Epigenetic Changes in the Regulation of Nicotiana tabacum Response to Cucumber Mosaic Virus Infection and Symptom Recovery through Single-Base Resolution Methylomes


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Plant Growth and Virus Inoculation
2.2. VIGS Assay
2.3. DNA and RNA Extraction
2.4. Reduced Representation Bisulfite Sequencing (RRBS) Library Construction and Sequencing
2.5. Identification and Analysis of Differential Methylated Regions (DMRs)
2.6. mRNA Sequencing and Analysis
2.7. Analysis of Correlation of DMRs and DEGs
2.8. Functional Enrichment Analysis
2.9. Small RNA-seq and Data Analysis
2.10. qRT-PCR
2.11. Detection of Hydrogen Peroxide
3. Results
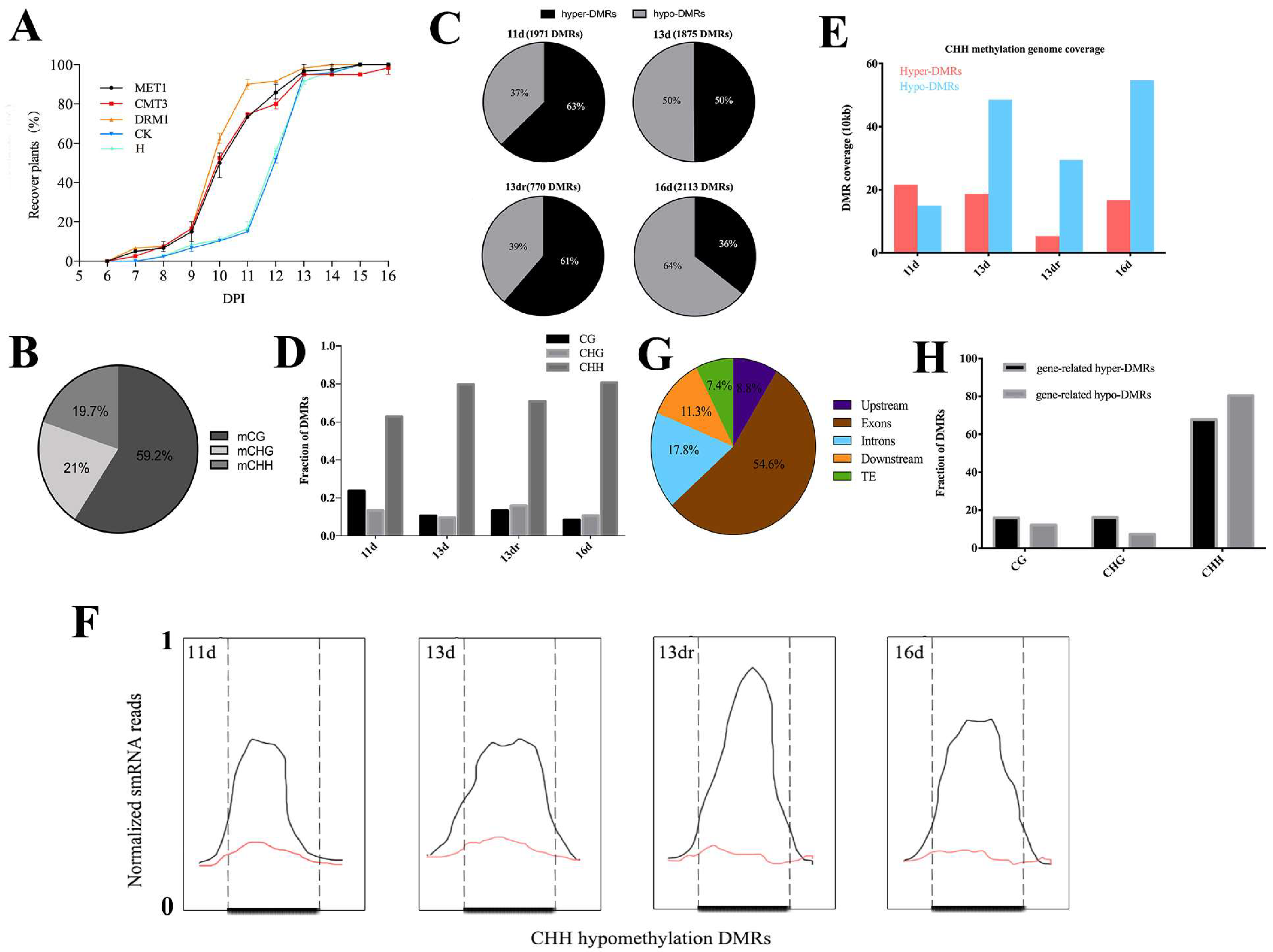
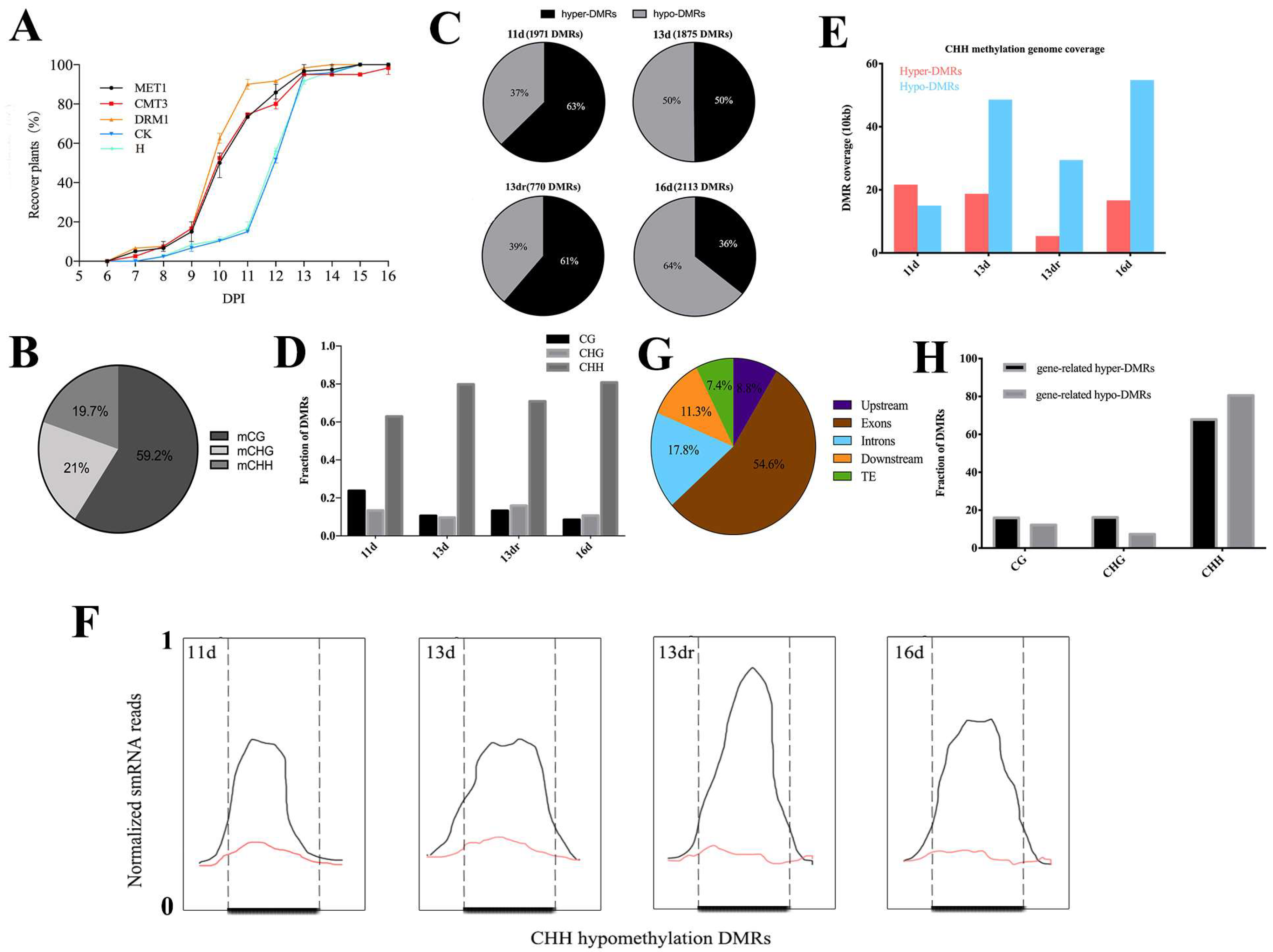
3.1. Loss of Methyltransferase Expression Activates Antiviral Defense
3.2. Viral Infection Is Associated with CHH Methylation
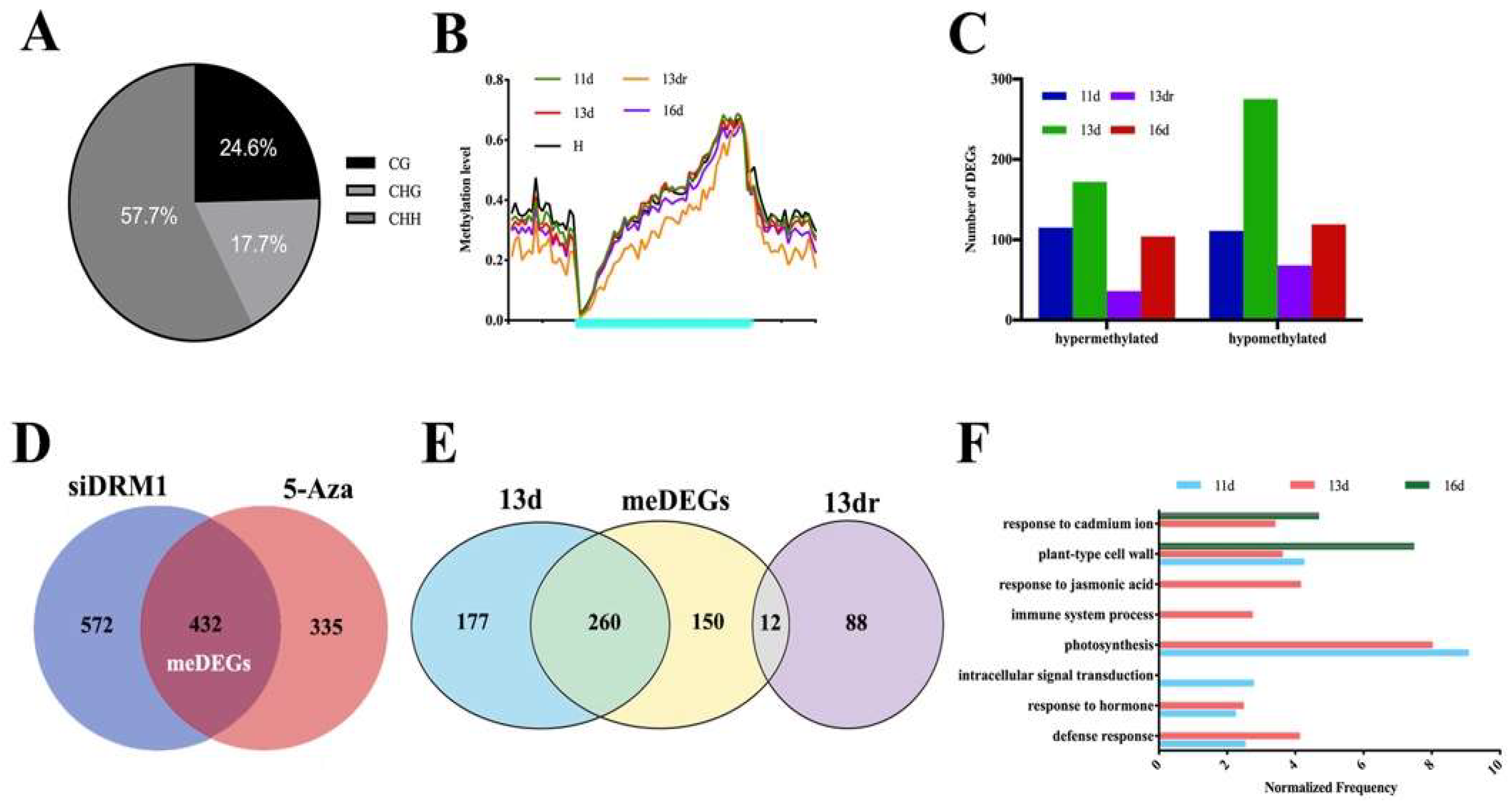
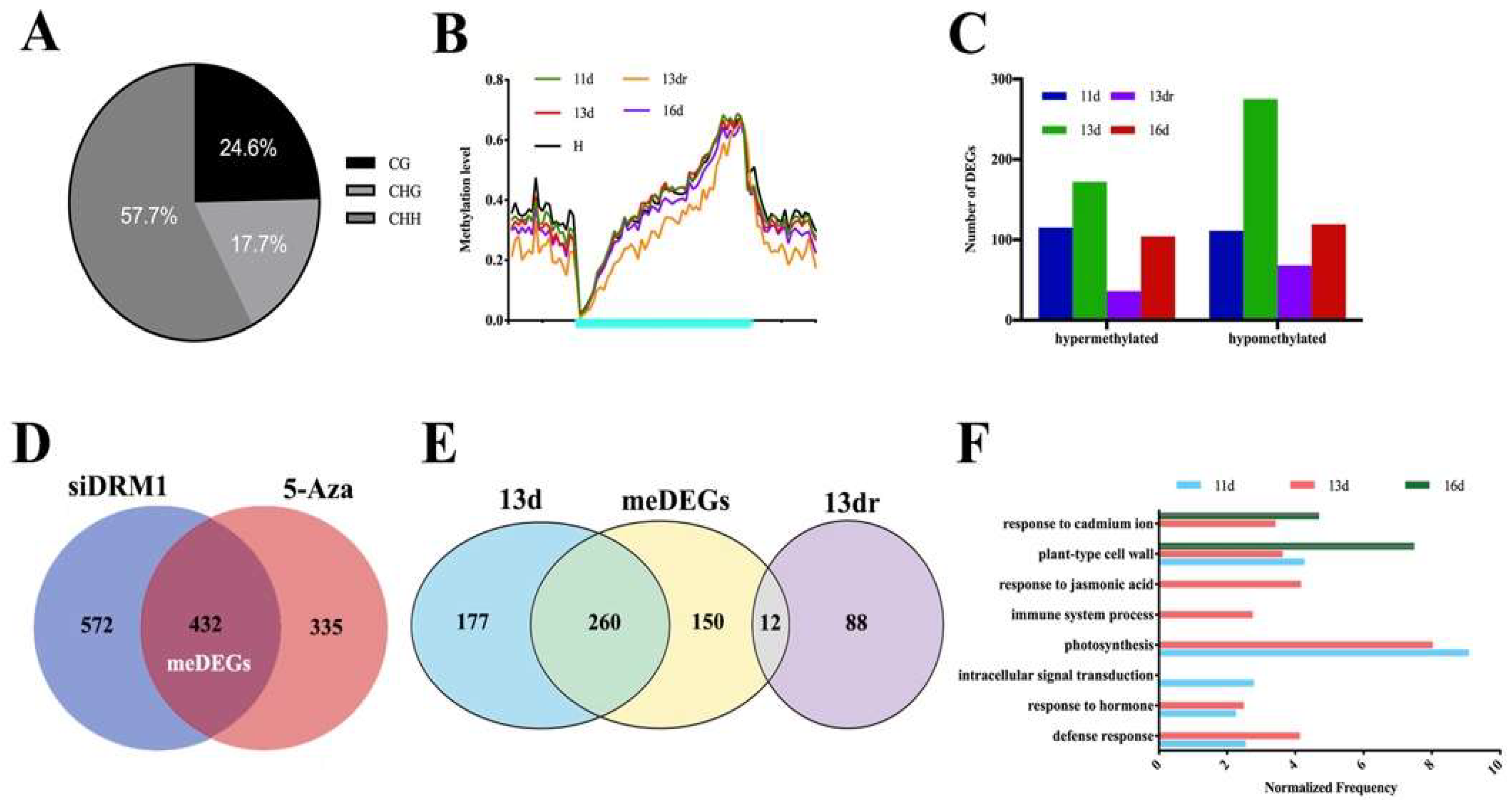
3.3. Methylation Contexts in Gene Body Correlate with Gene Expression
3.4. Functional Analysis of DMRs Located in Coding Genes in a Temporal Regulation
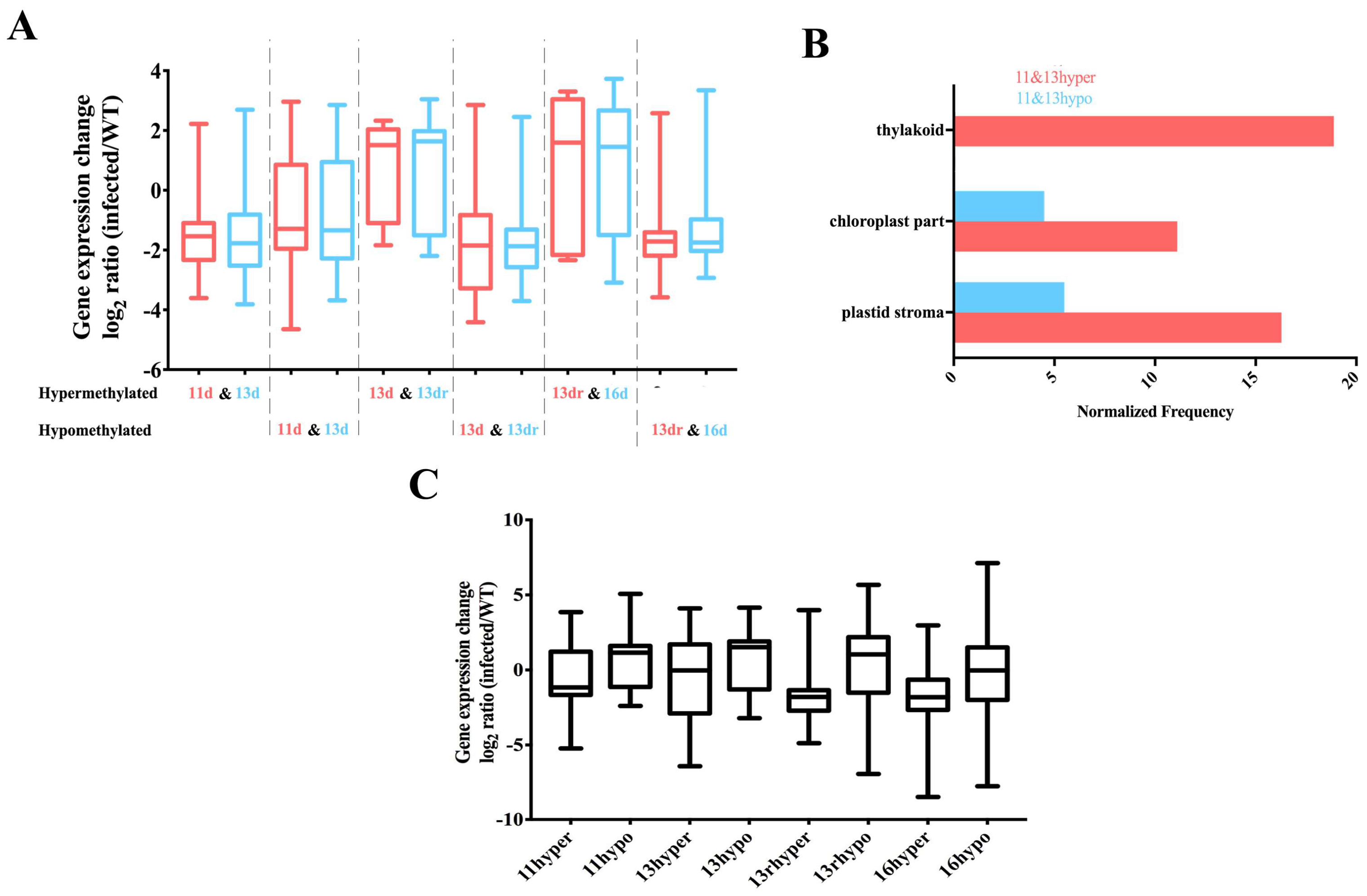
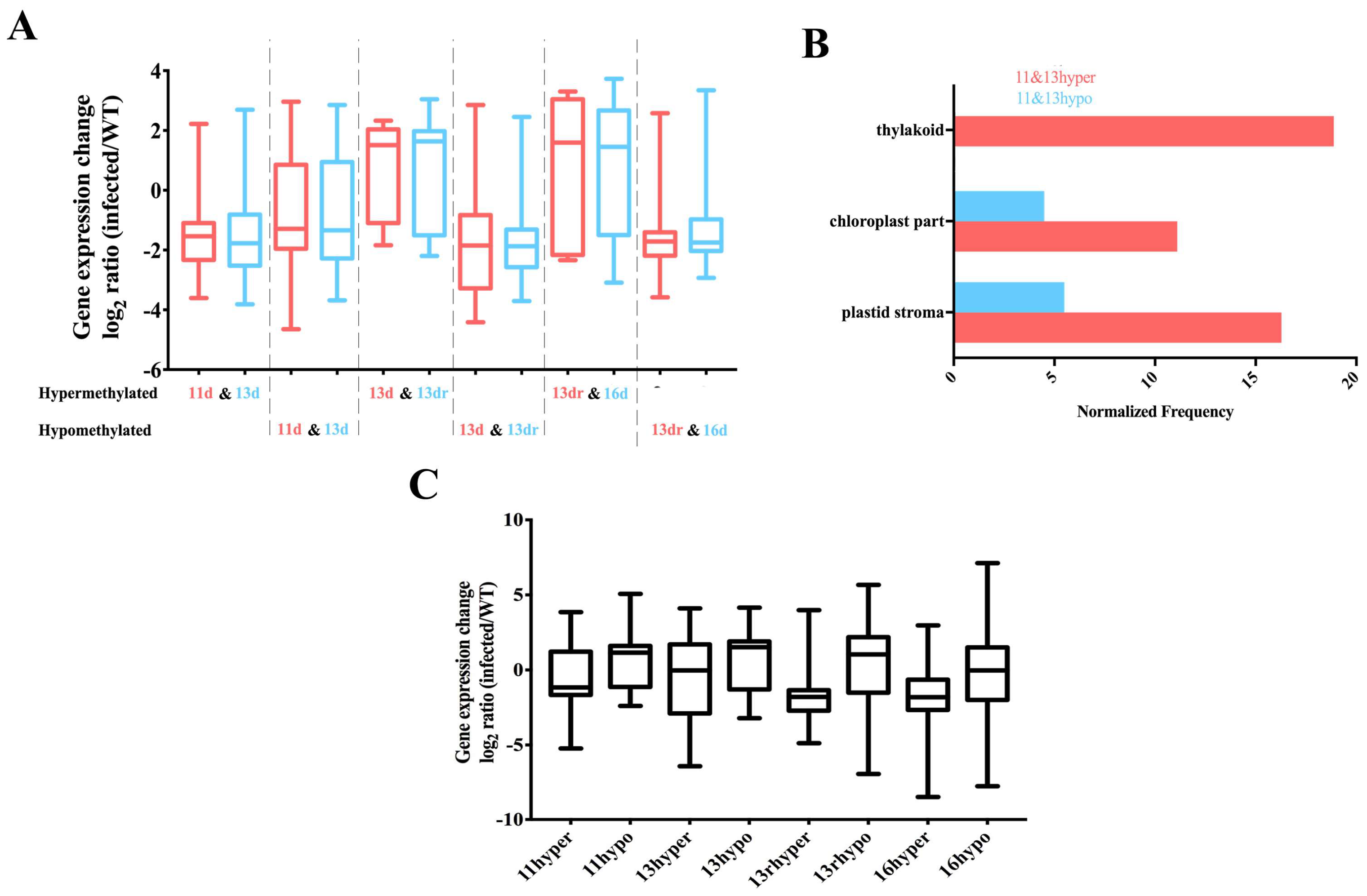
3.5. Stability of Methylated and Demethylated DMRs across the Developmental Stages
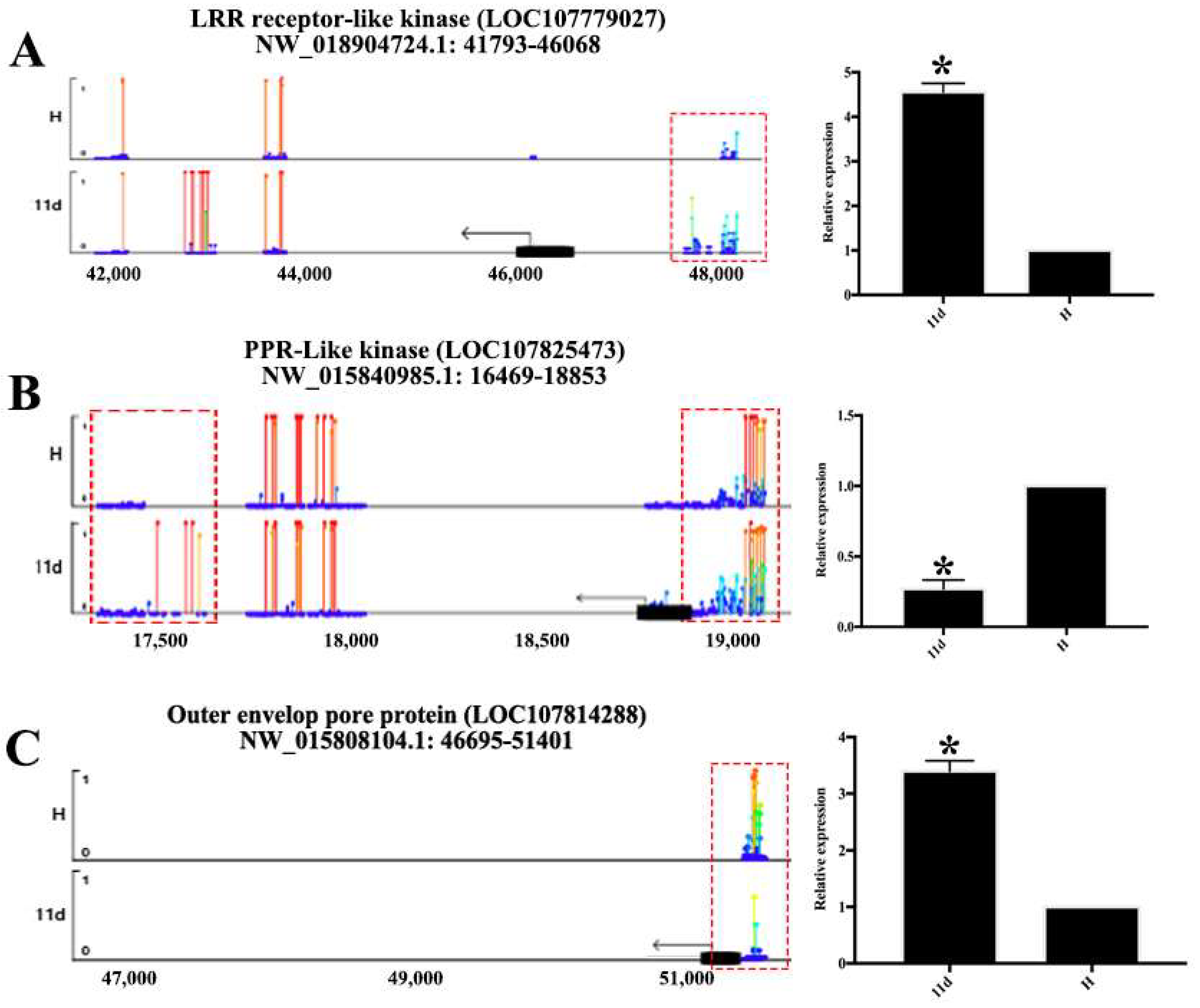
3.6. DMRs in Promoter Alters Gene Expression
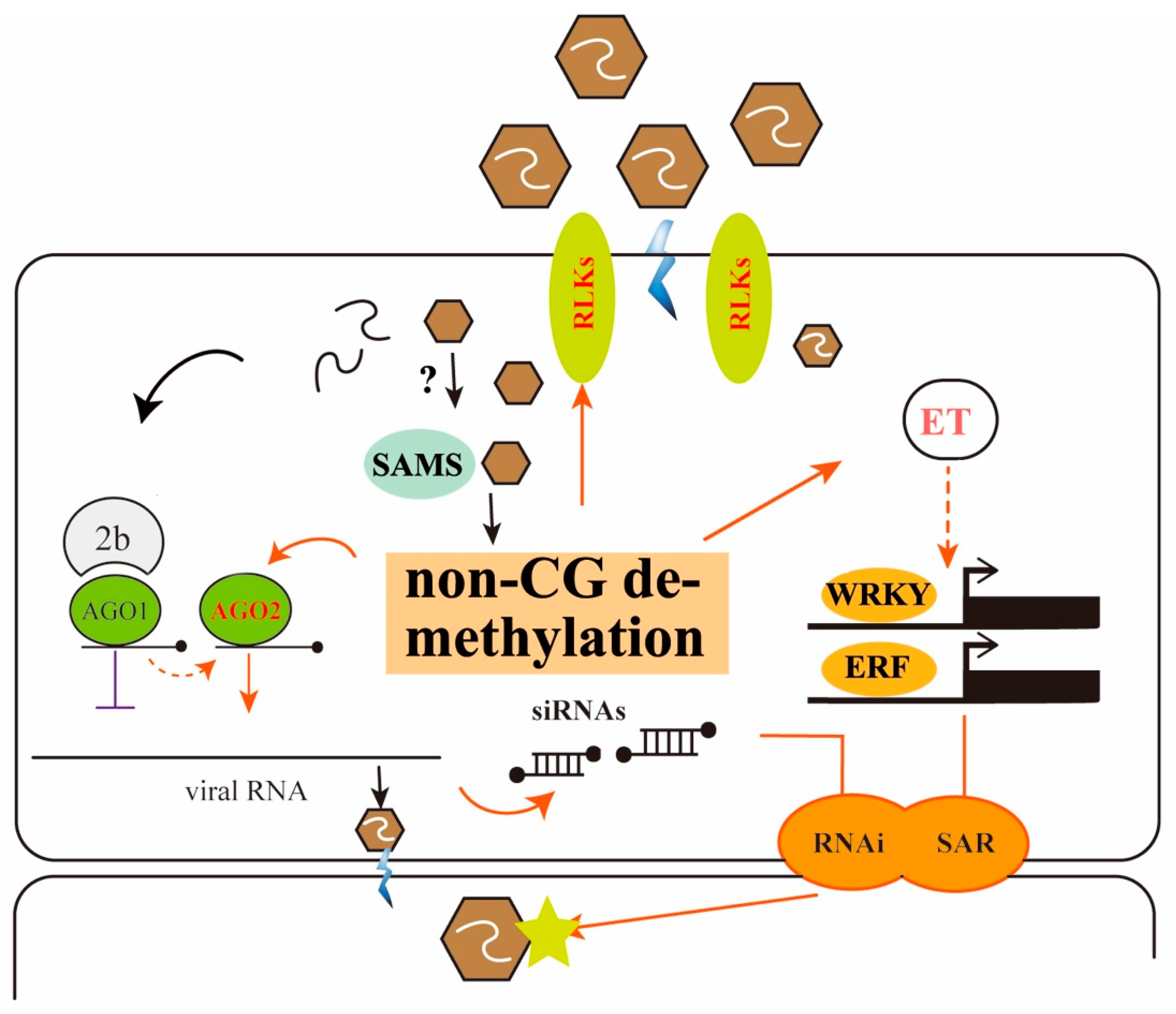
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Albacete, A.A.; Martínez-Andújar, C.; Pérez-Alfocea, F. Hormonal and metabolic regulation of source-sink relations under salinity and drought: From plant survival to crop yield stability. Biotechnol. Adv. 2014, 32, 12–30. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.W. Rna-based antiviral immunity. Nat. Rev. Immunol. 2010, 10, 632–644. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.Y.; Huang, T.K.; Yang, S.Y.; Hong, Y.T.; Huang, S.M.; Wang, F.N.; Su-Fen, C.; Shang-Yueh, T.; Lu, W.C.; Tzyy-Jen, C. Identification of plant vacuolar transporters mediating phosphate storage. Nat. Commun. 2016, 7, 11095. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, C.; Hagmann, J.; Müller, J.; Koenig, D.; Stegle, O.; Borgwardt, K.; Weigel, D. Spontaneous epigenetic variation in the arabidopsis thaliana methylome. Nature 2011, 480, 245–249. [Google Scholar] [CrossRef] [PubMed]
