Molecular Determinants and the Regulation of Human Cytomegalovirus Latency and Reactivation


1
BIO5 Institute, University of Arizona, Tucson, AZ 85721, USA
2
Department of Immunobiology, University of Arizona, Tucson, AZ 85721, USA
*
Author to whom correspondence should be addressed.
Viruses 2018, 10(8), 444; https://doi.org/10.3390/v10080444
Submission received: 19 July 2018
/
Revised: 16 August 2018
/
Accepted: 17 August 2018
/
Published: 20 August 2018
(This article belongs to the Special Issue Recent Advances in Cytomegalovirus Research)
Abstract
:Human cytomegalovirus (HCMV) is a beta herpesvirus that establishes a life-long persistence in the host, like all herpesviruses, by way of a latent infection. During latency, viral genomes are maintained in a quieted state. Virus replication can be reactivated from latency in response to changes in cellular signaling caused by stress or differentiation. The past decade has brought great insights into the molecular basis of HCMV latency. Here, we review the complex persistence of HCMV with consideration of latent reservoirs, viral determinants and their host interactions, and host signaling and the control of cellular and viral gene expression that contributes to the establishment of and reactivation from latency.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
Human cytomegalovirus (HCMV) remains a serious global health burden and has been assigned the highest priority for vaccine development by the National Vaccine Advisory Committee [1]. Infection with HCMV is very common, with seroprevalence among the world’s population ranging from 40–99% depending on geographical location and socioeconomic status [2]. HCMV infection of healthy individuals routinely goes undiagnosed, as symptoms are ambiguous and relatively mild; however, in the absence of adequate host adaptive immunity, infection poses a life-threatening disease risk [3]. HCMV is an important opportunistic pathogen among acquired immune deficiency syndrome (AIDS) patients [4] and those undergoing immunosuppressive therapies for the treatment of cancer [5]. HCMV is also the leading cause of infectious complications following solid organ [6] or stem cell transplantation [7,8], manifesting as interstitial pneumonia, gastroenteritis, retinitis, hepatitis, graft failure, and death [9]. Additionally, HCMV is the most common infectious cause of birth defects and is also more prevalent than some well-known non-infectious congenital conditions, such as Down syndrome or fetal alcohol syndrome [10]. Congenital HCMV infection of the immune naïve fetus may result in mild to severe hearing loss, cognitive impairment, microcephaly, and/or cerebral palsy [10].
Much of the pathogenesis associated with HCMV infection can be attributed to the virus’s ability to establish a persistent life-long infection in the host through latency. In its latent state, the viral genome is maintained in the host cell without active replication or the production of new viral progeny but with the capacity to reactivate viral replication from latency in response to changes in the host cell. Thus, HCMV has evolved a sophisticated persistence strategy whereby the virus reaches an armistice with the host by limiting viral replication and pathogenesis to avoid immune clearance. This strategy includes periodic reactivation events that, despite being well-controlled by the immune system, still allow for the production of viral progeny and their spread to additional hosts. Serious disease arises when an uncontrolled reactivation event occurs as a result of immunosuppression in an otherwise healthy host or, in the case of congenital infection, leads to transmission of the virus to an immune naïve host [11]. Indeed, a recent analysis of congenital HCMV infections in the United States from 1988 to 1994 revealed that only one in four resulted from primary infection of the mother during pregnancy [12], highlighting the importance of reactivation from latency (or re-infection) in seropositive pregnant women. In addition, mounting evidence suggests that the health cost of asymptomatic persistence in otherwise healthy individuals increases as we age, with HCMV emerging as a risk factor for the development of age-related pathologies, such as cardiovascular disease [13,14,15,16], immune dysfunction [17,18], and frailty [19,20,21].
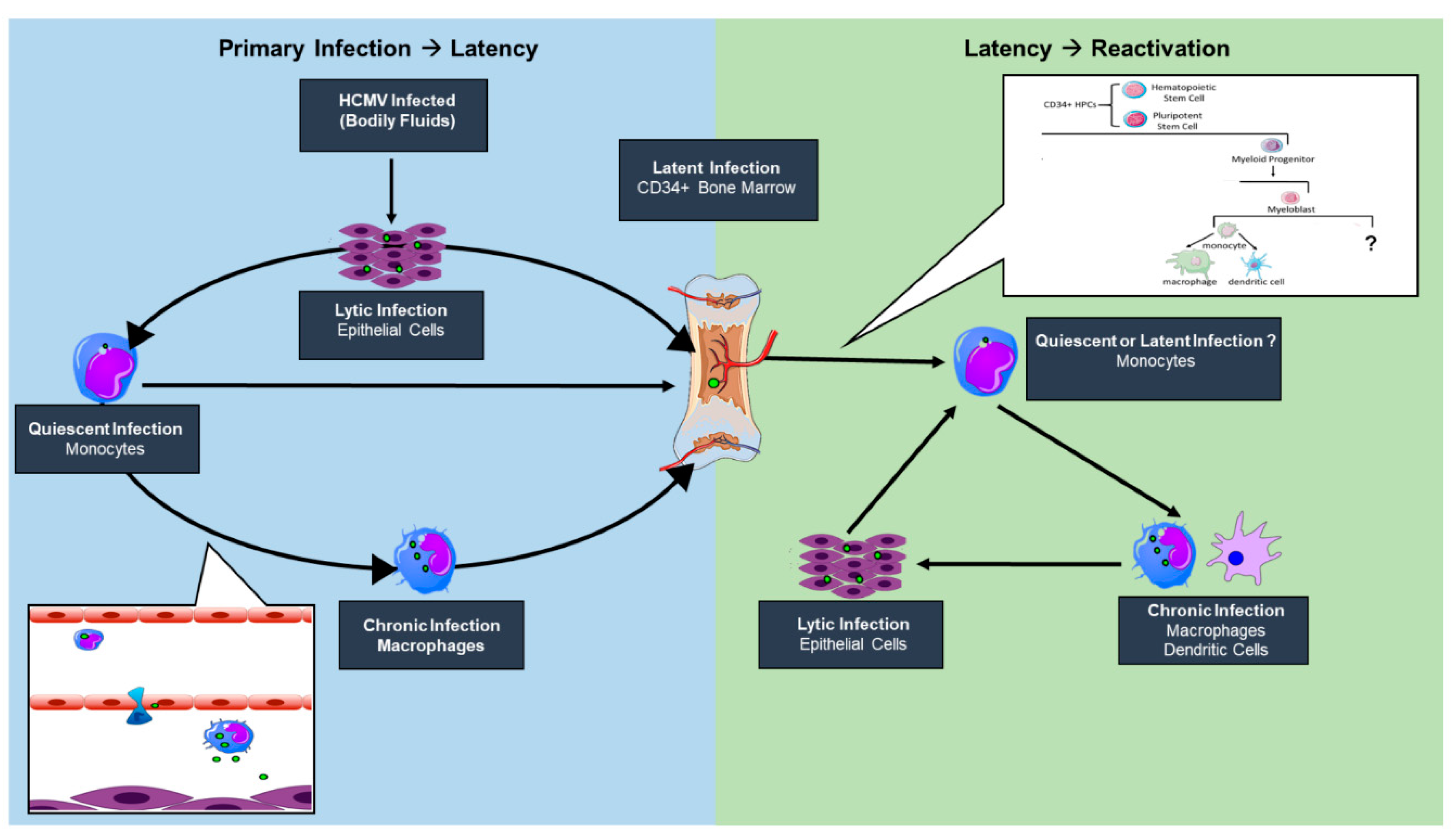
HCMV has a broad cellular tropism and infects a number of cell types during primary infection; however, the outcome of infection varies widely and is largely cell type-dependent. Some cell types, such as fibroblasts and smooth muscle cells, support robust viral replication and provide a platform for high levels of viral proliferation from which the virus is likely eventually cleared [22]. Epithelial and endothelial cells support a chronic or “smoldering” infection that presumably elicits a subtler immune response, resulting in low-level virus shedding that facilitates both interhost (e.g., oral epithelial cells) and intrahost (e.g., endothelial lining of the blood vessels) transmission of the virus [22,23]. Undifferentiated hematopoietic cells, such as CD34+ human progenitor cells (HPCs) and CD14+ monocytes, do not support a replicative infection and serve as a reservoir for HCMV latency as well as a platform for viral dissemination throughout the host [24,25,26,27,28,29,30,31]. Latent HCMV can then reactivate gene expression when the infected cell differentiates along the myeloid lineage toward a macrophage or a dendritic cell [32,33,34,35,36].
2. HCMV Latency Reservoirs
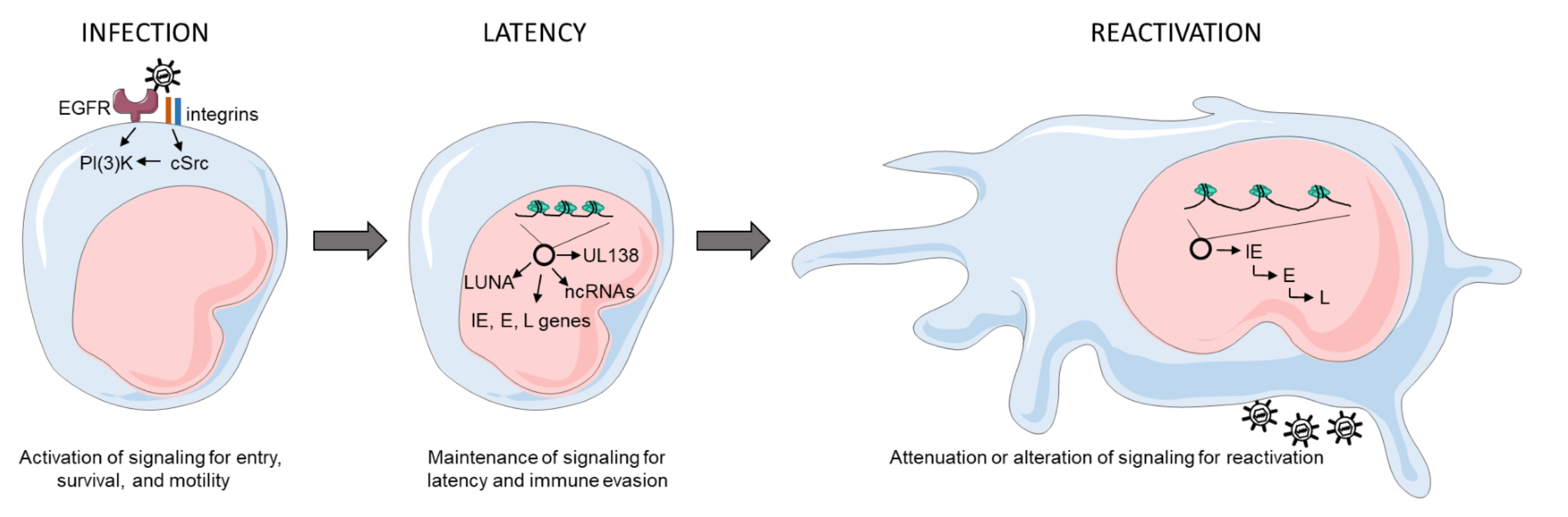
HCMV is highly species-specific but its cell tropism within the human host is vast, infecting endothelial, epithelial, fibroblasts, neuronal, monocytes/macrophages, granulocytes, and smooth muscle cells [37]. This broad cell tropism allows for the establishment of persistent infection, with dissemination beginning when a seronegative individual comes into contact with bodily fluids, such as tears, saliva, urine, semen, or breast milk, of an infected individual [38,39,40]. An initial round of lytic replication occurs in mucosal epithelial cells, where circulating peripheral blood CD14+ monocytes (PBM) in contact with infected epithelial cells may become infected (Figure 1). HCMV induces monocyte-to-inflammatory macrophage differentiation and survival of newly infected PBMs by activating EGFR/PI3K/Akt and integrin/src cellular signaling pathways during entry in the absence of de novo viral gene expression [41,42,43,44,45,46,47]. HCMV primes differentiating PBMs via the activation of the PI3K/NF-kappaB pathway to extravasate from the circulation to peripheral organ sites [48], establishing a “chronic infection” or a low-grade lytic infection reservoir in tissue-resident macrophages and neighboring epithelial layers. Further, monocytes can travel directly to the bone marrow and infect CD34+ hematopoietic stem cells, a major latency reservoir, establishing a life-long latent infection [49,50]. A direct infection of circulating CD34+ cells by mucosal epithelial cells then a subsequent migration to and seeding of the bone marrow is possible but remains elusive. HCMV can remain latent for extended periods of time within the hematopoietic cells; however, reactivation can occur following immune insult, allogenic stimulation, or differentiation signals [34] (Figure 1).
The state of knowledge in the field supports the notion that reactivation has no singular signal but is a multifaceted event where the virus is an active participant. We propose that a nexus of viral microRNAs and proteins, including ULb’ or tegument proteins, and cellular signaling play a role in the switch from latent to lytic infection in the bone marrow myeloid niche [51,52,53,54]. The relative contributions of viral and host factors in initiating or sustaining reactivation are far from clear. However, it appears that changes in the immune, homeostatic, or stress state at the level of the host or infected cell can result in changes in signaling that stimulate changes in viral gene expression, forging the path towards successful replication. In addition to altering viral activity, changes in host signaling associated with either latency or reactivation from latency have profound implications for hematopoietic differentiation. HCMV is known to be myelosuppressive following stem cell transplantation. Preferential differentiation of primary CD34+ hematopoietic progenitors into immunosuppressive monocytes following reactivation in a STAT-3-dependent manner has been demonstrated in vitro [55]. Whether these immunosuppressive monocytes are a “secondary latency” state or a “quiescent infection” still remains to be determined, as viral gene expression and the requirement of an external stimulus to promote differentiation has yet to be determined. Allogenic stimulation of monocytes [34] or monocyte-to-macrophage differentiation [35,36] stimulates HCMV reactivation, strongly suggesting that myeloid lineage differentiation and activation pathways are connected with HCMV reactivation. Further, differentiation of CD34+ progenitor cells to mature dendritic cells results in reactivated viral lytic gene expression and infectious virus production [56]. Defining alterations in differentiation between cells infected as progenitor cells or circulating monocytes would contribute to our understanding of viral persistence and the potential for reseeding peripheral tissue sites following bone marrow mobilization (Figure 1). However, there has been no study to date that comprehensively addresses the possibility of other cellular latency reservoirs within the host. While these may exist, our review will focus on work done to understand establishment and maintenance of or reactivation from latency in hematopoietic cell model systems.
3. HCMV Latency Models
In vitro systems have been vital for the study of HCMV latency for two reasons. First, the number of cells harboring latent virus in the infected host is extremely low (between 1:10,000 to 1:100,000 cells carry viral genomes) [57], and there is currently no way to identify and enrich for these cells. Second, CMVs are highly restricted in their tropism, so the human virus is challenging to study outside of cultured cells. While a number of cell culture models exist, these models present their own challenges associated with the difficulty in culturing primary hematopoietic cells and the limited utility of immortalized or transformed cells for the study of HCMV.
3.1. Primary Progenitor Cells
CD34+ HPCs are a heterogeneous subpopulation of cells ranging from pluripotent stem cells to cells in the earliest stages of commitment to myeloid or lymphoid lineage. Ex vivo culture should be carefully considered given that our understanding of the complexity of molecular events that induce or block differentiation is still very limited. This limitation creates a risk that ex vivo culture may result in artefact given that CD34+ HPCs are so prone to differentiate and may do so aberrantly given their culture conditions. To address this, stromal cell co-culture and stromal-free systems have been developed in an attempt to maintain the physiological relevance of cultured CD34+ HPCs. In the case of stromal cell support cultures, stromal cell clones recapitulate the hematopoietic microenvironment in vitro. These stromal cell cultures release a physiologically balanced milieu of cytokines and have been demonstrated to maintain hematopoietic progenitor cell phenotype and support stem cell self-renewal such that they retain their ability to repopulate the hematopoietic system in mice following xenotransplantation [58]. We have demonstrated the ability of these cultures to maintain latently infected cells [24,59,60,61]. Cell-free culture systems for CD34+ HPCs are also widely used that require the addition of cytokine cocktails to maintain cell phenotype and viability or promote differentiation down a specific lineage [62,63,64,65]. In all in vitro systems, reactivation is stimulated by inducing myeloid differentiation, often with fibroblast co-culture to detect virus produced from reactivation [24,56].
