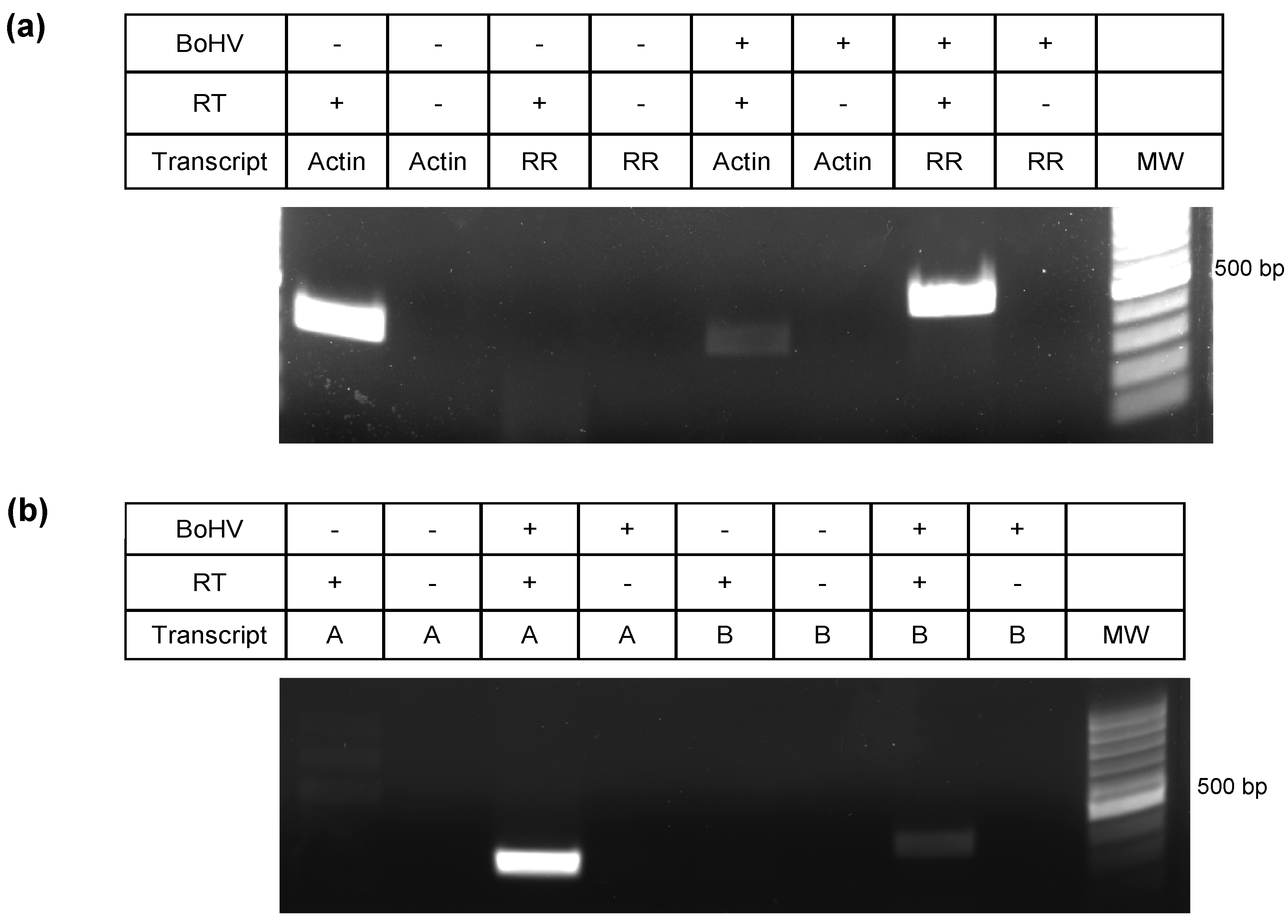
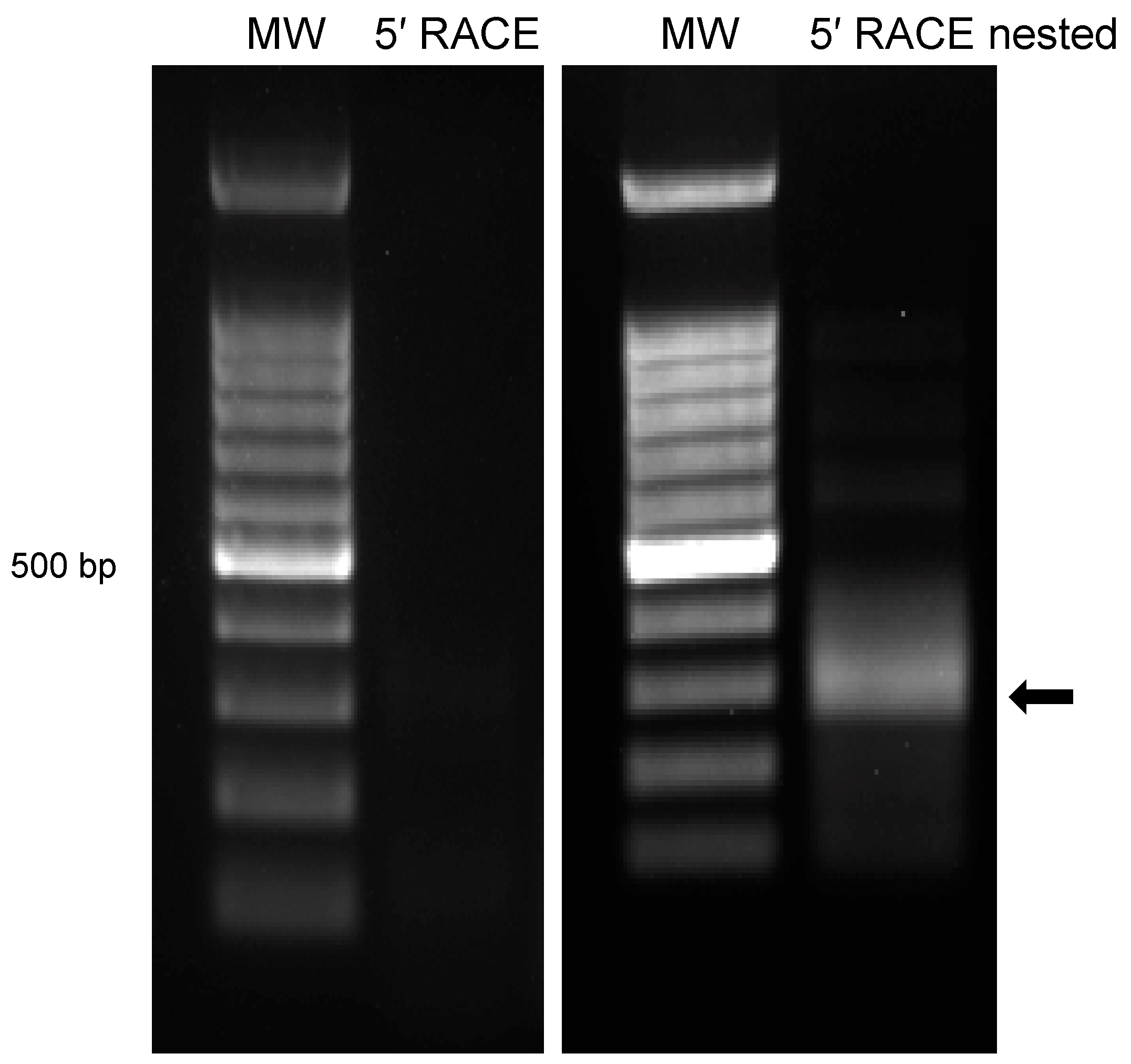
3.3. Characterization of 5′ Terminus of ORF-A via Rapid Amplification of cDNA Ends (RACE)
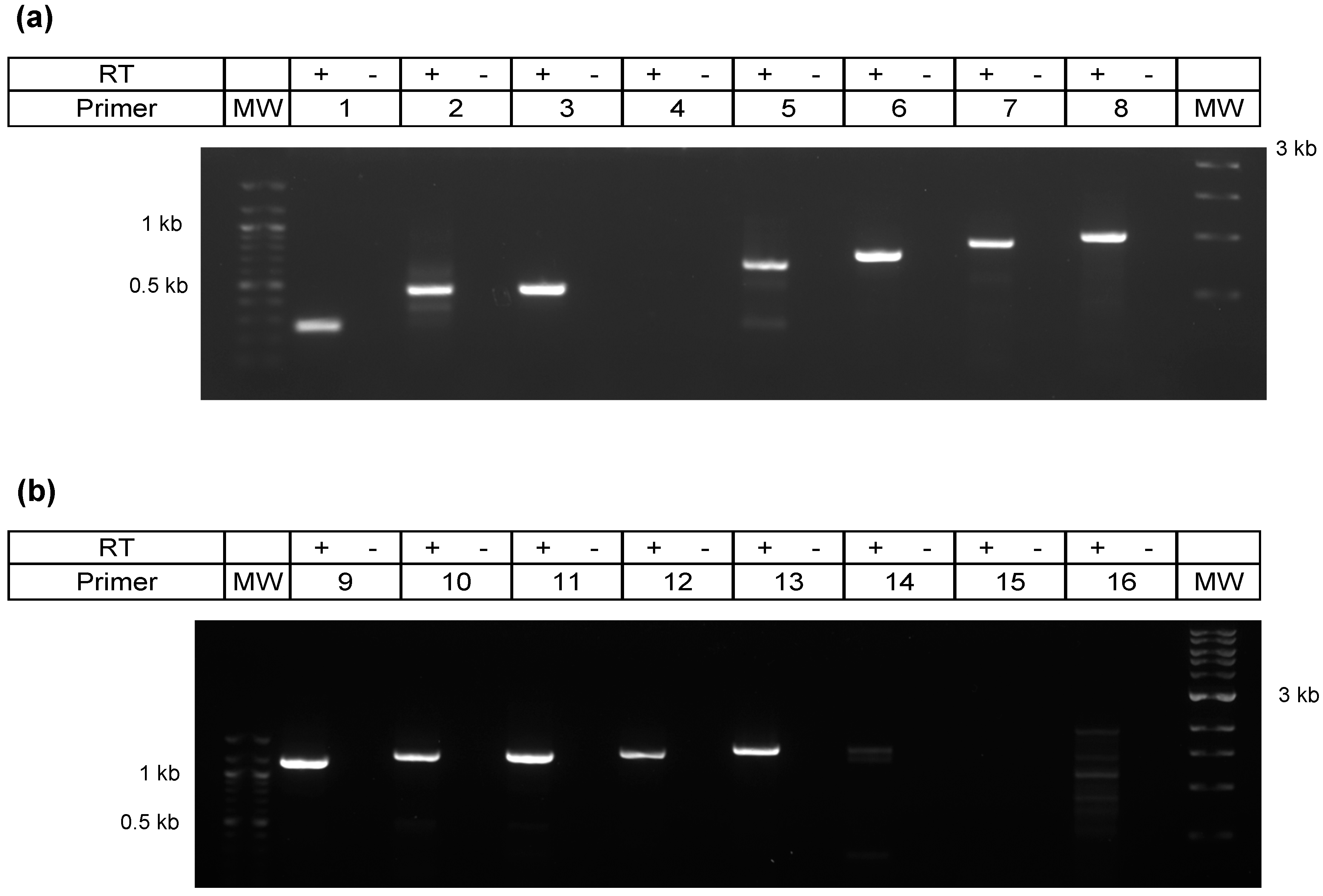
3.4. Characterization of 3′ Terminus of the A Transcript by Primer Walking
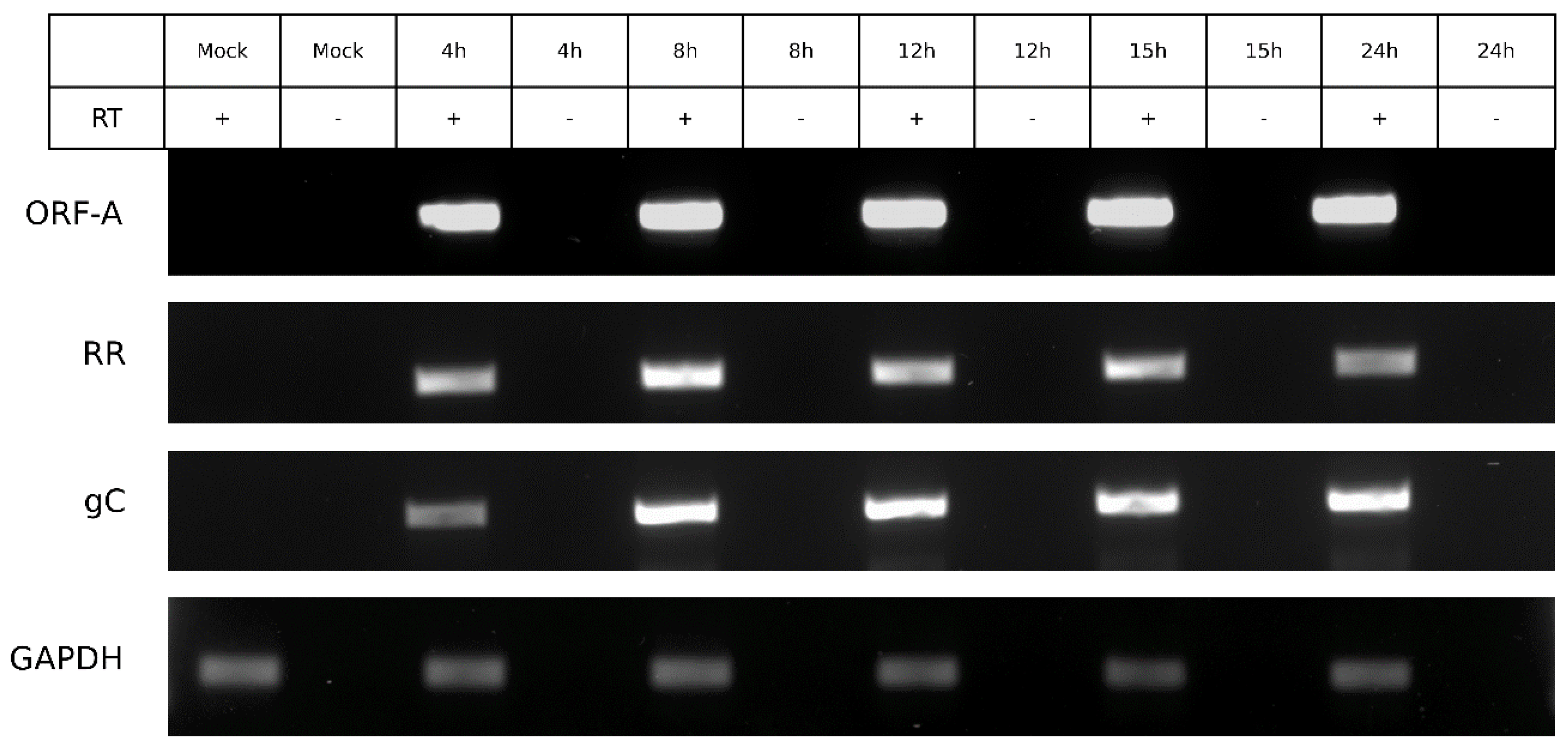
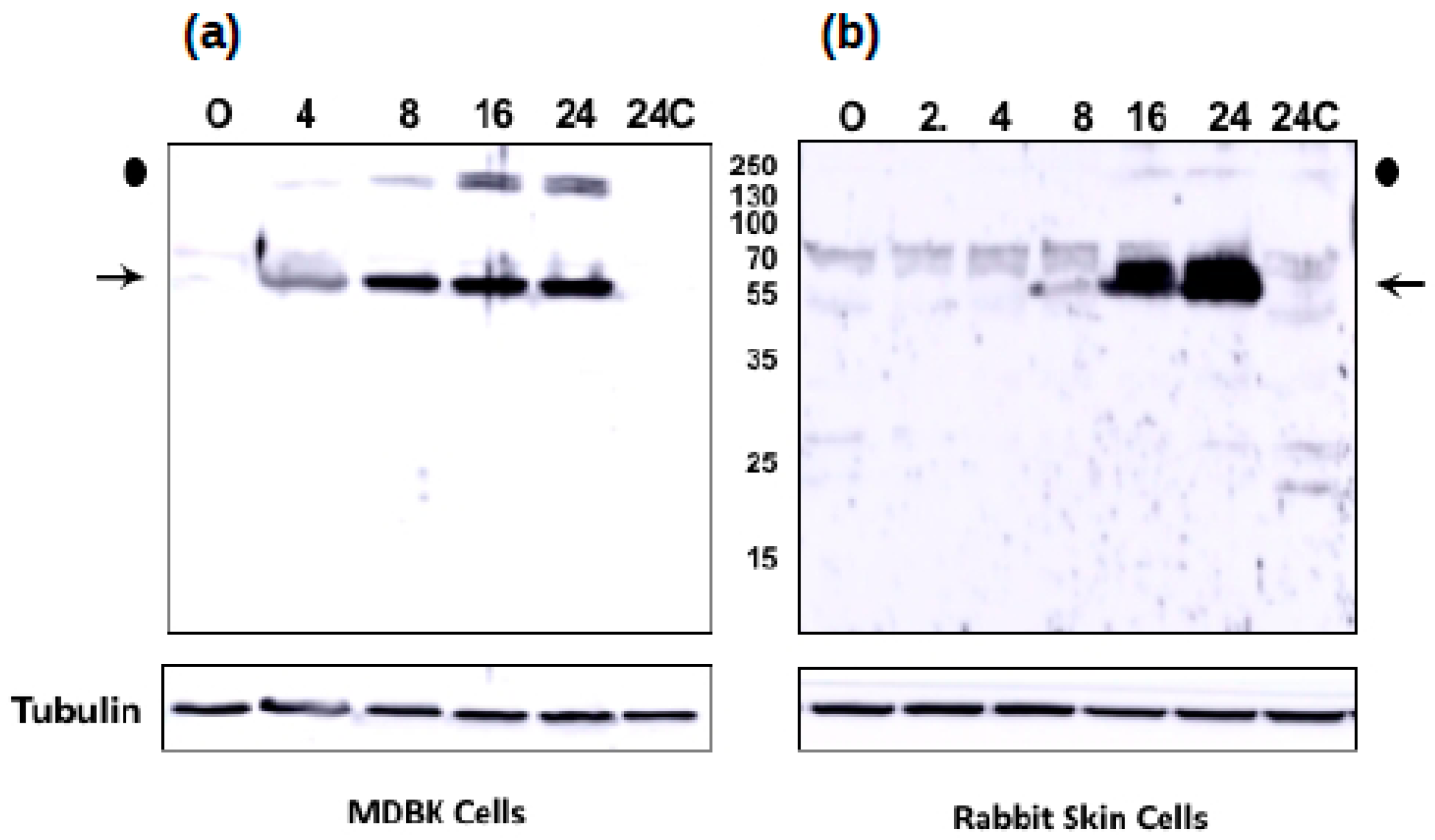
3.5. The Novel ORF-A Expresses a 55 KDa Protein in Bovine Cells
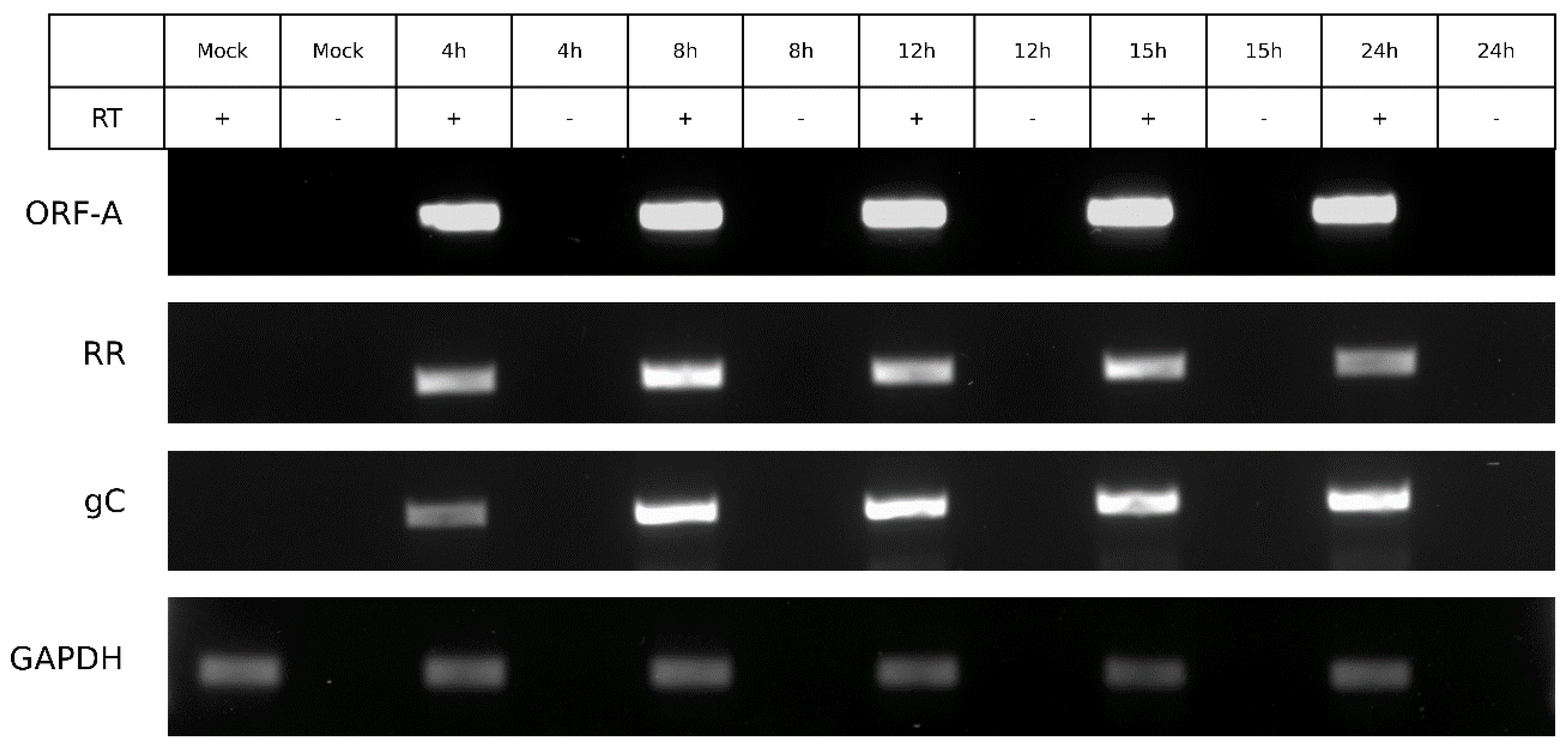
3.6. ORF-A Transcription Profile is Consistent with Protein Synthesis
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Thiry, J.; Keuser, V.; Muylkens, B.; Meurens, F.; Gogev, S.; Vanderplasschen, A.; Thiry, E. Ruminant alphaherpesviruses related to bovine herpesvirus 1. Vet. Res. 2006, 37, 169–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Metzler, A.E.; Schudel, A.A.; Engels, M. Bovine Herpesvirus 1: Molecular and antigenic characteristics of variant viruses isolated from calves with neurological disease. Arch. Virol. 1986, 87, 205–217. [Google Scholar] [CrossRef] [PubMed]
- Engels, M.; Giuliani, C.; Wild, P.; Beck, T.M.; Loepfe, E.; Wyler, R. The genome of bovine herpesvirus 1 (BHV-1) strains exhibiting a neuropathogenic potential compared to known BHV-1 strains by restriction site mapping and cross-hybridization. Virus Res. 1986, 6, 57–73. [Google Scholar] [CrossRef]
- Jones, C.; Chowdhury, S. Bovine herpesvirus type 1 (BHV-1) is an important cofactor in the bovine respiratory disease complex. Vet. Clin. N. Am. Food Anim. Pract. 2010, 26, 303–321. [Google Scholar] [CrossRef] [PubMed]
- D’Arce, R.C.; Almeida, R.S.; Silva, T.C.; Franco, A.C.; Spilki, F.; Roehe, P.M.; Arns, C.W. Restriction endonuclease and monoclonal antibody analysis of Brazilian isolates of bovine herpesviruses types 1 and 5. Vet. Microbiol. 2002, 88, 315–324. [Google Scholar] [CrossRef]
