Remnants of an Ancient Deltaretrovirus in the Genomes of Horseshoe Bats (Rhinolophidae)

 , ,
, ,  , , , and
, , , and
Abstract
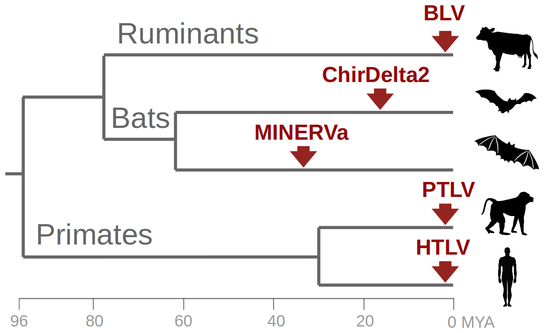
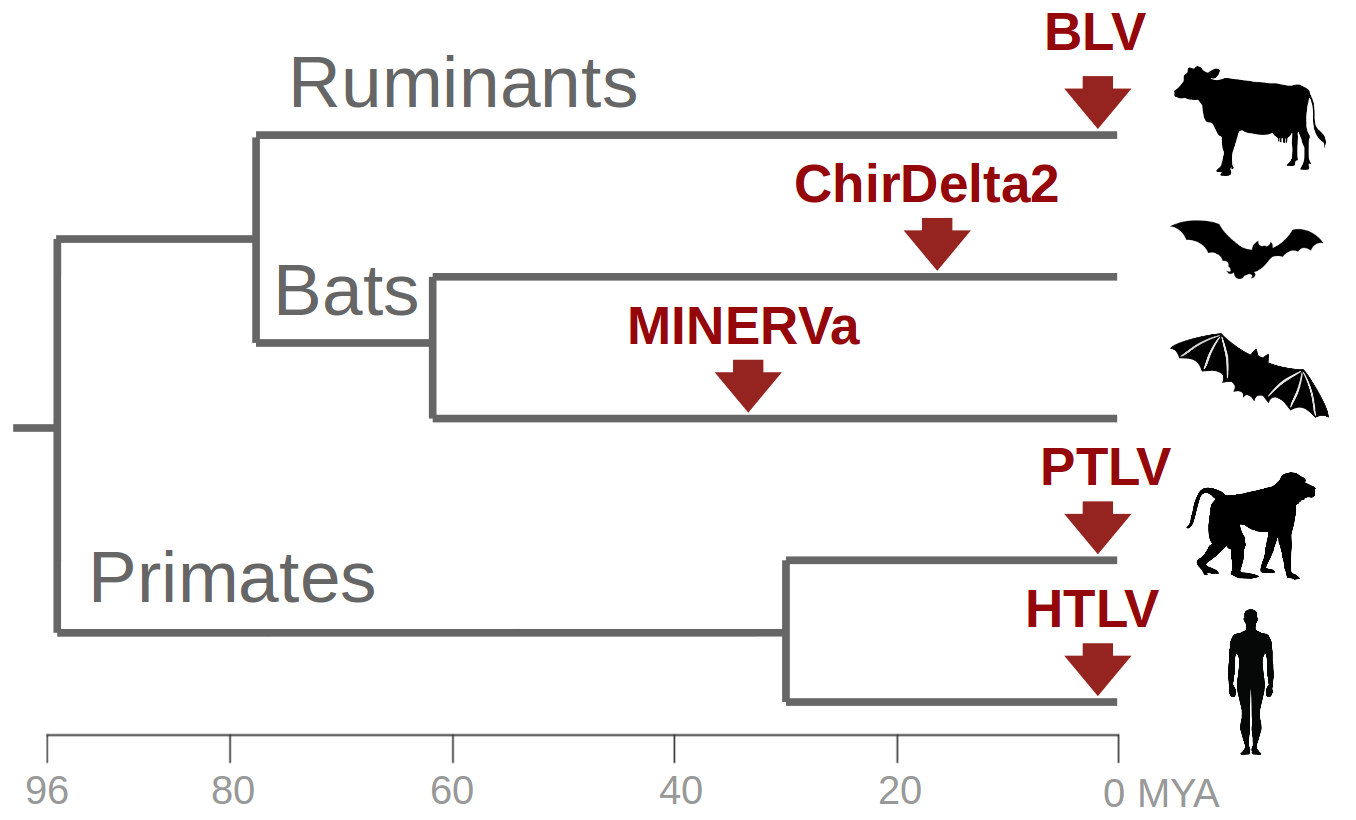