- Du, J.; Johnson, L.M.; Jacobsen, S.E.; Patel, D.J. DNA methylation pathways and their crosstalk with histone methylation. Nat. Rev. Mol. Cell Boil. 2015, 16, 519–532. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, Z.D.; Meissner, A. DNA methylation: Roles in mammalian development. Nat. Rev. Genet. 2013, 14, 204–220. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Zhu, P.; Yan, L.; Li, R.; Hu, B.; Lian, Y.; Yan, J.; Ren, X.; Lin, S.; Li, J. The DNA methylation landscape of human early embryos. Nature 2014, 511, 606–610. [Google Scholar] [CrossRef] [PubMed]
- Cokus, S.J.; Feng, S.; Zhang, X.; Chen, Z.; Merriman, B.; Haudenschild, C.D.; Pradhan, S.; Nelson, S.F.; Pellegrini, M.; Jacobsen, S.E. Shotgun bisulfite sequencing of the arabidopsis genome reveals DNA methylation patterning. Nature 2008, 452, 215–219. [Google Scholar] [CrossRef] [PubMed]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cao, X.; Jacobsen, S.E. Locus-specific control of asymmetric and cpnpg methylation by the drm and cmt3 methyltransferase genes. Proc. Natl. Acad. Sci. USA 2002, 99 (Suppl. S4), 16491–16498. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Du, J.; Hale, C.; Gallego-Bartolome, J.; Feng, S.; Vashisht, A.; Chory, J.; Wohlschlegel, J.; Patel, D.; Jacobsen, S. Molecular mechanism of action of plant drm de novo DNA methyltransferases. Cell 2014, 157, 1050–1060. [Google Scholar] [CrossRef] [PubMed]
- Stroud, H.; Do, T.; Du, J.; Zhong, X.; Feng, S.; Johnson, L.; Patel, D.J.; Jacobsen, S.E. Non-cg methylation patterns shape the epigenetic landscape in arabidopsis. Nat. Struct. Mol. Boil. 2014, 21, 64–72. [Google Scholar] [CrossRef] [PubMed]
- Matzke, M.A.; Mosher, R.A. Rna-directed DNA methylation: An epigenetic pathway of increasing complexity. Nat. Rev. Genet. 2014, 15, 394–408. [Google Scholar] [CrossRef] [PubMed]
- Blevins, T.; Podicheti, R.; Mishra, V.; Marasco, M.; Wang, J.; Rusch, D.; Tang, H.; Pikaard, C.S. Identification of Pol IV and RDR2-dependent precursors of 24 nt siRNAs guiding de novo DNA methylation in arabidopsis. eLife 2015, 4, e09591. [Google Scholar] [CrossRef] [PubMed]
- Ye, R.; Wang, W.; Iki, T.; Liu, C.; Wu, Y.; Ishikawa, M.; Zhou, X.; Qi, Y. Cytoplasmic assembly and selective nuclear import of arabidopsis argonaute4/siRNA complexes. Mol. Cell 2012, 46, 859. [Google Scholar] [CrossRef] [PubMed]