- Van Oirschot, J.T. Bovine herpesvirus 1 in semen of bulls and the risk of transmission: A brief review. Vet Q. 1995, 17, 29–33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, C.; Chowdhury, S. A review of the biology of bovine herpesvirus type 1 (BHV-1), its role as a cofactor in the bovine respiratory disease complex and development of improved vaccines. Anim. Health Res. Rev. 2007, 8, 187–205. [Google Scholar] [CrossRef] [PubMed]
- Kirchhoff, J.; Uhlenbruck, S.; Goris, K.; Keil, G.M.; Herrler, G. Three viruses of the bovine respiratory disease complex apply different strategies to initiate infection. Vet. Res. 2014, 45, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Yates, W.D. A review of infectious bovine rhinotracheitis, shipping fever pneumonia and viral-bacterial synergism in respiratory disease of cattle. Can. J. Comp. Med. 1982, 46, 225–263. [Google Scholar] [PubMed]
- Neibergs, H.; Seabury, C.; Wojtowicz, A.J.; Wang, Z.; Scraggs, E.; Kiser, J.N.; Neupane, M.; Womack, J.E.; Van Eenennaam, A.; Hagevortm, G.R.; et al. Susceptibility loci revealed for bovine respiratory disease complex in pre-weaned holstein calves. BMC Genomics 2014, 15, 1–19. [Google Scholar] [CrossRef] [PubMed]
- Nataraj, C.; Eidmann, S.; Hariharan, M.J.; Sur, J.H.; Perry, G.A.; Srikumaran, S. Bovine Herpesvirus 1 downregulates the expression of bovine MHC class I molecules. Viral. Immunol. 1997, 10, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Winkler, M.T.C.; Doster, A.; Jones, C. Bovine Herpesvirus 1 can infect CD4+ T lymphocytes and induce programmed cell death during acute infection of cattle. J. Virol. 1999, 73, 8657–8668. [Google Scholar] [PubMed]
- Jones, C. Herpes Simplex Virus Type 1 and Bovine Herpesvirus 1 Latency. Clin. Microbiol. Rev. 2003, 16, 79–95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turin, L.; Russo, S.; Poli, G. BHV-1: New Molecular Approaches to Control Common and Widespread Infection. Mol. Med. 1999, 5, 261–284. [Google Scholar] [CrossRef] [PubMed]
