Redox Biology of Respiratory Viral Infections


1
Engelhardt Institute of Molecular Biology, Russian Academy of Sciences, Vavilov str, 32, 119991 Moscow, Russia
2
Inserm U1052, Cancer Research Center Lyon, University of Lyon, 69000 Lyon, France
3
DevWeCan Laboratories of Excellence Network (Labex), 69003 Lyon, France
*
Authors to whom correspondence should be addressed.
Viruses 2018, 10(8), 392; https://doi.org/10.3390/v10080392
Submission received: 13 June 2018
/
Revised: 17 July 2018
/
Accepted: 24 July 2018
/
Published: 26 July 2018
(This article belongs to the Special Issue What’s New with Flu?)
Abstract
:Respiratory viruses cause infections of the upper or lower respiratory tract and they are responsible for the common cold—the most prevalent disease in the world. In many cases the common cold results in severe illness due to complications, such as fever or pneumonia. Children, old people, and immunosuppressed patients are at the highest risk and require fast diagnosis and therapeutic intervention. However, the availability and efficiencies of existing therapeutic approaches vary depending on the virus. Investigation of the pathologies that are associated with infection by respiratory viruses will be paramount for diagnosis, treatment modalities, and the development of new therapies. Changes in redox homeostasis in infected cells are one of the key events that is linked to infection with respiratory viruses and linked to inflammation and subsequent tissue damage. Our review summarizes current knowledge on changes to redox homeostasis, as induced by the different respiratory viruses.
1. Introduction
Redox biology embraces events involving shift of balance between reactive oxygen or nitrogen species (ROS and RNS, respectively) production and their scavenging. Changes of redox status take place during various cellular processes, including proliferation, differentiation, signaling, and metabolism [1]. Redox homeostasis also plays important roles in pathology. Accumulation of ROS or RNS or/and depletion of scavenging systems leads to the development of oxidative stress, chronic activation of immune responses, and inflammation [2]. Due to the ability of ROS to react with almost any kind of biological molecules, including proteins, lipids, and nucleic acids, their chronic elevation is generally associated with genome instability, dysfunction of organelles, and apoptosis [3].
The term “reactive oxygen species” covers short-lived oxygen-containing intermediates with high reactivity. The most extensively studied ROS include superoxide anion (O2•−), hydroxyl radical (HO•), and hydrogen peroxide (H2O2) [4]. The consequences of their production and corresponding antioxidant pathway, activated in response to it, depend on the location in a cell. An example of a well characterized source of O2•− is the electron transfer to oxygen that occurs during respiration in mitochondria. A predominant ROS-scavenging enzyme that is implicated in neutralization of O2•− in mitochondria is superoxide dismutase 2 (SOD2 or MnSOD) [5]. SOD2 converts O2•− to H2O2, which in turn, can be metabolized by different enzymes, including catalase (CAT), peroxiredoxins (Prdx), or glutathione peroxidases (GPx) [6]. An extensive description of the various cellular ROS sources and scavenging enzymes and pathways are described further below and in the following comprehensive reviews [7,8].
Markers of redox misbalance in blood and tissues are often taken into account in the pathology of various diseases. Chronic viral hepatitis B and C are generally associated with oxidative stress, and levels of ROS and oxidized metabolites correlate with the severity of liver damage and with the risk of the development of related pathologies—fibrosis and hepatocellular carcinoma [9]. Moreover, currently redox biology pays much attention to localization of ROS sources, since local changes in ROS levels may influence signaling pathways by activating “redox switches” [10,11]. In lungs, redox homeostasis is crucial in the pathology of asthma [12]. Lung infection with respiratory viruses is, in general, associated with cytokine production, inflammation, cell death, and other pathological processes, which could be triggered by enhanced ROS production. Investigation of the influence of these infections on ROS-producing and ROS-scavenging enzymes and systems may allow for the identification of those that are crucial for replication of the pathogens and occurrence of virus-associated disease. Our review aims to summarize the known data on the role of redox biology in the pathologies that are associated with infection with respiratory viruses.
2. Respiratory Viruses
Respiratory infections comprise a group of diseases that affect millions of people worldwide and pose threat to many of them, especially for kids and elderly. Respiratory viruses cause infections of the upper or lower respiratory tract. The group of respiratory viruses comprises influenza (IV), human respiratory syncytial (HRSV), human rhino (HRV), human metapneumo (HMPV), parainfluenza, and adeno- and corona-viruses (severe acute respiratory syndrome coronavirus SARS-CoV). Many of them cause common clinical syndromes, including nasal congestion, cough, sore throat, and fever, although some of these pathogens may also display more specific clinical manifestations, such as bronchiolitis, pneumonia, etc. (discussed below in this section).
2.1. Influenza Viruses
Influenza viruses are RNA viruses belonging to the Orthomyxoviridae family [13]. Three IVs are infectious for humans, namely types A, B, and C. The IV genome consists of eight segments (seven for influenza C virus) encoding 9–11 proteins, depending on the type of virus. These proteins include the surface proteins hemagglutinin (HA) and neuraminidase (NA), three subunits of RNA polymerase (PA, PB1, and PB2), two non-structural proteins (NS1 and NEP), and two matrix proteins (M1 and a selective proton channel protein M2). Various subtypes of the most common influenza A viruses are classified based on the diversity in the structure of HA and NA proteins. Influenza A and B viruses cause epidemics, whereas influenza C virus is less wide spread and tends to cause infections with less severe symptoms. According to the World Health Organization (WHO), seasonal epidemics that are associated with these infections result in 3 to 5 million cases of severe illness each year, and 250 to 500 thousands deaths worldwide [14]. Currently, safe and effective vaccines are available. However, their specificity is limited to a particular subtype of the virus due to the diversity of circulating influenza A strains and ability of IV to rapidly accumulate mutations (viral antigenic drift). So, often these vaccines do not perfectly match the circulating subtype or become ineffective within short amounts of time due to viral antigenic drift, thus requiring the annual re-fabrication of vaccines and revaccination. Treatment options for IV infections are limited to inhibitors of neuraminidase, which prevent virion release and spread, but not replication [15] and blockers of the M2 proton channel [16]. However, the development of drug resistance is an important issue. Due to the high prevalence of IV serotypes that are resistant to adamantane antiviral drugs, the WHO recommends neuraminidase inhibitors instead of M2 proton channel blockers (amantadine and rimantadine) [14].
2.2. Human Respiratory Syncytial Virus
Human respiratory syncytial virus belongs to the Pneumoviridae family of RNA viruses [17]. The viral genome is represented by a single-stranded negative-sense 15.2 kb RNA encoding 11 proteins. Similarly to IV, HRSVs are classified as A or B genotypes, according to the structure of fusion (F) and attachment (G) proteins. The rest of the viral proteome includes proteins N, M, P, SH, L (polymerase), M2-1, M2-2, and NS1, NS2. HRSV is a leading cause of bronchiolitis and pneumonia in children, especially in pre-mature infants and children with cardiac or pulmonary diseases. It was also shown that HRSV significance in adults is comparable to nonpandemic IV [18]. The group of risk includes the elderly and immunocompromised people. Approximately 33 million cases of infection are reported annually worldwide [19,20]. No HRSV vaccine is available yet. The only prophylactic treatment option, palivizumab (Synagis®)—a monoclonal antibody against the F viral protein is recommended for HRSV prevention and the treatment of infants at high risk of severe disease and complications. Other therapeutic approaches, including ribavirin administration, are discussed in detail elsewhere [21].
2.3. Human Rhinovirus
Human rhinovirus, the major and most prevalent cause of the common cold, belongs to the Picornaviridae family of RNA viruses [13]. The HRV genomic single-stranded positive-sense 7.2 kb RNA encodes 11 proteins. Viral capsid is formed by proteins VP1–4; non-structural proteins include polymerase three-dimensional (3D), two proteases 2A and 3C, proteins 2B, 2C, 3A, and 3B. HRV replicates at 33–35 °C, thus it mostly infects nose epithelial cells. Therefore, direct HRV-induced pathologies manifest as nasal congestion, rhinitis, and sore throat. However, sometimes rhinoviral infection leads to more severe disease, including bronchiolitis, pneumonia, and exacerbations of chronic pulmonary disease. Therapy of infection is mostly supportive [22]. Several antivirals with preventive mode of action (e.g., capsid binding for abrogation of viral RNA release into cells) were in clinical trials, including pleconaril, vapendavir, and rupintrivir, however, to the best of our knowledge, none of them were approved and recommended for HRV therapy. Supportive treatment may include Echinacea, vitamine C, and zinc.
Parainfluenza virus belongs to the Paramyxoviridae family, with its genome being represented by a single-stranded RNA. Parainfluenza virus infects both upper and lower respiratory tracts and usually causes common cold-like symptoms. The virus is known to be dangerous for young children; however, it also infects adults. Sendai virus (SeV) known as former murine parainfluenza virus belongs to the same Paramyxoviridae family. SeV is negative sense, single-stranded RNA virus, which is capable of infecting rodents and other animals. It is accepted as a model of respiratory infection. Adenoviruses are members of Adenoviridae family and have DNA genome. Adenovirus infections are usually mild, and only symptomatic treatment may be required.
3. Enhanced ROS Production during Viral Respiratory Infections
Many lines of evidence suggest that marked signs of increased production of ROS accompany all respiratory viral infections. However, none of the published data are based on direct measurement of ROS levels using electron paramagnetic resonance technique. In all existing papers levels of ROS were assessed indirectly either by using redox-sensitive dyes (2′,7′-dichlorodihydrofluorescein diacetate, DCFHDA; dihydroethidium, DHE, etc.) that are oxidized by ROS into quantifiable fluorescent products or via quantification of cellular oxidation products. In patients that are infected with IV marked increases in DNA (8-hydroxydeoxyguanosine), lipid (malondialdehyde, F2-isoprostane, 7-ketocholesterol, and 7β-hydroxycholesterol), and protein (carbonyl content) oxidation products in blood plasma and urine were reported [23,24,25]. Elevated levels of sterol oxidation products were detected not only during the infection but also three months after IV clearance [24]. Increased levels of ROS as well as of nitric oxide synthase 2 (iNOS) and nitrotyrosine content representing markers of nitrosative stress were also reported in lung tissues of patients that died in the fatal IV pandemics [26]. IV-infected mice and cell lines also exhibit an enhanced production of ROS and disturbance of antioxidant defense [27,28,29,30] and are, therefore relevant models of IV infection for investigation of the changes in redox homeostasis induced by the virus.
Augmented ROS production is not only a feature of IV. Other respiratory viruses also promote ROS production. Elevation of total ROS levels in A549 airway cells is also triggered by HRSV [31,32] and in various cell lines by SeV [33,34], as was shown with DCFHDA. Consistently, the accumulation of lipid peroxidation products and oxidized glutathione (GSH) were reported in plasma of infants with HRSV-induced acute bronchiolitis [35]. SeV, as shown above for IV, also triggers the induction of iNOS, enhanced production of nitric oxide, and accumulation of nitroguanosine [36]. A reduced antioxidant capacity, another marker of oxidative stress, was detected in HRSV-infected infants, mice, and cells [37,38,39,40]. Reduced levels of antioxidant enzymes were also reported in airway cells and in mice replicating human HMPV [37,41]. HRV was shown to induce production of ROS in airway cells by enhancing O2•− production and by depleting intracellular GSH [42,43,44].
4. Sources of ROS in the Infected Cells
Respiratory viruses are known to induce ROS-generating enzymes, including nicotinamide adenine dinucleotide phosphate oxidases (NADPH oxidases, Nox) and xanthine oxidase (XO) and to disturb antioxidant defenses. Increased activities of the Nox and Dual oxidase (Duox) family were observed both in vitro and in vivo. Treatment of infected cell cultures and laboratory animals (mice) with the pan-Nox inhibitor dibenziodolium chloride (DPI) profoundly attenuated ROS production induced by IV [29], HRSV [45,46], and HRV [44]. Although DPI has low specificity for Nox in comparison to other flavoproteins [47], a detailed analysis with other approaches revealed that several NADPH oxidases are implicated in generation of ROS: Nox1, Nox2, Nox4, and Duox2 (Figure 1).
Nox2 is a phagocytic enzyme that contributes to virus-induced ROS production during in vitro and in vivo infection with IV [30,48,49,50], HRSV [50,51], HRV [44,50], and SeV [50,51]. In Nox2−/− knockout mice, the IV-induced ROS and RNS increase was less pronounced [48]. Moreover, virus titer and virus-induced inflammation were also lower in these mice, and were correlated with improved lung functions [52]. It is generally acknowledged that Nox2 activation requires phosphorylation of its regulatory subunit p47phox/NCF1 (neutrophil cytosolic factor 1) [53]. In line with this, Nox-derived ROS production during infection with inactivated H5N1 IV is attenuated in NCF−/− mice when compared to wild type littermates, and this effect is accompanied by improved lung functions [54]. Similarly, HRV infection in cells lacking NCF1 is accompanied by significantly lower levels of O2•− and H2O2 [44]. Noteworthy that Nox2 is expressed in macrophages, monocytes, neutrophils, and, to a lesser extent, in epithelial cells [53,55,56], see also a review of Grandvaux et al. [57] for detailed analysis of expression of Nox isoforms in different respiratory tract sections and cell lines. To et al. recently reported that respiratory RNA viruses induce Nox2-mediated ROS production in endosomes of alveolar macrophages [50]. In addition, recruitment of neutrophils and monocytes from the bloodstream to the infection site may contribute significantly to the enhanced production of superoxide anions by Nox2 during the infection. Indeed, influenza [58,59] and HRSV [60] cause a pronounced migration of neutrophils to the respiratory tract. However, to the best of our knowledge, input of macrophages or neutrophils into Nox2-derived production of O2•− has not been assessed for any of these viruses.
Another NADPH oxidase, Nox4, was also shown to be a source of ROS in lung cancer cells or in primary airway epithelial cells infected with IV [29,61]. Amatore et al. described induction of Nox4 and reduction of Nox2 in in vitro IV infections. However, in the context of other respiratory virus infections, the activity of Nox4 has not been studied. Interesting data exist on the possible involvement of Nox1 in the regulation of ROS production in IV-infected mice [62]. O2•− production in BALF (bronchoalveolar wash fluid) cells of Nox1 knockout mice infected with IV subtype H3N2 is similar to that of wild-type littermates during the early stages of infection (day 3), but suppressed at late stages of infection (day 7). This suggests that Nox1 plays no or only a marginal role in the production of O2•−. Nox1 knockout furthermore significantly augments production of proinflammatory cytokines and chemokines and overall inflammation in mice at three days post-infection with no effect on viral titer, but it suppresses cytokine production at later stages of disease when the viral titer significantly decreases, indicating the resolution of the infection. These findings suggest a protective role of Nox1 during IV infection, which is not limited to changes in total ROS production [62]. However, recent work from another group pointed to a role for Nox1 in the induction of ROS. Survival of IV-infected mice with inactive Nox1 was prolonged [63], suggesting that Nox1 is a source of ROS that is hazardous. In HRV-infected polarized airway epithelial cells Nox1-derived ROS are responsible for triggering barrier dysfunction, a dangerous process leading to decreased transepithelial resistance, thus making the host susceptible to bacterial pathogens [64,65]. Interestingly, ROS generation and disruption of the barrier during HRV infection also depend on one of the double-stranded RNA (dsRNA) receptors, Nod-like receptor X-1 (NLRX-1) [65]. This receptor has mitochondrial location and is able to activate nuclear factor kappa B (NFκB) via ROS [66]. However, direct mechanisms underlying this effect remain unknown. In the case of HRSV, down-regulation of Nox2, but not of Nox1 by RNA interference affects NFκB signaling [51], even though NFκB is one of the ROS-dependent transcription factors [67] responsible for cytokine and chemokine gene expression. Thus, involvement of Nox1 in direct induction of oxidative stress during respiratory viral infection remains controversial. Another source of ROS during IV infection that belongs to the Nox/Duox family is Duox2 [61,68,69]. IV triggers, both in vitro and in vivo, a pronounced induction of Duox2 and DuoxA2 and a moderate down-regulation of Duox1 [69,70]. Noteworthy, Duox2/DuoxA2 induction is induced by H1N1 but not by the H3N2 subtype [69]. Duox2-mediated production of ROS is required in the redox regulation of type 1 and 3 interferon (IFN) antiviral response pathways in airway cells (see Section 6 below).