- Wierzbicki, A.T.; Ream, T.; Haag, J.R.; Pikaard, C.S. RNA polymerase V transcription guides ARGONAUTE4 to chromatin. Nat. Genet. 2009, 41, 630–634. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, H.; Zhu, P.; Wu, X.; Li, X.; Wen, L.; Tang, F. Single-cell methylome landscapes of mouse embryonic stem cells and early embryos analyzed using reduced representation bisulfite sequencing. Genome Res. 2013, 23, 2126–2135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Li, L.; Yan, J. Dissecting meiotic recombination based on tetrad analysis by single-microspore sequencing in maize. Nat. Commun. 2015, 6, 6648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, F.; Yan, L.; Guo, H.; Li, L.; Hu, B.; Zhao, Y.; Yong, J.; Hu, Y.; Wang, X.; Wei, Y. The transcriptome and DNA methylome landscapes of human primordial germ cells. Cell 2015, 161, 1437–1452. [Google Scholar] [CrossRef] [PubMed]
- Dong, Q.; Li, X.; Wang, C.-Z.; Xu, S.; Yuan, G.; Shao, W.; Liu, B.; Zheng, Y.; Wang, H.; Lei, X. Roles of the cse1l-mediated nuclear import pathway in epigenetic silencing. Proc. Natl. Acad. Sci. USA 2018, 115, E4013–E4022. [Google Scholar] [CrossRef] [PubMed]
- Regulski, M.; Lu, Z.; Kendall, J.; Donoghue, M.T.; Reinders, J.; Llaca, V.; Deschamps, S.; Smith, A.; Levy, D.; McCombie, W.R. The maize methylome influences mrna splice sites and reveals widespread paramutation-like switches guided by small RNA. Genome Res. 2013, 23, 1651–1662. [Google Scholar] [CrossRef] [PubMed]
- Stroud, H.; Ding, B.; Simon, S.A.; Feng, S.; Bellizzi, M.; Pellegrini, M.; Wang, G.-L.; Meyers, B.C.; Jacobsen, S.E. Plants regenerated from tissue culture contain stable epigenome changes in rice. eLife 2013, 2, e00354. [Google Scholar] [CrossRef] [PubMed]
- Standage, D.S.; Berens, A.J.; Glastad, K.M.; Severin, A.J.; Brendel, V.P.; Toth, A.L. Genome, transcriptome and methylome sequencing of a primitively eusocial wasp reveal a greatly reduced DNA methylation system in a social insect. Mol. Ecol. 2016, 25, 1769–1784. [Google Scholar] [CrossRef] [PubMed]
- Zhong, S.; Fei, Z.; Chen, Y.-R.; Zheng, Y.; Huang, M.; Vrebalov, J.; McQuinn, R.; Gapper, N.; Liu, B.; Xiang, J. Single-base resolution methylomes of tomato fruit development reveal epigenome modifications associated with ripening. Nat. Biotechnol. 2013, 31, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Xing, M.-Q.; Zhang, Y.; Zhou, S.-R.; Hu, W.-Y.; Wu, X.-T.; Ye, Y.-J.; Xiao, Y.-P.; Li, X.; Xue, H.-W. Global analysis reveals the crucial roles of DNA methylation during rice seed development. Plant Physiol. 2015, 00414. [Google Scholar] [CrossRef] [PubMed]
- Chodavarapu, R.K.; Feng, S.; Ding, B.; Simon, S.A.; Lopez, D.; Jia, Y.; Wang, G.-L.; Meyers, B.C.; Jacobsen, S.E.; Pellegrini, M. Transcriptome and methylome interactions in rice hybrids. Proc. Natl. Acad. Sci. USA 2012, 109, 12040–12045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, J.; Zhou, S.; Gong, X.; Song, Y.; van Nocker, S.; Ma, F.; Guan, Q. Single-Base methylome analysis reveals dynamic epigenomic differences associated with water deficit in apple. Plant Biotechnol. J. 2018, 16, 672–687. [Google Scholar] [CrossRef] [PubMed]