- Van Drunen Littel-van den Hurk, S. Rationale and perspectives on the success of vaccination against bovine herpesvirus-1. Vet. Microbiol. 2006, 113, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Khattar, S.K.; van Drunen Littel-van den Hurk, S.; Babiuk, L.A.; Tikoo, S.K. Identification and transcriptional analysis of a 3′-coterminal gene cluster containing UL1, UL2, UL3, and UL3.5 open reading frames of bovine herpesvirus-1. Virology 1995, 213, 28–37. [Google Scholar] [CrossRef] [PubMed]
- Leung-Tack, P.; Audonnet, J.-C.; Riviere, M. The Complete DNA Sequence and the Genetic Organization of the Short Unique Region (US) of the Bovine Herpesvirus Type 1 (ST Strain). Virology 1994, 199, 409–421. [Google Scholar] [CrossRef] [PubMed]
- Pidone, C.L.; Galosi, C.M.; Echeverria, M.G.; Nosetto, E.O.; Etcheverrigaray, M.E. Restriction Endonuclease Analysis of BHV-1 and BHV-5 Strains Isolated in Argentina. Zentralbl. Veterinarmed. B 1999, 46, 453–456. [Google Scholar] [CrossRef] [PubMed]
- Simard, C.; Langlois, I.; Styger, D.; Vogt, B.; Vlcek, C.; Chalifour, A.; Trudel, M.; Schwyzer, M. Sequence Analysis of the UL39, UL38, and UL37 Homologues of Bovine Herpesvirus 1 and Expression Studies of UL40 and UL39, the Subunits of Ribonucleotide Reductase. Virology 1995, 212, 734–740. [Google Scholar] [CrossRef] [PubMed]
- Vlček, Č.; Beneš, V.; Lu, Z.; Kutish, G.F.; Pačes, V.; Rock, D.; Letchworth, G.J.; Schwyzer, M. Nucleotide Sequence Analysis of a 30-kb Region of the Bovine Herpesvirus 1 Genome Which Exhibits a Colinear Gene Arrangement with the UL21 to UL4 Genes of Herpes Simplex Virus. Virology 1995, 210, 100–108. [Google Scholar]
- Schwyzer, M.; Ackermann, M. Molecular virology of ruminant herpesviruses. Vet. Microbiol. 1996, 53, 17–29. [Google Scholar] [CrossRef]
- D’Offay, J.M.; Fulton, R.W.; Eberle, R. Complete genome sequence of the NVSL BoHV-1.1 Cooper reference strain. Arch. Virol. 2013, 158, 1109–1113. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Galperin, M.Y. Genome Annotation and Analysis. In Sequence-Evolution-Function: Computational Approaches in Comparative Genomics; Eugene, V.K., Michael, Y.G., Eds.; Kluwer Academic: Boston, MA, USA, 2003; pp. 193–226, ISBN-10: 1-40207-274-0. [Google Scholar]