Similar data also exist for other viruses. Fink et al. described the elevation of the transcripts encoding Duox2, Nox4, and a regulatory subunit of Nox2-p67phos in cells infected with SeV and HRSV [51]. This paper also shows absence of notable changes in transcription of other NADPH oxidases. They did not assess contribution of Nox/Duox to ROS production during SeV or HRSV infection. Instead, they showed that two pan-Nox inhibitors (DPI and apocynin) and knockdown of Nox2 but not of Nox1 or Nox5 prevented activation of the NFκB pathway by these viruses. However, in their subsequent paper, it was shown that SeV does induce Duox2 at protein level and increases extracellular levels of H2O2, whereas HRSV almost does not [57]. In the case of SeV, the mechanism of induction involves production and secretion of two cytokines: IFNβ and tumor necrosis factor alpha (TNFα), the accumulation of which activates Duox2 gene transcription through signal transducer and activator of transcription protein/interferon regulatory factor 9 (Stat2/IRF9)-dependent Stat1 independent pathway. In contrast, HRSV blocks this pathway and thus does not allow for the induction of Duox2.
HRV-infected human primary tracheobronchial epithelial cells also trigger induction of Duox2 [71]. Patients with chronic obstructive pulmonary disease (COPD) have increased the susceptibility to HRV and HRV-infected airway epithelial cells from COPD patients show elevated levels of Duox1 and Duox2 [72].
Xanthine oxidase is another ROS-generating enzyme that is induced by IV [73,74]. XO catalyzes the conversion of hypoxanthine to xanthine and further to uric acid thus being implicated in the catabolism of purine nucleic bases [75]. Elevation of XO levels was observed in serum, lung tissues, and in bronchoalveolar wash fluid of IV-infected mice [73,74]. Effective inhibition of O2•− production with allopurinol, a classical XO inhibitor, validated the significant contribution of this enzyme to virus-induced ROS production [73]. Up-regulation of XO was also shown for HRV [43], although Kaul et al. reported the absence of the inhibitory effect of allopurinol towards HRV-induced ROS production and subsequent interleukin 8 (IL8) induction [44]. Whether XO contributes to HRSV-mediated enhancement of ROS generation is unknown.
Finally, mitochondria are known to be an important source of O2•− in IV infection due to the virus-induced leakage of electrons from the respiratory chain [61,76]. However, this is unlikely due to a direct interaction of viral proteins with the electron transport chain, indeed data supporting mitochondrial localization of viral proteins have not been reported so far. Enhanced production of ROS in mitochondria may rather be mediated by Duox2 [61] or Nox2 (reviewed in [77,78]), both of which have been shown to induce mitochondrial dysfunction. So far, there is no data concerning the disruption of the mitochondrial electron transport by other respiratory viruses, and this area of research clearly requires further studies. For SeV there is indirect evidence that this virus does not affect production of ROS in mitochondria: activation of caspase 9, a key event in SeV-induced apoptosis, is insensitive to ROS scavengers and is not accompanied by cytochrome c release, a frequent consequence of severe mitochondrial dysfunction [79].
Finally, another interesting mechanism of O2•− production was shown for IV and SeV [36]. It involves the induction of iNOS, production of nitric oxide, formation of nitroguanosine, and the latter together with iNOS and P450 reductases leads to the production of this type of ROS. Indeed, the same group described a mechanism of O2•− formation that involves single-electron reduction of a 8-nitro-guanosine into the respective anion-radical with NADPH-cytochrome P450 reductase (P450 reductase), any isoform of NO-synthase (NOS) or even xanthine oxidase, and the subsequent transfer of the electron to molecular oxygen [80]. Interestingly, such data are missing for any other virus that triggers oxidative stress.
Investigations of the molecular mechanisms by which respiratory viruses induce massive ROS production and in the particular identification of the viral proteins that are responsible for these effects are scarce in comparison to viruses, such as human immunodeficiency virus (HIV) and hepatitis B and C viruses (HBV and HCV, respectively) [9,81]. Amongst the few reports covering this topic are reports on the IV M2 [82] and PB1-F2 [83] proteins. Overexpression of M2, a proton channel protein, activates protein kinase C (PKC) and increases the production of ROS that was detected using DCFH2DA and MitoSOX as readouts [82]. PB1-F2 decreases SOD1 expression resulting in impaired ROS scavenging [83]. These data are corroborated by a study of Flory et al. who pointed at M, HA, and NP proteins as activators of NFκB via IκB kinase (IKKβ), whose effects can be suppressed by antioxidants [84]. In respect to HRSV infection, one single paper suggests that the generation of ROS is augmented by the viral fusion (F) protein [45]. Treatment with DPI abrogates ROS production in F-expressing cells suggesting the involvement of NADPH oxidase(s). However, since DPI also inhibits xanthine oxidase, proteins of mitochondrial electron transport chain, and other flavin-dependent enzymes [47], the role of NADPH-oxidases remain to be explored. Finally, the overexpression of SARS-CoV 3CL-Pro (protease) protein induced the generation of ROS and NFκB activation; however, the molecular mechanisms are unknown [85].
5. Respiratory Viruses and Antioxidant Defense Pathways
Respiratory viruses not only enhance ROS production but also affect cellular defense systems against ROS. Despite the important role of physiological levels of ROS in signaling, chronically elevated levels cause severe oxidative injury. The antioxidant defense system is comprised of a variety of enzymes and transcription factors, as well as an array of low molecular weight molecules that are often referred to as antioxidants. These latter compounds directly scavenge ROS, participate in recycling of the defense enzymes, or regulate redox-sensitive transcription factors.
The key transcription factor that controls expression of an array of defense enzymes is nuclear factor E2-related factor 2 (Nrf2) [86]. During normal levels of ROS generation, Nrf2 is retained in the cytoplasm by kelch-like ECH-associated protein 1 (Keap1), which targets Nrf2 to ubiquitin-dependent degradation. When ROS production is enhanced, Nrf2 dissociates from Keap1 and translocates into the nucleus, where it binds the antioxidant response element (ARE) in the promoter regions of target genes encoding predominantly antioxidant enzymes. Noteworthy, the Nrf2 gene itself also contains ARE-like sequences in its promoter [87], thus amplifying its expression in a positive feedback loop. Amongst the Nrf2-regulated target genes are superoxide dismutases, catalase, peroxiredoxins, and glutathione peroxidases. O2•− are converted into H2O2 by three SOD isoforms in mammalian cells: cytoplasmic SOD1, mitochondrial SOD2, and extracellular SOD3 (EC-SOD). Soluble peroxides are eliminated by catalase, peroxiredoxins, and glutathione peroxidases. The peroxiredoxin family comprises six different members with redox-active cysteine residues. Peroxiredoxins differ by localization and mechanism of re-activation. The eight members of the GPx family display different localization patterns and activities in the cell and they have varying affinities for different types of peroxides. Noteworthy, GPx4 and Prdx6 are capable of neutralizing lipid peroxides in addition to soluble peroxides [88,89]. Finally, many antioxidant defense enzymes are involved in the biosynthesis of natural antioxidants, such as glutathione and the maintenance of a reduced pool of GSH. These include glutathione synthetase, reductase and peroxidase, thioredoxin reductase, and others.
IV strains are thought to activate the Nrf2/ARE defense pathway in vitro and in mice by inducing nuclear translocation and transcriptional activity of Nrf2 because transcription of the Nrf2 target gene heme oxygenase 1 (HO-1) was shown to be augmented [90,91]. Interestingly, differences between seasonal and especially pandemic H1N1 subtypes versus highly pathogenic H7N9 and H5N1 strains in their capacities to promote Nrf2 phosphorylation and the subsequent translocation/activation have been reported [92]. Jacoby et al. did not find any changes to SOD1 levels in IAV-infected cells [93], however, other groups observed a decrease of SOD1 as mediated by proteasomal degradation of specificity protein 1 (Sp1), a transcription factor that drives the expression of the SOD1 gene [94,95]. An increase in SOD1 expression was found in patients with asymptomatic influenza A infection [96]. Decline in total SOD activity was reported in children infected with H1N1 subtype [25]. So, SOD1 status during IV infection is still questionable.
Similar controversy exists over other antioxidant enzymes, such as CAT and indoleamine 2,3-dioxygenase (IDO). The latter scavenges O2•− and uses them for oxidation or conversion of tryptophan into kinurenin [97]. Levels of IDO were shown to be unaltered and levels of catalase decreased in IAV-infected cells in vitro, [93], whereas in infected mice, IDO and HO-1 were induced and CAT levels remained unchanged [98]. Yamada et al. reported depletion of CAT and Prdx6 in mice infected with subtype H1N1, however, this was due to depletion of the IV permissive bronchial Clara and/or alveolar type 2 (AT2) cells in which these enzymes are predominantly expressed [91]. In addition, the same study also reported the pronounced induction of several other antioxidant enzymes, namely, GPx3 and HO-1.
Unlike IV, HRSV reduces mRNA levels and levels of Nrf2 in nuclei of airway epithelial cells [38]. Indeed, HRSV is capable of inducing Nrf2 deacetylation and subsequent proteasomal degradation, which leads to the down-regulation of expression of antioxidant enzymes [99]. However, the residual Nrf2/ARE pathway activity does play an important role in protection against HRSV-induced ROS production: Nrf2−/− infected mice show more severe oxidative stress and inflammation when compared to their wild-type littermates [100]. Moreover, Nrf2 deficiency leads to the activation of the NFκB pathway and subsequent cytokine production. This Nrf2-NFκB crosstalk may play pivotal roles in in the associated pathogenesis, and there is clear lack of information about it for respiratory viruses. HRSV exhibits different effects on the antioxidant defense system depending on the duration of the infection. A transient activation has been reported during the first several hours post infection in lung carcinoma cells in vitro, as exemplified by induction of SOD1, SOD2, GST (glutathione S-transferase), catalase, and GPx. With progression of disease in cell culture, only SOD2 remained up-regulated resulting in enhanced H2O2 production, whereas other antioxidant enzymes, including those that are responsible for neutralization of H2O2, were suppressed [38,101]. In mice and children, no signs of induction of antioxidant enzymes were reported; instead, their levels fell down during infection [37]. HRSV infection also induced the accumulation of oxidized forms of several members of the Prdx family, including cytoplasmic Prdx1, mitochondrial Prdx3, and endoplasmic reticulum-residing Prdx4, without changes to their total protein levels [102]. This indicates that massive ROS production occurs in the respective organelles of the infected cells, since peroxiredoxins scavenge peroxides in their close proximity. Moreover, down-regulation of Prdx1 and 4 by RNA interference (RNAi) led to a more pronounced increase of ROS levels [102].
Information on the role of individual viral proteins in changes to the redox status and redox-associated processes is very scarce and is based on one article, which reports an NS1-mediated, Nrf2-independent suppression of SOD2 [103]. In this paper, HRSV NS1 protein prevented the activation of the JAK/STAT1 pathway (Janus kinases/Signal Transducer and Activator of Transcription proteins) and decreased expression of genes bearing GAS and ISG promoter sequences (interferon-gamma activated sequence and interferon stimulated genes, respectively), such as SOD2 [103]. However, SOD2 gene expression is normally induced by NFκB rather than by IFNγ signaling [104]. Therefore, the above-mentioned effect might be explained by possible alterations of the NFκB pathway by the viral NS1 protein.
Few studies have investigated the antioxidant defense system of the host cell in human HMPV and HRV infections. HMPV has been shown to increase levels of SOD2 but to down-regulate SOD3, catalase, glutathione S-transferases, and peroxiredoxins 1, 3, and 6 [41]. In contrast, HRV does not affect activity of SOD2, catalase, and GPx, but increase levels and activity of SOD1 [105].
For SeV, we did not find any literature on interference of the virus with expression of antioxidant defense enzymes. However, a fluctuating reduction of the total GSH content has been reported in SeV-infected cells [106]. Noteworthy, this decrease was observed during the first hour post-infection and then again 24 h post-infection. Initial reduction of GSH content was due to damage of the host cell membrane during virus penetration, whereas the secondary reduction resulted from massive incorporation of cysteine into the viral proteome. However, in neither case, any changes in levels of GSSG were observed.
In addition to Nrf2, there are other transcription factors that belong to this family: Nrf1 and Nrf3. To date very little is known about functions of Nrf3 in the absence of infection. Functions of Nrf1 are defined. Nrf1 can also bind to ARE in the promoters of antioxidant defense genes, though to a lesser extent than Nrf2 (reviewed in [107]). Nrf2 and Nrf1 knockout animals have distinct phenotype meaning that functions of these two factors do not fully overlap. But again, there are no data about status of Nrf1 during respiratory as well as other viruses. So, the investigation of their effect on Nrf1 and study of a role of the latter in viral replication and pathology seems to be a nice direction for future studies.
6. Role of ROS in the Life Cycle and Propagation of Respiratory Viruses
So far, the evidence for a direct impact of ROS on the life cycles of respiratory viruses is very scarce. ROS-induced cell death and lysis can favor release and spread of virions and thus stimulate the replication of those respiratory viruses with a lytic life cycle. At the same time, ROS are known to contribute to suppression of some respiratory infections through the induction of innate immune responses. On the other hand, antioxidant therapies are known to ameliorate and improve disease outcome.
Overexpression of SOD1 or treatment with antioxidants reduce intracellular levels of IV polymerase thus providing a possible mechanism of the viral titer reduction in response to antioxidant treatment [94]. The observation that content of GSH negatively correlates with IV replication in infected cells or mice further corroborates this finding [108,109]. Similar results were obtained upon the inhibition of additional ROS-producing enzymes or Nox2 knockout [29,48]. However, ROS were also shown to have antiviral effect but due to modulation of immune responses.
Activation of immune responses during IV infection is achieved partly through Duox2-mediated induction of viral dsRNA sensors RIG-I and MDA5 (retinoic acid-inducible gene I and melanoma differentiation-associated protein 5, respectively) [68,70], pattern recognition receptors that trigger type I IFN responses. IV also enhances production of interferons λ1 (IL29), and λ2/3 (IL28A/IL28B), again via ROS [61]. ROS scavenging or suppression of ROS production by Duox2 or mitochondria leads to the inhibition of IFNλ synthesis and secretion, and thus the enhancement of viral replication. Induction of type I and III interferons is likely to be achieved through the Nrf2 pathway, since activation of the latter in response to IV infection or chemical inducers (i.e., sulforaphane and epigallocatechin gallate) is accompanied by an increased expression of antiviral mediators RIG-I, IFNβ, and MxA [110]. Nrf2 overexpression negatively affects the replication of IV, whereas knockdown leads to enhanced virus entry and replication [90,110]. However, this effect is unlikely to be mediated through interferon-stimulated genes, such as MX1 and OAS1 (2′,5′-oligoadenylate synthetase 1), since their levels are not altered during Nrf2 overexpression in the absence of the infection and are decreased in infected cells [90].
One of the mechanisms by which IV interferes with the interferon pathway is via the activation of the ROS-sensitive transcriptional factor NFκB, which controls proinflammatory cytokine production. Indeed, in IV-infected cells SOCS3 (suppressor of cytokine signaling 3) is induced through the activation of NFκB [111], whereas prevention of NFκB activation leads to the reduction of viral titers, but cytokine production also decreased [112]. Another proviral role of NFκB is the inhibition of β- and γ-catenin-dependent transcription of interferon-stimulated genes [113]. However, NFκB can also exhibit antiviral properties by activating interferon β gene transcription. Viral protein NS1 is able to prevent NFκB activation by interacting with IKK, which is required for NFκB signaling [114]. Overall, the balance between pro- and anti-viral activities of NFκB remains to be analyzed.
An interesting piece of evidence came from an investigation of SeV and its effects on the expression of Duox2 [115]. It showed that the replication of this virus is controlled by Duox2 and by the H2O2 Duox2 produces. Induction of Duox2 in SeV-infected cells is achieved through a combined action of IFNβ and TNFα that are synthesized and secreted from the infected cells through IRF3 and NFκB, respectively. In turn, Duox2 contributes to secretion of type I and III interferons. However, it should be mentioned that the detailed mechanism underlying the antiviral effect of Duox2 remains vague, and several possible assumptions have been made (see [57]).