- Yong-Villalobos, L.; González-Morales, S.I.; Wrobel, K.; Gutiérrez-Alanis, D.; Cervantes-Peréz, S.A.; Hayano-Kanashiro, C.; Oropeza-Aburto, A.; Cruz-Ramírez, A.; Martínez, O.; Herrera-Estrella, L. Methylome analysis reveals an important role for epigenetic changes in the regulation of the arabidopsis response to phosphate starvation. Proc. Natl. Acad. Sci. USA 2015, 112, E7293–E7302. [Google Scholar] [CrossRef] [PubMed]
- Dowen, R.H.; Pelizzola, M.; Schmitz, R.J.; Lister, R.; Dowen, J.M.; Nery, J.R.; Dixon, J.E.; Ecker, J.R. Widespread dynamic DNA methylation in response to biotic stress. Proc. Natl. Acad. Sci. USA 2012, 109, E2183–E2191. [Google Scholar] [CrossRef] [PubMed]
- Yu, A.; Lepère, G.; Jay, F.; Wang, J.; Bapaume, L.; Wang, Y.; Abraham, A.-L.; Penterman, J.; Fischer, R.L.; Voinnet, O. Dynamics and biological relevance of DNA demethylation in arabidopsis antibacterial defense. Proc. Natl. Acad. Sci. USA 2013, 110, 2389–2394. [Google Scholar] [CrossRef] [PubMed]
- Deleris, A.; Halter, T.; Navarro, L. DNA methylation and demethylation in plant immunity. Annu. Rev. Phytopathol. 2016, 54, 579–603. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Chen, H.; Huang, X.; Xia, R.; Zhao, Q.; Lai, J.; Teng, K.; Li, Y.; Liang, L.; Du, Q. BSCTV C2 attenuates the degradation of SAMDC1 to suppress DNA methylation-mediated gene silencing in arabidopsis. Plant Cell 2011, 23, 273–288. [Google Scholar] [CrossRef] [PubMed]
- Mandadi, K.K.; Scholthof, K.B.G. Plant immune responses against viruses: How does a virus cause disease? Plant Cell 2013, 25, 1489–1505. [Google Scholar] [CrossRef] [PubMed]
- Palukaitis, P.; García-Arenal, F. Cucumoviruses. Adv. Virus Res. 2003, 62, 241–323. [Google Scholar] [PubMed]
- Lu, J.; Du, Z.X.; Kong, J.; Chen, L.N.; Qiu, Y.H.; Li, G.F.; Meng, X.H.; Zhu, S.F. Transcriptome analysis of nicotiana tabacum infected by cucumber mosaic virus during systemic symptom development. PLoS ONE 2012, 7, e43447. [Google Scholar] [CrossRef] [PubMed]
- Ghoshal, B.; Sanfaçon, H. Symptom recovery in virus-infected plants: Revisiting the role of RNA silencing mechanisms. Virology 2015, 479, 167–179. [Google Scholar] [CrossRef] [PubMed]