- Gupta, N.; Benhamida, J.; Bhargava, V.; Goodman, D.; Kain, E.; Kerman, I.; Nguyen, N.; Ollikainen, N.; Rodriguez, J.; Wang, J.; et al. Comparative Proteogenomics: Combining Mass Spectrometry and Comparative Genomics to Analyze Multiple Genomes. Genome Res. 2008, 18, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Kunec, D.; Nanduri, B.; Burgess, S.C. Experimental annotation of channel catfish virus by probabilistic proteogenomic mapping. Proteomics 2009, 9, 2634–2647. [Google Scholar] [CrossRef] [PubMed]
- Jaffe, J.D.; Berg, H.C.; Church, G.M. Proteogenomic mapping as a complementary method to perform genome annotation. Proteomics 2004, 4, 59–77. [Google Scholar] [CrossRef] [PubMed]
- Bechtel, J.T.; Winant, R.C.; Ganem, D. Host and Viral Proteins in the Virion of Kaposi’s Sarcoma-Associated Herpesvirus Host and Viral Proteins in the Virion of Kaposi’s Sarcoma-Associated Herpesvirus. Society 2005, 79, 4952–4964. [Google Scholar]
- Engel, E.A.; Song, R.; Koyuncu, O.O.; Enquist, L.W. Investigating the biology of alpha herpesviruses with MS-based proteomics. Proteomics 2015, 15, 1943–1956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Johannsen, E.; Luftig, M.; Chase, M.R.; Weicksel, S.; Cahir-McFarland, E.; Illanes, D.; Sarracino, D.; Kieff, E. Proteins of purified Epstein-Barr virus. Proc. Natl. Acad. Sci. USA 2004, 101, 16286–16291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vidick, S.; Leroy, B.; Palmeira, L.; Machiels, B.; Mast, J.; François, S.; Wattiez, R.; Vanderplasschen, A.; Gillet, L. Proteomic characterization of murid herpesvirus 4 extracellular virions. PLoS ONE 2013, 8, e83842. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.X.; Chong, J.M.; Wu, L.; Yuan, Y. Virion Proteins of Kaposi’s Sarcoma-Associated Herpesvirus. 2005, 79, 800–811. J. virol. 2005, 79, 800–811. [Google Scholar] [CrossRef] [PubMed]