HRSV is known to evade the antiviral effect of Duox2. Fink et al. showed that HRSV triggers the production and secretion of functional IFNβ and TNFα to a level comparable to that known to induce Duox2 in SeV infection [115]. However, HRSV blocks the Stat2/IRF9 pathway and thus controls Duox2 gene expression. Other groups found that activation of IFNs by HRSV is achieved through enhanced ROS production and the consequent STAT signaling [116]. Among the various ROS-sources, NADPH oxidases are the major triggers of STAT signaling, since treatment with Nox inhibitors abrogates STAT and RANTES (regulated on activation, normal T cell expressed, and secreted) induction [116]. Specifically, HRSV- and SeV-induced activation of NFκB and IRF3 signaling and downstream proinflammatory and antiviral signaling is initiated by sensing viral RNA by Rig-I and MDA5 helicase receptors with subsequent involvement of mitochondrial antiviral-signaling protein (MAVS) and TNF receptor-associated factor 6 (TRAF6) adaptors [56,117,118]. All of these processes depend in Nox2 [117,118], which controls the expression of MAVS [56]. Not much is known about the mechanisms of viral interference with the above mentioned pathway, however nonstructural viral proteins NS1 and NS2 are thought to be implied, albeit in an opposing way—these proteins inhibit IFN, IRF-3, and NFκB activation [119,120,121,122,123]. Thus, the virus antagonizes IFN signaling.
The role of ROS in the life cycle of HRV remains mostly unexplored. However, ROS have been reported to stimulate expression of intercellular adhesion molecule-1 (ICAM1), a major receptor for entry of this virus [124]. In line with this, treatment with reduced GSH decreased virus-mediated ICAM1 activation.
Finally, an interesting piece of evidence comes from investigation of SeV. Ciriolo et al. showed that this virus causes a pronounced decrease in GSH content in infected cells [106]. Supplementation with exogenous GSH inhibits the replication of the virus [125], whereas depletion, as caused by buthionine sulfoximine (BSO) that interferes with biosynthesis of this tripeptide, in contrast—increases virus titers [106]. However, the exact mechanism by which GSH content is linked to virus replication remains unknown. It definitely does not involve modulation of mitochondrial status, as mitochondria-derived ROS do not affect SeV propagation [126]. Finally, such investigations are missing for other respiratory viral infections.
7. ROS in Respiratory Virus Pathology
Alterations of ROS-producing and scavenging pathways that are caused by respiratory viral infections are implicated in inflammation, lung epithelial disruption, and tissue damage, and, in some cases, even pulmonary fibrosis. These events are at least partially interregulated: inflammation can contribute to lung damage and epithelial dysfunction and vice versa. While there is a clear correlation between markers of oxidative stress and severity of the disease in chronic hepatitis B and C [9,127,128,129], for respiratory viruses, such clinical data are scarce. The observation that ROS are implicated in the pathology of these viruses is mainly based on experimental infection models. Death of lung epithelial cells that are infected with IV in vitro occurs through enhanced ROS production, whereas antioxidants prevent cell death and also reduce damage in lungs of IV-infected mice [90,130,131,132]. GPx1 was shown to play a particular role in the pathogenicity of IV infection. In mice lacking H2O2-scavenging enzyme, GPx1, IV infection results in a more severe and longer BALF inflammation as compared to their wild type littermates [133]. Moreover, these animals exhibit higher levels of proinflammatory tumor necrosis factor alpha (TNFα), macrophage inflammatory proteins MIP-1a and MIP-2 expression in lung and elevated number of IV-specific CD8+ T cells in spleen. Such decrease of GPx1 can also result from selenium deficiency. Selenium deficiency leads to down-regulation of GPx1 and catalase and increased production of mucosa and altered epithelial morphology [134]. However, there are no direct data showing that IV does interfere with selenium metabolism and expression of the key Se-metabolizing proteins.
Similar data that point to an important role of ROS in the associated pathology also exist for SARS-CoV [85,135] and HRSV [32]. DNA damage and a proliferation arrest with consequent activation of cellular senescence program was observed in HRSV-infected immortalized epithelial cells and in the respiratory epithelia of HRSV-infected mice even after viral clearance [32]. Overexpression of catalase, treatment with N-acetylcysteine (NAC) or with cell-permeable reduced glutathione ethyl ester (GSHee) reversed these effects, strongly implying the accumulation of ROS in these events [32]. However, contradictory data also exist. For example, Olsen et al. demonstrated that influenza A and B-induced apoptosis of Madin-Darby Canine Kidney (MDCK) cells cannot be blocked by antioxidants or compounds protecting mitochondria [136]; moreover, the cell permeable calcium chelator BAPTA-AM severely augmented cell death, whereas ionomycin, a calcium ionophore, blocked apoptosis. For SeV the data are contradictory: Gao et al. reported that virus-induced apoptosis can be prevented by treatment with NAC [33], whereas Bitzer et al. found that the activation of caspases in SeV-infected cells is not sensitive to antioxidants [79].
One of the key events of inflammation is the interaction of airway epithelial and endothelial cells with leukocytes. It is mediated by a set of cell adhesion molecules, such as VCAM1 (vascular cell adhesion protein 1), ICAM1, and E-selectin, which trigger and augment infiltration with leukocytes [137,138]. At least for HRV it was demonstrated that infection leads to induction of ICAM1. ICAM1 induction was successfully blocked by addition of reduced GSH [124] showing that ROS play a particular role in endothelial cell mediated leukocyte recruitment.
It is well known that IV, HRSV, and other viral infections in vivo trigger massive production of proinflammatory cytokines and chemokines, such as TNFα, IL6, and IL8 [139,140], referred to as cytokine storm and thus trigger cell death [141]. The cytokine storm is responsible for lung tissue damage during viral respiratory infections. Activation of immune cells is also necessary for infection elimination, but it should be taken into account that in many cases not viruses themselves, but associated host immune response is more fatal for tissue integrity and functionality. One of the key mediators of induction of cytokines and chemokines is NFκB. NFκB signaling is directly activated by ROS or/and by certain proinflammatory cytokines, such as TNFα and IL1β, and drives in turn expression of a wide spectrum of other cytokines and chemokines [142]. These include interleukins 2, 6m and 8, MIP1a, MCP-1 (monocyte chemoattractant protein 1), and RANTES [142]. Consequently, NFκB is a key player that coordinates initiation of innate and inflammatory responses as well as maturation of lymphocytes within the adaptive immune system [142]. Respiratory viruses induce NFκB signaling both in vitro and in vivo in a ROS-dependent fashion [42,51,54,84,139]. Several IV proteins, including M, HA, and NP activate NFκB [84]. In IV-infected mice, NFκB activation is accompanied by the increased production of IL6 and IL8, TNFα, CCL5/RANTES, CXCL10 (C-X-C motif chemokine 10) [139]. Treatment with NAC abolishes NFκB activation and cytokine production in infected cells [143]. However, contrary data also exist: Mastronarde et al. noticed that although induction of IL8 by RSV is sensitive to antioxidants such as NAC, the latter inhibits binding of AP-1 and NF-IL6 factors to the promoter region of IL8 gene, but it does not affect the binding of NFκB [144]. So, a precise action of ROS in induction of proinflammatory cytokines merits further studies.
Mechanisms of cytokine induction by HRV infection are less clear. On the one hand, low molecular weight antioxidants including NAC and reduced GSH abolish both NFκB activation and cytokine production during HRV-infection, thus pointing to ROS as key mediators of both effects [42]. Moreover, XO has a significant input in NFκB, since its inhibitor allopurinol effectively prevents the activation of this transcription factor in a cell culture model [43]. Several other studies pointed to Nox2-generated ROS as triggers of NFκB activation and concomitant production of proinflammatory cytokines, including TNFα and RANTES [51,117]. However, Kim et al. reported that HRV infection can induce IL6 and IL8 in an NFκB-independent manner [145]. Distinct mechanisms seem to activate NFκB during HRV infection, and they imply NLRX-1-mediated ROS generation [65,66]. Not much is known about HMPV-mediated cytokine induction. Although HMPV activates NFκB, just like other respiratory viruses, its SH protein, when being expressed by itself, is capable of preventing this activation [146]. Disturbance of the redox balance in HRSV-infected airway epithelial cells such as A549 cells, induces IL8 [140], MCP-1, and interferon regulatory factor 3 (IRF-3). The latter binds the promoter of the chemokine RANTES [31,147,148]. RANTES activates different types of immune cells in lung inflammatory infiltrates during HRSV infection. It was recently shown that other proteins of the inflammatory response, namely cytokine High Mobility Group Box 1 (HMGB1), were also upregulated by HRSV infection, in an at least partially ROS-dependent manner [149]. Another possible mechanism of HRSV-associated ROS production and proinflammatory cytokines is activation of immune responsive gene 1 (IRG1), since its inhibition/suppression during infection abrogates immune cell infiltration and reduces ROS production and lung injury in infected mice [150]. This enzyme catalyzes production of itaconic acid from cis-aconitate, an intermediate of tricarbonic acid (TCA) cycle [151]. It is well described that IRG1 induction leads both to enhanced ROS production presumably through promotion of the pentose phosphate pathway [152] and through controlling succinate levels [153] as well as to activation of the Nrf2 pathway through direct alkylation of the Nrf2 partner—Keap1 [154]. A recent paper also showed that IRG1 can be induced by type I interferons [154]. NFκB pathway activation during HRSV infection is at least partly mediated by the ROS-dependent activation of MSK1 (mitogen- and stress-activated protein kinase 1), one of the kinases responsible for NFκB serine phosphorylation [39]. Also, as described above, phosphorylation of NFκB p65 Ser536 requires the induction of Nox2 that triggers MAVS expression, thus allowing for RigI- and MDA5-mediated sensing of viral RNA and concomitant TRAF6-IKKβ-NFκB signaling [117] (see Section 6 above).
Production of cytokines is controlled not only transcriptionally, but also by proteolytic cleavage of precursor polypeptides by cellular complexes, referred to as inflammasomes into active cytokines [155,156]. Inflammasomes contain sensor molecules that are connected to caspase 1 via a specialized adaptor protein. A majority of complexes contain NOD-like receptor (NLR) sensor molecules, among which the best characterized is NLRP3. Induction of this sensor is not sufficient for the activation of NLRP3 inflammasomes. To date, three possible mechanisms of NLRP3 inflammasome activation have been proposed. One of them involves mitogen-activated protein kinase-mediated ROS signaling [157]. Respiratory (IV, HRSV) as well as non-respiratory (encephalomyocarditis, measles) viruses all activate NLRP3 inflammasomes via ROS [46,158]. Mechanistically, ROS production by NADPH oxidases has been implied [46], however mitochondrial ROS were also shown to play a role [159]. Data on HRV- and SARS-CoV-mediated NLRP3 inflammasome activation is scarce and it just shows that disturbance of calcium homeostasis plays a role [160,161]. The endoplasmic reticulum (ER) is the main storage of calcium ions in a cell. Calcium signaling can be induced in many cases by events referred to as ER overload response (EOR) that occurs during accumulation of some viral and several host cell proteins in the ER [162,163]. One of the major consequences of EOR is an activation of the mitogen-activated protein kinase MAPK/NFκB cascade, which plays crucial roles in inflammation regulation [164,165]. So far, IV has been shown to induce ER stress and concomitant classical unfolded protein response (UPR) [166]. At the same time, the induction of EOR in infected cells and its role in the activation of inflammasomes remains to be elucidated.
Another pathological consequence of enhanced production of ROS in the presence of respiratory viruses is a barrier dysfunction, which leads to an increased susceptibility of the host to bacterial pathogens (Figure 2). HRV e.g., disrupts airway epithelial barrier function with a consequent increase of barrier leakage and susceptibility to pathogens [64,65,167]. Suppression of Nox1 using pharmacological inhibitors or small interfering RNAs (siRNAs) prevented barrier disruption, revealing the role of this oxidase in the development of HRV-associated tissue damage [64]. HRSV also deteriorates barrier function through a mechanism involving the activation of protein kinase D (PKD) [168]. Although a causative role of enhanced generation of ROS was not investigated in that study, the fact that PKD is redox regulated is well known [169,170]. Interestingly, discrepant data were reported for IV infection. IV-infected differentiated primary porcine airway epithelial cells maintained barrier functions despite the loss of ciliated cells [171]. However, in other publications, IV was shown to disrupt cellular tight junctions both in vitro and in infected mice [172,173]. Recently, a connection between IV-induced Nox2-mediated ROS production and enhanced sensibility to post-IV Staphylococcus aureus pneumonia was revealed [174]. Concomitant treatment of co-infected mice with both antibiotics and a Nox2 inhibitor increased the survival of animals. Bacterial pneumonia is the most dangerous consequence for IV patients, and this work may help to improve treatment strategies. However, the authors did not investigate the link between the observed sensitivity to bacterial infection and the disruption of epithelial resistance. So, the clinical importance of epithelial barrier dysfunction in the settings of IV infection remains to be elucidated.
8. Antioxidant Therapy of Respiratory Viruses
Despite the antiviral role of cytokines, their “over” production during the cytokine storm is more fatal for lung tissues in respiratory infections than the viruses themselves [175]. Therefore, various agents have been evaluated and they are used as remedies to target not only the viruses but also the virus-associated inflammation. Much attention has been paid to antioxidants due to the correlation between severity of lung injury and the markers of oxidative stress in lung and blood of patients that are infected with HRSV [35,37] or IV [24,54]. Here, we briefly summarize the major findings for treatment of respiratory infections with antioxidants. More detailed data can be found in the following reviews [101,176,177,178].
The most studied agents targeting ROS that were evaluated for treatment of IV and HRSV infections are listed in Table 1. Noteworthy, almost no data exist regarding efficacy of antioxidant therapies in humans. Instead, the most studied antioxidants—NAC, ascorbic acid and vitamin E were shown to have positive effects such as decrease of virus replication and inflammation in cells and mice infected with IV and/or HRSV.
It should be noted that there is no strong link between HRSV viral titer and the severity of disease during antioxidant treatment. For example, treatment with an inhibitor of NO production ameliorates inflammation during HRSV infection, but it increases viral replication both in cell culture and in a mouse model [179,180]. Another interesting approach for antioxidant treatment proposed recently is usage of hydrogen sulfide donors [181]. It is based on the fact that HRSV infection is accompanied by depletion of H2S due to decreased expression of H2S-generating enzyme cystathionine γ-liase (CSE) [182]. Since H2S and its donors are able to prevent mitochondrial dysfunction, oxidative stress, and reduce inflammation, this fact provides another piece of evidence that respiratory viruses interfere with cellular redox-sensitive pathways.
An additional strategy is inhibition of ROS-generating enzymes such as Nox2 in the case of IV. Detailed analysis of such an approach is presented in several excellent reviews [183,184]. Briefly, the most studied inhibitors, gp91dstat and apocynin, inhibit the association of Nox2 with p47phox and prevent the assembly of the active complex of the oxidase. Both of the inhibitors were shown to reduce inflammation in IV-infected mice thus validating perspectiveness of antioxidant therapy of IV [48,50].
Data on the treatment of HRV with antioxidants are scarce and they have only been produced in in vitro systems. Pyrrolidine dithiocarbamate (PDTC), an antioxidant and inhibitor of NFκB, inhibits HRV replication and the processing of the viral polyprotein in cells [211,212]. Other well studied antioxidants, such as ascorbic acid, Trolox, 2-mercaptoethanol, and NAC, do not display any promising therapeutic activity [211]. Carbocisteine and ambroxol reduce HRV titers at least partly via down-regulation of the viral receptor ICAM1 [213,214].
9. Conclusions and Future Perspectives
Respiratory viruses cause millions of cases of severe illness and thousands deaths each year. So far, no efficient measures for prevention and treatment exist. Future research in these areas will be vital. These infections are associated with pronounced inflammation. Importantly, the virus-induced ROS production and disturbance of the host’s redox balance are important triggers of inflammation. Oxidative stress is triggered in many distinct ways, including the induction of ROS-generating enzymes and disturbance of antioxidant defense. However, not enough is known about mechanisms of virus-associated oxidative stress and the subsequent consequences for cells, tissue, and the organism. Many conflicting data on the antioxidant defense status and role of ROS in viral propagation exist and need to be resolved based on in vitro as well as clinical studies. In particular, techniques that allow for localizing ROS production and scavenging within the cell, and to trace exact oxidative reactions of ROS with host cell proteins will be required to resolve these questions.