3.2. Primary Monocytes
Monocytes are a clear reservoir of latent virus and are critical to viral dissemination in the host. However, it remains unclear if direct infection of monocytes in vitro results in a bona fide latent infection [26,64] or a “quiescent infection” during which viral genomes are carried, but expression of viral genes is delayed and is associated with cytokine stimulation (e.g., granulocyte macrophage-colony stimulating factor, GM-CSF) and macrophage differentiation [32,34]. The Yurochko group demonstrated that monocyte-to-macrophage differentiation occurs in vitro at approximately 3 weeks post infection without the addition of GMCSF or MCSF as survival signals [66]. During this 3-week period, no lytic gene expression is detected [67]. It remains to be determined whether HCMV induces a de novo cellular signal to drive monocyte-to-macrophage differentiation in the absence of an external stimulus. If so, then monocytes that become infected in the circulation may exist as a distinct latency reservoir from monocytes that were infected as CD34+ HPCs and simply retained viral genomes through their differentiation. This primary “quiescent infection” model in monocytes has been used to study early entry and signaling events required for dissemination during HCMV infection, which is not possible in cells lines [41,42,67,68,69,70,71].
3.3. Cell Lines
Cell lines have long been sought as a model for HCMV latency as they offer the advantages of homogeneity, ease of cost and culture, and enhanced availability relative to primary cell systems. However, many cell lines do not faithfully recapitulate all the hallmarks of latency (e.g., the ability to produce progeny virus upon differentiation into a permissive cell type), leaving primary cell cultures as the gold standard for studying HCMV latency. Human monocytic leukemia cells (THP-1) have long been used to model the latent infection. In these cells, transcriptional reactivation from latency is achieved following treatment with phorbol ester [72], as subsequent viral immediate early (IE) genes [73] and early gene products (DNA polymerase) are expressed [74,75]. THP-1 cells have been successfully utilized for the study of viral gene expression [76,77], viral manipulation of cellular transcription factors [78,79], histone modification [80,81], and chromatin remodeling [73]. The CD34+ Kasumi 3 leukemic cell line and embryonic cell lines (ESC) are alternative in vitro models that harbor latent genomes and reactivate producing viral progeny [82,83]. Kasumi 3 have been used to study FAS ligand-mediated apoptosis through cellular IL-10 [84]. ESCs are typically differentiated into a neural precursor differentiation to study HCMV infection during neural development [85].
The NTERA-2 (NT2) cell line is a pluripotent human embryonal carcinoma cell line that can be differentiated into neurons following treatment with retinoic acid (RA) [86]. These cells have been used as a model for HCMV latency and reactivation, as HCMV establishes a latent infection in NT2 neuronal precursors that is reactivated following RA-stimulated differentiation [87]. While these cells do not model latency in myeloid lineage cells, they may serve as a model for latency in neuronal progenitor cells in vivo [88,89,90].
3.4. Animal Models
Human CMV is restricted in tropism to primary human cells. This barrier has complicated understanding CMV latency and the immune responses to infection. Murine CMV has been an invaluable tool for understanding latency, but it differs from the human virus in a number of significant ways. Namely, the smaller murine CMV lacks the ULb’ region of the genome that has been shown to encode a number of viral proteins important to latency and reactivation [51,91,92,93,94,95,96]. A humanized mouse model (huNSG) has been developed that supports a latent infection with the human virus in human hematopoietic cells, and recapitulates many of the phenotypes demonstrated in vitro [97]. UL136, a ULb’ gene, expresses 5 isoforms that have both replication-repressive (23- and 19-kDa isoforms) and -promoting (33- and 26-kDa) functions and contribute to latency and reactivation in CD34+ HPCs, respectively [94,95]. The in vitro phenotypes of the UL136-mutant viruses lacking specific isoforms are largely recapitulated in huNSG mice [94]. The 25-kDa UL136 isoform differs in its in vitro and in vivo phenotypes; mutation of this isoform results in a virus more prone to reactivate in vitro but fails to reactivate in vivo. This finding suggests that these systems may allow differentiation of phenotypes important for infection in the host. Further, binding of HCMV UL7 to Fms-like tyrosine kinase 3 receptor (Flt-3R) promotes differentiation in the huNSG mouse model, and is required for reactivation in CD34+ HPC and CD14+ monocyte models as well as in huNSG mice [98]. These cornerstone studies establish the importance of the humanized mouse model in defining viral or host factors important to latency and in understanding the impact of HCMV latency on signaling, immune responses, and hematopoiesis.
4. CMV-Mediated Control of Host Signaling for Latency/Reactivation
CMV is a master manipulator of the cellular environment with the seemingly simple goal of persisting within its host. While the virus infects a broad range of cells, one commonality across cell types is that CMV manipulates a variety of signaling pathways to reconfigure the homeostatic environment for viral objectives. However, our understanding of CMV-mediated manipulation of host signaling has been largely generated through studies in fibroblasts during active viral replication, while a significantly smaller portion of work has been targeted toward cell types that support latency. For the purposes of this review, our focus will be on the body of work pertaining only to latency models to highlight the importance of signaling during latency and the necessity for future work in this area.
4.1. CMV Initiation of EGFR and Integrin Signaling: Starting off on the Right Receptor
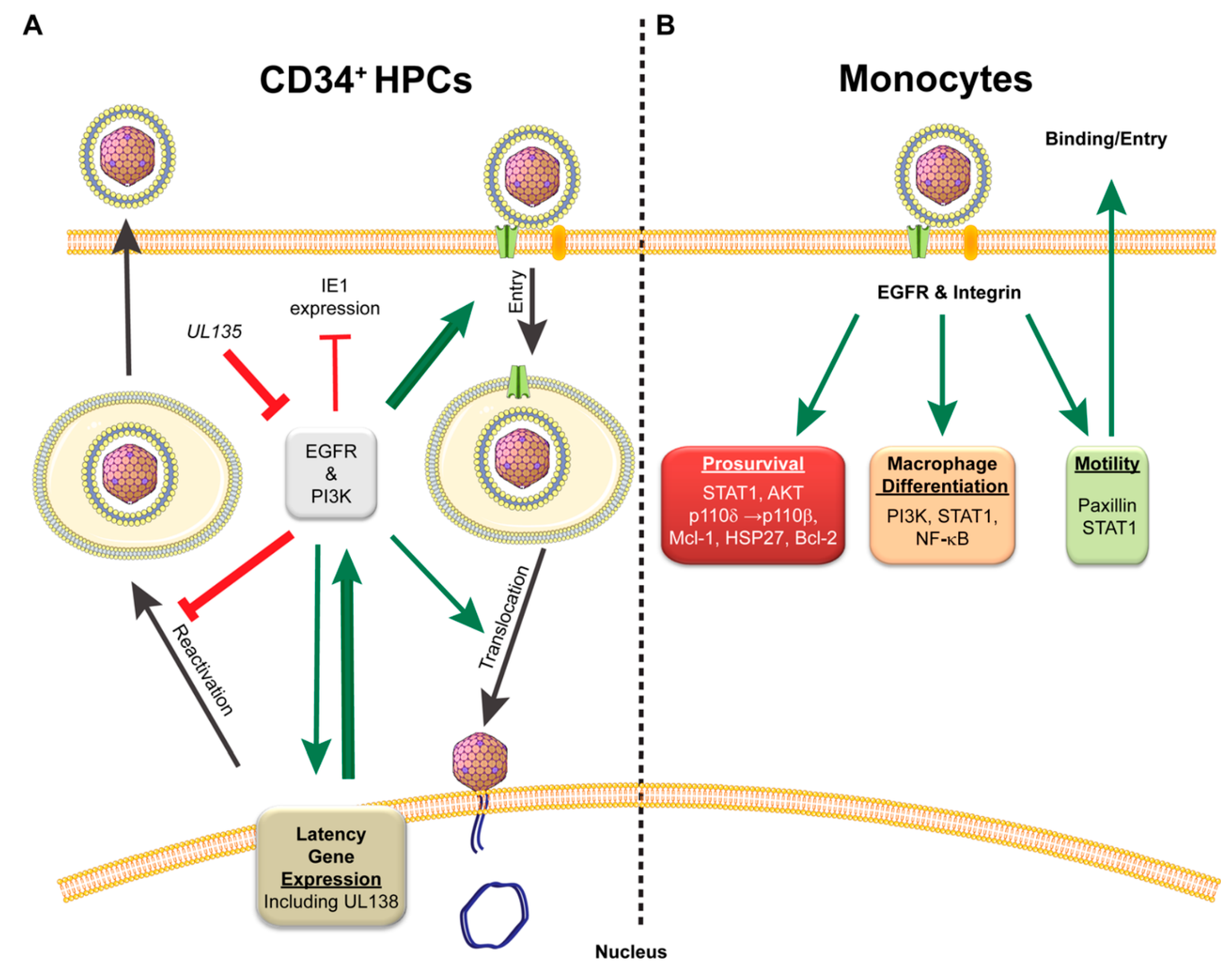
Cytomegalovirus infection is a well-choreographed series of events to take over a cell from initial binding and entry to successful virus progeny production or the establishment of latency, all of which contributes to life-long persistence within the host. To achieve early control of cells destined for latency, CMV binds signaling receptors, such as epidermal growth factor receptor (EGFR), platelet-derived growth factor receptor (PDGFR), and integrins, on the surface of the cell to facilitate viral entry and to initiate cellular signaling [42,52,99,100]. While CMV does enter through PDGFR in fibroblasts, endothelial cells, and epithelial cells, little is known about its role in signaling during infection in latency cell types, so focus will be given to EGFR and integrins for this section. The use of both EGFR and integrins as entry receptors initiates downstream signaling events that can prime the cells for the establishment of a latent infection prior to the virus even entering the cell (Figure 2). The activation of EGFR allows the virus to manipulate cellular proliferation, differentiation, migration, survival, innate immunity, and DNA repair [101,102,103,104,105], while integrin signaling regulates many of the above functions but also interacts with and influences intra- and intercellular homeostatic signaling by tyrosine kinase receptors, such as EGFR (Reviewed in [106,107,108]).
In CD34+ hematopoietic progenitor cells (HPCs), EGFR and integrin signaling is integral for CMV entry and the establishment of latency [52,68,69,70]. Kim et al. demonstrated that blocking EGFR signaling, either through chemical inhibition or with an interfering antibody, retains virions at the cell surface with EGFR and diminishes entry into CD34+ HPCs [52]. Inhibition of phosphatidylinositol-4,5-bisphosphate 3-kinase (PI3K), downstream of EGFR, also inhibits entry of CMV into CD34+ HPCs. EGFR trafficking is required for translocation of the viral genome to the nucleus to initiate CMV infection, and inhibition of EGFR signaling shortly after entry significantly delays gene expression. Interestingly, CMV infection of CD34+ HPCs results in a transient increase in surface expression of EGFR early on in infection [51]. This is in sharp contrast to infection in fibroblasts, where EGFR levels are reduced 50–70% compared to uninfected controls, and inhibition of EGFR kinase activity further enhances virus replication [51]. This cell-type-dependent regulation of EGFR supports a possible requirement for EGFR signaling during CMV infection of CD34+ HPCs for the establishment of latency, and loss of EGFR or PI3K signaling enhances reactivation potential during infection [51]. Similar results were seen in HSV-1 and pseudorabies virus infection when PI3K signaling was inhibited [109,110]. Further, inhibition of EGFR by AG1478, a chemical inhibitor of EGFR kinase activity, increases IE1 gene expression while decreasing the UL138 latency determinant in CD34+ HPCs [52]. The changes in EGFR signaling in CD34+ HPCs may explain some of the infection-induced alterations in myeloid differentiation, as levels of the inflammatory cytokine interleukin-12 are increased during CMV infection and further exacerbated by inhibition of EGFR signaling [52]. Collectively, this work demonstrates a pivotal role for EGFR and virus-mediated control of EGFR signaling in regulating replicative and latent states during HCMV infection.
The intertwining of integrin, specifically β1 and β3, and EGFR signaling has been shown to be critical for HCMV infection of monocytes [69,70]. Virus entry into monocytes also requires the pUL128–131 pentameric complex proteins, which activate the integrin-c-Src signaling pathway to alter the actin cytoskeleton [70]. Within 5 min following entry, infection further alters EGFR-dependent Stat1 signaling to generate pro-survival and motility signals [99]. CMV maintenance of pro-survival signaling and promotion of cellular motility is driven through AKT and STAT1 pathways and represents a major viral target during monocyte infection [41,42,67,68,99]. Within 15 min of gB binding to EGFR on a monocyte, CMV increases AKT signaling in a PI3K-dependent manner [41,67]. The activation of Akt occurs through shifting PI3-kinase preference from p110δ to p110β and by stimulating SH2-domain-containing inositol 5-phosphatase 1 (SHIP1)-mediated conversion of PI3P to PI2P. These alterations to signaling result in the maintenance of AKT activation for up 72 h, allowing for the monocytes to survive beyond the typical 48-h survival gate [41]. This non-canonical activation of AKT signaling during HCMV infection preferentially activates mTOR to promote the expression of myeloid cell leukemia sequence 1 (Mcl-1) and heat shock protein 27 (HSP27), which prevents the cleavage of caspases and the initiation of apoptosis [42]. As CMV infection progresses, Bcl-2 is required to maintain the anti-apoptotic state to counter an upregulation in Bax expression [111]. These changes in signaling restructure the cytokine environment to increase motility and migration [69]. Enhanced survival and motility of infected monocytes aids dissemination of CMV throughout the infected host to tissues [112]. Additionally, CMV-mediated activation of PI3K and STAT1, as well as NF-κB, also promotes monocyte-to-macrophage differentiation, which is necessary to promote viral replication and dissemination within and between hosts [44,99]. Together, the body of work demonstrates that CMV manipulates EGFR, integrins, and their downstream pathways for infection, persistence, and dissemination in the host.
4.2. CMV Manipulation of the Cellular Environment for Establishment and Maintenance of Latent Population
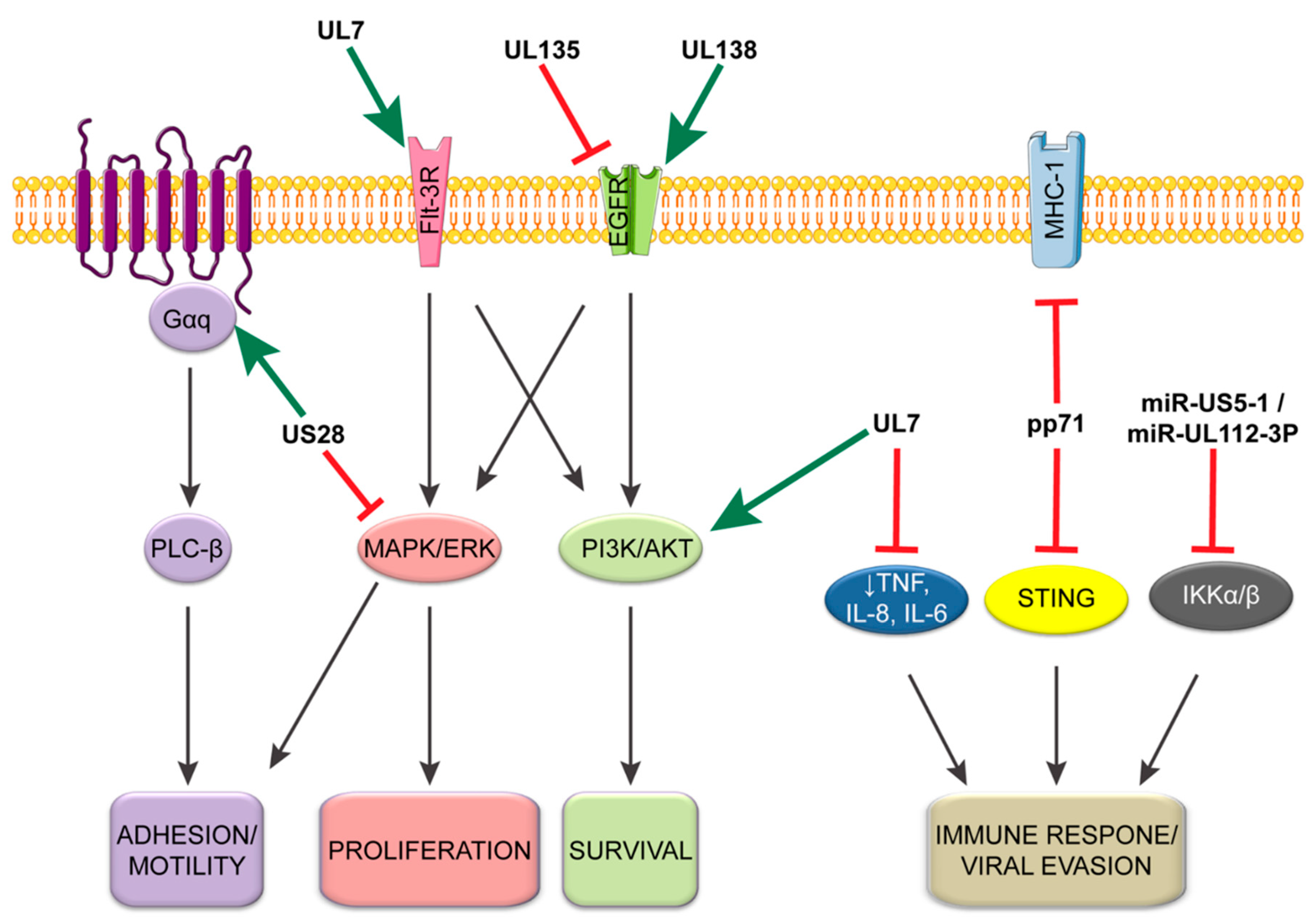
Herpesvirus latency is associated with profound changes in cell signaling to modulate survival, sensing, and differentiation [53,113,114]. In the case of HCMV infection in monocytes, these changes include increasing MCP-1 to enhance cellular migration, promotion of immune response genes, increase protein and lipid metabolism, and a dramatic shift in the differentiation profile toward macrophages [115,116,117]. Some of these changes are the result of signaling outcomes initiated during entry, as described above, but many other mechanisms exist to stabilize and maintain a latently infected population. This subsection will focus on those other pathways (Figure 3).