- Butterbach, P.; Verlaan, M.G.; Dullemans, A.; Lohuis, D.; Visser, R.G.; Bai, Y.; Kormelink, R. Tomato yellow leaf curl virus resistance by Ty-1 involves increased cytosine methylation of viral genomes and is compromised by cucumber mosaic virus infection. Proc. Natl. Acad. Sci. USA 2014, 111, 12942–12947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, C.-G.; Fang, Y.-Y.; Zhou, B.-J.; Zhao, J.-H.; Hou, W.-N.; Zhu, H.; Ding, S.-W.; Guo, H.-S. Suppression of arabidopsis ARGONAUTE1-mediated slicing, transgene-induced RNA silencing, and DNA methylation by distinct domains of the cucumber mosaic virus 2b protein. Plant Cell 2012, 24, 259–274. [Google Scholar] [CrossRef] [PubMed]
- Rodríguez-Negrete, E.; Lozano-Durán, R.; Piedra-Aguilera, A.; Cruzado, L.; Bejarano, E.R.; Castillo, A.G. Geminivirus Rep protein interferes with the plant DNA methylation machinery and suppresses transcriptional gene silencing. New Phytol. 2013, 199, 464–475. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Y.; Schiff, M.; Dinesh-Kumar, S. Virus-induced gene silencing in tomato. Plant J. 2002, 31, 777–786. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smallwood, S.A.; Lee, H.J.; Angermueller, C.; Krueger, F.; Saadeh, H.; Peat, J.; Andrews, S.R.; Stegle, O.; Reik, W.; Kelsey, G. Single-cell genome-wide bisulfite sequencing for assessing epigenetic heterogeneity. Nat. Methods 2014, 11, 817–820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guo, W.; Fiziev, P.; Yan, W.; Cokus, S.; Sun, X.; Zhang, M.Q.; Chen, P.-Y.; Pellegrini, M. Bs-seeker2: A versatile aligning pipeline for bisulfite sequencing data. BMC Genomics 2013, 14, 774. [Google Scholar] [CrossRef] [PubMed]
- Guo, W.; Zhu, P.; Pellegrini, M.; Zhang, M.Q.; Wang, X.; Ni, Z. Cgmaptools improves the precision of heterozygous SNV calls and supports allele-specific methylation detection and visualization in bisulfite-sequencing data. Bioinformatics 2017, 34, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Sierro, N.; Battey, J.N.; Ouadi, S.; Bakaher, N.; Bovet, L.; Willig, A.; Goepfert, S.; Peitsch, M.C.; Ivanov, N.V. The tobacco genome sequence and its comparison with those of tomato and potato. Nat. Commun. 2014, 5, 3833. [Google Scholar] [CrossRef] [PubMed]
- Mandadi, K.K.; Scholthof, K.B. Characterization of a viral synergism in the monocot brachypodium distachyon reveals distinctly altered host molecular processes associated with disease. Plant Physiol. 2012, 160, 1432–1452. [Google Scholar] [CrossRef] [PubMed]