Production of ROS can induce cell death and the release of virions representing possible proviral role of enhanced ROS production and altered redox balance. On the other hand, one of the important roles of oxidative stress is the triggering of an antiviral immune response. However, too strong immune responses lead to a cytokine storm and severe inflammation, which is very dangerous for tissue and may disturb lung function. From this point of view, antioxidant supplementation is expected to ameliorate the consequences of infection. Many studies showed the positive role of antioxidant therapy in infected cells and animals. At the same time, almost no relevant clinical data exist even for popular antioxidants, such as NAC, ascorbic acid, and vitamin E. This gap needs to be filled in by new research.
Funding
This manuscript was funded by the Russian Science Foundation (project 14-14-01021, Section 3, Section 4, Section 5 and Section 6), the Program of fundamental research for state academies for 2013–2020 years (No. 01201363818) (Section 7), and the French National Agency for AIDS and Viral Hepatitis Research (14370), Comité de Saône-et-Loire de la Ligue contre le cancer, Agence Nationale de Recherche, the Region Rhone-Alpes, DevWeCan French Laboratories of Excellence Network (Labex, Grant ANR-10-LABX-61) and the OpeRa IHU program (GRANT ANR-10-IBHU-004) (Section 1, Section 2, Section 8 and Section 9).
Conflicts of Interest
The authors declare no conflict of interest.
References
- Van der Vliet, A.; Janssen-Heininger, Y.M. Hydrogen peroxide as a damage signal in tissue injury and inflammation: Murderer, mediator, or messenger? J. Cell. Biochem. 2014, 115, 427–435. [Google Scholar] [CrossRef] [PubMed]
- Mittal, M.; Siddiqui, M.R.; Tran, K.; Reddy, S.P.; Malik, A.B. Reactive oxygen species in inflammation and tissue injury. Antioxid. Redox Signal. 2014, 20, 1126–1167. [Google Scholar] [CrossRef] [PubMed]
- Circu, M.L.; Aw, T.Y. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic. Biol. Med. 2010, 48, 749–762. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birben, E.; Sahiner, U.M.; Sackesen, C.; Erzurum, S.; Kalayci, O. Oxidative stress and antioxidant defense. World Allergy Organ. J. 2012, 5, 9–19. [Google Scholar] [CrossRef] [PubMed]
- Fukai, T.; Ushio-Fukai, M. Superoxide dismutases: Role in redox signaling, vascular function, and diseases. Antioxid. Redox Signal. 2011, 15, 1583–1606. [Google Scholar] [CrossRef] [PubMed]
- Cox, A.G.; Winterbourn, C.C.; Hampton, M.B. Mitochondrial peroxiredoxin involvement in antioxidant defence and redox signalling. Biochem. J. 2009, 425, 313–325. [Google Scholar] [CrossRef] [PubMed]
- Winterbourn, C.C. Reconciling the chemistry and biology of reactive oxygen species. Nat. Chem. Biol. 2008, 4, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Trachootham, D.; Lu, W.; Ogasawara, M.A.; Nilsa, R.D.; Huang, P. Redox regulation of cell survival. Antioxid. Redox Signal. 2008, 10, 1343–1374. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Valuev-Elliston, V.T.; Tyurina, D.A.; Ivanova, O.N.; Kochetkov, S.N.; Bartosch, B.; Isaguliants, M.G. Oxidative stress, a trigger of hepatitis C and B virus-induced liver carcinogenesis. Oncotarget 2017, 8, 3895–3932. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.P.; Sies, H. The Redox Code. Antioxid. Redox Signal. 2015, 23, 734–746. [Google Scholar] [CrossRef] [PubMed]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [PubMed]
- Hoffman, S.; Nolin, J.; McMillan, D.; Wouters, E.; Janssen-Heininger, Y.; Reynaert, N. Thiol redox chemistry: Role of protein cysteine oxidation and altered redox homeostasis in allergic inflammation and asthma. J. Cell. Biochem. 2015, 116, 884–892. [Google Scholar] [CrossRef] [PubMed]
- Knipe, D.M.; Howley, P.M. Fields Virology, 6th ed.; Wolters Kluwer/Lippincott Williams & Wilkins Health: Philadelphia, PA, USA, 2013; 2 volumes. [Google Scholar]
- World Health Organization. Influenza (Seasonal) Fact Sheet No 211; World Health Organization: Geneva, Switzerland, 2016. [Google Scholar]
- Colman, P.M. Influenza virus neuraminidase: Structure, antibodies, and inhibitors. Protein Sci. 1994, 3, 1687–1696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pielak, R.M.; Schnell, J.R.; Chou, J.J. Mechanism of drug inhibition and drug resistance of influenza A M2 channel. Proc. Natl. Acad. Sci. USA 2009, 106, 7379–7384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Afonso, C.L.; Amarasinghe, G.K.; Banyai, K.; Bao, Y.; Basler, C.F.; Bavari, S.; Bejerman, N.; Blasdell, K.R.; Briand, F.X.; Briese, T.; et al. Taxonomy of the order Mononegavirales: Update 2016. Arch. Virol. 2016, 161, 2351–2360. [Google Scholar] [CrossRef] [PubMed]
- Falsey, A.R.; Walsh, E.E. Respiratory syncytial virus infection in adults. Clin. Microbiol. Rev. 2000, 13, 371–384. [Google Scholar] [CrossRef] [PubMed]
- Nair, H.; Nokes, D.J.; Gessner, B.D.; Dherani, M.; Madhi, S.A.; Singleton, R.J.; O’Brien, K.L.; Roca, A.; Wright, P.F.; Bruce, N.; et al. Global burden of acute lower respiratory infections due to respiratory syncytial virus in young children: A systematic review and meta-analysis. Lancet 2010, 375, 1545–1555. [Google Scholar] [CrossRef]
- Bont, L.; Checchia, P.A.; Fauroux, B.; Figueras-Aloy, J.; Manzoni, P.; Paes, B.; Simoes, E.A.; Carbonell-Estrany, X. Defining the Epidemiology and Burden of Severe Respiratory Syncytial Virus Infection Among Infants and Children in Western Countries. Infect. Dis. Ther. 2016, 5, 271–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Turner, T.L.; Kopp, B.T.; Paul, G.; Landgrave, L.C.; Hayes, D., Jr.; Thompson, R. Respiratory syncytial virus: Current and emerging treatment options. Clinicoecon. Outcomes Res. 2014, 6, 217–225. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, S.E.; Lamson, D.M.; St George, K.; Walsh, T.J. Human rhinoviruses. Clin. Microbiol. Rev. 2013, 26, 135–162. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.Y.; Oh, E.; Kim, Y.; Jung, W.W.; Kim, H.S.; Lee, J.; Sul, D. Enhanced oxidative damage to DNA, lipids, and proteins and levels of some antioxidant enzymes, cytokines, and heat shock proteins in patients infected with influenza H1N1 virus. Acta Virol. 2014, 58, 253–260. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ng, M.P.; Lee, J.C.; Loke, W.M.; Yeo, L.L.; Quek, A.M.; Lim, E.C.; Halliwell, B.; Seet, R.C. Does influenza A infection increase oxidative damage? Antioxid. Redox Signal. 2014, 21, 1025–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Erkekoglu, P.; Asci, A.; Ceyhan, M.; Kizilgun, M.; Schweizer, U.; Atas, C.; Kara, A.; Kocer Giray, B. Selenium levels, selenoenzyme activities and oxidant/antioxidant parameters in H1N1-infected children. Turk. J. Pediatr. 2013, 55, 271–282. [Google Scholar] [PubMed]
- Nin, N.; Sanchez-Rodriguez, C.; Ver, L.S.; Cardinal, P.; Ferruelo, A.; Soto, L.; Deicas, A.; Campos, N.; Rocha, O.; Ceraso, D.H.; et al. Lung histopathological findings in fatal pandemic influenza A (H1N1). Med. Intensiv. 2012, 36, 24–31. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buffinton, G.D.; Christen, S.; Peterhans, E.; Stocker, R. Oxidative stress in lungs of mice infected with influenza A virus. Free Radic. Res. Commun. 1992, 16, 99–110. [Google Scholar] [CrossRef] [PubMed]
- Hennet, T.; Peterhans, E.; Stocker, R. Alterations in antioxidant defences in lung and liver of mice infected with influenza A virus. J. Gen. Virol. 1992, 73 Pt 1, 39–46. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amatore, D.; Sgarbanti, R.; Aquilano, K.; Baldelli, S.; Limongi, D.; Civitelli, L.; Nencioni, L.; Garaci, E.; Ciriolo, M.R.; Palamara, A.T. Influenza virus replication in lung epithelial cells depends on redox-sensitive pathways activated by NOX4-derived ROS. Cell. Microbiol. 2015, 17, 131–145. [Google Scholar] [CrossRef] [PubMed]
- Ye, S.; Lowther, S.; Stambas, J. Inhibition of reactive oxygen species production ameliorates inflammation induced by influenza A viruses via upregulation of SOCS1 and SOCS3. J. Virol. 2015, 89, 2672–2683. [Google Scholar] [CrossRef] [PubMed]
- Casola, A.; Burger, N.; Liu, T.; Jamaluddin, M.; Brasier, A.R.; Garofalo, R.P. Oxidant tone regulates RANTES gene expression in airway epithelial cells infected with respiratory syncytial virus. Role in viral-induced interferon regulatory factor activation. J. Biol. Chem. 2001, 276, 19715–19722. [Google Scholar] [CrossRef] [PubMed]
- Martinez, I.; Garcia-Carpizo, V.; Guijarro, T.; Garcia-Gomez, A.; Navarro, D.; Aranda, A.; Zambrano, A. Induction of DNA double-strand breaks and cellular senescence by human respiratory syncytial virus. Virulence 2016, 7, 427–442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, H.; Li, L.Y.; Zhang, M.; Zhang, Q. Inactivated Sendai virus induces apoptosis mediated by reactive oxygen species in murine melanoma cells. Biomed. Environ. Sci. 2016, 29, 877–884. [Google Scholar] [PubMed]
- Qian, M.; Tan, H.M.; Yu, N.; Wang, T.; Zhang, Q. Inactivated Sendai Virus Induces ROS-dependent Apoptosis and Autophagy in Human Prostate Cancer Cells. Biomed. Environ. Sci. 2018, 31, 280–289. [Google Scholar] [PubMed]
- Moreno-Solis, G.; de la Torre-Aguilar, M.J.; Torres-Borrego, J.; Llorente-Cantarero, F.J.; Fernandez-Gutierrez, F.; Gil-Campos, M.; Tunez-Finana, I.; Perez-Navero, J.L. Oxidative Stress and Inflamatory Plasma Biomarkers in Respiratory Syncytial Virus Bronchiolitis. Clin. Respir. J. 2015, 11, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Akaike, T.; Okamoto, S.; Sawa, T.; Yoshitake, J.; Tamura, F.; Ichimori, K.; Miyazaki, K.; Sasamoto, K.; Maeda, H. 8-nitroguanosine formation in viral pneumonia and its implication for pathogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 685–690. [Google Scholar] [CrossRef] [PubMed]
- Hosakote, Y.M.; Jantzi, P.D.; Esham, D.L.; Spratt, H.; Kurosky, A.; Casola, A.; Garofalo, R.P. Viral-mediated inhibition of antioxidant enzymes contributes to the pathogenesis of severe respiratory syncytial virus bronchiolitis. Am. J. Respir. Crit. Care Med. 2011, 183, 1550–1560. [Google Scholar] [CrossRef] [PubMed]
- Hosakote, Y.M.; Liu, T.; Castro, S.M.; Garofalo, R.P.; Casola, A. Respiratory syncytial virus induces oxidative stress by modulating antioxidant enzymes. Am. J. Respir. Cell. Mol. Biol. 2009, 41, 348–357. [Google Scholar] [CrossRef] [PubMed]
- Jamaluddin, M.; Tian, B.; Boldogh, I.; Garofalo, R.P.; Brasier, A.R. Respiratory syncytial virus infection induces a reactive oxygen species-MSK1-phospho-Ser-276 RelA pathway required for cytokine expression. J. Virol. 2009, 83, 10605–10615. [Google Scholar] [CrossRef] [PubMed]
- Mochizuki, H.; Todokoro, M.; Arakawa, H. RS virus-induced inflammation and the intracellular glutathione redox state in cultured human airway epithelial cells. Inflammation 2009, 32, 252–264. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Sinha, M.; Liu, T.; Hong, C.; Luxon, B.A.; Garofalo, R.P.; Casola, A. Identification of human metapneumovirus-induced gene networks in airway epithelial cells by microarray analysis. Virology 2008, 374, 114–127. [Google Scholar] [CrossRef] [PubMed]
- Biagioli, M.C.; Kaul, P.; Singh, I.; Turner, R.B. The role of oxidative stress in rhinovirus induced elaboration of IL-8 by respiratory epithelial cells. Free Radic. Biol. Med. 1999, 26, 454–462. [Google Scholar] [CrossRef]
- Papi, A.; Contoli, M.; Gasparini, P.; Bristot, L.; Edwards, M.R.; Chicca, M.; Leis, M.; Ciaccia, A.; Caramori, G.; Johnston, S.L.; et al. Role of xanthine oxidase activation and reduced glutathione depletion in rhinovirus induction of inflammation in respiratory epithelial cells. J. Biol. Chem. 2008, 283, 28595–28606. [Google Scholar] [CrossRef] [PubMed]
- Kaul, P.; Biagioli, M.C.; Singh, I.; Turner, R.B. Rhinovirus-induced oxidative stress and interleukin-8 elaboration involves p47-phox but is independent of attachment to intercellular adhesion molecule-1 and viral replication. J. Infect. Dis. 2000, 181, 1885–1890. [Google Scholar] [CrossRef] [PubMed]
- Funchal, G.A.; Jaeger, N.; Czepielewski, R.S.; Machado, M.S.; Muraro, S.P.; Stein, R.T.; Bonorino, C.B.; Porto, B.N. Respiratory syncytial virus fusion protein promotes TLR-4-dependent neutrophil extracellular trap formation by human neutrophils. PLoS ONE 2015, 10, e0124082. [Google Scholar] [CrossRef] [PubMed]
- Segovia, J.; Sabbah, A.; Mgbemena, V.; Tsai, S.Y.; Chang, T.H.; Berton, M.T.; Morris, I.R.; Allen, I.C.; Ting, J.P.; Bose, S. TLR2/MyD88/NF-κB pathway, reactive oxygen species, potassium efflux activates NLRP3/ASC inflammasome during respiratory syncytial virus infection. PLoS ONE 2012, 7, e29695. [Google Scholar] [CrossRef] [PubMed]
- Altenhofer, S.; Radermacher, K.A.; Kleikers, P.W.; Wingler, K.; Schmidt, H.H. Evolution of NADPH Oxidase Inhibitors: Selectivity and Mechanisms for Target Engagement. Antioxid. Redox Signal. 2015, 23, 406–427. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vlahos, R.; Stambas, J.; Bozinovski, S.; Broughton, B.R.; Drummond, G.R.; Selemidis, S. Inhibition of Nox2 oxidase activity ameliorates influenza A virus-induced lung inflammation. PLoS Pathog. 2011, 7, e1001271. [Google Scholar] [CrossRef] [PubMed]
- To, E.E.; Broughton, B.R.; Hendricks, K.S.; Vlahos, R.; Selemidis, S. Influenza A virus and TLR7 activation potentiate NOX2 oxidase-dependent ROS production in macrophages. Free Radic. Res. 2014, 48, 940–947. [Google Scholar] [CrossRef] [PubMed]
- To, E.E.; Vlahos, R.; Luong, R.; Halls, M.L.; Reading, P.C.; King, P.T.; Chan, C.; Drummond, G.R.; Sobey, C.G.; Broughton, B.R.S.; et al. Endosomal NOX2 oxidase exacerbates virus pathogenicity and is a target for antiviral therapy. Nat. Commun. 2017, 8, 69. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fink, K.; Duval, A.; Martel, A.; Soucy-Faulkner, A.; Grandvaux, N. Dual role of NOX2 in respiratory syncytial virus- and sendai virus-induced activation of NF-κB in airway epithelial cells. J. Immunol. 2008, 180, 6911–6922. [Google Scholar] [CrossRef] [PubMed]
- Snelgrove, R.J.; Edwards, L.; Rae, A.J.; Hussell, T. An absence of reactive oxygen species improves the resolution of lung influenza infection. Eur. J. Immunol. 2006, 36, 1364–1373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedard, K.; Krause, K.H. The NOX family of ROS-generating NADPH oxidases: Physiology and pathophysiology. Physiol. Rev. 2007, 87, 245–313. [Google Scholar] [CrossRef] [PubMed]