Like all viruses, CMV must evade the cellular intrinsic detection of foreign DNA. To accommodate this, pp71, a tegument protein encoded by CMV UL82, binds to the stimulator of interferon genes (STING) to inhibit its antiviral response [118]. The interaction between the pp71 tegument protein and STING prevents the translocation of STING from the ER to microsomes, subsequently preventing the recruitment and phosphorylation of IRF3 and TBK1. Loss of pp71 results in increased IFNβ1, ISG56, TNF, and IL-6 in THP-1, macrophage, and dendritic cells as well as an increase in ISG15 and IL-1β in THP-1 cells. This pp71-mediated evasion is also supported by an alternate function of the proteins, which decreases cell surface expression of major histocompatibility complex 1 (MHC-I) during CMV infection to prevent detection by the adaptive immune system [119]. Together, these results suggest that pp71 plays an important role in modulating CMV immune evasion following entry across cell types and might be important for establishing latency.
The UL133–UL138 locus encoded within the ULb’ region of the HCMV genome is dispensable for replication in fibroblasts but required for latency in hematopoietic cells. The ULb’ region is present in clinical and low-passage strains of the virus (e.g., Merlin, TB40/E, FIX), but lost during serial passage of all laboratory strains (e.g., AD169, Towne short) [120,121,122]. Four genes are encoded from this locus: UL133, UL135, UL136, and UL138 [91,123]. Replication-suppressing functions important for the establishment or maintenance of latency have been attributed to UL133 and UL138 [91,92,93]. By contrast, UL135 is important for reactivation from latency and functions, in part, by overcoming the growth-suppressive function of UL138 [124]. UL136 is more complicated in that it is expressed as five protein isoforms differing only in their amino terminal ends [95]. UL136 isoforms are expressed with later kinetics relative to the other proteins encoded by the UL133–UL138 locus, at least in fibroblasts. The two large membrane-bound UL136 isoforms are required for reactivation in CD34+ HPCs and in humanized mice, whereas the two small soluble isoforms suppress replication for latency [94]. Given the later expression kinetics of UL136 and the seemingly overlapping function of the isoforms with UL136 and UL135 in latency, a model can be envisioned whereby UL138 and UL135 contribute to the establishment of replicative or latent states, respectively. UL136 follows, perhaps in response to reactivation cues, and interplay between the UL136 isoforms facilitates maintenance of latency or reactivation. This model, while intriguing, has yet to be validated and much remains to be understood about the mechanisms by which UL136 isoforms mediate latent versus replicative states.
While much remains to be understood about the mechanisms by which UL133–UL138 proteins work to regulate states of latency and replication, the opposing functions of UL138 and UL135 in mediating latency and reactivation, respectively, have been attributed to their opposing regulation of epidermal growth factor signaling (EGFR) [51]. EGFR or downstream PI3K signaling pathways inhibit replication in fibroblasts following viral entry, and HCMV infection results in the downregulation of EGFR early in infection [51,125]. Consistent with this relationship, inhibition of EGFR and PI3K facilitates reactivation when combined with a differentiation stimulus. UL138 has been shown to upregulate EGFR at the cell surface, whereas UL135 stimulates the turnover of EGFR from the cell surface [51]. UL135 stimulates EGFR turnover by interacting with the host adapter proteins CIN85 and Abi-1, which are important for the intracellular trafficking of EGFR toward the lysosome [96]. It will be important to determine how CMV modulation of EGFR trafficking affects downstream signaling to affect latency and replication and how the function of UL133 and the UL136 isoforms contribute to this regulation. Receptor tyrosine kinase signaling, and specifically EGFR or analogous pathways, has been shown to be important for latency in other herpesvirus infections, including HSV-1, EBV, and KSHV [126,127,128].
The viral G-protein-coupled receptor (vGPCR) US28 also plays an important role in modulating the host environment in latency. During lytic infection in fibroblasts, US28 promotes MAPK, NF-κB, and CREB signaling to drive MIEP activity, which is facilitated by US28 binding a variety of chemokines and G proteins [129,130,131,132,133]. In the context of latency in primary CD34+ HPCs or the Kasumi-3 cell line, US28 is highly expressed relative to the immediate early IE1 gene and is required for the establishment of latency [134]. However, in the context of latency and in contrast to lytic infection, US28 is reported to attenuate NF-κB and MAPK signaling [134,135]. In the absence of US28, infected CD34+ HPCs have increased IE1 expression prior to and following a reactivation stimulus [134]. Additionally, US28 promotes cellular adhesion of monocytes on endothelial cells by activating Gαq/PLC-β signaling, which is dependent on G-protein coupling and independent of chemokine binding [136]. These findings demonstrate a dichotomy in US28 function and cell-type-specific infection outcome. It will be important to determine how the function of US28 switches to support latent versus replicative states in the context of reactivation from latency.
Another important aspect of persistence is the ability of viruses to assuage inflammatory and immune responses. CMV induces expression of the anti-inflammatory cytokine interleukin-10 (IL-10) to block FAS-mediated apoptosis of latently infected CD34+ HPCs [84]. By increasing IL-10, CMV promotes the expression of the potent FAS/TNFR1 PEA-15 [84,137]. Knockdown of either IL-10 or PEA-15 increased apoptotic susceptibility of latently infected CD34+ HPCs [84]. The anti-inflammatory state is further reinforced by the CMV miRNAs miR-US5-1 and miR-UL112-3P targeting IKKα and IKKβ to decrease IL-1β and TNF-α responsiveness [138]. These viral miRNAs target the 3’ UTR of IKKα and IKKβ in response to NF-κB signaling. Loss of miR-US5-1 and miR-UL112-3P results in increased NF-κB signaling and increased IL-6, CCL5, and TNF-α in THP-1 cells. Such increase in IL-6 particularly has been demonstrated to increase CMV reactivation from latently infected dendritic cells through ERK-MAPK pathways by promoting IE gene expression [139].
In addition to UL135, CMV UL7 also functions to regulate signaling and is required for reactivation. UL7 is secreted and functions as a ligand, sharing many similarities to the signaling lymphocyte-activation molecule (SLAM) CD229 [140]. Like many SLAM family members, UL7 is able promote leukocyte adhesion, which it does independent of SLAM family member interaction. UL7 activates PI3K/AKT and MAPK/ERK pathways by serving as a ligand for Fms-like tyrosine kinase 3 receptor (Flt-3R) in CD34+ HPCs [98]. UL7 suppresses the proinflammatory response by decreasing TNF, IL-8, and IL-6 expression in myeloid cells and promotes myelopoiesis and differentiation of monocytes and CD34+ HPCs. Further, in HCMV infection UL7 is required for HCMV reactivation from latency in both in vitro CD34+ cells and in vivo humanized mice as well as G-CSF-induced myelopoiesis in humanized mice. These results suggest that UL7 is a potent viral autocrine regulator of reactivation and hematopoietic differentiation. It will be interesting to understand how the UL7 promotion of PI3K/AKT and MAPK/ERK signaling contributes to reactivation in light of other studies which show that EGFR and PI3K signaling contribute to latency. These studies may indicate the importance of timing in the regulation of host signaling.
The perversion of cellular signaling pathways during CMV infection is critical to ensure cell survival and establish an intracellular environment conducive to latency and to avoid immune detection. There is still a substantial body of work that needs to be performed to fully define the degree to which CMV controls latently infected cells. This includes understanding the subtle signaling differences in the various latency models, such as monocytes versus dendritic cells versus CD34+ HPCs, and how these may change over time during latent infection. Additionally, there are many signaling pathways that lack sufficient data in latency cell types to determine how CMV interferes with their normal function.
In going forward, WNT is an important signaling pathway to consider in CMV latency as it is critical to the control of latency in other herpesviruses. Herpesviruses typically inhibit WNT signaling during replication and promote WNT signaling for the establishment of latency (Reviewed in [141]). Both EBV and KHSV target Glycogen synthase kinase 3 α/β (GSK-3) of the WNT destruction complex to prevent the degradation of β-catenin during latency [142,143,144]. The EBV LMP2A promotes AKT signaling to inhibit GSK-3 destruction of β-catenin [142], while the KSHV LANA interacts with the GSK-3 and sequesters it in the nucleus [143,144]. Additionally, the EBV LMP-1 increases β-catenin levels by blocking its ubiquitination [145,146,147]. The multifaceted approach evolved by γ-herpesviruses to maintain WNT signaling underscores the importance of this pathway to viral latency. Of the α-herpesviruses, the bovine herpesvirus 1 (BHV1) ORF2 protein promotes WNT signaling through interactions with high-mobility group AT-hook protein (HMGA1), mastermind-like protein 1 (MAML1), and AKT3 to promote WNT signaling [148,149,150]. Interestingly, reactivation of BHV1 results in the production of Dickkopf-1 (DKK1) and secreted Frizzled-related protein 2 (SFRP2), which antagonize WNT signaling [148]. This suggests that controlling WNT signaling can serve as a molecular switch between latency and reactivation that may be important across herpesviridae. In CMV infection, lytic replication inhibits WNT signaling by increasing β-catenin turnover or sequestration in the viral assembly compartment [151,152]. However, despite this, inhibition of WNT signaling has been shown to inhibit immediate early gene expression and replication [153]. It will be important to define the role of WNT signaling in modulating states of replication and latency in CMV infection.
5. Cellular Control of the Viral Gene Expression Program for Latency/Reactivation
During replicative infection, such as in fibroblasts, the HCMV genome is transcribed in a temporal cascade of three kinetic gene classes designated as immediate early (IE), early (E), and late (L) [154,155]. The major immediate early (MIE) genes are expressed first following activation of the major immediate early promoter (MIEP) by cellular transcription factors and viral tegument components [156]. The MIE gene products (IE1-72 kDa and IE2-86 kDa) transactivate viral early gene promoters [157,158], leading to viral DNA replication and expression of the viral late genes [159]. Because initial activation of the MIEP occurs without a requirement for de novo synthesis of viral proteins, it represents a crucial control point where the cellular environment can exert control over the viral replicative cycle. As such, regulation of the MIEP has been the focus of numerous studies aimed at unravelling the virus–host interactions that govern viral gene expression during latency and reactivation. The MIEP is chromatinized and silenced during latency [56,81,160]. This section reviews our current knowledge of how the MIEP is regulated to control the transition between latent and replicative states of infection.
5.1. Cellular Transcription Factors
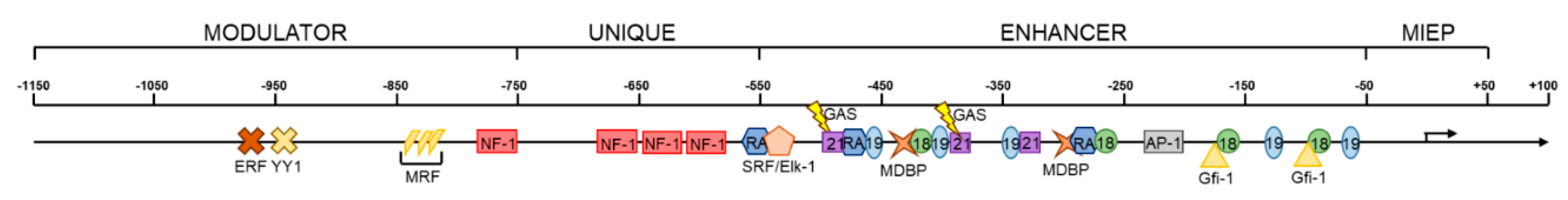
The MIEP consists of a core promoter (−65 to +3 nucleotides from the MIE transcription start site), an upstream enhancer (−520 to −65 nucleotides), a unique region (−780 to −610 nucleotides), and a modulator (−1145 to −750 nucleotides) (Figure 4). The core promoter is sufficient for low-level transcription of MIE genes [161]. The enhancer element, which augments transcription from the MIE locus [162,163,164], contains a number of small repeat sequences (18-bp, 19-bp, and 21-bp) that function as binding sites for activating cellular transcription factors [161,162,163,164,165,166]. The 18-bp repeats bind NF-κB/rel [167], the 19-bp repeats bind CREB/ATF [168], and the 21-bp repeats bind Sp-1 [169]. Although the cyclic AMP (cAMP) response element binding (CREB) protein sites and the NF-κB sites have received the most attention, additional binding sites for activators of transcription have been identified in the enhancer, including sites for AP-1 [170], retinoic acid (RA) [171,172,173], serum response factor (SRF)/Elk-1 [174], and gamma-interferon activating sequence (GAS) [175]. Binding sites for repressive transcription factors methylate DNA binding protein (MDBP) [176] and growth factor independence 1 (Gfi-1) [177] have also been identified in the enhancer, and subsequent studies found that the 21-bp repeats also bind the repressive transcription factors Yin Yang-1 (YY1) [178] and Ets-2 repressor factor (ERF) [179], which likely reflects the repressive effect of this region in undifferentiated hematopoietic cells [180]. The unique region contains a cluster of binding sites for the activating transcription factor nuclear factor-1 (NF-1) [181,182]. The modulator contains binding sites for the repressors ERF [179] and YY1 [178], formerly identified as modulator binding factors (MBFs) 1 and 4, respectively [180,183], and modulator recognition factor (MRF) [184]. The enormous number of cellular transcription factors potentially impacting MIEP activity makes for a complex landscape of regulation.
In transient transfection assays, the modulator was shown to alter transcription from the MIE locus in a differentiation-dependent manner. The modulator’s effects are repressive in undifferentiated THP-1 and NT2 cells; however, the modulator stimulates MIE gene expression in permissive fibroblasts [183,184,185,186]. Because undifferentiated progenitor cells harbor a latent HCMV infection, whereas their differentiated counterparts support viral replication, the cellular transcription factors binding the modulator appeared to be ideal candidates for determining whether the virus enters a replicative or latent state of infection. In addition, a number of these studies in non-permissive cell types seemed to indicate that the 21-bp repeats in the enhancer region, in addition to the modulator, are responsible for the inhibition of MIEP activity in undifferentiated cells [180,185,186], consistent with binding of the repressive factors YY1 [178] and ERF [179] to the 21-bp repeats. Given these findings and the discovery that the modulator element showed changes in DNAseI hypersensitivity following differentiation to a cell type that is permissive for viral replication [187], it was hypothesized that differential binding of cellular transcription factors in latency-associated versus permissive cells played a key role in determining IE expression in each cell type. Emphasis was put on examining the balance of activating versus repressive transcription factors present in replication-permissive cells compared to latency-associated cell types, with unclear results.
Differentiation of non-permissive cells into a permissive phenotype correlates with increases in the expression of transcription factors that stimulate transcription from the MIEP by binding the enhancer region. For example, stimulation of the cAMP/PKA signaling pathway [188,189] or addition of phorbol 12-myristate 13-acetate (PMA) [190], which would activate CREB [188,189] or CREB and NF-κB [190], increases IE expression in latently infected NT2 cells, and upregulation of NF-kB during myeloid differentiation also contributes to increased IE expression [191]. However, the stimuli used to activate cell signaling pathways in these assays would also induce additional cellular changes that accompany differentiation and could trigger viral reactivation by mechanisms other than upregulation of activating transcription factors. Transcription factors that repress the MIEP in transfection assays (e.g., modulator binding factor 1 (MBF1) or ERF [179,180,183], MRF [184], YY1 [178], and Gfi-1 [177]) appear to be expressed at higher levels in undifferentiated latency-associated cell types when compared to cell types that are permissive for viral replication; however, differentiation of these cells results in little change in the mRNA expression of either YY1 or ERF [179]. It remains possible that the differences in YY1 and ERF expression levels following differentiation are the result of post-translational regulation and not upregulation of the genes following cell differentiation. Despite similar expression levels of YY1 and ERF both prior to and following differentiation, YY1-mediated [178] and ERF-mediated [179] repression of the MIEP are decreased following differentiation stimulus. Importantly, in contrast to the transient transfection studies, deletion of the modulator in recombinant viruses does not have a measurable effect on IE1 and IE2 transcription during experimental infection of permissive fibroblasts or undifferentiated NT2 or THP-1 cells [192], suggesting that additional viral factors may also play a role in repressing MIE viral gene expression. Going forward, it will be important to understand how cellular transcription factors regulate the viral genome to affect states of latency or reactivation. This will be important to understand not only in the major immediate early region, but also throughout the genome, and particularly with regard to latency-associated genes.