- Tian, T.; Liu, Y.; Yan, H.; You, Q.; Yi, X.; Du, Z.; Xu, W.; Su, Z. Agrigo v2.0: A go analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 2017, 45, W122–W129. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- Lei, R.; Du, Z.; Qiu, Y.; Zhu, S. The detection of hydrogen peroxide involved in plant virus infection by fluorescence spectroscopy. Luminescence 2016, 31, 1158–1165. [Google Scholar] [CrossRef] [PubMed]
- Lister, R.; O’Malley, R.C.; Tonti-Filippini, J.; Gregory, B.D.; Berry, C.C.; Millar, A.H.; Ecker, J.R. Highly integrated single-base resolution maps of the epigenome in arabidopsis. Cell 2008, 133, 523. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; He, H.; Li, J.; Chen, W.; Wang, X.; Guo, L.; Peng, Z.; He, G.; Zhong, S.; Qi, Y. Genome-Wide analysis of DNA methylation and gene expression changes in two arabidopsis ecotypes and their reciprocal hybrids. Plant Cell 2012, 24, 875–892. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Zhu, J.; Hu, F.; Ge, S.; Ye, M.; Xiang, H.; Zhang, G.; Zheng, X.; Zhang, H.; Zhang, S. Single-Base resolution maps of cultivated and wild rice methylomes and regulatory roles of DNA methylation in plant gene expression. BMC Genom. 2012, 13, 300. [Google Scholar] [CrossRef] [PubMed]
- Song, Q.-X.; Lu, X.; Li, Q.-T.; Chen, H.; Hu, X.-Y.; Ma, B.; Zhang, W.-K.; Chen, S.-Y.; Zhang, J.-S. Genome-Wide analysis of DNA methylation in soybean. Mol. Plant 2013, 6, 1961–1974. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Han, H.; De Carvalho, D.D.; Lay, F.D.; Jones, P.A.; Liang, G. Gene body methylation can alter gene expression and is a therapeutic target in cancer. Cancer Cell 2014, 26, 577–590. [Google Scholar] [CrossRef] [PubMed]
- Kumar, G.S.; Kumar, R.A.; Singh, K.S.; Sharma, T.R. Comparative analysis of zinc finger proteins involved in plant disease resistance. PLoS ONE 2012, 7, e42578. [Google Scholar]
- Le, T.-N.; Schumann, U.; Smith, N.A.; Tiwari, S.; Au, P.C.K.; Zhu, Q.-H.; Taylor, J.M.; Kazan, K.; Llewellyn, D.J.; Zhang, R. DNA demethylases target promoter transposable elements to positively regulate stress responsive genes in arabidopsis. Genome Boil. 2014, 15, 458. [Google Scholar] [CrossRef] [PubMed]
- Bruno, H.; Milagros, G.; Salas, M.L.; Germán, A. African swine fever virus undergoes outer envelope disruption, capsid disassembly and inner envelope fusion before core release from multivesicular endosomes. Plos Pathog. 2016, 12, e1005595. [Google Scholar]
- Caplan, J.L.; Kumar, A.S.; Park, E.; Padmanabhan, M.S.; Hoban, K.; Modla, S.; Czymmek, K.; Dinesh-Kumar, S.P. Chloroplast stromules function during innate immunity. Dev. Cell 2015, 34, 45. [Google Scholar] [CrossRef] [PubMed]