- Imai, Y.; Kuba, K.; Neely, G.G.; Yaghubian-Malhami, R.; Perkmann, T.; van Loo, G.; Ermolaeva, M.; Veldhuizen, R.; Leung, Y.H.; Wang, H.; et al. Identification of oxidative stress and Toll-like receptor 4 signaling as a key pathway of acute lung injury. Cell 2008, 133, 235–249. [Google Scholar] [CrossRef] [PubMed]
- Trocme, C.; Deffert, C.; Cachat, J.; Donati, Y.; Tissot, C.; Papacatzis, S.; Braunersreuther, V.; Pache, J.C.; Krause, K.H.; Holmdahl, R.; et al. Macrophage-specific NOX2 contributes to the development of lung emphysema through modulation of SIRT1/MMP-9 pathways. J. Pathol. 2015, 235, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Soucy-Faulkner, A.; Mukawera, E.; Fink, K.; Martel, A.; Jouan, L.; Nzengue, Y.; Lamarre, D.; Vande Velde, C.; Grandvaux, N. Requirement of NOX2 and reactive oxygen species for efficient RIG-I-mediated antiviral response through regulation of MAVS expression. PLoS Pathog. 2010, 6, e1000930. [Google Scholar] [CrossRef] [PubMed]
- Grandvaux, N.; Mariani, M.; Fink, K. Lung epithelial NOX/DUOX and respiratory virus infections. Clin. Sci. (Lond.) 2015, 128, 337–347. [Google Scholar] [CrossRef] [PubMed]
- Van den Brand, J.M.; Stittelaar, K.J.; van Amerongen, G.; Reperant, L.; de Waal, L.; Osterhaus, A.D.; Kuiken, T. Comparison of temporal and spatial dynamics of seasonal H3N2, pandemic H1N1 and highly pathogenic avian influenza H5N1 virus infections in ferrets. PLoS ONE 2012, 7, e42343. [Google Scholar] [CrossRef] [PubMed]
- Camp, J.V.; Bagci, U.; Chu, Y.K.; Squier, B.; Fraig, M.; Uriarte, S.M.; Guo, H.; Mollura, D.J.; Jonsson, C.B. Lower Respiratory Tract Infection of the Ferret by 2009 H1N1 Pandemic Influenza A Virus Triggers Biphasic, Systemic, and Local Recruitment of Neutrophils. J. Virol. 2015, 89, 8733–8748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geerdink, R.J.; Pillay, J.; Meyaard, L.; Bont, L. Neutrophils in respiratory syncytial virus infection: A target for asthma prevention. J. Allergy Clin. Immunol. 2015, 136, 838–847. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, C.H.; Ryu, J.H.; Kim, M.J.; Park, C.Y.; Lee, J.M.; Holtzman, M.J.; Yoon, J.H. Reactive oxygen species induce antiviral innate immune response through IFN-λ regulation in human nasal epithelial cells. Am. J. Respir. Cell. Mol. Biol. 2013, 49, 855–865. [Google Scholar] [CrossRef] [PubMed]
- Selemidis, S.; Seow, H.J.; Broughton, B.R.; Vinh, A.; Bozinovski, S.; Sobey, C.G.; Drummond, G.R.; Vlahos, R. NOX1 oxidase suppresses influenza a virus-induced lung inflammation and oxidative stress. PLoS ONE 2013, 8, e60792. [Google Scholar] [CrossRef] [PubMed]
- Hofstetter, A.R.; De La Cruz, J.A.; Cao, W.; Patel, J.; Belser, J.A.; McCoy, J.; Liepkalns, J.S.; Amoah, S.; Cheng, G.; Ranjan, P.; et al. NADPH Oxidase 1 Is Associated with Altered Host Survival and T Cell Phenotypes after Influenza A Virus Infection in Mice. PLoS ONE 2016, 11, e0149864. [Google Scholar] [CrossRef] [PubMed]
- Comstock, A.T.; Ganesan, S.; Chattoraj, A.; Faris, A.N.; Margolis, B.L.; Hershenson, M.B.; Sajjan, U.S. Rhinovirus-induced barrier dysfunction in polarized airway epithelial cells is mediated by NADPH oxidase 1. J. Virol. 2011, 85, 6795–6808. [Google Scholar] [CrossRef] [PubMed]
- Unger, B.L.; Ganesan, S.; Comstock, A.T.; Faris, A.N.; Hershenson, M.B.; Sajjan, U.S. Nod-like receptor X-1 is required for rhinovirus-induced barrier dysfunction in airway epithelial cells. J. Virol. 2014, 88, 3705–3718. [Google Scholar] [CrossRef] [PubMed]
- Tattoli, I.; Carneiro, L.A.; Jehanno, M.; Magalhaes, J.G.; Shu, Y.; Philpott, D.J.; Arnoult, D.; Girardin, S.E. NLRX1 is a mitochondrial NOD-like receptor that amplifies NF-κB and JNK pathways by inducing reactive oxygen species production. EMBO Rep. 2008, 9, 293–300. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surh, Y.J.; Kundu, J.K.; Na, H.K.; Lee, J.S. Redox-sensitive transcription factors as prime targets for chemoprevention with anti-inflammatory and antioxidative phytochemicals. J. Nutr. 2005, 135, S2993–S3001. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.J.; Kim, C.H.; Kim, M.J.; Ryu, J.H.; Seong, S.Y.; Kim, S.; Lim, S.J.; Holtzman, M.J.; Yoon, J.H. The Induction of Pattern-Recognition Receptor Expression against Influenza A Virus through Duox2-Derived Reactive Oxygen Species in Nasal Mucosa. Am. J. Respir. Cell Mol. Biol. 2015, 53, 525–535. [Google Scholar] [CrossRef] [PubMed]
- Strengert, M.; Jennings, R.; Davanture, S.; Hayes, P.; Gabriel, G.; Knaus, U.G. Mucosal reactive oxygen species are required for antiviral response: Role of Duox in influenza a virus infection. Antioxid. Redox Signal. 2014, 20, 2695–2709. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.N.; Kim, J.Y.; Kim, H.; Kim, D.Y.; Won, T.B.; Han, D.H.; Rhee, C.S.; Kim, H.J. Duox2 is required for the transcription of pattern recognition receptors in acute viral lung infection: An interferon-independent regulatory mechanism. Antivir. Res. 2016, 134, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Harper, R.W.; Xu, C.; Eiserich, J.P.; Chen, Y.; Kao, C.Y.; Thai, P.; Setiadi, H.; Wu, R. Differential regulation of dual NADPH oxidases/peroxidases, Duox1 and Duox2, by Th1 and Th2 cytokines in respiratory tract epithelium. FEBS Lett. 2005, 579, 4911–4917. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schneider, D.; Ganesan, S.; Comstock, A.T.; Meldrum, C.A.; Mahidhara, R.; Goldsmith, A.M.; Curtis, J.L.; Martinez, F.J.; Hershenson, M.B.; Sajjan, U. Increased cytokine response of rhinovirus-infected airway epithelial cells in chronic obstructive pulmonary disease. Am. J. Respir. Crit. Care Med. 2010, 182, 332–340. [Google Scholar] [CrossRef] [PubMed]
- Akaike, T.; Ando, M.; Oda, T.; Doi, T.; Ijiri, S.; Araki, S.; Maeda, H. Dependence on O2− generation by xanthine oxidase of pathogenesis of influenza virus infection in mice. J. Clin. Investig. 1990, 85, 739–745. [Google Scholar] [CrossRef] [PubMed]
- Oda, T.; Akaike, T.; Hamamoto, T.; Suzuki, F.; Hirano, T.; Maeda, H. Oxygen radicals in influenza-induced pathogenesis and treatment with pyran polymer-conjugated SOD. Science 1989, 244, 974–976. [Google Scholar] [CrossRef] [PubMed]
- Cantu-Medellin, N.; Kelley, E.E. Xanthine oxidoreductase-catalyzed reactive species generation: A process in critical need of reevaluation. Redox Biol. 2013, 1, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Kim, M.J.; Park, D.Y.; Chung, H.J.; Kim, C.H.; Yoon, J.H.; Kim, H.J. Mitochondrial reactive oxygen species modulate innate immune response to influenza A virus in human nasal epithelium. Antivir. Res. 2015, 119, 78–83. [Google Scholar] [CrossRef] [PubMed]
- Dikalov, S. Cross talk between mitochondria and NADPH oxidases. Free Radic. Biol. Med. 2011, 51, 1289–1301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daiber, A.; Di Lisa, F.; Oelze, M.; Kroller-Schon, S.; Steven, S.; Schulz, E.; Munzel, T. Crosstalk of mitochondria with NADPH oxidase via reactive oxygen and nitrogen species signalling and its role for vascular function. Br. J. Pharmacol. 2017, 174, 1670–1689. [Google Scholar] [CrossRef] [PubMed]
- Bitzer, M.; Armeanu, S.; Prinz, F.; Ungerechts, G.; Wybranietz, W.; Spiegel, M.; Bernlohr, C.; Cecconi, F.; Gregor, M.; Neubert, W.J.; et al. Caspase-8 and Apaf-1-independent caspase-9 activation in Sendai virus-infected cells. J. Biol. Chem. 2002, 277, 29817–29824. [Google Scholar] [CrossRef] [PubMed]
- Sawa, T.; Akaike, T.; Ichimori, K.; Akuta, T.; Kaneko, K.; Nakayama, H.; Stuehr, D.J.; Maeda, H. Superoxide generation mediated by 8-nitroguanosine, a highly redox-active nucleic acid derivative. Biochem. Biophys. Res. Commun. 2003, 311, 300–306. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, A.V.; Valuev-Elliston, V.T.; Ivanova, O.N.; Kochetkov, S.N.; Starodubova, E.S.; Bartosch, B.; Isaguliants, M.G. Oxidative Stress during HIV Infection: Mechanisms and Consequences. Oxid. Med. Cell. Longev. 2016, 2016, 8910396. [Google Scholar] [CrossRef] [PubMed]
- Lazrak, A.; Iles, K.E.; Liu, G.; Noah, D.L.; Noah, J.W.; Matalon, S. Influenza virus M2 protein inhibits epithelial sodium channels by increasing reactive oxygen species. FASEB J. 2009, 23, 3829–3842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, N.; Pyo, C.W.; Jung, K.I.; Choi, S.Y. Influenza A virus PB1-F2 is involved in regulation of cellular redox state in alveolar epithelial cells. Biochem. Biophys. Res. Commun. 2015, 459, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Flory, E.; Kunz, M.; Scheller, C.; Jassoy, C.; Stauber, R.; Rapp, U.R.; Ludwig, S. Influenza virus-induced NF-κB-dependent gene expression is mediated by overexpression of viral proteins and involves oxidative radicals and activation of IκB kinase. J. Biol. Chem. 2000, 275, 8307–8314. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.W.; Lin, K.H.; Hsieh, T.H.; Shiu, S.Y.; Li, J.Y. Severe acute respiratory syndrome coronavirus 3C-like protease-induced apoptosis. FEMS Immunol. Med. Microbiol. 2006, 46, 375–380. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.; Nioi, P.; Pickett, C.B. The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress. J. Biol. Chem. 2009, 284, 13291–13295. [Google Scholar] [CrossRef] [PubMed]
- Kwak, M.K.; Itoh, K.; Yamamoto, M.; Kensler, T.W. Enhanced expression of the transcription factor Nrf2 by cancer chemopreventive agents: Role of antioxidant response element-like sequences in the nrf2 promoter. Mol. Cell. Biol. 2002, 22, 2883–2892. [Google Scholar] [CrossRef] [PubMed]
- Manevich, Y.; Shuvaeva, T.; Dodia, C.; Kazi, A.; Feinstein, S.I.; Fisher, A.B. Binding of peroxiredoxin 6 to substrate determines differential phospholipid hydroperoxide peroxidase and phospholipase A(2) activities. Arch. Biochem. Biophys. 2009, 485, 139–149. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.S.; Stockwell, B.R. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016, 26, 165–176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kosmider, B.; Messier, E.M.; Janssen, W.J.; Nahreini, P.; Wang, J.; Hartshorn, K.L.; Mason, R.J. Nrf2 protects human alveolar epithelial cells against injury induced by influenza A virus. Respir. Res. 2012, 13, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, Y.; Limmon, G.V.; Zheng, D.; Li, N.; Li, L.; Yin, L.; Chow, V.T.; Chen, J.; Engelward, B.P. Major shifts in the spatio-temporal distribution of lung antioxidant enzymes during influenza pneumonia. PLoS ONE 2012, 7, e31494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simon, P.F.; McCorrister, S.; Hu, P.; Chong, P.; Silaghi, A.; Westmacott, G.; Coombs, K.M.; Kobasa, D. Highly Pathogenic H5N1 and Novel H7N9 Influenza A Viruses Induce More Profound Proteomic Host Responses than Seasonal and Pandemic H1N1 Strains. J. Proteome Res. 2015, 14, 4511–4523. [Google Scholar] [CrossRef] [PubMed]
- Jacoby, D.B.; Choi, A.M. Influenza virus induces expression of antioxidant genes in human epithelial cells. Free Radic. Biol. Med. 1994, 16, 821–824. [Google Scholar] [CrossRef]
- Pyo, C.W.; Shin, N.; Jung, K.I.; Choi, J.H.; Choi, S.Y. Alteration of copper-zinc superoxide dismutase 1 expression by influenza A virus is correlated with virus replication. Biochem. Biophys. Res. Commun. 2014, 450, 711–716. [Google Scholar] [CrossRef] [PubMed]
- Lin, X.; Wang, R.; Zou, W.; Sun, X.; Liu, X.; Zhao, L.; Wang, S.; Jin, M. The Influenza Virus H5N1 Infection Can Induce ROS Production for Viral Replication and Host Cell Death in A549 Cells Modulated by Human Cu/Zn Superoxide Dismutase (SOD1) Overexpression. Viruses 2016, 8, 13. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Zaas, A.K.; Rao, A.; Dobigeon, N.; Woolf, P.J.; Veldman, T.; Oien, N.C.; McClain, M.T.; Varkey, J.B.; Nicholson, B.; et al. Temporal dynamics of host molecular responses differentiate symptomatic and asymptomatic influenza a infection. PLoS Genet. 2011, 7, e1002234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soliman, H.; Mediavilla-Varela, M.; Antonia, S. Indoleamine 2,3-dioxygenase: Is it an immune suppressor? Cancer J. 2010, 16, 354–359. [Google Scholar] [CrossRef] [PubMed]
- Choi, A.M.; Knobil, K.; Otterbein, S.L.; Eastman, D.A.; Jacoby, D.B. Oxidant stress responses in influenza virus pneumonia: Gene expression and transcription factor activation. Am. J. Physiol. 1996, 271, L383–L391. [Google Scholar] [CrossRef] [PubMed]
- Komaravelli, N.; Tian, B.; Ivanciuc, T.; Mautemps, N.; Brasier, A.R.; Garofalo, R.P.; Casola, A. Respiratory syncytial virus infection down-regulates antioxidant enzyme expression by triggering deacetylation-proteasomal degradation of Nrf2. Free Radic. Biol. Med. 2015, 88, 391–403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cho, H.Y.; Imani, F.; Miller-DeGraff, L.; Walters, D.; Melendi, G.A.; Yamamoto, M.; Polack, F.P.; Kleeberger, S.R. Antiviral activity of Nrf2 in a murine model of respiratory syncytial virus disease. Am. J. Respir. Crit. Care Med. 2009, 179, 138–150. [Google Scholar] [CrossRef] [PubMed]
- Garofalo, R.P.; Kolli, D.; Casola, A. Respiratory syncytial virus infection: Mechanisms of redox control and novel therapeutic opportunities. Antioxid. Redox Signal. 2013, 18, 186–217. [Google Scholar] [CrossRef] [PubMed]
- Jamaluddin, M.; Wiktorowicz, J.E.; Soman, K.V.; Boldogh, I.; Forbus, J.D.; Spratt, H.; Garofalo, R.P.; Brasier, A.R. Role of peroxiredoxin 1 and peroxiredoxin 4 in protection of respiratory syncytial virus-induced cysteinyl oxidation of nuclear cytoskeletal proteins. J. Virol. 2010, 84, 9533–9545. [Google Scholar] [CrossRef] [PubMed]
- Hastie, M.L.; Headlam, M.J.; Patel, N.B.; Bukreyev, A.A.; Buchholz, U.J.; Dave, K.A.; Norris, E.L.; Wright, C.L.; Spann, K.M.; Collins, P.L.; et al. The human respiratory syncytial virus nonstructural protein 1 regulates type I and type II interferon pathways. Mol. Cell. Proteom. 2012, 11, 108–127. [Google Scholar] [CrossRef] [PubMed]
- Darville, M.I.; Ho, Y.S.; Eizirik, D.L. NF-κB is required for cytokine-induced manganese superoxide dismutase expression in insulin-producing cells. Endocrinology 2000, 141, 153–162. [Google Scholar] [CrossRef] [PubMed]
- Kaul, P.; Singh, I.; Turner, R.B. Effect of rhinovirus challenge on antioxidant enzymes in respiratory epithelial cells. Free Radic. Res. 2002, 36, 1085–1089. [Google Scholar] [CrossRef] [PubMed]