5.2. Chromatinization
YY1 [178] and ERF [179] binding to the MIE enhancer and modulator regions and their repression of MIE gene expression has been attributed to histone deacetylase (HDAC)-mediated chromatin remodeling of the MIEP enhancer region to silence IE gene expression [193,194]. An opposite function been shown for CREB, which acts cooperatively with mitogen and stress-activated kinases (MSKs) to initiate chromatin remodeling at the MIEP required for expression of IE genes and viral reactivation [195]. Therefore, a major area of focus in latency and reactivation across the herpesvirus field has become regulation of viral gene expression through epigenetic modifications by the cellular machinery [196].
CMV gene expression is controlled by histone modifications (acetylation or methylation), and the CMV genome itself is not known to be methylated [197,198]. The CMV genome is not associated with histones when packaged in the virion [199,200], but becomes associated with histones within 30 min of entering a permissive fibroblast [201]. Some post-translational modifications to histones bound to DNA, such as acetylation, result in a more open chromatin structure for gene expression [202,203]. Methylation is more complex, with a single methylation of most residues serving as an activating mark, and accumulation of additional methyl groups tipping the balance toward repression on some histone residues but not others. Modifications to some residues are constitutively repressive whereas others are facultatively repressive and thus more readily reversible. The pattern of histone association during infection in permissive fibroblasts correlates with what is known about the dynamics of promoter activation and lytic gene expression. During immediate early times of infection, early and late promoters are associated with repressive histone marks; as the infection cycle progresses, early and then late promoters gradually become associated with histones carrying activating marks [204].
Because of its central role in initiation and progression of the viral replication cycle, much of the focus on chromatinization has centered on the MIEP. The MIEP is associated with relatively high levels of histone deacetylases (HDACs) and hypoacetylated histones during latent infection, consistent with a closed chromatin formation and less transcription from the locus [56,160,198]. Following differentiation into a permissive macrophage or dendritic cell, there is less association with HDACs and higher levels of histone acetylation [56,160,198]. In comparison to the transcriptionally repressed MIEP, the promoter that drives latency unique nuclear antigen (LUNA) expression is euchromatic during latent infection, indicating transcriptional activity [49]. Ioudinkova et al. demonstrated that the MIEP and representative early, early-late, and late promoters were preferentially associated with acetylated histone 3 at lysine 9 (H3K9) (a marker of activation for cellular genes) and not dimethylated H3K9 (a marker of repression) during lytic infection of differentiated THP-1 cells [81]. This correlated with detectable accumulation of IE mRNA and protein in these cells. However, during latent infection of THP-1 cells, the MIEP enhancer region was associated with neither activating acetylated H3K9 nor repressive dimethylated H3K9 histone modifications, although the early, early-late, and late promoters were associated with repressive dimethylation at H3K9 [81]. The finding that the MIEP enhancer is associated with neither activating nor repressive histone modifications while early and late promoters were repressed is consistent with reports that additional blocks downstream of IE expression exist as a barrier for viral reactivation [76]. This finding may also reflect a need for epigenetic modifications in the MIEP region to be readily reversible in response to changes in the cellular environment to allow viral reactivation to be triggered. This notion is consistent with studies in THP-1 and NT2 cells, which show that polycomb repressive complex 2 (PRC2), which is known to catalyze the formation of reversibly silenced facultative heterochromatin domains marked by histone H3 trimethylated at lysine 27 (H3K27me3), plays a role in regulating viral gene expression during latency [205].
It is presumed that the MIEP is activated due to a reversal in repressive chromatin following differentiation along the myeloid lineage into a permissive macrophage or dendritic cell. Treatment of quiescently infected NT2 cells with an HDAC inhibitor permits viral replication [192], and this same treatment results in disruption of repressive heterochromatin at the MIEP [198]. In latently infected NT2 cells, use of an HDAC inhibitor in combination with activation of the cAMP/PKA signaling pathway results in a synergistic increase in IE expression [188], indicating that a complex mechanism involving multiple cellular (and viral) factors is required to trigger viral reactivation. These changes correlate with increased levels of IE mRNA and protein, typically used as a proxy for MIEP activity. However, MIEP activity has not been explicitly demonstrated following reactivation. While it is clear that epigenetic regulation of the viral genome contributes to the establishment of, maintenance of, and reactivation from latency, further studies are needed to fully decipher the complex mechanisms that regulate the transition between latent and replicative states of infection.
5.3. Viral Factors and Chromatinization
During replicative infection, the MIEP is initially silenced following formation of a repressive chromatin structure by the nuclear domain 10/promyelocytic leukemia nuclear body (PML-NB) proteins Daxx and ATRX [200,204,206,207,208]. Early during infection in fibroblasts, the viral tegument protein pp71 traffics to the PML-NBs in the nucleus and displaces ATRX [209], then induces sumoylation [210] and ubiquitin-dependent degradation [207,211] of Daxx, resulting in IE expression [212,213,214,215,216,217]. After IE gene products are made, they further neutralize repression by PML-NB proteins and HDACs [200,206,207,218,219,220]. The PML-NB defense is also present in latency-associated NT2 and THP-1 cells, and could contribute to the establishment of latency [65,77]. In THP-1 cells, pp71 is sequestered in the cytoplasm, resulting in high levels of Daxx and repression of IEs [65]. IE and E gene expression is rescued following use of an HDAC inhibitor (VPA) or Daxx knockdown in NT2s and THP-1s infected with the AD169 lab strain of HCMV. However, the reactivation is ultimately aborted since viral L gene expression was not detected [65]. IE expression is not rescued by VPA treatment or Daxx knockdown during infection with clinical strains, suggesting a redundant mechanism in the ULb’ region for silencing IE expression during the establishment of latency [65]. One such factor is likely the viral latency determinant UL138, as its expression enhances the formation of repressive chromatin around the MIEP by inhibiting recruitment of lysine-specific demethylases (KDMs) [221] and represses viral replication for latency [91,92,222]. Incidentally, one of the KDMs whose function is blocked by UL138 has a specific binding activity to reverse facultative H3K27me3 histone modifications [223], suggesting a potential mechanism by which UL138 contributes to the maintenance of latency.
5.4. Viral Gene Expression during Latency
During latent infection in hematopoietic cells, it has been presumed that the viral genome is largely transcriptionally silent, particularly the MIEP, as early studies did not detect IE transcripts or proteins in latently infected cells [30,36,224]. This assertion is predominantly supported by transient transfection assays in undifferentiated NT2 and THP-1 cells [183,225] and by gene therapy studies where attempts to use the CMV MIE promoter have been unsuccessful [226]. More recent studies using highly sensitive techniques that can detect lower levels and a greater breadth of gene expression have shown that IE genes are transiently expressed at low levels following initial infection, and then silenced as latency is established [24,26,227,228]. While the gene expression detected could be attributed to infection in vitro and the heterogeneity of infected cells, these findings distinctly raise the possibility that low-level gene expression might exist in cells which are not productively replicating the virus [229]. Further blurring the lines of distinction between replicative and latent states of infection, binding of RNA polymerase II to the MIE region has been reported in latently infected cells [230] and transcripts originating from the MIE locus have been detected during both the establishment and maintenance of latency [27,64,229,231,232]. The true nature of this seemingly more dynamic latency is still somewhat obscured by the limitations inherent in using heterogeneous populations of hematopoietic cells that could potentially reflect different latency programs, such as those that are hallmarks of EBV latency [233].
These observations pose a series of new and important questions: if the MIEP is not completely silent during the latent infection, is transcription from the MIEP considerably reduced such that it sometimes falls below our limits of detection? Are there transient bursts of viral gene expression from the MIEP that only rarely reach a threshold required for the full reactivation of viral gene expression? Although IE transcripts are detected in the context of latent infection, there is no evidence that IE1 and IE2 proteins are synthesized or that this results in the production of progeny virions. Indeed, a recent study showed that ectopic expression of IE1 and IE2 in undifferentiated THP-1 cells is enough to activate early gene expression, but does not result in production of viral progeny [76], indicating that there are multiple blocks that must be navigated before full reactivation is triggered. An important one appears to be cellular differentiation, suggesting that a complex interaction of both viral and cellular factors is ultimately required for successful viral reactivation, as is seen in the alpha herpesvirus family [109,234].
The established model where reactivation of latent HCMV is triggered by differentiation along the myeloid lineage rests on the assumption that differentiation-dependent changes in the cellular environment trigger reactivation of the MIEP as a first step in initiating the viral gene expression program, although this has not been explicitly shown. A non-canonical IE1 transcript that includes exon 4, but not exons 1 and 2, was recently discovered in latently infected CD34+ cells [64]. This transcript arises from a cryptic promoter in the MIE locus, other than the MIEP, and encodes a smaller IE1 protein (IE1 exon 4) that is also expressed in latently infected CD34+ cells and is proposed to tether the latent viral genome to cellular chromatin to promote viral genome maintenance and amplification in dividing CD34+ cells [230]. This finding introduces the possibility that chromatin rearrangement during latency promotes silencing of the MIEP, but leaves alternative MIE promoters open and active to produce a latency-specific transcriptional profile. Further, other promoters have been identified to reside within intron A of the MIE gene locus that contribute to IE1 and IE2 expression in fibroblasts [235] and could contribute to reactivation in a cell-type-dependent manner. It remains to be determined if there is a true latent transcriptome or if gene expression is simply maintained at very low levels that do not support virus replication [236].
6. Final Thoughts
CMV latency is remarkably complex and has eluded definition due to the diversity of cell types infected in the host, the heterogeneity of cells harboring latent virus, the challenges of in vitro models, and the paucity of in vivo animal models. Going forward, it will be important for the field to embrace these complexities to understand how a multitude of virus–host interactions and the control of viral gene expression impact latency and reactivation. These include, but are not limited to, host pathways targeted by viral factors expressed during latency and the changes that accompany and, indeed, facilitate the transition from latent to active states of replication. As a field, we are well-poised with advancement of both in vitro and in vivo models and -omics technologies to make unprecedented leaps in our understanding of CMV latency and reactivation.
Funding
This work was supported by Public Health Service Grants AI079059 (to F.G.). D.C.-M. is supported by an American Heart Association Fellowship and J.B. is supported by an American Cancer Society Fellowship.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Arvin, A.M.; Fast, P.; Myers, M.; Plotkin, S.; Rabinovich, R. Vaccine development to prevent cytomegalovirus disease: Report from the national vaccine advisory committee. Clin. Infect. Dis. 2004, 39, 233–239. [Google Scholar] [PubMed]
- Cannon, M.J.; Schmid, D.S.; Hyde, T.B. Review of cytomegalovirus seroprevalence and demographic characteristics associated with infection. Rev. Med. Virol. 2010, 20, 202–213. [Google Scholar] [CrossRef] [PubMed]
- Nogalski, M.T.; Collins-McMillen, D.; Yurochko, A.D. Overview of human cytomegalovirus pathogenesis. In Human Cytomegaloviruses: Methods and Protocols; Yurochko, A.D., Miller, W.E., Eds.; Humana Press: New York, NY, USA, 2014; pp. 15–28. [Google Scholar]
- Christensen-Quick, A.; Vanpouille, C.; Lisco, A.; Gianella, S. Cytomegalovirus and HIV persistence: Pouring gas on the fire. AIDS Res. Hum. Retroviruses 2017, 33, S23–S30. [Google Scholar] [CrossRef] [PubMed]
- Schlick, K.; Grundbichler, M.; Auberger, J.; Kern, J.M.; Hell, M.; Hohla, F.; Hopfinger, G.; Greil, R. Cytomegalovirus reactivation and its clinical impact in patients with solid tumors. Infect. Agent. Cancer 2015, 10, 45. [Google Scholar] [CrossRef] [PubMed]
- Ramanan, P.; Razonable, R.R. Cytomegalovirus infections in solid organ transplantation: A review. Infect. Chemother. 2013, 45, 260–271. [Google Scholar] [CrossRef] [PubMed]
- Ljungman, P. The role of cytomegalovirus serostatus on outcome of hematopoietic stem cell transplantation. Curr. Opin. Hematol. 2014, 21, 466–469. [Google Scholar] [CrossRef] [PubMed]
- Bhat, V.; Joshi, A.; Sarode, R.; Chavan, P. Cytomegalovirus infection in the bone marrow transplant patient. World J. Transplant. 2015, 5, 287–291. [Google Scholar] [CrossRef] [PubMed]
- Ljungman, P.; Griffiths, P.; Paya, C. Definitions of cytomegalovirus infection and disease in transplant recipients. Clin. Infect. Dis. 2002, 34, 1094–1097. [Google Scholar] [CrossRef] [PubMed]
- Thackeray, R.; Wright, A.; Chipman, K. Congenital cytomegalovirus reference material: A content analysis of coverage and accuracy. Matern. Child Health J. 2014, 18, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Styczynski, J. Who is the patient at risk of CMV recurrence: A review of the current scientific evidence with a focus on hematopoietic cell transplantation. Infect. Dis. Ther. 2018, 7, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Lanzieri, T.M.; Dollard, S.C.; Bialek, S.R.; Grosse, S.D. Systematic review of the birth prevalence of congenital cytomegalovirus infection in developing countries. Int. J. Infect. Dis. 2014, 22, 44–48. [Google Scholar] [CrossRef] [PubMed]
- Adam, E.; Melnick, J.L.; DeBakey, M.E. Cytomegalovirus infection and atherosclerosis. Cent. Eur. J. Public Health 1997, 5, 99–106. [Google Scholar] [PubMed]