- Lété, C.; Palmeira, L.; Leroy, B.; Mast, J.; Machiels, B.; Wattiez, R.; Vanderplasschen, A.; Gillet, L. Proteomic Characterization of Bovine Herpesvirus 4 Extracellular Virions. J Virol. 2012, 86, 11567–11580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Connor, C.M.; Kedes, D.H. Mass Spectrometric Analyses of Purified Rhesus Monkey Rhadinovirus Reveal 33 Virion-Associated Proteins. J. Virol. 2006, 80, 1574–1583. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kunec, D. Proteomics applied to avian herpesviruses. Avian Dis. 2013, 57, 351–359. [Google Scholar] [CrossRef] [PubMed]
- Flores, E.F.; Donis, R.O. Isolation of a Mutant MDBK Cell Line Resistant to Bovine Viral Diarrhea Virus Infection Due to a Block in Viral Entry. Virology 1995, 208, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European molecular biology open software suite. Trends. Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Geer, L.Y.; Markey, S.P.; Kowalak, J.A.; Wagner, L.; Xu, M.; Maynard, D.M.; Yang, X.; Shi, W.; Bryant, S.H. Open Mass Spectrometry Search Algorithm. J. Proteome Res. 2004, 3, 958–964. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinformatics 2009, 10, 421. [Google Scholar] [CrossRef] [PubMed]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics. 2010, 26, 841–842. [Google Scholar] [CrossRef] [PubMed]
- Abd-Alla, A.M.M.; Kariithi, H.M.; Cousserans, F.; Parker, N.J.; İnce, İ.A.; Scully, E.D.; Boeren, S.; Geib, S.M.; Mekonnen, S.; Vlak, J.M.; et al. Comprehensive annotation of Glossina pallidipes salivary gland hypertrophy virus from Ethiopian tsetse flies: A proteogenomics approach. J. Gen. Virol. 2016, 97, 1010–1031. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Engstrom, P.G.; Tellgren-Roth, C.; Baudo, C.D.; Kennell, J.C.; Sun, S.; Blake Billmyre, R.; Sch oder, M.S.; Andersson, A.; Holm, T.; et al. Proteogenomics produces comprehensive and highly accurate protein-coding gene annotation in a complete genome assembly of Malassezia sympodialis. Nucleic Acids Res. 2017, 45, 2629–2643. [Google Scholar] [PubMed]