- Ciriolo, M.R.; Palamara, A.T.; Incerpi, S.; Lafavia, E.; Bue, M.C.; De Vito, P.; Garaci, E.; Rotilio, G. Loss of GSH, oxidative stress, and decrease of intracellular pH as sequential steps in viral infection. J. Biol. Chem. 1997, 272, 2700–2708. [Google Scholar] [CrossRef] [PubMed]
- Biswas, M.; Chan, J.Y. Role of Nrf1 in antioxidant response element-mediated gene expression and beyond. Toxicol. Appl. Pharmacol. 2010, 244, 16–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, J.; Chen, Y.; Seth, S.; Furukawa, S.; Compans, R.W.; Jones, D.P. Inhibition of influenza infection by glutathione. Free Radic. Biol. Med. 2003, 34, 928–936. [Google Scholar] [CrossRef]
- Nencioni, L.; Iuvara, A.; Aquilano, K.; Ciriolo, M.R.; Cozzolino, F.; Rotilio, G.; Garaci, E.; Palamara, A.T. Influenza A virus replication is dependent on an antioxidant pathway that involves GSH and Bcl-2. FASEB J. 2003, 17, 758–760. [Google Scholar] [CrossRef] [PubMed]
- Kesic, M.J.; Simmons, S.O.; Bauer, R.; Jaspers, I. Nrf2 expression modifies influenza A entry and replication in nasal epithelial cells. Free Radic. Biol. Med. 2011, 51, 444–453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pauli, E.K.; Schmolke, M.; Wolff, T.; Viemann, D.; Roth, J.; Bode, J.G.; Ludwig, S. Influenza A virus inhibits type I IFN signaling via NF-κB-dependent induction of SOCS-3 expression. PLoS Pathog. 2008, 4, e1000196. [Google Scholar] [CrossRef] [PubMed]
- Pinto, R.; Herold, S.; Cakarova, L.; Hoegner, K.; Lohmeyer, J.; Planz, O.; Pleschka, S. Inhibition of influenza virus-induced NF-κB and Raf/MEK/ERK activation can reduce both virus titers and cytokine expression simultaneously in vitro and in vivo. Antivir. Res. 2011, 92, 45–56. [Google Scholar] [CrossRef] [PubMed]
- Hillesheim, A.; Nordhoff, C.; Boergeling, Y.; Ludwig, S.; Wixler, V. β-catenin promotes the type I IFN synthesis and the IFN-dependent signaling response but is suppressed by influenza A virus-induced RIG-I/NF-κB signaling. Cell Commun. Signal. 2014, 12, 29. [Google Scholar] [CrossRef] [PubMed]
- Gao, S.; Song, L.; Li, J.; Zhang, Z.; Peng, H.; Jiang, W.; Wang, Q.; Kang, T.; Chen, S.; Huang, W. Influenza A virus-encoded NS1 virulence factor protein inhibits innate immune response by targeting IKK. Cell. Microbiol. 2012, 14, 1849–1866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fink, K.; Martin, L.; Mukawera, E.; Chartier, S.; De Deken, X.; Brochiero, E.; Miot, F.; Grandvaux, N. IFNβ/TNFalpha synergism induces a non-canonical STAT2/IRF9-dependent pathway triggering a novel DUOX2 NADPH oxidase-mediated airway antiviral response. Cell. Res. 2013, 23, 673–690. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Castro, S.; Brasier, A.R.; Jamaluddin, M.; Garofalo, R.P.; Casola, A. Reactive oxygen species mediate virus-induced STAT activation: Role of tyrosine phosphatases. J. Biol. Chem. 2004, 279, 2461–2469. [Google Scholar] [CrossRef] [PubMed]
- Yoboua, F.; Martel, A.; Duval, A.; Mukawera, E.; Grandvaux, N. Respiratory syncytial virus-mediated NF-κB p65 phosphorylation at serine 536 is dependent on RIG-I, TRAF6, and IKKβ. J. Virol. 2010, 84, 7267–7277. [Google Scholar] [CrossRef] [PubMed]
- Grandvaux, N.; Guan, X.; Yoboua, F.; Zucchini, N.; Fink, K.; Doyon, P.; Martin, L.; Servant, M.J.; Chartier, S. Sustained activation of interferon regulatory factor 3 during infection by paramyxoviruses requires MDA5. J. Innate Immun. 2014, 6, 650–662. [Google Scholar] [CrossRef] [PubMed]
- Spann, K.M.; Tran, K.C.; Collins, P.L. Effects of nonstructural proteins NS1 and NS2 of human respiratory syncytial virus on interferon regulatory factor 3, NF-κB, and proinflammatory cytokines. J. Virol. 2005, 79, 5353–5362. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Liu, T.; Pang, L.; Li, K.; Garofalo, R.P.; Casola, A.; Bao, X. A novel mechanism for the inhibition of interferon regulatory factor-3-dependent gene expression by human respiratory syncytial virus NS1 protein. J. Gen. Virol. 2011, 92, 2153–2159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barik, S. Respiratory syncytial virus mechanisms to interfere with type 1 interferons. Curr. Top. Microbiol. Immunol. 2013, 372, 173–191. [Google Scholar] [PubMed]
- Boyapalle, S.; Wong, T.; Garay, J.; Teng, M.; San Juan-Vergara, H.; Mohapatra, S.; Mohapatra, S. Respiratory syncytial virus NS1 protein colocalizes with mitochondrial antiviral signaling protein MAVS following infection. PLoS ONE 2012, 7, e29386. [Google Scholar] [CrossRef] [PubMed]
- Swedan, S.; Musiyenko, A.; Barik, S. Respiratory syncytial virus nonstructural proteins decrease levels of multiple members of the cellular interferon pathways. J. Virol. 2009, 83, 9682–9693. [Google Scholar] [CrossRef] [PubMed]
- Papi, A.; Papadopoulos, N.G.; Stanciu, L.A.; Bellettato, C.M.; Pinamonti, S.; Degitz, K.; Holgate, S.T.; Johnston, S.L. Reducing agents inhibit rhinovirus-induced up-regulation of the rhinovirus receptor intercellular adhesion molecule-1 (ICAM-1) in respiratory epithelial cells. FASEB J. 2002, 16, 1934–1936. [Google Scholar] [CrossRef] [PubMed]
- Palamara, A.T.; Di Francesco, P.; Ciriolo, M.R.; Bue, C.; Lafavia, E.; Rotilio, G.; Garaci, E. Cocaine increases Sendai virus replication in cultured epithelial cells: Critical role of the intracellular redox status. Biochem. Biophys. Res. Commun. 1996, 228, 579–585. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Dosal, R.; Horan, K.A.; Paludan, S.R. Mitochondria-derived reactive oxygen species negatively regulates immune innate signaling pathways triggered by a DNA virus, but not by an RNA virus. Biochem Biophys. Res. Commun. 2012, 418, 806–810. [Google Scholar] [CrossRef] [PubMed]
- Vendemiale, G.; Grattagliano, I.; Portincasa, P.; Serviddio, G.; Palasciamo, G.; Altomare, E. Oxidative stress in symptom-free HCV carriers: Relation with ALT flare-up. Eur. J. Clin. Investig. 2001, 31, 54–63. [Google Scholar] [CrossRef]
- Jain, S.K.; Pemberton, P.W.; Smith, A.; McMahon, R.F.; Burrows, P.C.; Aboutwerat, A.; Warnes, T.W. Oxidative stress in chronic hepatitis C: Not just a feature of late stage disease. J. Hepatol. 2002, 36, 805–811. [Google Scholar] [CrossRef]
- Bolukbas, C.; Bolukbas, F.F.; Horoz, M.; Aslan, M.; Celik, H.; Erel, O. Increased oxidative stress associated with the severity of the liver disease in various forms of hepatitis B virus infection. BMC Infect. Dis. 2005, 5, 95. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Parrish, M.; Chan, T.K.; Yin, L.; Rai, P.; Yoshiyuki, Y.; Abolhassani, N.; Tan, K.B.; Kiraly, O.; Chow, V.T.; et al. Influenza infection induces host DNA damage and dynamic DNA damage responses during tissue regeneration. Cell. Mol. Life Sci. 2015, 72, 2973–2988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kash, J.C.; Xiao, Y.; Davis, A.S.; Walters, K.A.; Chertow, D.S.; Easterbrook, J.D.; Dunfee, R.L.; Sandouk, A.; Jagger, B.W.; Schwartzman, L.M.; et al. Treatment with the reactive oxygen species scavenger EUK-207 reduces lung damage and increases survival during 1918 influenza virus infection in mice. Free Radic. Biol. Med. 2014, 67, 235–247. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uchide, N.; Ohyama, K. Antiviral function of pyrrolidine dithiocarbamate against influenza virus: The inhibition of viral gene replication and transcription. J. Antimicrob. Chemother. 2003, 52, 8–10. [Google Scholar] [CrossRef] [PubMed]
- Yatmaz, S.; Seow, H.J.; Gualano, R.C.; Wong, Z.X.; Stambas, J.; Selemidis, S.; Crack, P.J.; Bozinovski, S.; Anderson, G.P.; Vlahos, R. Glutathione peroxidase-1 reduces influenza A virus-induced lung inflammation. Am. J. Respir. Cell Mol. Biol. 2013, 48, 17–26. [Google Scholar] [CrossRef] [PubMed]
- Jaspers, I.; Zhang, W.; Brighton, L.E.; Carson, J.L.; Styblo, M.; Beck, M.A. Selenium deficiency alters epithelial cell morphology and responses to influenza. Free Radic. Biol. Med. 2007, 42, 1826–1837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, L.; Wei, L.; Jiang, D.; Wang, J.; Cong, X.; Fei, R. SARS-CoV nucleocapsid protein induced apoptosis of COS-1 mediated by the mitochondrial pathway. Artif. Cells Blood Substit Immobil. Biotechnol. 2007, 35, 237–253. [Google Scholar] [CrossRef] [PubMed]
- Olsen, C.W.; Dybdahl-Sissoko, N.; Hinshaw, V.S. The influence of calcium and reactive oxygen species on influenza virus-induced apoptosis. Cell Death Differ. 1996, 3, 191–197. [Google Scholar] [PubMed]
- Blume, C.; Reale, R.; Held, M.; Loxham, M.; Millar, T.M.; Collins, J.E.; Swindle, E.J.; Morgan, H.; Davies, D.E. Cellular crosstalk between airway epithelial and endothelial cells regulates barrier functions during exposure to double-stranded RNA. Immun. Inflamm. Dis. 2017, 5, 45–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moldoveanu, B.; Otmishi, P.; Jani, P.; Walker, J.; Sarmiento, X.; Guardiola, J.; Saad, M.; Yu, J. Inflammatory mechanisms in the lung. J. Inflamm. Res. 2009, 2, 1–11. [Google Scholar] [PubMed]
- Go, Y.M.; Kang, S.M.; Roede, J.R.; Orr, M.; Jones, D.P. Increased inflammatory signaling and lethality of influenza H1N1 by nuclear thioredoxin-1. PLoS ONE 2011, 6, e18918. [Google Scholar] [CrossRef] [PubMed]
- Mastronarde, J.G.; Monick, M.M.; Hunninghake, G.W. Oxidant tone regulates IL-8 production in epithelium infected with respiratory syncytial virus. Am. J. Respir. Cell. Mol. Biol. 1995, 13, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Tisoncik, J.R.; Korth, M.J.; Simmons, C.P.; Farrar, J.; Martin, T.R.; Katze, M.G. Into the eye of the cytokine storm. Microbiol. Mol. Biol. Rev. 2012, 76, 16–32. [Google Scholar] [CrossRef] [PubMed]
- Wong, E.T.; Tergaonkar, V. Roles of NF-κB in health and disease: Mechanisms and therapeutic potential. Clin. Sci. (Lond.) 2009, 116, 451–465. [Google Scholar] [CrossRef] [PubMed]
- Mata, M.; Morcillo, E.; Gimeno, C.; Cortijo, J. N-acetyl-l-cysteine (NAC) inhibit mucin synthesis and pro-inflammatory mediators in alveolar type II epithelial cells infected with influenza virus A and B and with respiratory syncytial virus (RSV). Biochem. Pharmacol. 2011, 82, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Mastronarde, J.G.; Monick, M.M.; Mukaida, N.; Matsushima, K.; Hunninghake, G.W. Activator protein-1 is the preferred transcription factor for cooperative interaction with nuclear factor-κB in respiratory syncytial virus-induced interleukin-8 gene expression in airway epithelium. J. Infect. Dis. 1998, 177, 1275–1281. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Sanders, S.P.; Siekierski, E.S.; Casolaro, V.; Proud, D. Role of NF-κB in cytokine production induced from human airway epithelial cells by rhinovirus infection. J. Immunol. 2000, 165, 3384–3392. [Google Scholar] [CrossRef] [PubMed]
- Bao, X.; Kolli, D.; Liu, T.; Shan, Y.; Garofalo, R.P.; Casola, A. Human metapneumovirus small hydrophobic protein inhibits NF-κB transcriptional activity. J. Virol. 2008, 82, 8224–8229. [Google Scholar] [CrossRef] [PubMed]
- Carpenter, L.R.; Moy, J.N.; Roebuck, K.A. Respiratory syncytial virus and TNF alpha induction of chemokine gene expression involves differential activation of Rel A and NF-κB1. BMC Infect. Dis. 2002, 2, 5. [Google Scholar] [CrossRef] [Green Version]
- Indukuri, H.; Castro, S.M.; Liao, S.M.; Feeney, L.A.; Dorsch, M.; Coyle, A.J.; Garofalo, R.P.; Brasier, A.R.; Casola, A. Ikkepsilon regulates viral-induced interferon regulatory factor-3 activation via a redox-sensitive pathway. Virology 2006, 353, 155–165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosakote, Y.M.; Brasier, A.R.; Casola, A.; Garofalo, R.P.; Kurosky, A. Respiratory Syncytial Virus Infection Triggers Epithelial HMGB1 Release as a Damage-Associated Molecular Pattern Promoting a Monocytic Inflammatory Response. J. Virol. 2016, 90, 9618–9631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, K.; Lv, Y.; Zhuo, Y.; Chen, C.; Shi, H.; Guo, L.; Yang, G.; Hou, Y.; Tan, R.X.; Li, E. Suppression of IRG-1 Reduces Inflammatory Cell Infiltration and Lung Injury in Respiratory Syncytial Virus Infection by Reducing Production of Reactive Oxygen Species. J. Virol. 2016, 90, 7313–7322. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Michelucci, A.; Cordes, T.; Ghelfi, J.; Pailot, A.; Reiling, N.; Goldmann, O.; Binz, T.; Wegner, A.; Tallam, A.; Rausell, A.; et al. Immune-responsive gene 1 protein links metabolism to immunity by catalyzing itaconic acid production. Proc. Natl. Acad. Sci. USA 2013, 110, 7820–7825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, X.; Wu, X.P.; Zhu, X.L.; Li, T.; Liu, Y. IRG1 increases MHC class I level in macrophages through STAT-TAP1 axis depending on NADPH oxidase mediated reactive oxygen species. Int. Immunopharmacol. 2017, 48, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Lampropoulou, V.; Sergushichev, A.; Bambouskova, M.; Nair, S.; Vincent, E.E.; Loginicheva, E.; Cervantes-Barragan, L.; Ma, X.; Huang, S.C.; Griss, T.; et al. Itaconate Links Inhibition of Succinate Dehydrogenase with Macrophage Metabolic Remodeling and Regulation of Inflammation. Cell Metab. 2016, 24, 158–166. [Google Scholar] [CrossRef] [PubMed]
- Mills, E.L.; Ryan, D.G.; Prag, H.A.; Dikovskaya, D.; Menon, D.; Zaslona, Z.; Jedrychowski, M.P.; Costa, A.S.H.; Higgins, M.; Hams, E.; et al. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 2018, 556, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Latz, E.; Xiao, T.S.; Stutz, A. Activation and regulation of the inflammasomes. Nat. Rev. Immunol. 2013, 13, 397–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schroder, K.; Tschopp, J. The inflammasomes. Cell 2010, 140, 821–832. [Google Scholar] [CrossRef] [PubMed]
- Harijith, A.; Ebenezer, D.L.; Natarajan, V. Reactive oxygen species at the crossroads of inflammasome and inflammation. Front. Physiol. 2014, 5, 352. [Google Scholar] [CrossRef] [PubMed]
- Allen, I.C.; Scull, M.A.; Moore, C.B.; Holl, E.K.; McElvania-TeKippe, E.; Taxman, D.J.; Guthrie, E.H.; Pickles, R.J.; Ting, J.P. The NLRP3 inflammasome mediates in vivo innate immunity to influenza A virus through recognition of viral RNA. Immunity 2009, 30, 556–565. [Google Scholar] [CrossRef] [PubMed]
- Ichinohe, T.; Yamazaki, T.; Koshiba, T.; Yanagi, Y. Mitochondrial protein mitofusin 2 is required for NLRP3 inflammasome activation after RNA virus infection. Proc. Natl. Acad. Sci. USA 2013, 110, 17963–17968. [Google Scholar] [CrossRef] [PubMed]