- Streblow, D.N.; Orloff, S.L.; Nelson, J.A. Do pathogens accelerate atherosclerosis? J. Nutr. 2001, 131, 2798S–2804S. [Google Scholar] [CrossRef] [PubMed]
- Caposio, P.; Orloff, S.L.; Streblow, D.N. The role of cytomegalovirus in angiogenesis. Virus Res. 2011, 157, 204–211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Streblow, D.N.; Dumortier, J.; Moses, A.V.; Orloff, S.L.; Nelson, J.A. Mechanisms of cytomegalovirus-accelerated vascular disease: Induction of paracrine factors that promote angiogenesis and wound healing. Curr. Top Microbiol. Immunol. 2008, 325, 397–415. [Google Scholar] [PubMed]
- Sylwester, A.W.; Mitchell, B.L.; Edgar, J.B.; Taormina, C.; Pelte, C.; Ruchti, F.; Sleath, P.R.; Grabstein, K.H.; Hosken, N.A.; Kern, F.; et al. Broadly targeted human cytomegalovirus-specific CD4+ and CD8+ t cells dominate the memory compartments of exposed subjects. J. Exp. Med. 2005, 202, 673–685. [Google Scholar] [CrossRef] [PubMed]
- Pawelec, G. Immunosenenescence: Role of cytomegalovirus. Exp. Gerontol. 2014, 54, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Schmaltz, H.N.; Fried, L.P.; Xue, Q.L.; Walston, J.; Leng, S.X.; Semba, R.D. Chronic cytomegalovirus infection and inflammation are associated with prevalent frailty in community-dwelling older women. J. Am. Geriatr. Soc. 2005, 53, 747–754. [Google Scholar] [CrossRef] [PubMed]
- High, K.P. Chronic infection and frailty: Surrogate markers, associations, and causality. J. Am. Geriatr. Soc. 2005, 53, 906–908. [Google Scholar] [CrossRef] [PubMed]
- Wang, G.C.; Kao, W.H.; Murakami, P.; Xue, Q.L.; Chiou, R.B.; Detrick, B.; McDyer, J.F.; Semba, R.D.; Casolaro, V.; Walston, J.D.; et al. Cytomegalovirus infection and the risk of mortality and frailty in older women: A prospective observational cohort study. Am. J. Epidemiol. 2010, 171, 1144–1152. [Google Scholar] [CrossRef] [PubMed]
- Sinzger, C.; Digel, M.; Jahn, G. Cytomegalovirus cell tropism. Curr. Top. Microbiol. Immunol. 2008, 325, 63–83. [Google Scholar] [PubMed]
- Bentz, G.L.; Jarquin-Pardo, M.; Chan, G.; Smith, M.S.; Sinzger, C.; Yurochko, A.D. Human cytomegalovirus (HCMV) infection of endothelial cells promotes naive monocyte extravasation and transfer of productive virus to enhance hematogenous dissemination of HCMV. J. Virol. 2006, 80, 11539–11555. [Google Scholar] [CrossRef] [PubMed]
- Goodrum, F.D.; Jordan, C.T.; High, K.; Shenk, T. Human cytomegalovirus gene expression during infection of primary hematopoietic progenitor cells: A model for latency. Proc. Natl. Acad. Sci. USA 2002, 99, 16255–16260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mendelson, M.; Monard, S.; Sissons, P.; Sinclair, J. Detection of endogenous human cytomegalovirus in CD34+ bone marrow progenitors. J. Gen. Virol. 1996, 77, 3099–3102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hargett, D.; Shenk, T. Experimental human cytomegalovirus latency in CD14+ monocytes. Proc. Natl. Acad. Sci. USA 2010, 107, 20039–20044. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kondo, K.; Kaneshima, H.; Mocarski, E.S. Human cytomegalovirus latent infection of granulocyte-macrophage progenitors. Proc. Natl. Acad. Sci. USA 1994, 91, 11879–11883. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Wiedeman, J.; Sissons, J.G.; Borysiewicz, L.K.; Sinclair, J. Monocytes are a major site of persistence of human cytomegalovirus in peripheral blood mononuclear cells. J. Gen. Virol. 1991, 72, 2059–2064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuravskaya, T.; Maciejewski, J.P.; Netski, D.M.; Bruening, E.; Mackintosh, F.R.; St Jeor, S. Spread of human cytomegalovirus (HCMV) after infection of human hematopoietic progenitor cells: Model of HCMV latency. Blood 1997, 90, 2482–2491. [Google Scholar] [PubMed]
- Hahn, G.; Jores, R.; Mocarski, E.S. Cytomegalovirus remains latent in a common precursor of dendritic and myeloid cells. Proc. Natl. Acad. Sci. USA 1998, 95, 3937–3942. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sindre, H.; Tjoonnfjord, G.E.; Rollag, H.; Ranneberg-Nilsen, T.; Veiby, O.P.; Beck, S.; Degre, M.; Hestdal, K. Human cytomegalovirus suppression of and latency in early hematopoietic progenitor cells. Blood 1996, 88, 4526–4533. [Google Scholar] [PubMed]
- Ibanez, C.E.; Schrier, R.; Ghazal, P.; Wiley, C.; Nelson, J.A. Human cytomegalovirus productively infects primary differentiated macrophages. J. Virol. 1991, 65, 6581–6588. [Google Scholar] [PubMed]
- Reeves, M.B.; Sinclair, J.H. Circulating dendritic cells isolated from healthy seropositive donors are sites of human cytomegalovirus reactivation in vivo. J. Virol. 2013, 87, 10660–10667. [Google Scholar] [CrossRef] [PubMed]
- Soderberg-Naucler, C.; Fish, K.N.; Nelson, J.A. Reactivation of latent human cytomegalovirus by allogeneic stimulation of blood cells from healthy donors. Cell 1997, 91, 119–126. [Google Scholar] [CrossRef]
- Soderberg-Naucler, C.; Streblow, D.N.; Fish, K.N.; Allan-Yorke, J.; Smith, P.P.; Nelson, J.A. Reactivation of latent human cytomegalovirus in CD14(+) monocytes is differentiation dependent. J. Virol. 2001, 75, 7543–7554. [Google Scholar] [CrossRef] [PubMed]
- Taylor-Wiedeman, J.; Sissons, P.; Sinclair, J. Induction of endogenous human cytomegalovirus gene expression after differentiation of monocytes from healthy carriers. J. Virol. 1994, 68, 1597–1604. [Google Scholar] [PubMed]
- Mocarski, E.S.; Courcelle, C.T. Cytomegaloviruses and their replication. In Fields Virology, 4th ed.; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2001; Volume 2, pp. 2629–2673. [Google Scholar]
- Cannon, M.J.; Hyde, T.B.; Schmid, D.S. Review of cytomegalovirus shedding in bodily fluids and relevance to congenital cytomegalovirus infection. Rev. Med. Virol. 2011, 21, 240–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halwachs-Baumann, G.; Genser, B.; Pailer, S.; Engele, H.; Rosegger, H.; Schalk, A.; Kessler, H.H.; Truschnig-Wilders, M. Human cytomegalovirus load in various body fluids of congenitally infected newborns. J. Clin. Virol. 2002, 25 (Suppl. 3), S81–S87. [Google Scholar] [CrossRef]
- Gianella, S.; Massanella, M.; Richman, D.D.; Little, S.J.; Spina, C.A.; Vargas, M.V.; Lada, S.M.; Daar, E.S.; Dube, M.P.; Haubrich, R.H.; et al. Cytomegalovirus replication in semen is associated with higher levels of proviral HIV DNA and CD4+ T cell activation during antiretroviral treatment. J. Virol. 2014, 88, 7818–7827. [Google Scholar] [CrossRef] [PubMed]
- Cojohari, O.; Peppenelli, M.A.; Chan, G.C. Human cytomegalovirus induces an atypical activation of Akt to stimulate the survival of short-lived monocytes. J. Virol. 2016, 90, 6443–6452. [Google Scholar] [CrossRef] [PubMed]
- Peppenelli, M.A.; Arend, K.C.; Cojohari, O.; Moorman, N.J.; Chan, G.C. Human cytomegalovirus stimulates the synthesis of select Akt-dependent antiapoptotic proteins during viral entry to promote survival of infected monocytes. J. Virol. 2016, 90, 3138–3147. [Google Scholar] [CrossRef] [PubMed]
- Chan, G.; Bivins-Smith, E.R.; Smith, M.S.; Smith, P.M.; Yurochko, A.D. Transcriptome analysis reveals human cytomegalovirus reprograms monocyte differentiation toward an m1 macrophage. J. Immunol. 2008, 181, 698–711. [Google Scholar] [CrossRef] [PubMed]
- Chan, G.; Bivins-Smith, E.R.; Smith, M.S.; Yurochko, A.D. NF-kappaB and phosphatidylinositol 3-kinase activity mediates the HCMV-induced atypical M1/M2 polarization of monocytes. Virus Res. 2009, 144, 329–333. [Google Scholar] [CrossRef] [PubMed]
- Chan, G.; Nogalski, M.T.; Stevenson, E.V.; Yurochko, A.D. Human cytomegalovirus induction of a unique signalsome during viral entry into monocytes mediates distinct functional changes: A strategy for viral dissemination. J. Leukoc. Biol. 2012, 92, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Chan, G.; Nogalski, M.T.; Yurochko, A.D. Human cytomegalovirus stimulates monocyte-to-macrophage differentiation via the temporal regulation of caspase 3. J. Virol. 2012, 86, 10714–10723. [Google Scholar] [CrossRef] [PubMed]
- Yurochko, A.D. Human cytomegalovirus modulation of signal transduction. Curr. Top. Microbiol. Immunol. 2008, 325, 205–220. [Google Scholar] [PubMed]
- Smith, M.S.; Bivins-Smith, E.R.; Tilley, A.M.; Bentz, G.L.; Chan, G.; Minard, J.; Yurochko, A.D. Roles of phosphatidylinositol 3-kinase and NF-kappaB in human cytomegalovirus-mediated monocyte diapedesis and adhesion: Strategy for viral persistence. J. Virol. 2007, 81, 7683–7694. [Google Scholar] [CrossRef] [PubMed]
- Reeves, M.B.; Sinclair, J.H. Analysis of latent viral gene expression in natural and experimental latency models of human cytomegalovirus and its correlation with histone modifications at a latent promoter. J. Gen. Virol. 2010, 91, 599–604. [Google Scholar] [CrossRef] [PubMed]
- Goodrum, F.; Caviness, K.; Zagallo, P. Human cytomegalovirus persistence. Cell. Microbiol. 2012, 14, 644–655. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buehler, J.; Zeltzer, S.; Reitsma, J.; Petrucelli, A.; Umashankar, M.; Rak, M.; Zagallo, P.; Schroeder, J.; Terhune, S.; Goodrum, F. Opposing regulation of the EGF receptor: A molecular switch controlling cytomegalovirus latency and replication. PLoS Pathog. 2016, 12, e1005655. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Collins-McMillen, D.; Buehler, J.C.; Goodrum, F.D.; Yurochko, A.D. Human cytomegalovirus requires epidermal growth factor receptor signaling to enter and initiate the early steps in the establishment of latency in CD34+ human progenitor cells. J. Virol. 2017, 91, e01206. [Google Scholar] [CrossRef] [PubMed]
- Goodrum, F. Human cytomegalovirus latency: Approaching the gordian knot. Annu. Rev. Virol. 2016, 3, 333–357. [Google Scholar] [CrossRef] [PubMed]
- Poole, E.; Avdic, S.; Hodkinson, J.; Jackson, S.; Wills, M.; Slobedman, B.; Sinclair, J. Latency-associated viral interleukin-10 (IL-10) encoded by human cytomegalovirus modulates cellular IL-10 and CCL8 secretion during latent infection through changes in the cellular microRNA hsa-miR-92a. J. Virol. 2014, 88, 13947–13955. [Google Scholar] [CrossRef] [PubMed]
- Zhu, D.; Pan, C.; Sheng, J.; Liang, H.; Bian, Z.; Liu, Y.; Trang, P.; Wu, J. Human cytomegalovirus reprogrammes haematopoietic progenitor cells into immunosuppressive monocytes to achieve latency. Nat. Microbiol. 2018, 3, 503–513. [Google Scholar] [CrossRef] [PubMed]
- Reeves, M.B.; Lehner, P.J.; Sissons, J.G.; Sinclair, J.H. An in vitro model for the regulation of human cytomegalovirus latency and reactivation in dendritic cells by chromatin remodelling. J. Gen. Virol. 2005, 86, 2949–2954. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsson, S.; Soderberg-Naucler, C.; Wang, F.Z.; Moller, E. Cytomegalovirus DNA can be detected in peripheral blood mononuclear cells from all seropositive and most seronegative healthy blood donors over time. Transfusion 1998, 38, 271–278. [Google Scholar] [CrossRef] [PubMed]
- Miller, P.H.; Cheung, A.M.; Beer, P.A.; Knapp, D.J.; Dhillon, K.; Rabu, G.; Rostamirad, S.; Humphries, R.K.; Eaves, C.J. Enhanced normal short-term human myelopoiesis in mice engineered to express human-specific myeloid growth factors. Blood 2013, 121, e1-4. [Google Scholar] [CrossRef] [PubMed]
- Hogge, D.E.; Lansdorp, P.M.; Reid, D.; Gerhard, B.; Eaves, C.J. Enhanced detection, maintenance, and differentiation of primitive human hematopoietic cells in cultures containing murine fibroblasts engineered to produce human steel factor, interleukin-3, and granulocyte colony-stimulating factor. Blood 1996, 88, 3765–3773. [Google Scholar] [PubMed]
- Miller, C.L.; Eaves, C.J. Long-term culture-initiating cell assays for human and murine cells. Methods Mol. Med. 2002, 63, 123–141. [Google Scholar] [PubMed]
- Umashankar, M.; Goodrum, F. Hematopoietic long-term culture (hLTC) for human cytomegalovirus latency and reactivation. Methods Mol. Biol. 2014, 1119, 99–112. [Google Scholar] [PubMed]
- Coronel, R.; Takayama, S.; Juwono, T.; Hertel, L. Dynamics of human cytomegalovirus infection in CD34+ hematopoietic cells and derived langerhans-type dendritic cells. J. Virol. 2015, 89, 5615–5632. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, C.M.; Vanicek, J.; Murphy, E.A. Host microRNA regulation of human cytomegalovirus immediate early protein translation promotes viral latency. J. Virol. 2014, 88, 5524–5532. [Google Scholar] [CrossRef] [PubMed]
- Rossetto, C.C.; Tarrant-Elorza, M.; Pari, G.S. Cis and trans acting factors involved in human cytomegalovirus experimental and natural latent infection of CD14 (+) monocytes and CD34 (+) cells. PLoS Pathog. 2013, 9, e1003366. [Google Scholar] [CrossRef] [PubMed]
- Saffert, R.T.; Penkert, R.R.; Kalejta, R.F. Cellular and viral control over the initial events of human cytomegalovirus experimental latency in CD34+ cells. J. Virol. 2010, 84, 5594–5604. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.S.; Bentz, G.L.; Alexander, J.S.; Yurochko, A.D. Human cytomegalovirus induces monocyte differentiation and migration as a strategy for dissemination and persistence. J. Virol. 2004, 78, 4444–4453. [Google Scholar] [CrossRef] [PubMed]
- Chan, G.; Nogalski, M.T.; Bentz, G.L.; Smith, M.S.; Parmater, A.; Yurochko, A.D. PI3K-dependent upregulation of Mcl-1 by human cytomegalovirus is mediated by epidermal growth factor receptor and inhibits apoptosis in short-lived monocytes. J. Immunol. 2010, 184, 3213–3222. [Google Scholar] [CrossRef] [PubMed]
- Chan, G.; Nogalski, M.T.; Yurochko, A.D. Activation of EGFR on monocytes is required for human cytomegalovirus entry and mediates cellular motility. Proc. Natl. Acad. Sci. USA 2009, 106, 22369–22374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nogalski, M.T.; Chan, G.; Stevenson, E.V.; Gray, S.; Yurochko, A.D. Human cytomegalovirus-regulated paxillin in monocytes links cellular pathogenic motility to the process of viral entry. J. Virol. 2011, 85, 1360–1369. [Google Scholar] [CrossRef] [PubMed]
- Nogalski, M.T.; Chan, G.C.; Stevenson, E.V.; Collins-McMillen, D.K.; Yurochko, A.D. The HCMV gH/gL/UL128-131 complex triggers the specific cellular activation required for efficient viral internalization into target monocytes. PLoS Pathog. 2013, 9, e1003463. [Google Scholar] [CrossRef] [PubMed]
- Smith, M.S.; Bentz, G.L.; Smith, P.M.; Bivens, E.R.; Yurochko, A.D. HCMV activates PI(3)K in monocytes and promotes monocyte motility and transendothelial migration in a PI(3)K-dependent manner. J. Leukoc. Biol. 2004, 76, 65–76. [Google Scholar] [CrossRef] [PubMed]
- Arcangeletti, M.C.; Vasile Simone, R.; Rodighiero, I.; De Conto, F.; Medici, M.C.; Maccari, C.; Chezzi, C.; Calderaro, A. Human cytomegalovirus reactivation from latency: Validation of a “switch” model in vitro. Virol. J. 2016, 13, 179. [Google Scholar] [CrossRef] [PubMed]
- Wagenknecht, N.; Reuter, N.; Scherer, M.; Reichel, A.; Muller, R.; Stamminger, T. Contribution of the major ND10 proteins PML, hDaxx and Sp100 to the regulation of human cytomegalovirus latency and lytic replication in the monocytic cell line THP-1. Viruses 2015, 7, 2884–2907. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Lee, G.C.; Chan, Y.J.; Chiou, C.J.; Ahn, J.H.; Hayward, G.S. Factors affecting human cytomegalovirus gene expression in human monocyte cell lines. Mol. Cells 1999, 9, 37–44. [Google Scholar] [PubMed]
- Beisser, P.S.; Laurent, L.; Virelizier, J.L.; Michelson, S. Human cytomegalovirus chemokine receptor gene US28 is transcribed in latently infected thp-1 monocytes. J. Virol. 2001, 75, 5949–5957. [Google Scholar] [CrossRef] [PubMed]