- Zhu, Y.; Orre, L.M.; Johansson, H.J.; Huss, M.; Boekel, J.; Vesterlund, M.; Fernandez-Woodbridge, A.; Branca, R.M.M.; Lehtiö, J. Discovery of coding regions in the human genome by integrated proteogenomics analysis workflow. Nat. Commun. 2018, 9, 903. [Google Scholar] [CrossRef] [PubMed]
- Devireddy, L.R.; Jones, C. Alternative splicing of the latency-related transcript of bovine herpesvirus 1 yields RNAs containing unique open reading frames. J. Virol. 1998, 72, 7294–7301. [Google Scholar] [PubMed]
- Devireddy, L.R.; Zhang, Y.; Jones, C.J. Cloning and initial characterization of an alternatively spliced transcript encoded by the bovine herpes virus 1 latency-related gene. J. Neurovirol. 2003, 9, 612–622. [Google Scholar] [CrossRef] [PubMed]
- Wirth, U.V.; Vogt, B.; Schwyzer, M. The three major immediate-early transcripts of bovine herpesvirus 1 arise from two divergent and spliced transcription units. J. Virol. 1991, 65, 195–205. [Google Scholar] [PubMed]
- Firth, A.E.; Brierley, I. Non-canonical translation in RNA viruses. J. Gen. Virol. 2012, 93, 1385–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sasaki, J. Nakashima, N. Translation initiation at the CUU codon is mediated by the internal ribosome entry site of an insect picorna-like virus in vitro. J. Virol. 1999, 73, 1219–1226. [Google Scholar] [PubMed]
- Schneider, R.; Agol, V.I.; Andino, R.; Bayard, F.; Cavener, D.R.; Chappell, S.A.; Chen, J.J.; Darlix, J.L.; Dasgupta, A.; Donzé, O.; et al. New ways of initiating translation in eukaryotes? Mol. Cell Biol. 2001, 21, 8238–8246. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.-J.; Lin, G.; Chang, K.-J.; Yeh, L.-S.; Wang, C.-C. Translational efficiency of a non-AUG initiation codon is significantly affected by its sequence context in yeast. J. Biol. Chem. 2008, 283, 3173–3180. [Google Scholar] [CrossRef] [PubMed]