- Triantafilou, K.; Kar, S.; van Kuppeveld, F.J.; Triantafilou, M. Rhinovirus-induced calcium flux triggers NLRP3 and NLRC5 activation in bronchial cells. Am. J. Respir. Cell Mol. Biol. 2013, 49, 923–934. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Torres, J.L.; Verdia-Baguena, C.; Jimenez-Guardeno, J.M.; Regla-Nava, J.A.; Castano-Rodriguez, C.; Fernandez-Delgado, R.; Torres, J.; Aguilella, V.M.; Enjuanes, L. Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology 2015, 485, 330–339. [Google Scholar] [CrossRef] [PubMed]
- Mekahli, D.; Bultynck, G.; Parys, J.B.; De Smedt, H.; Missiaen, L. Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a004317. [Google Scholar] [CrossRef] [PubMed]
- Marciniak, S.J.; Ron, D. Endoplasmic reticulum stress signaling in disease. Physiol. Rev. 2006, 86, 1133–1149. [Google Scholar] [CrossRef] [PubMed]
- Darling, N.J.; Cook, S.J. The role of MAPK signalling pathways in the response to endoplasmic reticulum stress. Biochim. Biophys. Acta 2014, 1843, 2150–2163. [Google Scholar] [CrossRef] [PubMed]
- Newton, K.; Dixit, V.M. Signaling in innate immunity and inflammation. Cold Spring Harb. Perspect. Biol. 2012, 4, a006049. [Google Scholar] [CrossRef] [PubMed]
- Roberson, E.C.; Tully, J.E.; Guala, A.S.; Reiss, J.N.; Godburn, K.E.; Pociask, D.A.; Alcorn, J.F.; Riches, D.W.; Dienz, O.; Janssen-Heininger, Y.M.; et al. Influenza induces endoplasmic reticulum stress, caspase-12-dependent apoptosis, and c-Jun N-terminal kinase-mediated transforming growth factor-β release in lung epithelial cells. Am. J. Respir. Cell Mol. Biol. 2012, 46, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Sajjan, U.; Wang, Q.; Zhao, Y.; Gruenert, D.C.; Hershenson, M.B. Rhinovirus disrupts the barrier function of polarized airway epithelial cells. Am. J. Respir. Crit. Care Med. 2008, 178, 1271–1281. [Google Scholar] [CrossRef] [PubMed]
- Rezaee, F.; DeSando, S.A.; Ivanov, A.I.; Chapman, T.J.; Knowlden, S.A.; Beck, L.A.; Georas, S.N. Sustained protein kinase D activation mediates respiratory syncytial virus-induced airway barrier disruption. J. Virol. 2013, 87, 11088–11095. [Google Scholar] [CrossRef] [PubMed]
- Waldron, R.T.; Rey, O.; Zhukova, E.; Rozengurt, E. Oxidative stress induces protein kinase C-mediated activation loop phosphorylation and nuclear redistribution of protein kinase D. J. Biol. Chem. 2004, 279, 27482–27493. [Google Scholar] [CrossRef] [PubMed]
- Waldron, R.T.; Rozengurt, E. Oxidative stress induces protein kinase D activation in intact cells. Involvement of Src and dependence on protein kinase C. J. Biol. Chem. 2000, 275, 17114–17121. [Google Scholar] [CrossRef] [PubMed]
- Wu, N.H.; Yang, W.; Beineke, A.; Dijkman, R.; Matrosovich, M.; Baumgartner, W.; Thiel, V.; Valentin-Weigand, P.; Meng, F.; Herrler, G. The differentiated airway epithelium infected by influenza viruses maintains the barrier function despite a dramatic loss of ciliated cells. Sci. Rep. 2016, 6, 39668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Short, K.R.; Kasper, J.; van der Aa, S.; Andeweg, A.C.; Zaaraoui-Boutahar, F.; Goeijenbier, M.; Richard, M.; Herold, S.; Becker, C.; Scott, D.P.; et al. Influenza virus damages the alveolar barrier by disrupting epithelial cell tight junctions. Eur. Respir. J. 2016, 47, 954–966. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gotts, J.E.; Abbott, J.; Matthay, M.A. Influenza causes prolonged disruption of the alveolar-capillary barrier in mice unresponsive to mesenchymal stem cell therapy. Am. J. Physiol. Lung Cell. Mol. Physiol. 2014, 307, L395–L406. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, K.; Yajjala, V.K.; Bauer, C.; Talmon, G.A.; Fischer, K.J.; Kielian, T.; Metzger, D.W. Nox2-derived oxidative stress results in inefficacy of antibiotics against post-influenza S. aureus pneumonia. J. Exp. Med. 2016, 213, 1851–1864. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Elia, R.V.; Harrison, K.; Oyston, P.C.; Lukaszewski, R.A.; Clark, G.C. Targeting the “cytokine storm” for therapeutic benefit. Clin. Vaccine Immunol. 2013, 20, 319–327. [Google Scholar] [CrossRef] [PubMed]
- Komaravelli, N.; Casola, A. Respiratory Viral Infections and Subversion of Cellular Antioxidant Defenses. J. Pharmacogenomics Pharmacoproteomics 2014, 5, 1000141. [Google Scholar] [PubMed]
- Uchide, N.; Toyoda, H. Antioxidant therapy as a potential approach to severe influenza-associated complications. Molecules 2011, 16, 2032–2052. [Google Scholar] [CrossRef] [PubMed]
- Sgarbanti, R.; Amatore, D.; Celestino, I.; Marcocci, M.E.; Fraternale, A.; Ciriolo, M.R.; Magnani, M.; Saladino, R.; Garaci, E.; Palamara, A.T.; et al. Intracellular redox state as target for anti-influenza therapy: Are antioxidants always effective? Curr. Top. Med. Chem. 2014, 14, 2529–2541. [Google Scholar] [CrossRef] [PubMed]
- Ali-Ahmad, D.; Bonville, C.A.; Rosenberg, H.F.; Domachowske, J.B. Replication of respiratory syncytial virus is inhibited in target cells generating nitric oxide in situ. Front. Biosci. 2003, 8, a48–a53. [Google Scholar] [PubMed]
- Stark, J.M.; Khan, A.M.; Chiappetta, C.L.; Xue, H.; Alcorn, J.L.; Colasurdo, G.N. Immune and functional role of nitric oxide in a mouse model of respiratory syncytial virus infection. J. Infect. Dis. 2005, 191, 387–395. [Google Scholar] [CrossRef] [PubMed]
- Bazhanov, N.; Ansar, M.; Ivanciuc, T.; Garofalo, R.P.; Casola, A. Hydrogen Sulfide: A Novel Player in Airway Development, Pathophysiology of Respiratory Diseases, and Antiviral Defenses. Am. J. Respir. Cell. Mol. Biol. 2017, 57, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Ma, Y.; Escaffre, O.; Ivanciuc, T.; Komaravelli, N.; Kelley, J.P.; Coletta, C.; Szabo, C.; Rockx, B.; Garofalo, R.P.; et al. Role of hydrogen sulfide in paramyxovirus infections. J. Virol. 2015, 89, 5557–5568. [Google Scholar] [CrossRef] [PubMed]
- Vlahos, R.; Selemidis, S. NADPH oxidases as novel pharmacologic targets against influenza A virus infection. Mol. Pharmacol. 2014, 86, 747–759. [Google Scholar] [CrossRef] [PubMed]
- Vlahos, R.; Stambas, J.; Selemidis, S. Suppressing production of reactive oxygen species (ROS) for influenza A virus therapy. Trends Pharmacol. Sci. 2012, 33, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Geiler, J.; Michaelis, M.; Naczk, P.; Leutz, A.; Langer, K.; Doerr, H.W.; Cinatl, J., Jr. N-acetyl-l-cysteine (NAC) inhibits virus replication and expression of pro-inflammatory molecules in A549 cells infected with highly pathogenic H5N1 influenza A virus. Biochem. Pharmacol. 2010, 79, 413–420. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, P.; Ungheri, D. Synergistic combination of N-acetylcysteine and ribavirin to protect from lethal influenza viral infection in a mouse model. Int. J. Immunopathol. Pharmacol. 2004, 17, 99–102. [Google Scholar] [CrossRef] [PubMed]
- Garozzo, A.; Tempera, G.; Ungheri, D.; Timpanaro, R.; Castro, A. N-acetylcysteine synergizes with oseltamivir in protecting mice from lethal influenza infection. Int. J. Immunopathol. Pharmacol. 2007, 20, 349–354. [Google Scholar] [CrossRef] [PubMed]
- De Flora, S.; Grassi, C.; Carati, L. Attenuation of influenza-like symptomatology and improvement of cell-mediated immunity with long-term N-acetylcysteine treatment. Eur. Respir. J. 1997, 10, 1535–1541. [Google Scholar] [CrossRef] [PubMed]
- Mata, M.; Sarrion, I.; Armengot, M.; Carda, C.; Martinez, I.; Melero, J.A.; Cortijo, J. Respiratory syncytial virus inhibits ciliagenesis in differentiated normal human bronchial epithelial cells: Effectiveness of N-acetylcysteine. PLoS ONE 2012, 7, e48037. [Google Scholar] [CrossRef] [PubMed]
- Wiesener, N.; Zimmer, C.; Jarasch-Althof, N.; Wutzler, P.; Henke, A. Therapy of experimental influenza virus infection with pyrrolidine dithiocarbamate. Med. Microbiol. Immunol. 2011, 200, 115–126. [Google Scholar] [CrossRef] [PubMed]
- Castro, S.M.; Guerrero-Plata, A.; Suarez-Real, G.; Adegboyega, P.A.; Colasurdo, G.N.; Khan, A.M.; Garofalo, R.P.; Casola, A. Antioxidant treatment ameliorates respiratory syncytial virus-induced disease and lung inflammation. Am. J. Respir. Crit. Care Med. 2006, 174, 1361–1369. [Google Scholar] [CrossRef] [PubMed]
- Serkedjieva, J.; Roeva, I.; Angelova, M.; Dolashka, P.; Voelter, W.G. Combined protective effect of a fungal Cu/Zn-containing superoxide dismutase and rimantadine hydrochloride in experimental murine influenza a virus infection. Acta Virol. 2003, 47, 53–56. [Google Scholar] [PubMed]
- Wyde, P.R.; Moore, D.K.; Pimentel, D.M.; Gilbert, B.E.; Nimrod, R.; Panet, A. Recombinant superoxide dismutase (SOD) administered by aerosol inhibits respiratory syncytial virus infection in cotton rats. Antivir. Res. 1996, 31, 173–184. [Google Scholar] [CrossRef]
- Sidwell, R.W.; Huffman, J.H.; Bailey, K.W.; Wong, M.H.; Nimrod, A.; Panet, A. Inhibitory effects of recombinant manganese superoxide dismutase on influenza virus infections in mice. Antimicrob. Agents Chemother. 1996, 40, 2626–2631. [Google Scholar] [PubMed]
- Suliman, H.B.; Ryan, L.K.; Bishop, L.; Folz, R.J. Prevention of influenza-induced lung injury in mice overexpressing extracellular superoxide dismutase. Am. J. Physiol. Lung Cell. Mol. Physiol. 2001, 280, L69–L78. [Google Scholar] [CrossRef] [PubMed]
- Mata, M.; Martinez, I.; Melero, J.A.; Tenor, H.; Cortijo, J. Roflumilast inhibits respiratory syncytial virus infection in human differentiated bronchial epithelial cells. PLoS ONE 2013, 8, e69670. [Google Scholar] [CrossRef] [PubMed]
- Yamaya, M.; Nishimura, H.; Shinya, K.; Hatachi, Y.; Sasaki, T.; Yasuda, H.; Yoshida, M.; Asada, M.; Fujino, N.; Suzuki, T.; et al. Inhibitory effects of carbocisteine on type A seasonal influenza virus infection in human airway epithelial cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2010, 299, L160–168. [Google Scholar] [CrossRef] [PubMed]
- Asada, M.; Yoshida, M.; Hatachi, Y.; Sasaki, T.; Yasuda, H.; Deng, X.; Nishimura, H.; Kubo, H.; Nagatomi, R.; Yamaya, M. l-carbocisteine inhibits respiratory syncytial virus infection in human tracheal epithelial cells. Respir. Physiol. Neurobiol. 2012, 180, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Furuya, A.; Uozaki, M.; Yamasaki, H.; Arakawa, T.; Arita, M.; Koyama, A.H. Antiviral effects of ascorbic and dehydroascorbic acids in vitro. Int. J. Mol. Med. 2008, 22, 541–545. [Google Scholar] [PubMed]
- Kim, Y.; Kim, H.; Bae, S.; Choi, J.; Lim, S.Y.; Lee, N.; Kong, J.M.; Hwang, Y.I.; Kang, J.S.; Lee, W.J. Vitamin C Is an Essential Factor on the Anti-viral Immune Responses through the Production of Interferon-α/β at the Initial Stage of Influenza A Virus (H3N2) Infection. Immune Netw. 2013, 13, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Li, Y.F.; Tang, L.P.; Tsoi, B.; Chen, M.; Chen, H.; Chen, X.M.; Tan, R.R.; Kurihara, H.; He, R.R. A new mechanism of vitamin C effects on A/FM/1/47(H1N1) virus-induced pneumonia in restraint-stressed mice. Biomed. Res. Int. 2015, 2015, 675149. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Yao, D.F.; Ohuchi, M.; Ide, M.; Yano, M.; Okumura, Y.; Kido, H. Ambroxol suppresses influenza-virus proliferation in the mouse airway by increasing antiviral factor levels. Eur. Respir. J. 2002, 19, 952–958. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palamara, A.T.; Nencioni, L.; Aquilano, K.; De Chiara, G.; Hernandez, L.; Cozzolino, F.; Ciriolo, M.R.; Garaci, E. Inhibition of influenza A virus replication by resveratrol. J. Infect. Dis. 2005, 191, 1719–1729. [Google Scholar] [CrossRef] [PubMed]
- Xie, X.H.; Zang, N.; Li, S.M.; Wang, L.J.; Deng, Y.; He, Y.; Yang, X.Q.; Liu, E.M. Resveratrol Inhibits respiratory syncytial virus-induced IL-6 production, decreases viral replication, and downregulates TRIF expression in airway epithelial cells. Inflammation 2012, 35, 1392–1401. [Google Scholar] [CrossRef] [PubMed]
- Zang, N.; Xie, X.; Deng, Y.; Wu, S.; Wang, L.; Peng, C.; Li, S.; Ni, K.; Luo, Y.; Liu, E. Resveratrol-mediated gamma interferon reduction prevents airway inflammation and airway hyperresponsiveness in respiratory syncytial virus-infected immunocompromised mice. J. Virol. 2011, 85, 13061–13068. [Google Scholar] [CrossRef] [PubMed]
- Uchide, N.; Ohyama, K.; Bessho, T.; Yuan, B.; Yamakawa, T. Effect of antioxidants on apoptosis induced by influenza virus infection: Inhibition of viral gene replication and transcription with pyrrolidine dithiocarbamate. Antivir. Res. 2002, 56, 207–217. [Google Scholar] [CrossRef]
- Mileva, M.; Bakalova, R.; Tancheva, L.; Galabov, A.; Ribarov, S. Effect of vitamin E supplementation on lipid peroxidation in blood and lung of influenza virus infected mice. Comp. Immunol. Microbiol. Infect. Dis. 2002, 25, 1–11. [Google Scholar] [CrossRef]
- Hayek, M.G.; Taylor, S.F.; Bender, B.S.; Han, S.N.; Meydani, M.; Smith, D.E.; Eghtesada, S.; Meydani, S.N. Vitamin E supplementation decreases lung virus titers in mice infected with influenza. J. Infect. Dis. 1997, 176, 273–276. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.; Shi, Z.; Huang, H.; Zhu, H.; Zhou, P.; Zhu, H.; Ju, D. Ability of recombinant human catalase to suppress inflammation of the murine lung induced by influenza A. Inflammation 2014, 37, 809–817. [Google Scholar] [CrossRef] [PubMed]
- Shi, X.L.; Shi, Z.H.; Huang, H.; Zhu, H.G.; Zhou, P.; Ju, D. Therapeutic effect of recombinant human catalase on H1N1 influenza-induced pneumonia in mice. Inflammation 2010, 33, 166–172. [Google Scholar] [CrossRef] [PubMed]
- Gaudernak, E.; Seipelt, J.; Triendl, A.; Grassauer, A.; Kuechler, E. Antiviral effects of pyrrolidine dithiocarbamate on human rhinoviruses. J. Virol. 2002, 76, 6004–6015. [Google Scholar] [CrossRef] [PubMed]
- Krenn, B.M.; Holzer, B.; Gaudernak, E.; Triendl, A.; van Kuppeveld, F.J.; Seipelt, J. Inhibition of polyprotein processing and RNA replication of human rhinovirus by pyrrolidine dithiocarbamate involves metal ions. J. Virol. 2005, 79, 13892–13899. [Google Scholar] [CrossRef] [PubMed]
- Yasuda, H.; Yamaya, M.; Sasaki, T.; Inoue, D.; Nakayama, K.; Yamada, M.; Asada, M.; Yoshida, M.; Suzuki, T.; Nishimura, H.; et al. Carbocisteine inhibits rhinovirus infection in human tracheal epithelial cells. Eur. Respir. J. 2006, 28, 51–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamaya, M.; Nishimura, H.; Nadine, L.K.; Ota, C.; Kubo, H.; Nagatomi, R. Ambroxol inhibits rhinovirus infection in primary cultures of human tracheal epithelial cells. Arch. Pharm. Res. 2014, 37, 520–529. [Google Scholar] [CrossRef] [PubMed]
Figure 1.