- Yee, L.F.; Lin, P.L.; Stinski, M.F. Ectopic expression of HCMV IE72 and IE86 proteins is sufficient to induce early gene expression but not production of infectious virus in undifferentiated promonocytic THP-1 cells. Virology 2007, 363, 174–188. [Google Scholar] [CrossRef] [PubMed]
- Saffert, R.T.; Kalejta, R.F. Human cytomegalovirus gene expression is silenced by Daxx-mediated intrinsic immune defense in model latent infections established in vitro. J. Virol. 2007, 81, 9109–9120. [Google Scholar] [CrossRef] [PubMed]
- Murayama, T.; Ohara, Y.; Obuchi, M.; Khabar, K.S.; Higashi, H.; Mukaida, N.; Matsushima, K. Human cytomegalovirus induces interleukin-8 production by a human monocytic cell line, THP-1, through acting concurrently on AP-1- and NF-kappaB-binding sites of the interleukin-8 gene. J. Virol. 1997, 71, 5692–5695. [Google Scholar] [PubMed]
- Geist, L.J.; Monick, M.M.; Stinski, M.F.; Hunninghake, G.W. The immediate early genes of human cytomegalovirus upregulate tumor necrosis factor-alpha gene expression. J. Clin. Investig. 1994, 93, 474–478. [Google Scholar] [CrossRef] [PubMed]
- Gan, X.; Wang, H.; Yu, Y.; Yi, W.; Zhu, S.; Li, E.; Liang, Y. Epigenetically repressing human cytomegalovirus lytic infection and reactivation from latency in THP-1 model by targeting H3K9 and H3K27 histone demethylases. PLoS ONE 2017, 12, e0175390. [Google Scholar] [CrossRef] [PubMed]
- Ioudinkova, E.; Arcangeletti, M.C.; Rynditch, A.; De Conto, F.; Motta, F.; Covan, S.; Pinardi, F.; Razin, S.V.; Chezzi, C. Control of human cytomegalovirus gene expression by differential histone modifications during lytic and latent infection of a monocytic cell line. Gene 2006, 384, 120–128. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, C.M.; Murphy, E.A. A myeloid progenitor cell line capable of supporting human cytomegalovirus latency and reactivation, resulting in infectious progeny. J. Virol. 2012, 86, 9854–9865. [Google Scholar] [CrossRef] [PubMed]
- Penkert, R.R.; Kalejta, R.F. Human embryonic stem cell lines model experimental human cytomegalovirus latency. mBio 2013, 4, e00298-13. [Google Scholar] [CrossRef] [PubMed]
- Poole, E.; Lau, J.C.; Sinclair, J. Latent infection of myeloid progenitors by human cytomegalovirus protects cells from FAS-mediated apoptosis through the cellular IL-10/PEA-15 pathway. J. Gen. Virol. 2015, 96, 2355–2359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berger, A.A.; Gil, Y.; Panet, A.; Weisblum, Y.; Oiknine-Djian, E.; Gropp, M.; Steiner, D.; Reubinoff, B.E.; Wolf, D.G. Transition toward human cytomegalovirus susceptibility in early human embryonic stem cell-derived neural precursors. J. Virol. 2015, 89, 11159–11164. [Google Scholar] [CrossRef] [PubMed]
- Andrews, P.W. Retinoic acid induces neuronal differentiation of a cloned human embryonal carcinoma cell line in vitro. Dev. Biol. 1984, 103, 285–293. [Google Scholar] [CrossRef]
- Gonczol, E.; Andrews, P.W.; Plotkin, S.A. Cytomegalovirus replicates in differentiated but not in undifferentiated human embryonal carcinoma cells. Science 1984, 224, 159–161. [Google Scholar] [CrossRef] [PubMed]
- Odeberg, J.; Wolmer, N.; Falci, S.; Westgren, M.; Seiger, A.; Soderberg-Naucler, C. Human cytomegalovirus inhibits neuronal differentiation and induces apoptosis in human neural precursor cells. J. Virol. 2006, 80, 8929–8939. [Google Scholar] [CrossRef] [PubMed]
- Luo, M.H.; Schwartz, P.H.; Fortunato, E.A. Neonatal neural progenitor cells and their neuronal and glial cell derivatives are fully permissive for human cytomegalovirus infection. J. Virol. 2008, 82, 9994–10007. [Google Scholar] [CrossRef] [PubMed]
- Belzile, J.P.; Stark, T.J.; Yeo, G.W.; Spector, D.H. Human cytomegalovirus infection of human embryonic stem cell-derived primitive neural stem cells is restricted at several steps but leads to the persistence of viral DNA. J. Virol. 2014, 88, 4021–4039. [Google Scholar] [CrossRef] [PubMed]
- Umashankar, M.; Petrucelli, A.; Cicchini, L.; Caposio, P.; Kreklywich, C.N.; Rak, M.; Bughio, F.; Goldman, D.C.; Hamlin, K.L.; Nelson, J.A.; et al. A novel human cytomegalovirus locus modulates cell type-specific outcomes of infection. PLoS Pathog. 2011, 7, e1002444. [Google Scholar] [CrossRef] [PubMed]
- Petrucelli, A.; Rak, M.; Grainger, L.; Goodrum, F. Characterization of a novel Golgi apparatus-localized latency determinant encoded by human cytomegalovirus. J. Virol. 2009, 83, 5615–5629. [Google Scholar] [CrossRef] [PubMed]
- Petrucelli, A.; Umashankar, M.; Zagallo, P.; Rak, M.; Goodrum, F. Interactions between proteins encoded within the human cytomegalovirus UL133-UL138 locus. J. Virol. 2012, 86, 8653–8662. [Google Scholar] [CrossRef] [PubMed]
- Caviness, K.; Bughio, F.; Crawford, L.B.; Streblow, D.N.; Nelson, J.A.; Caposio, P.; Goodrum, F. Complex interplay of the UL136 isoforms balances cytomegalovirus replication and latency. mBio 2016, 7, e01986. [Google Scholar] [CrossRef] [PubMed]
- Caviness, K.; Cicchini, L.; Rak, M.; Umashankar, M.; Goodrum, F.D. Complex expression of the UL136 gene of human cytomegalovirus results in multiple proteins isoforms with unique roles in replication. J. Virol. 2014, 88, 14412–14425. [Google Scholar] [CrossRef] [PubMed]
- Rak, M.; Buehler, J.; Zeltzer, S.; Reitsma, J.; Terhune, S.; Goodrum, F. Human cytomegalovirus UL135 interacts with host adaptor proteins to regulate epidermal growth factor receptor and reactivation from latency. J. Virol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Crawford, L.B.; Streblow, D.N.; Hakki, M.; Nelson, J.A.; Caposio, P. Humanized mouse models of human cytomegalovirus infection. Curr. Opin. Virol. 2015, 13, 86–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crawford, L.B.; Kim, J.H.; Collins-McMillen, D.; Lee, B.J.; Landais, I.; Held, C.; Nelson, J.A.; Yurochko, A.D. Human cytomegalovirus encodes a novel FLT3 receptor ligand necessary for hematopoietic cell differentiation and viral reactivation. mBio 2018, 9, e00682-18. [Google Scholar] [CrossRef] [PubMed]
- Collins-McMillen, D.; Stevenson, E.V.; Kim, J.H.; Lee, B.J.; Cieply, S.J.; Nogalski, M.T.; Chan, G.C.; Frost, R.W., 3rd; Spohn, C.R.; Yurochko, A.D. HCMV utilizes a non-traditional stat1 activation cascade via signaling through egfr and integrins to efficiently promote the motility, differentiation, and polarization of infected monocytes. J. Virol. 2017. [Google Scholar] [CrossRef] [PubMed]
- Kabanova, A.; Marcandalli, J.; Zhou, T.; Bianchi, S.; Baxa, U.; Tsybovsky, Y.; Lilleri, D.; Silacci-Fregni, C.; Foglierini, M. Platelet-derived growth factor-alpha receptor is the cellular receptor for human cytomegalovirus ghglgo trimer. Nat. Microbiol. 2016, 1, 16082. [Google Scholar] [CrossRef] [PubMed]
- Avraham, R.; Yarden, Y. Feedback regulation of EGFR signalling: Decision making by early and delayed loops. Nat. Rev. Mol. Cell. Biol. 2011, 12, 104–117. [Google Scholar] [CrossRef] [PubMed]
- Lindsey, S.; Langhans, S.A. Epidermal growth factor signaling in transformed cells. Int. Rev. Cell Mol. Biol. 2015, 314, 1–41. [Google Scholar] [PubMed]
- Lupberger, J.; Duong, F.H.; Fofana, I.; Zona, L.; Xiao, F.; Thumann, C.; Durand, S.C.; Pessaux, P.; Zeisel, M.B.; Heim, M.H.; et al. Epidermal growth factor receptor signaling impairs the antiviral activity of interferon-alpha. Hepatology 2013, 58, 1225–1235. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamashita, M.; Chattopadhyay, S.; Fensterl, V.; Saikia, P.; Wetzel, J.L.; Sen, G.C. Epidermal growth factor receptor is essential for Toll-like receptor 3 signaling. Sci. Signal. 2012, 5, ra50. [Google Scholar] [CrossRef] [PubMed]
- Ortega, J.; Li, J.Y.; Lee, S.; Tong, D.; Gu, L.; Li, G.M. Phosphorylation of PCNA by EGFR inhibits mismatch repair and promotes misincorporation during DNA synthesis. Proc. Natl. Acad. Sci. USA 2015, 112, 5667–5672. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luo, B.H.; Springer, T.A. Integrin structures and conformational signaling. Curr. Opin. Cell Biol. 2006, 18, 579–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berman, A.E.; Kozlova, N.I.; Morozevich, G.E. Integrins: Structure and signaling. Biochemistry 2003, 68, 1284–1299. [Google Scholar] [CrossRef] [PubMed]
- Caswell, P.T.; Vadrevu, S.; Norman, J.C. Integrins: Masters and slaves of endocytic transport. Nat. Rev. Mol. Cell. Biol. 2009, 10, 843–853. [Google Scholar] [CrossRef] [PubMed]
- Cliffe, A.R.; Arbuckle, J.H.; Vogel, J.L.; Geden, M.J.; Rothbart, S.B.; Cusack, C.L.; Strahl, B.D.; Kristie, T.M.; Deshmukh, M. Neuronal stress pathway mediating a histone Methyl/Phospho switch is required for herpes simplex virus reactivation. Cell. Host Microbe 2015, 18, 649–658. [Google Scholar] [CrossRef] [PubMed]
- Koyuncu, O.O.; MacGibeny, M.A. Compartmented neuronal cultures reveal two distinct mechanisms for alpha herpesvirus escape from genome silencing. PLoS Pathog. 2017, 13, e1006608. [Google Scholar] [CrossRef] [PubMed]
- Collins-McMillen, D.; Kim, J.H.; Nogalski, M.T.; Stevenson, E.V.; Chan, G.C.; Caskey, J.R.; Cieply, S.J.; Yurochko, A.D. Human Cytomegalovirus Promotes Survival of Infected Monocytes via a Distinct Temporal Regulation of Cellular Bcl-2 Family Proteins. J. Virol. 2015, 90, 2356–2371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maciejewski, J.P.; Bruening, E.E.; Donahue, R.E.; Sellers, S.E.; Carter, C.; Young, N.S.; St Jeor, S. Infection of mononucleated phagocytes with human cytomegalovirus. Virology 1993, 195, 327–336. [Google Scholar] [CrossRef] [PubMed]
- Roizman, B.; Whitley, R.J. An inquiry into the molecular basis of HSV latency and reactivation. Annu. Rev. Microbiol. 2013, 67, 355–374. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Banerjee, S. The regulatory role of protein phosphorylation in human gammaherpesvirus associated cancers. Virol. Sin. 2017, 32, 357–368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stern, J.L.; Slobedman, B. Human cytomegalovirus latent infection of myeloid cells directs monocyte migration by up-regulating monocyte chemotactic protein-1. J. Immunol. 2008, 180, 6577–6585. [Google Scholar] [CrossRef] [PubMed]
- Noriega, V.M.; Haye, K.K.; Kraus, T.A.; Kowalsky, S.R.; Ge, Y.; Moran, T.M.; Tortorella, D. Human cytomegalovirus modulates monocyte-mediated innate immune responses during short-term experimental latency in vitro. J. Virol. 2014, 88, 9391–9405. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Gao, Y.; Zhou, Q.; Zhang, Q.; Peng, Y.; Tian, K.; Wang, J.; Zheng, X. Human cytomegalovirus latent infection alters the expression of cellular and viral microRNA. Gene 2014, 536, 272–278. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.Z.; Su, S.; Gao, Y.Q.; Wang, P.P.; Huang, Z.F.; Hu, M.M.; Luo, W.W.; Li, S.; Luo, M.H.; Wang, Y.Y.; et al. Human cytomegalovirus tegument protein UL82 inhibits sting-mediated signaling to evade antiviral immunity. Cell. Host Microbe 2017, 21, 231–243. [Google Scholar] [CrossRef] [PubMed]
- Trgovcich, J.; Cebulla, C.; Zimmerman, P.; Sedmak, D.D. Human cytomegalovirus protein pp71 disrupts major histocompatibility complex class I cell surface expression. J. Virol. 2006, 80, 951–963. [Google Scholar] [CrossRef] [PubMed]
- Cha, T.A.; Tom, E.; Kemble, G.W.; Duke, G.M.; Mocarski, E.S.; Spaete, R.R. Human cytomegalovirus clinical isolates carry at least 19 genes not found in laboratory strains. J. Virol. 1996, 70, 78–83. [Google Scholar] [PubMed]
- Murphy, E.; Yu, D.; Grimwood, J.; Schmutz, J.; Dickson, M.; Jarvis, M.A.; Hahn, G.; Nelson, J.A.; Myers, R.M.; Shenk, T.E. Coding potential of laboratory and clinical strains of human cytomegalovirus. Proc. Natl. Acad. Sci. USA 2003, 100, 14976–14981. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dolan, A.; Cunningham, C.; Hector, R.D.; Hassan-Walker, A.F.; Lee, L.; Addison, C.; Dargan, D.J.; McGeoch, D.J.; Gatherer, D.; Emery, V.C.; et al. Genetic content of wild-type human cytomegalovirus. J. Gen. Virol. 2004, 85, 1301–1312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grainger, L.; Cicchini, L.; Rak, M.; Petrucelli, A.; Fitzgerald, K.D.; Semler, B.L.; Goodrum, F. Stress-inducible alternative translation initiation of human cytomegalovirus latency protein pUL138. J. Virol. 2010, 84, 9472–9486. [Google Scholar] [CrossRef] [PubMed]
- Umashankar, M.; Rak, M.; Bughio, F.; Zagallo, P.; Caviness, K.; Goodrum, F.D. Antagonistic determinants controlling replicative and latent states of human cytomegalovirus infection. J. Virol. 2014, 88, 5987–6002. [Google Scholar] [CrossRef] [PubMed]
- Jafferji, I.; Bain, M.; King, C.; Sinclair, J.H. Inhibition of epidermal growth factor receptor (EGFR) expression by human cytomegalovirus correlates with an increase in the expression and binding of Wilms’ Tumour 1 protein to the EGFR promoter. J. Gen. Virol. 2009, 90, 1569–1574. [Google Scholar] [CrossRef] [PubMed]
- Camarena, V.; Kobayashi, M.; Kim, J.Y.; Roehm, P.; Perez, R.; Gardner, J.; Wilson, A.C.; Mohr, I.; Chao, M.V. Nature and duration of growth factor signaling through receptor tyrosine kinases regulates HSV-1 latency in neurons. Cell. Host Microbe 2010, 8, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.; Wu, T.T.; Tchieu, J.H.; Feng, J.; Brown, H.J.; Feng, J.; Li, X.; Qi, J.; Deng, H.; Vivanco, I.; et al. Inhibition of the phosphatidylinositol 3-kinase-Akt pathway enhances gamma-2 herpesvirus lytic replication and facilitates reactivation from latency. J. Gen. Virol. 2010, 91, 463–469. [Google Scholar] [CrossRef] [PubMed]
- Kung, C.P.; Meckes, D.G., Jr.; Raab-Traub, N. Epstein-Barr virus LMP1 activates EGFR, STAT3, and ERK through effects on PKCdelta. J. Virol. 2011, 85, 4399–4408. [Google Scholar] [CrossRef] [PubMed]
- Moepps, B.; Tulone, C.; Kern, C.; Minisini, R.; Michels, G.; Vatter, P.; Wieland, T.; Gierschik, P. Constitutive serum response factor activation by the viral chemokine receptor homologue pUS28 is differentially regulated by Galpha(q/11) and Galpha(16). Cell. Signal. 2008, 20, 1528–1537. [Google Scholar] [CrossRef] [PubMed]
- Miller, W.E.; Zagorski, W.A.; Brenneman, J.D.; Avery, D.; Miller, J.L.; O’Connor, C.M. US28 is a potent activator of phospholipase c during hcmv infection of clinically relevant target cells. PLoS ONE 2012, 7, e50524. [Google Scholar] [CrossRef] [PubMed]
- Slinger, E.; Maussang, D.; Schreiber, A.; Siderius, M.; Rahbar, A.; Fraile-Ramos, A.; Lira, S.A.; Soderberg-Naucler, C.; Smit, M.J. HCMV-encoded chemokine receptor US28 mediates proliferative signaling through the Il-6-STAT3 axis. Sci. Signal. 2010, 3, ra58. [Google Scholar] [CrossRef] [PubMed]
- Boomker, J.M.; The, T.H.; de Leij, L.F.; Harmsen, M.C. The human cytomegalovirus-encoded receptor US28 increases the activity of the major immediate-early promoter/enhancer. Virus Res. 2006, 118, 196–200. [Google Scholar] [CrossRef] [PubMed]
- Wen, D.Q.; Zhang, Y.Y.; Lv, L.P.; Zhou, X.P.; Yan, F.; Ma, P.; Xu, J.B. Human cytomegalovirus-encoded chemokine receptor homolog US28 stimulates the major immediate early gene promoter/enhancer via the induction of creb. J. Recept. Signal Transduct. Res. 2009, 29, 266–273. [Google Scholar] [CrossRef] [PubMed]
- Humby, M.S.; O’Connor, C.M. Human cytomegalovirus US28 is important for latent infection of hematopoietic progenitor cells. J. Virol. 2015, 90, 2959–2970. [Google Scholar] [CrossRef] [PubMed]
- Krishna, B.A.; Poole, E.L.; Jackson, S.E. Latency-associated expression of human cytomegalovirus US28 attenuates cell signaling pathways to maintain latent infection. mBio 2017, 8, e01754-17. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.E.; Miller, W.E. The HCMV US28 vGPCR induces potent Gαq/PLC-β signaling in monocytes leading to increased adhesion to endothelial cells. Virology 2016, 497, 233–243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Todaro, M.; Zerilli, M.; Ricci-Vitiani, L.; Bini, M.; Perez Alea, M.; Maria Florena, A.; Miceli, L.; Condorelli, G.; Bonventre, S.; Di Gesu, G.; et al. Autocrine production of interleukin-4 and interleukin-10 is required for survival and growth of thyroid cancer cells. Cancer Res. 2006, 66, 1491–1499. [Google Scholar] [CrossRef] [PubMed]