- Tombácz, D.; Balázs, Z.; Csabai, Z.; Snyder, M.; Boldogkői, Z. Long-Read Sequencing Revealed an Extensive Transcript Complexity in Herpesviruses. Front. Genet. 2018, 9, 259. [Google Scholar] [CrossRef] [PubMed]
- Yoshinaka, Y.; Katoh, I.; Copeland, T.D.; Oroszlan, S. Murine leukemia virus protease is encoded by the gag-pol gene and is synthesized through suppression of an amber termination codon. Proc. Natl. Acad. Sci. USA 1985, 82, 1618–1622. [Google Scholar] [CrossRef] [PubMed]
- Feng, Y.X.; Levin, J.G.; Hatfield, D.L.; Schaefer, T.S.; Gorelick, R.J.; Rein, A. Suppression of UAA and UGA termination codons in mutant murine leukemia viruses. J. Virol. 1989, 63, 2870–2873. [Google Scholar] [PubMed]
- Dreher, T.W.; Miller, W.A. Translational control in positive strand RNA plant viruses. Virology 2006, 344, 185–197. [Google Scholar] [CrossRef] [PubMed]
- Jungreis, I.; Chan, C.S.; Waterhouse, R.M.; Fields, G.; Lin, M.F.; Kellis, M. Evolutionary Dynamics of Abundant Stop Codon Readthrough. Mol. Biol. Evol. 2016, 33, 3108–3132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacks, T.; Varmus, H.E. Expression of the Rous sarcoma virus pol gene by ribosomal frameshifting. Science 1985, 230, 1237–1242. [Google Scholar] [CrossRef] [PubMed]
- Liljeqvist, J.-Å.; Svennerholm, B.; Bergström, T. Herpes Simplex Virus Type 2 Glycoprotein G-Negative Clinical Isolates Are Generated by Single Frameshift Mutations. J. Virol. 1999, 73, 9796–9802. [Google Scholar] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (a) | ||
|---|---|---|
| Target | Forward | Reverse |
| A | ATGTGCTTTGTTTTACAGCCCGGT | ACGTGCGGCTGCCCCGTAG |
| B | ATGGCCGCGATCATGTACGGGT | CGCTGGCTGCTGTGGTTCATGGA |
| Actin | CGTGACATTAAGGAGAAGCTGTGC | CTCAGGAGGAGCAATGATCTTGAT |
| GAPDH | CCATGGAGAAGGCTGGGG | CAAAGTTGTCATGGATGACC |
| RR | TTTTACGAGACCGAGTGCCC | GACGAAAAGGTTGTGGGTGC |
| gC | TGATCGCAGCTATTTTCGCC | TTCTGGGCTACGAACAGCAG |
| 5′ RACE | provided by Invitrogen | CGTGGCGCAGCTGCTTCTGCTGG |