Sources of reactive oxygen species (ROS) in airway epithelial cells infected with influenza virus (IV, the left cell) or human respiratory syncytial virus (HRSV) and rhinovirus (HRV, the right cell). Cellular events, triggered by ROS production, are detailed in the text. The sources are mainly represented by nicotinamide adenine dinucleotide phosphate oxidases (NADPH oxidases, Nox), Dual oxidase (Duox) and xanthine oxidase (XO). Respiratory viruses also contribute to the production of superoxide anion by Nox2, an enzyme expressed in macrophages and, to a lesser extent, in epithelial cells.
Figure 1.
Sources of reactive oxygen species (ROS) in airway epithelial cells infected with influenza virus (IV, the left cell) or human respiratory syncytial virus (HRSV) and rhinovirus (HRV, the right cell). Cellular events, triggered by ROS production, are detailed in the text. The sources are mainly represented by nicotinamide adenine dinucleotide phosphate oxidases (NADPH oxidases, Nox), Dual oxidase (Duox) and xanthine oxidase (XO). Respiratory viruses also contribute to the production of superoxide anion by Nox2, an enzyme expressed in macrophages and, to a lesser extent, in epithelial cells.

Figure 2.
Mechanisms of cytokine production (cytokine storm) and epithelial barrier disruption by respiratory viruses. Infection leads to the enhanced ROS production that may trigger cell death and subsequent macrophage activation. This activation is accompanied by cytokine production leading to the inflammation and destruction of epithelial cell contacts. Proinflammatory cytokines could also be produced by infected cells via activation of redox-sensitive nuclear factor kappa B (NFκB) pathway that drives transcription of their genes and via activation of NLRP3 inflammasome in ROS-dependent manner that mediates maturation and secretion of cytokines. Disruption of epithelial barrier results in the increased susceptibility to bacterial infection.
Figure 2.
Mechanisms of cytokine production (cytokine storm) and epithelial barrier disruption by respiratory viruses. Infection leads to the enhanced ROS production that may trigger cell death and subsequent macrophage activation. This activation is accompanied by cytokine production leading to the inflammation and destruction of epithelial cell contacts. Proinflammatory cytokines could also be produced by infected cells via activation of redox-sensitive nuclear factor kappa B (NFκB) pathway that drives transcription of their genes and via activation of NLRP3 inflammasome in ROS-dependent manner that mediates maturation and secretion of cytokines. Disruption of epithelial barrier results in the increased susceptibility to bacterial infection.


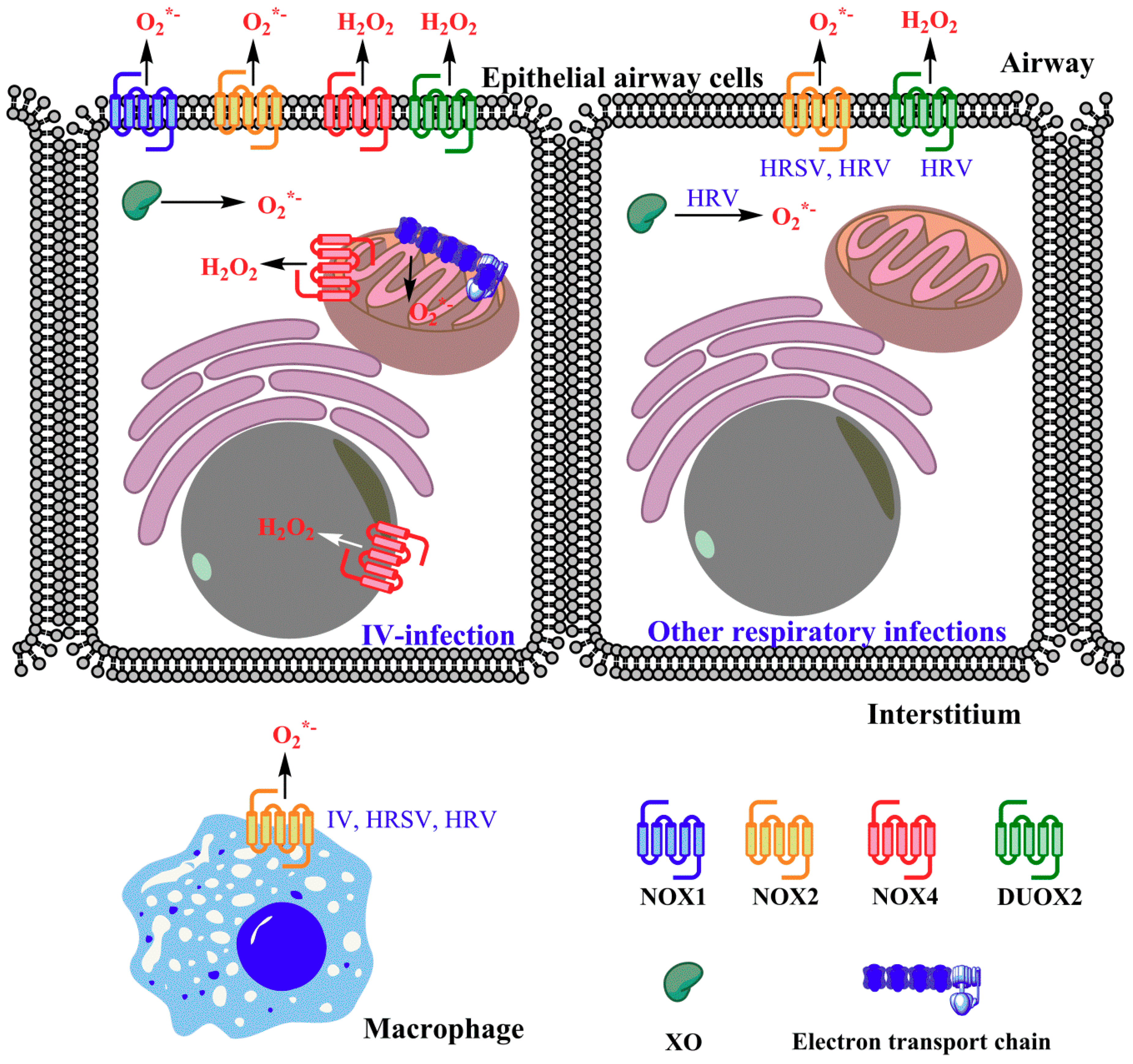{kind=link}
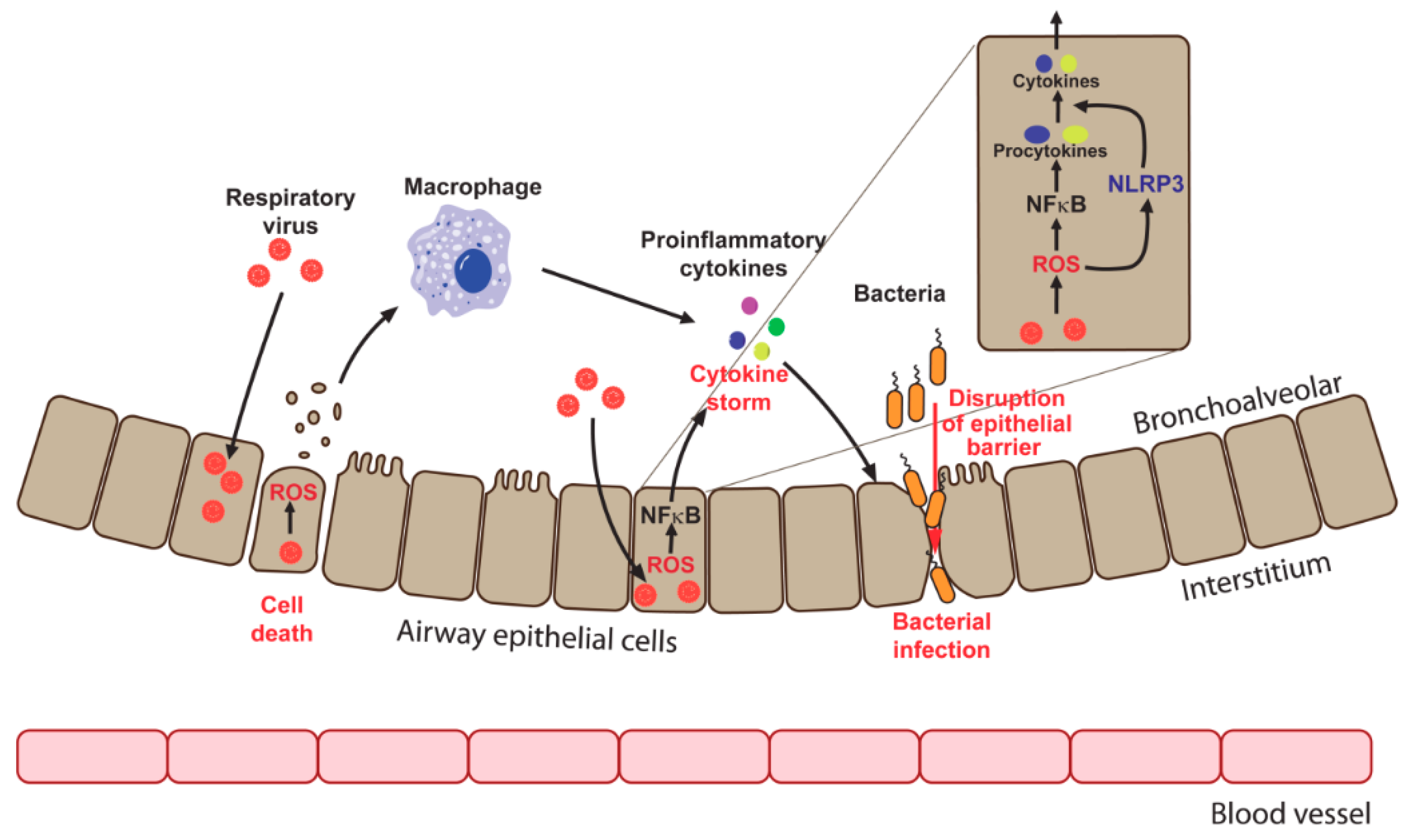{kind=link}
Table 1.
Antiviral activity of antioxidants and antioxidant enzymes towards respiratory viral infections.
Table 1.
Antiviral activity of antioxidants and antioxidant enzymes towards respiratory viral infections.
| Antioxidant | Influenza Virus | Human Respiratory Syncytial Virus | ||||||
|---|---|---|---|---|---|---|---|---|
| Model System | Antiviral Activity | Synergism with | Ref | Model System | Antiviral Activity | Synergism with | Ref | |
| N-acetylcysteine (NAC) | Cells Mice Human | Yes | Ribavirin, oseltamivir | [185,186,187,188] | Cells | ? * | NA | [31,32,143,189] |
| Pyrrolidine dithiocarbamate (PDTC) | Cells Mice | Yes | ND ** | [132,190] | ND | ND | ND | ND |
| Glutathione | Cells Mice | Yes | ND | [108,109] | Cells | Yes | ND | [32] |
| Butylated hydroxyanisole (BHA) | - | - | - | - | Cells Mice | No | ND | [31,191] |
| SOD1 | Cells Mice | No | Rimantadine | [74,95,192] | Rats | Yes | ND | [193] |
| SOD2 | Mice | No | Ribavirin | [194] | Rats | Yes | ND | [193] |
| SOD3 | Mice | No | ND | [195] | ND | ND | ND | ND |
| Roflumilast | - | - | - | - | Cells | Yes | NA | [196] |
| Carbocisteine | Cells | Yes | ND | [197] | Cells | Yes | NA | [198] |
| Ascorbic acid | Cells Mice | Yes | ND | [199,200,201] | ND | ND | ND | ND |
| Ambroxol | Mice | Yes | ND | [202] | ND | ND | ND | ND |
| Resveratrol | Cells Mice | Yes | ND | [203] | Cells Mice | Yes | ND | [204,205] |
| Vitamin E or Trolox | Cells Mice | ? *** | ND | [206,207,208] | ND | ND | ND | ND |
| Catalase | Mice | Yes | ND | [209,210] | Cells | No | ND | [32] |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Khomich, O.A.; Kochetkov, S.N.; Bartosch, B.; Ivanov, A.V. Redox Biology of Respiratory Viral Infections. Viruses 2018, 10, 392. https://doi.org/10.3390/v10080392
AMA Style
Khomich OA, Kochetkov SN, Bartosch B, Ivanov AV. Redox Biology of Respiratory Viral Infections. Viruses. 2018; 10(8):392. https://doi.org/10.3390/v10080392
Chicago/Turabian StyleKhomich, Olga A., Sergey N. Kochetkov, Birke Bartosch, and Alexander V. Ivanov. 2018. "Redox Biology of Respiratory Viral Infections" Viruses 10, no. 8: 392. https://doi.org/10.3390/v10080392
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.