- Hancock, M.H.; Hook, L.M.; Mitchell, J.; Nelson, J.A. Human Cytomegalovirus MicroRNAs miR-US5-1 and miR-UL112-3p Block Proinflammatory Cytokine Production in Response to NF-κB-Activating Factors through Direct Downregulation of IKKα and IKKβ. mBio 2017, 8, e00109-17. [Google Scholar] [CrossRef] [PubMed]
- Reeves, M.B.; Compton, T. Inhibition of inflammatory interleukin-6 activity via extracellular signal-regulated kinase-mitogen-activated protein kinase signaling antagonizes human cytomegalovirus reactivation from dendritic cells. J. Virol. 2011, 85, 12750–12758. [Google Scholar] [CrossRef] [PubMed]
- Engel, P.; Perez-Carmona, N.; Alba, M.M.; Robertson, K.; Ghazal, P.; Angulo, A. Human cytomegalovirus UL7, a homologue of the SLAM-family receptor CD229, impairs cytokine production. Immunol. Cell Biol. 2011, 89, 753–766. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zwezdaryk, K.J.; Combs, J.A.; Morris, C.A.; Sullivan, D.E. Regulation of Wnt/beta-catenin signaling by herpesviruses. World J. Virol. 2016, 5, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Morrison, J.A.; Klingelhutz, A.J.; Raab-Traub, N. Epstein-Barr virus latent membrane protein 2A activates beta-catenin signaling in epithelial cells. J. Virol. 2003, 77, 12276–12284. [Google Scholar] [CrossRef] [PubMed]
- Fujimuro, M.; Hayward, S.D. The latency-associated nuclear antigen of kaposi’s sarcoma-associated herpesvirus manipulates the activity of glycogen synthase kinase-3beta. J. Virol. 2003, 77, 8019–8030. [Google Scholar] [CrossRef] [PubMed]
- Fujimuro, M.; Wu, F.Y.; ApRhys, C.; Kajumbula, H.; Young, D.B.; Hayward, G.S.; Hayward, S.D. A novel viral mechanism for dysregulation of beta-catenin in kaposi’s sarcoma-associated herpesvirus latency. Nat. Med. 2003, 9, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Shackelford, J.; Maier, C.; Pagano, J.S. Epstein-barr virus activates beta-catenin in type III latently infected B lymphocyte lines: Association with deubiquitinating enzymes. Proc. Natl. Acad. Sci. USA 2003, 100, 15572–15576. [Google Scholar] [CrossRef] [PubMed]
- Everly, D.N., Jr.; Kusano, S.; Raab-Traub, N. Accumulation of cytoplasmic beta-catenin and nuclear glycogen synthase kinase 3beta in epstein-barr virus-infected cells. J. Virol. 2004, 78, 11648–11655. [Google Scholar] [CrossRef] [PubMed]
- Jang, K.L.; Shackelford, J.; Seo, S.Y.; Pagano, J.S. Up-regulation of beta-catenin by a viral oncogene correlates with inhibition of the seven in absentia homolog 1 in b lymphoma cells. Proc. Natl. Acad. Sci. USA 2005, 102, 18431–18436. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hancock, M.; Workman, A.; Doster, A.; Jones, C. Beta-catenin, a transcription factor activated by canonical Wnt signaling, is expressed in sensory neurons of calves latently infected with bovine herpesvirus 1. J. Virol. 2016, 90, 3148–3159. [Google Scholar] [CrossRef] [PubMed]
- Workman, A.; Zhu, L.; Keel, B.N.; Smith, T.P.L.; Jones, C. The Wnt Signaling Pathway Is Differentially Expressed during the Bovine Herpesvirus 1 Latency-Reactivation Cycle: Evidence That Two Protein Kinases Associated with Neuronal Survival, Akt3 and BMPR2, Are Expressed at Higher Levels during Latency. J. Virol. 2018, 92. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Workman, A.; Jones, C. Potential role for a beta-catenin coactivator (high-mobility group at-hook 1 protein) during the latency-reactivation cycle of bovine herpesvirus 1. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Angelova, M.; Zwezdaryk, K.; Ferris, M.; Shan, B.; Morris, C.A.; Sullivan, D.E. Human cytomegalovirus infection dysregulates the canonical wnt/beta-catenin signaling pathway. PLoS Pathog. 2012, 8, e1002959. [Google Scholar] [CrossRef] [PubMed]
- Roy, S.; Liu, F.; Arav-Boger, R. Human Cytomegalovirus Inhibits the PARsylation Activity of Tankyrase—A Potential Strategy for Suppression of the Wnt Pathway. Viruses 2016, 8, 8. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, A.; He, R.; Venkatadri, R.; Forman, M.; Arav-Boger, R. Wnt modulating agents inhibit human cytomegalovirus replication. Antimicrob. Agents Chemother. 2013, 57, 2761–2767. [Google Scholar] [CrossRef] [PubMed]
- Stinski, M.F. Sequence of protein synthesis in cells infected by human cytomegalovirus: Early and late virus-induced polypeptides. J. Virol. 1978, 26, 686–701. [Google Scholar] [PubMed]
- Wathen, M.W.; Stinski, M.F. Temporal patterns of human cytomegalovirus transcription: Mapping the viral rnas synthesized at immediate early, early, and late times after infection. J. Virol. 1982, 41, 462–477. [Google Scholar] [PubMed]
- Stinski, M.F.; Meier, J.L. Immediate-early viral gene regulation and function. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Pizzorno, M.C.; O’Hare, P.; Sha, L.; LaFemina, R.L.; Hayward, G.S. Trans-activation and autoregulation of gene expression by the immediate-early region 2 gene products of human cytomegalovirus. J. Virol. 1988, 62, 1167–1179. [Google Scholar] [PubMed]
- Malone, C.L.; Vesole, D.H.; Stinski, M.F. Transactivation of a human cytomegalovirus early promoter by gene products from the immediate-early gene IE2 and augmentation by IE1: Mutational analysis of the viral proteins. J. Virol. 1990, 64, 1498–1506. [Google Scholar] [PubMed]
- Anders, D.G.; Kerry, J.A.; Pari, G.S. DNA synthesis and late viral gene expression. In Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Arvin, A., Campadelli-Fiume, G., Mocarski, E., Moore, P.S., Roizman, B., Whitley, R., Yamanishi, K., Eds.; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Reeves, M.B.; MacAry, P.A.; Lehner, P.J.; Sissons, J.G.; Sinclair, J.H. Latency, chromatin remodeling, and reactivation of human cytomegalovirus in the dendritic cells of healthy carriers. Proc. Natl. Acad. Sci. USA 2005, 102, 4140–4145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomsen, D.R.; Stenberg, R.M.; Goins, W.F.; Stinski, M.F. Promoter-regulatory region of the major immediate early gene of human cytomegalovirus. Proc. Natl. Acad. Sci. USA 1984, 81, 659–663. [Google Scholar] [CrossRef] [PubMed]
- Boshart, M.; Weber, F.; Jahn, G.; Dorsch-Hasler, K.; Fleckenstein, B.; Schaffner, W. A very strong enhancer is located upstream of an immediate early gene of human cytomegalovirus. Cell 1985, 41, 521–530. [Google Scholar] [CrossRef] [Green Version]
- Ghazal, P.; Lubon, H.; Fleckenstein, B.; Hennighausen, L. Binding of transcription factors and creation of a large nucleoprotein complex on the human cytomegalovirus enhancer. Proc. Natl. Acad. Sci. USA 1987, 84, 3658–3662. [Google Scholar] [CrossRef] [PubMed]
- Ghazal, P.; Lubon, H.; Hennighausen, L. Multiple sequence-specific transcription factors modulate cytomegalovirus enhancer activity in vitro. Mol. Cell. Biol. 1988, 8, 1809–1811. [Google Scholar] [CrossRef] [PubMed]
- Akrigg, A.; Wilkinson, G.W.; Oram, J.D. The structure of the major immediate early gene of human cytomegalovirus strain AD169. Virus Res. 1985, 2, 107–121. [Google Scholar] [CrossRef]
- Stinski, M.F.; Roehr, T.J. Activation of the major immediate early gene of human cytomegalovirus by cis-acting elements in the promoter-regulatory sequence and by virus-specific trans-acting components. J. Virol. 1985, 55, 431–441. [Google Scholar] [PubMed]
- Sambucetti, L.C.; Cherrington, J.M.; Wilkinson, G.W.; Mocarski, E.S. NF-kappa B activation of the cytomegalovirus enhancer is mediated by a viral transactivator and by T cell stimulation. EMBO J. 1989, 8, 4251–4258. [Google Scholar] [PubMed]
- Hunninghake, G.W.; Monick, M.M.; Liu, B.; Stinski, M.F. The promoter-regulatory region of the major immediate-early gene of human cytomegalovirus responds to T-lymphocyte stimulation and contains functional cyclic amp-response elements. J. Virol. 1989, 63, 3026–3033. [Google Scholar] [PubMed]
- Lang, D.; Fickenscher, H.; Stamminger, T. Analysis of proteins binding to the proximal promoter region of the human cytomegalovirus IE-1/2 enhancer/promoter reveals both consensus and aberrant recognition sequences for transcription factors Sp1 and CREB. Nucleic Acids Res. 1992, 20, 3287–3295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wade, E.J.; Klucher, K.M.; Spector, D.H. An AP-1 binding site is the predominant cis-acting regulatory element in the 1.2-kilobase early RNA promoter of human cytomegalovirus. J. Virol. 1992, 66, 2407–2417. [Google Scholar] [PubMed]
- LaFemina, R.; Hayward, G.S. Constitutive and retinoic acid-inducible expression of cytomegalovirus immediate-early genes in human teratocarcinoma cells. J. Virol. 1986, 58, 434–440. [Google Scholar] [PubMed]
- Angulo, A.; Suto, C.; Heyman, R.A.; Ghazal, P. Characterization of the sequences of the human cytomegalovirus enhancer that mediate differential regulation by natural and synthetic retinoids. Mol. Endocrinol. 1996, 10, 781–793. [Google Scholar] [PubMed]
- Ghazal, P.; DeMattei, C.; Giulietti, E.; Kliewer, S.A.; Umesono, K.; Evans, R.M. Retinoic acid receptors initiate induction of the cytomegalovirus enhancer in embryonal cells. Proc. Natl. Acad. Sci. USA 1992, 89, 7630–7634. [Google Scholar] [CrossRef] [PubMed]
- Chan, Y.J.; Chiou, C.J.; Huang, Q.; Hayward, G.S. Synergistic interactions between overlapping binding sites for the serum response factor and ELK-1 proteins mediate both basal enhancement and phorbol ester responsiveness of primate cytomegalovirus major immediate-early promoters in monocyte and T-lymphocyte cell types. J. Virol. 1996, 70, 8590–8605. [Google Scholar] [PubMed]
- Netterwald, J.; Yang, S.; Wang, W.; Ghanny, S.; Cody, M.; Soteropoulos, P.; Tian, B.; Dunn, W.; Liu, F.; Zhu, H. Two gamma interferon-activated site-like elements in the human cytomegalovirus major immediate-early promoter/enhancer are important for viral replication. J. Virol. 2005, 79, 5035–5046. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.Y.; Ni, Y.S.; Saifudeen, Z.; Asiedu, C.K.; Supakar, P.C.; Ehrlich, M. Increasing binding of a transcription factor immediately downstream of the cap site of a cytomegalovirus gene represses expression. Nucleic Acids Res. 1995, 23, 3026–3033. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zweidler-Mckay, P.A.; Grimes, H.L.; Flubacher, M.M.; Tsichlis, P.N. Gfi-1 encodes a nuclear zinc finger protein that binds DNA and functions as a transcriptional repressor. Mol. Cell. Biol. 1996, 16, 4024–4034. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Baillie, J.; Sissons, J.G.; Sinclair, J.H. The transcription factor YY1 binds to negative regulatory elements in the human cytomegalovirus major immediate early enhancer/promoter and mediates repression in non-permissive cells. Nucleic Acids Res. 1994, 22, 2453–2459. [Google Scholar] [CrossRef] [PubMed]
- Bain, M.; Mendelson, M.; Sinclair, J. Ets-2 repressor factor (ERF) mediates repression of the human cytomegalovirus major immediate-early promoter in undifferentiated non-permissive cells. J. Gen. Virol. 2003, 84, 41–49. [Google Scholar] [CrossRef] [PubMed]
- Kothari, S.; Baillie, J.; Sissons, J.G.; Sinclair, J.H. The 21bp repeat element of the human cytomegalovirus major immediate early enhancer is a negative regulator of gene expression in undifferentiated cells. Nucleic Acids Res. 1991, 19, 1767–1771. [Google Scholar] [CrossRef] [PubMed]
- Hennighausen, L.; Fleckenstein, B. Nuclear factor 1 interacts with five DNA elements in the promoter region of the human cytomegalovirus major immediate early gene. EMBO J. 1986, 5, 1367–1371. [Google Scholar] [PubMed]
- Jeang, K.T.; Rawlins, D.R.; Rosenfeld, P.J.; Shero, J.H.; Kelly, T.J.; Hayward, G.S. Multiple tandemly repeated binding sites for cellular nuclear factor 1 that surround the major immediate-early promoters of simian and human cytomegalovirus. J. Virol. 1987, 61, 1559–1570. [Google Scholar] [PubMed]
- Shelbourn, S.L.; Kothari, S.K.; Sissons, J.G.; Sinclair, J.H. Repression of human cytomegalovirus gene expression associated with a novel immediate early regulatory region binding factor. Nucleic Acids Res. 1989, 17, 9165–9171. [Google Scholar] [CrossRef] [PubMed]
- Huang, T.H.; Oka, T.; Asai, T.; Okada, T.; Merrills, B.W.; Gertson, P.N.; Whitson, R.H.; Itakura, K. Repression by a differentiation-specific factor of the human cytomegalovirus enhancer. Nucleic Acids Res. 1996, 24, 1695–1701. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.A.; Reynolds-Kohler, C.; Smith, B.A. Negative and positive regulation by a short segment in the 5′-flanking region of the human cytomegalovirus major immediate-early gene. Mol. Cell. Biol. 1987, 7, 4125–4129. [Google Scholar] [CrossRef] [PubMed]
- Lubon, H.; Ghazal, P.; Hennighausen, L.; Reynolds-Kohler, C.; Lockshin, C.; Nelson, J. Cell-specific activity of the modulator region in the human cytomegalovirus major immediate-early gene. Mol. Cell. Biol. 1989, 9, 1342–1345. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.A.; Groudine, M. Transcriptional regulation of the human cytomegalovirus major immediate-early gene is associated with induction of DNase I-hypersensitive sites. Mol. Cell. Biol. 1986, 6, 452–461. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.J.; Wu, A.W.; Andrews, J.I.; McGonagill, P.W.; Tibesar, E.E.; Meier, J.L. Reversal of human cytomegalovirus major immediate-early enhancer/promoter silencing in quiescently infected cells via the cyclic amp signaling pathway. J. Virol. 2007, 81, 6669–6681. [Google Scholar] [CrossRef] [PubMed]
- Yuan, J.; Liu, X.; Wu, A.W.; McGonagill, P.W.; Keller, M.J.; Galle, C.S.; Meier, J.L. Breaking human cytomegalovirus major immediate-early gene silence by vasoactive intestinal peptide stimulation of the protein kinase A-CREB-TORC2 signaling cascade in human pluripotent embryonal NTera2 cells. J. Virol. 2009, 83, 6391–6403. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Yuan, J.; Wu, A.W.; McGonagill, P.W.; Galle, C.S.; Meier, J.L. Phorbol ester-induced human cytomegalovirus major immediate-early (MIE) enhancer activation through PKC-delta, CREB, and NF-kappaB desilences MIE gene expression in quiescently infected human pluripotent NTera2 cells. J. Virol. 2010, 84, 8495–8508. [Google Scholar] [CrossRef] [PubMed]
- Prosch, S.; Wuttke, R.; Kruger, D.H.; Volk, H.D. NF-kappaB—A potential therapeutic target for inhibition of human cytomegalovirus (re)activation? Biol. Chem. 2002, 383, 1601–1609. [Google Scholar] [CrossRef] [PubMed]
- Meier, J.L. Reactivation of the human cytomegalovirus major immediate-early regulatory region and viral replication in embryonal ntera2 cells: Role of trichostatin a, retinoic acid, and deletion of the 21-base-pair repeats and modulator. J. Virol. 2001, 75, 1581–1593. [Google Scholar] [CrossRef] [PubMed]
- Wright, E.; Bain, M.; Teague, L.; Murphy, J.; Sinclair, J. Ets-2 repressor factor recruits histone deacetylase to silence human cytomegalovirus immediate-early gene expression in non-permissive cells. J. Gen. Virol. 2005, 86, 535–544. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.M.; Inouye, C.; Zeng, Y.; Bearss, D.; Seto, E. Transcriptional repression by YY1 is mediated by interaction with a mammalian homolog of the yeast global regulator RPD3. Proc. Natl. Acad. Sci. USA 1996, 93, 12845–12850. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kew, V.G.; Yuan, J.; Meier, J.; Reeves, M.B. Mitogen and stress activated kinases act co-operatively with creb during the induction of human cytomegalovirus immediate-early gene expression from latency. PLoS Pathog. 2014, 10, e1004195. [Google Scholar] [CrossRef] [PubMed]