- Carr, J.P.; Lewsey, M.G.; Palukaitis, P. Signaling in induced resistance. Adv. Virus Res. 2010, 76, 57. [Google Scholar] [PubMed]
- Takuno, S.; Gaut, B.S. Gene body methylation is conserved between plant orthologs and is of evolutionary consequence. Proc. Natl. Acad. Sci. USA 2013, 110, 1797–1802. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greaves, I.K.; Groszmann, M.; Ying, H.; Taylor, J.M.; Peacock, J.; Dennis, E.S. Trans chromosomal methylation in arabidopsis hybrids. Proc. Natl. Acad. Sci. USA 2012, 109, 3570–3575. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.P.; Fang, Y.Y.; An, C.P.; Dong, L.; Zhang, Z.H.; Chen, H.; Xie, Q.; Guo, H.S. C2-mediated decrease in DNA methylation, accumulation of sirnas, and increase in expression for genes involved in defense pathways in plants infected with beet severe curly top virus. Plant J. 2013, 73, 910–917. [Google Scholar] [CrossRef] [PubMed]
- Jeske, H.; Lütgemeier, M.; Preiss, W. DNA forms indicate rolling circle and recombination-dependent replication of abutilon mosaic virus. EMBO J. 2001, 20, 6158–6167. [Google Scholar] [CrossRef] [PubMed]
- Alberter, B.; Ali, R.M.; Jeske, H. Replicative intermediates of tomato leaf curl virus and its satellite DNAs. Virology 2005, 331, 441–448. [Google Scholar] [CrossRef] [PubMed]
- Buchmann, R.C.; Asad, S.; Wolf, J.N.; Mohannath, G.; Bisaro, D.M. Geminivirus AL2 and L2 proteins suppress transcriptional gene silencing and cause genome-wide reductions in cytosine methylation. J. Virol. 2009, 83, 5005–5013. [Google Scholar] [CrossRef] [PubMed]
- Raja, P.; Sanville, B.C.; Buchmann, R.C.; Bisaro, D.M. Viral genome methylation as an epigenetic defense against geminiviruses. J. Virol. 2008, 82, 8997–9007. [Google Scholar] [CrossRef] [PubMed]
- Martínezpérez, M.; Aparicio, F.; Lópezgresa, M.P.; Bellés, J.M.; Sáncheznavarro, J.A.; Pallás, V. Arabidopsis m6A demethylase activity modulates viral infection of a plant virus and the m6A abundance in its genomic RNAs. Proc. Natl. Acad. Sci. USA 2017, 114, 10755–10760. [Google Scholar] [CrossRef] [PubMed]
- Matzke, M.; Kanno, T.; Daxinger, L.; Huettel, B.; Matzke, A.J. RNA-Mediated chromatin-based silencing in plants. Curr. Opin. Cell Boil. 2009, 21, 367–376. [Google Scholar] [CrossRef] [PubMed]