| 5′ Nested RACE | provided by Invitrogen | GCTGTCAACGATACGCTACGTAACG |
| (b) | ||
| Reverse Primer | ||
| 1 | GCGAAACGTCACGCAGGA | |
| 2 | GCCCTACGTGTTTGGTCGAT | |
| 3 | GAGGACGCCGGGTACGATA | |
| 4 | ACGCTCGCGCGGCTAA | |
| 5 | GCTACTCGGGGATCTTGCG | |
| 6 | CCCGGCGGATATGCCATTA | |
| 7 | GTGAGCCCGACTCGCTG | |
| 8 | CGCAACGATGAGCGGCG | |
| 9 | CAGGGCCACGGCGACCATTA | |
| 10 | GCCGGGACCGCGAGCTT | |
| 11 | CGGGCCGAGTGCGAAAAAGG | |
| 12 | TTCGGAGGGCCTGTGGAACC | |
| 13 | CGGCGCGCAGCTCTTCGTAT | |
| 14 | GGTCGGCGGCAGCTC | |
| 15 | AGACGCAACAGCATTAGCACCGGGGG | |
| 16 | GACGGCCTCGCAAAAGATCGTTCGGT |
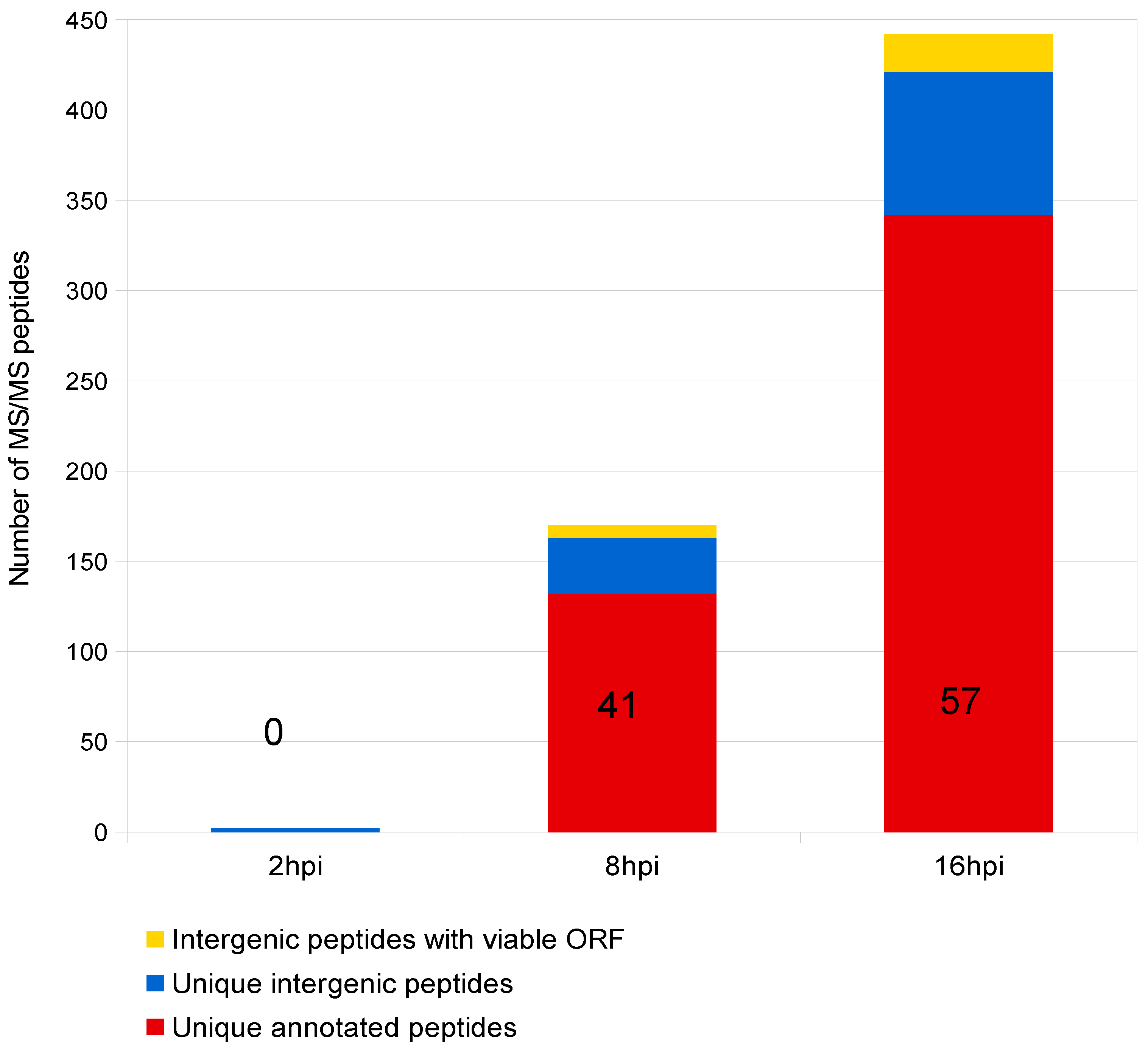
| 2 hpi | 8 hpi | 16 hpi | Total b | |
|---|---|---|---|---|
| Total viral peptides | 4 | 498 | 1991 | 2493 |
| Total annotated peptides | 0 | 394 | 1662 | 2056 |
| Unique a annotated peptides | 0 | 132 | 342 | 344 |
| Viral genes identified | 0 | 41 | 57 | 57 |
| Total intergenic peptides | 4 | 104 | 329 | 437 |
| Uniquea intergenic peptides | 2 | 31 | 79 | 92 |
| Intergenic peptides with viable ORF | 0 | 7 | 21 | 21 |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jefferson, V.A.; Barber, K.A.; El-mayet, F.S.; Jones, C.; Nanduri, B.; Meyer, F. Proteogenomic Identification of a Novel Protein-Encoding Gene in Bovine Herpesvirus 1 That Is Expressed during Productive Infection. Viruses 2018, 10, 499. https://doi.org/10.3390/v10090499
Jefferson VA, Barber KA, El-mayet FS, Jones C, Nanduri B, Meyer F. Proteogenomic Identification of a Novel Protein-Encoding Gene in Bovine Herpesvirus 1 That Is Expressed during Productive Infection. Viruses. 2018; 10(9):499. https://doi.org/10.3390/v10090499
Chicago/Turabian StyleJefferson, Victoria A., Kaley A. Barber, Fouad S. El-mayet, Clinton Jones, Bindu Nanduri, and Florencia Meyer. 2018. "Proteogenomic Identification of a Novel Protein-Encoding Gene in Bovine Herpesvirus 1 That Is Expressed during Productive Infection" Viruses 10, no. 9: 499. https://doi.org/10.3390/v10090499