- Lieberman, P.M. Epigenetics and genetics of viral latency. Cell. Host Microbe 2016, 19, 619–628. [Google Scholar] [CrossRef] [PubMed]
- Hummel, M.; Yan, S.; Li, Z.; Varghese, T.K.; Abecassis, M. Transcriptional reactivation of murine cytomegalovirus ie gene expression by 5-aza-2′-deoxycytidine and trichostatin A in latently infected cells despite lack of methylation of the major immediate-early promoter. J. Gen. Virol. 2007, 88, 1097–1102. [Google Scholar] [CrossRef] [PubMed]
- Murphy, J.C.; Fischle, W.; Verdin, E.; Sinclair, J.H. Control of cytomegalovirus lytic gene expression by histone acetylation. EMBO J. 2002, 21, 1112–1120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Varnum, S.M.; Streblow, D.N.; Monroe, M.E.; Smith, P.; Auberry, K.J.; Pasa-Tolic, L.; Wang, D.; Camp, D.G., 2nd; Rodland, K.; Wiley, S.; et al. Identification of proteins in human cytomegalovirus (HCMV) particles: The HCMV proteome. J. Virol. 2004, 78, 10960–10966. [Google Scholar] [CrossRef] [PubMed]
- Groves, I.J.; Reeves, M.B.; Sinclair, J.H. Lytic infection of permissive cells with human cytomegalovirus is regulated by an intrinsic ‘pre-immediate-early’ repression of viral gene expression mediated by histone post-translational modification. J. Gen. Virol. 2009, 90, 2364–2374. [Google Scholar] [CrossRef] [PubMed]
- Nitzsche, A.; Paulus, C.; Nevels, M. Temporal dynamics of cytomegalovirus chromatin assembly in productively infected human cells. J. Virol. 2008, 82, 11167–11180. [Google Scholar] [CrossRef] [PubMed]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [PubMed]
- Cuevas-Bennett, C.; Shenk, T. Dynamic histone H3 acetylation and methylation at human cytomegalovirus promoters during replication in fibroblasts. J. Virol. 2008, 82, 9525–9536. [Google Scholar] [CrossRef] [PubMed]
- Abraham, C.G.; Kulesza, C.A. Polycomb repressive complex 2 silences human cytomegalovirus transcription in quiescent infection models. J. Virol. 2013, 87, 13193–13205. [Google Scholar] [CrossRef] [PubMed]
- Tavalai, N.; Papior, P.; Rechter, S.; Leis, M.; Stamminger, T. Evidence for a role of the cellular ND10 protein PML in mediating intrinsic immunity against human cytomegalovirus infections. J. Virol. 2006, 80, 8006–8018. [Google Scholar] [CrossRef] [PubMed]
- Tavalai, N.; Papior, P.; Rechter, S.; Stamminger, T. Nuclear domain 10 components promyelocytic leukemia protein and hDaxx independently contribute to an intrinsic antiviral defense against human cytomegalovirus infection. J. Virol. 2008, 82, 126–137. [Google Scholar] [CrossRef] [PubMed]
- Woodhall, D.L.; Groves, I.J.; Reeves, M.B.; Wilkinson, G.; Sinclair, J.H. Human Daxx-mediated repression of human cytomegalovirus gene expression correlates with a repressive chromatin structure around the major immediate early promoter. J. Biol. Chem. 2006, 281, 37652–37660. [Google Scholar] [CrossRef] [PubMed]
- Lukashchuk, V.; McFarlane, S.; Everett, R.D.; Preston, C.M. Human cytomegalovirus protein PP71 displaces the chromatin-associated factor ATRX from nuclear domain 10 at early stages of infection. J. Virol. 2008, 82, 12543–12554. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Kalejta, R.F. Human cytomegalovirus protein PP71 induces Daxx sumoylation. J. Virol. 2009, 83, 6591–6598. [Google Scholar] [CrossRef] [PubMed]
- Hwang, J.; Kalejta, R.F. Proteasome-dependent, ubiquitin-independent degradation of Daxx by the viral PP71 protein in human cytomegalovirus-infected cells. Virology 2007, 367, 334–338. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Stinski, M.F. Human cytomegalovirus contains a tegument protein that enhances transcription from promoters with upstream ATF and AP-1 cis-acting elements. J. Virol. 1992, 66, 4434–4444. [Google Scholar] [PubMed]
- Cantrell, S.R.; Bresnahan, W.A. Interaction between the human cytomegalovirus UL82 gene product (pp71) and hdaxx regulates immediate-early gene expression and viral replication. J. Virol. 2005, 79, 7792–7802. [Google Scholar] [CrossRef] [PubMed]
- Cantrell, S.R.; Bresnahan, W.A. Human cytomegalovirus (HCMV) UL82 gene product (pp71) relieves hDaxx-mediated repression of HCMV replication. J. Virol. 2006, 80, 6188–6191. [Google Scholar] [CrossRef] [PubMed]
- Preston, C.M.; Nicholl, M.J. Role of the cellular protein hdaxx in human cytomegalovirus immediate-early gene expression. J. Gen. Virol. 2006, 87, 1113–1121. [Google Scholar] [CrossRef] [PubMed]
- Ishov, A.M.; Vladimirova, O.V.; Maul, G.G. Daxx-mediated accumulation of human cytomegalovirus tegument protein pp71 at ND10 facilitates initiation of viral infection at these nuclear domains. J. Virol. 2002, 76, 7705–7712. [Google Scholar] [CrossRef] [PubMed]
- Hofmann, H.; Sindre, H.; Stamminger, T. Functional interaction between the pp71 protein of human cytomegalovirus and the PML-interacting protein human Daxx. J. Virol. 2002, 76, 5769–5783. [Google Scholar] [CrossRef] [PubMed]
- Nevels, M.; Paulus, C.; Shenk, T. Human cytomegalovirus immediate-early 1 protein facilitates viral replication by antagonizing histone deacetylation. Proc. Natl. Acad. Sci. USA 2004, 101, 17234–17239. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Park, J.J.; Kim, Y.E.; Pham, H.T.; Kim, E.T.; Chung, Y.H.; Ahn, J.H. Functional interaction of the human cytomegalovirus IE2 protein with histone deacetylase 2 in infected human fibroblasts. J. Gen. Virol. 2007, 88, 3214–3223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishov, A.M.; Stenberg, R.M.; Maul, G.G. Human cytomegalovirus immediate early interaction with host nuclear structures: Definition of an immediate transcript environment. J. Cell Biol. 1997, 138, 5–16. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.H.; Albright, E.R.; Lee, J.H.; Jacobs, D. Cellular defense against latent colonization foiled by human cytomegalovirus UL138 protein. Sci. Adv. 2015, 1, e1501164. [Google Scholar] [CrossRef] [PubMed]
- Goodrum, F.; Reeves, M.; Sinclair, J.; High, K.; Shenk, T. Human cytomegalovirus sequences expressed in latently infected individuals promote a latent infection in vitro. Blood 2007, 110, 937–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, S.E.; Olsen, L. Structural basis of histone demethylase KDM6B histone 3 lysine 27 specificity. Biochemistry 2018, 57, 585–592. [Google Scholar] [CrossRef] [PubMed]
- Minton, E.J.; Tysoe, C.; Sinclair, J.H.; Sissons, J.G. Human cytomegalovirus infection of the monocyte/macrophage lineage in bone marrow. J. Virol. 1994, 68, 4017–4021. [Google Scholar] [PubMed]
- Sinclair, J.H.; Baillie, J.; Bryant, L.A.; Taylor-Wiedeman, J.A.; Sissons, J.G. Repression of human cytomegalovirus major immediate early gene expression in a monocytic cell line. J. Gen. Virol. 1992, 73 Pt 2, 433–435. [Google Scholar] [CrossRef]
- Stinski, M.F. Cytomegalovirus promoter for expression in mammalian cells. In Gene Expression Systems: Using Nature for the Art of Expression, 1st ed.; Fernandez, J.M., Hoeffler, J.P., Eds.; Elsevier-Academic Press: Cambridge, MA, USA, 1999; pp. 211–233. [Google Scholar]
- Goodrum, F.; Jordan, C.T.; Terhune, S.S.; High, K.; Shenk, T. Differential outcomes of human cytomegalovirus infection in primitive hematopoietic cell subpopulations. Blood 2004, 104, 687–695. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheung, A.K.; Abendroth, A.; Cunningham, A.L.; Slobedman, B. Viral gene expression during the establishment of human cytomegalovirus latent infection in myeloid progenitor cells. Blood 2006, 108, 3691–3699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, S.; Caviness, K.; Buehler, J.; Smithey, M.; Nikolich-Zugich, J.; Goodrum, F. Transcriptome-wide characterization of human cytomegalovirus in natural infection and experimental latency. Proc. Natl. Acad. Sci. USA 2017, 114. [Google Scholar] [CrossRef] [PubMed]
- Tarrant-Elorza, M.; Rossetto, C.C.; Pari, G.S. Maintenance and replication of the human cytomegalovirus genome during latency. Cell. Host Microbe 2014, 16, 43–54. [Google Scholar] [CrossRef] [PubMed]
- Kondo, K.; Xu, J.; Mocarski, E.S. Human cytomegalovirus latent gene expression in granulocyte-macrophage progenitors in culture and in seropositive individuals. Proc. Natl. Acad. Sci. USA 1996, 93, 11137–11142. [Google Scholar] [CrossRef] [PubMed]
- Maciejewski, J.P.; St Jeor, S.C. Human cytomegalovirus infection of human hematopoietic progenitor cells. Leuk. Lymphoma. 1999, 33, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Price, A.M.; Luftig, M.A. To be or not IIb: A multi-step process for epstein-barr virus latency establishment and consequences for b cell tumorigenesis. PLoS Pathog. 2015, 11, e1004656. [Google Scholar] [CrossRef] [PubMed]
- Cliffe, A.R.; Wilson, A.C. Restarting lytic gene transcription at the onset of herpes simplex virus reactivation. J. Virol. 2017, 91. [Google Scholar] [CrossRef] [PubMed]
- Arend, K.C.; Ziehr, B.; Vincent, H.A.; Moorman, N.J. Multiple transcripts encode full-length human cytomegalovirus IE1 and IE2 proteins during lytic infection. J. Virol. 2016, 90, 8855–8865. [Google Scholar] [CrossRef] [PubMed]
- Shnayder, M.; Nachshon, A.; Krishna, B.; Poole, E.; Boshkov, A.; Binyamin, A.; Maza, I.; Sinclair, J.; Schwartz, M.; Stern-Ginossar, N. Defining the transcriptional landscape during cytomegalovirus latency with single-cell RNA sequencing. mBio 2018, 9. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
A model of human cytomegalovirus (HCMV) infection reservoirs. An initial round of lytic replication occurs in mucosal epithelial cells following virus exposure. Circulating monocytes are infected through contact with an infected epithelial cell and induce monocyte-to-macrophage differentiation and extravasation to peripheral tissues, including the bone marrow where a latent infection is established. Upon a reactivation signal, HCMV can induce the differentiation and mobilization of latently infected CD34+ bone marrow cells. While not entirely defined, HCMV may stimulate CD34+ human progenitor cell (HPC) differentiation down a monocytic lineage with an eventual terminal differentiation state of an infected macrophage or dendritic cell. These terminally differentiated cells allow for the reseeding of epithelial cell layers within peripheral organ sites.
Figure 1.
A model of human cytomegalovirus (HCMV) infection reservoirs. An initial round of lytic replication occurs in mucosal epithelial cells following virus exposure. Circulating monocytes are infected through contact with an infected epithelial cell and induce monocyte-to-macrophage differentiation and extravasation to peripheral tissues, including the bone marrow where a latent infection is established. Upon a reactivation signal, HCMV can induce the differentiation and mobilization of latently infected CD34+ bone marrow cells. While not entirely defined, HCMV may stimulate CD34+ human progenitor cell (HPC) differentiation down a monocytic lineage with an eventual terminal differentiation state of an infected macrophage or dendritic cell. These terminally differentiated cells allow for the reseeding of epithelial cell layers within peripheral organ sites.

Figure 2.
Importance of epidermal growth factor receptor (EGFR) and integrin signaling during CMV latent infection. (A) EGFR signaling is important for CMV entry, translocation to the nucleus, and to establish latency. Attenuation of EGFR signaling by the UL135 proteins or inhibitors disrupts this pattern and contributes to reactivation of CMV replication. Thickness of arrows and lines depicts strength of supporting studies; (B) In monocytes, CMV binding and entry via EGFR and integrin receptors results in changes in signaling to promote cellular survival, macrophage differentiation, and motility to promote virus dissemination in the infected host. Red T bars indicate inhibition. Green arrows indicate activation.
Figure 2.
Importance of epidermal growth factor receptor (EGFR) and integrin signaling during CMV latent infection. (A) EGFR signaling is important for CMV entry, translocation to the nucleus, and to establish latency. Attenuation of EGFR signaling by the UL135 proteins or inhibitors disrupts this pattern and contributes to reactivation of CMV replication. Thickness of arrows and lines depicts strength of supporting studies; (B) In monocytes, CMV binding and entry via EGFR and integrin receptors results in changes in signaling to promote cellular survival, macrophage differentiation, and motility to promote virus dissemination in the infected host. Red T bars indicate inhibition. Green arrows indicate activation.

Figure 3.
Viral regulation of cellular signaling during latency. CMV utilizes a number of viral proteins and miRNAs during latent infection to alter the signaling environment within the cell to regulate the latent infection. This model demonstrates how each viral component either inhibits (red cross) or promotes (green arrow) cellular signaling events during a latent infection. As depicted, a number of viral factors target similar pathways demonstrating their necessity in the maintenance of and reactivation from latency.
Figure 3.
Viral regulation of cellular signaling during latency. CMV utilizes a number of viral proteins and miRNAs during latent infection to alter the signaling environment within the cell to regulate the latent infection. This model demonstrates how each viral component either inhibits (red cross) or promotes (green arrow) cellular signaling events during a latent infection. As depicted, a number of viral factors target similar pathways demonstrating their necessity in the maintenance of and reactivation from latency.

Figure 4.
Diagram of cellular transcription factor binding to the HCMV major immediate early promoter (MIEP). The major immediate early core promoter, enhancer, unique region, and modulator are shown. Relative locations of binding sites for activating and repressive cellular transcription factors are shown. Repeat elements are labeled with the number of base pairs repeated in each sequence. The 18-bp repeats bind NF-κB/rel, the 19-bp repeats bind CREB/ATF, and the 21-bp repeats bind SP-1, YY1, and ERF.
Figure 4.
Diagram of cellular transcription factor binding to the HCMV major immediate early promoter (MIEP). The major immediate early core promoter, enhancer, unique region, and modulator are shown. Relative locations of binding sites for activating and repressive cellular transcription factors are shown. Repeat elements are labeled with the number of base pairs repeated in each sequence. The 18-bp repeats bind NF-κB/rel, the 19-bp repeats bind CREB/ATF, and the 21-bp repeats bind SP-1, YY1, and ERF.

© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Collins-McMillen, D.; Buehler, J.; Peppenelli, M.; Goodrum, F. Molecular Determinants and the Regulation of Human Cytomegalovirus Latency and Reactivation. Viruses 2018, 10, 444. https://doi.org/10.3390/v10080444
AMA Style
Collins-McMillen D, Buehler J, Peppenelli M, Goodrum F. Molecular Determinants and the Regulation of Human Cytomegalovirus Latency and Reactivation. Viruses. 2018; 10(8):444. https://doi.org/10.3390/v10080444
Chicago/Turabian StyleCollins-McMillen, Donna, Jason Buehler, Megan Peppenelli, and Felicia Goodrum. 2018. "Molecular Determinants and the Regulation of Human Cytomegalovirus Latency and Reactivation" Viruses 10, no. 8: 444. https://doi.org/10.3390/v10080444
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.