- Cao, M.; Du, P.; Wang, X.; Yu, Y.; Qiu, Y.; Li, W.; Gal-On, A.; Zhou, C.; Li, Y.; Ding, S. Virus infection triggers widespread silencing of host genes by a distinct class of endogenous sirnas in arabidopsis. Proc. Natl. Acad. Sci. USA 2014, 111, 14613–14618. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.H.; Fang, Y.Y.; Duan, C.G.; Fang, R.X.; Ding, S.W.; Guo, H.S. Genome-Wide identification of endogenous RNA-directed DNA methylation loci associated with abundant 21-nucleotide siRNAs inarabidopsis. Sci. Rep. 2016, 6, 36247. [Google Scholar] [CrossRef] [PubMed]
- Sekine, K.T.; Nandi, A.; Ishihara, T.; Hase, S.; Ikegami, M.; Shah, J.; Takahashi, H. Enhanced resistance to cucumber mosaic virus in the arabidopsis thaliana ssi2 mutant is mediated via an SA-independent mechanism. Mol. Plant-Microbe Interact. 2004, 17, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Kachroo, A.; Lapchyk, L.; Fukushige, H.; Hildebrand, D.; Klessig, D.; Kachroo, P. Plastidial fatty acid signaling modulates salicylin acid- and jasmonic acid-mediated defense pathways in the arabidopsis ssi2 mutant. Plant Cell 2003, 15, 2952–2965. [Google Scholar] [CrossRef] [PubMed]
- Alazem, M.; Lin, N.S. Roles of plant hormones in the regulation of host–virus interactions. Mol. Plant Pathol. 2015, 16, 529–540. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Hong, W.; Wu, J.; Wang, Y.; Ji, S.; Zhu, S.; Wei, C.; Zhang, J.; Li, Y. A viral protein promotes host sams1 activity and ethylene production for the benefit of virus infection. eLife 2017, 6, e27529. [Google Scholar] [CrossRef] [PubMed]
- Harvey, J.J.; Lewsey, M.G.; Patel, K.; Westwood, J.; Heimstädt, S.; Carr, J.P.; Baulcombe, D.C. An antiviral defense role of AGO2 in plants. PLoS ONE 2011, 6, e14639. [Google Scholar] [CrossRef] [PubMed]


© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, C.; Wang, C.; Xu, W.; Zou, J.; Qiu, Y.; Kong, J.; Yang, Y.; Zhang, B.; Zhu, S. Epigenetic Changes in the Regulation of Nicotiana tabacum Response to Cucumber Mosaic Virus Infection and Symptom Recovery through Single-Base Resolution Methylomes. Viruses 2018, 10, 402. https://doi.org/10.3390/v10080402
Wang C, Wang C, Xu W, Zou J, Qiu Y, Kong J, Yang Y, Zhang B, Zhu S. Epigenetic Changes in the Regulation of Nicotiana tabacum Response to Cucumber Mosaic Virus Infection and Symptom Recovery through Single-Base Resolution Methylomes. Viruses. 2018; 10(8):402. https://doi.org/10.3390/v10080402
Chicago/Turabian StyleWang, Chenguang, Chaonan Wang, Wenjie Xu, Jingze Zou, Yanhong Qiu, Jun Kong, Yunshu Yang, Boyang Zhang, and Shuifang Zhu. 2018. "Epigenetic Changes in the Regulation of Nicotiana tabacum Response to Cucumber Mosaic Virus Infection and Symptom Recovery through Single-Base Resolution Methylomes" Viruses 10, no. 8: 402. https://doi.org/10.3390/v10080402