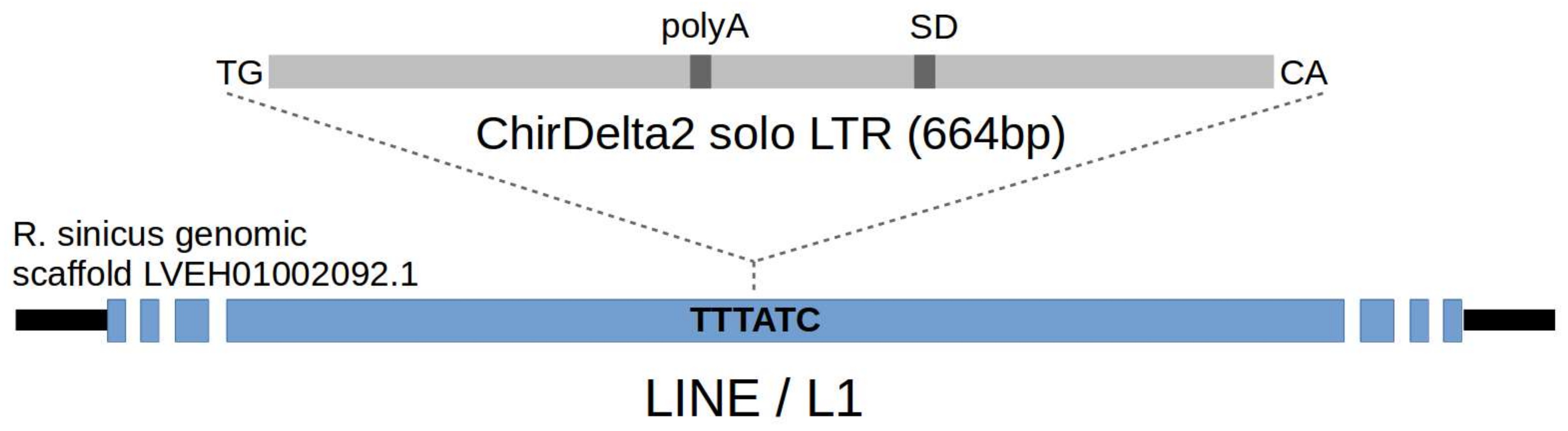
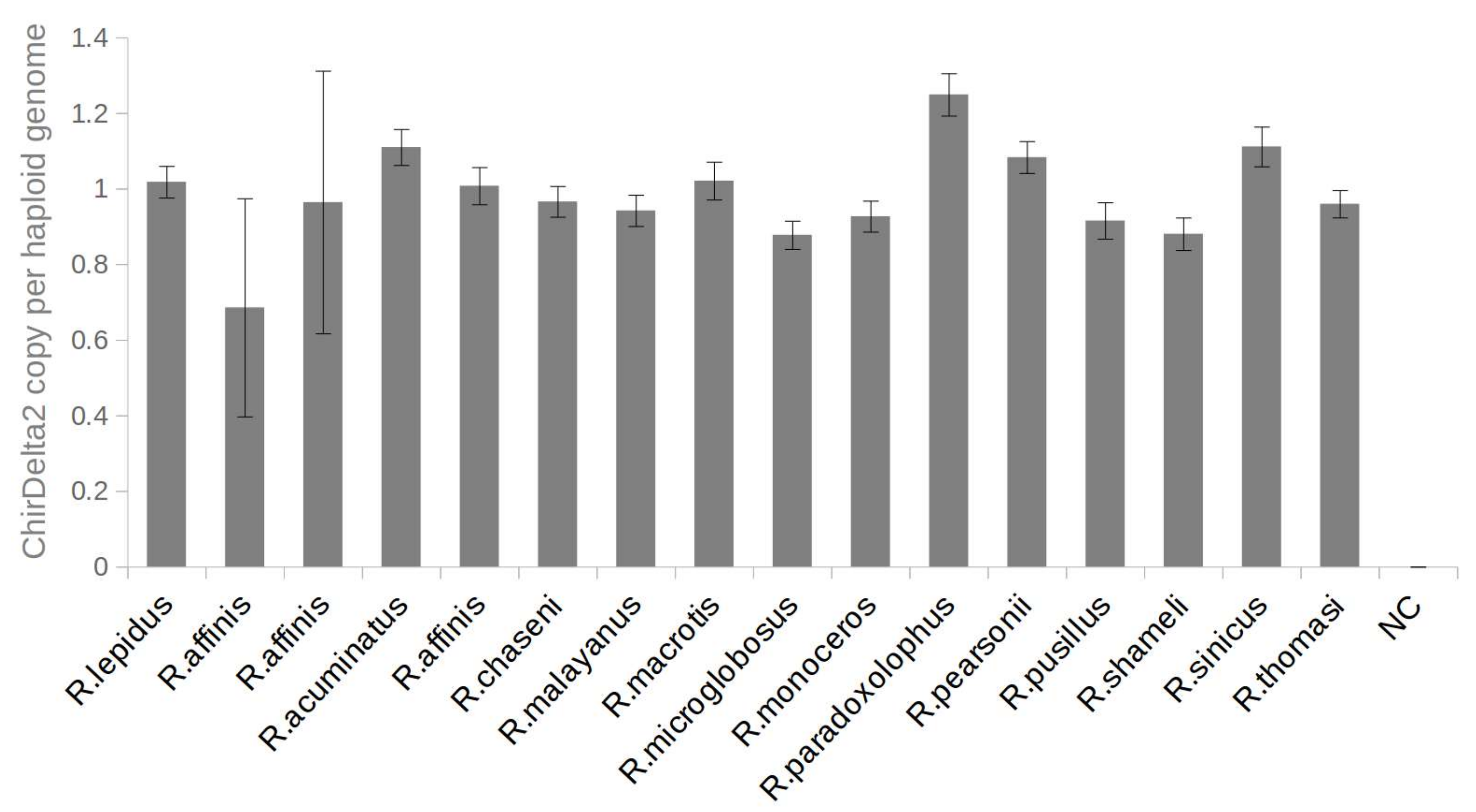
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Analysis of Sequencing Data
2.2. Genomic DNA Samples
2.3. PCR and Sequencing
2.4. Provirus Copy-Number Analysis
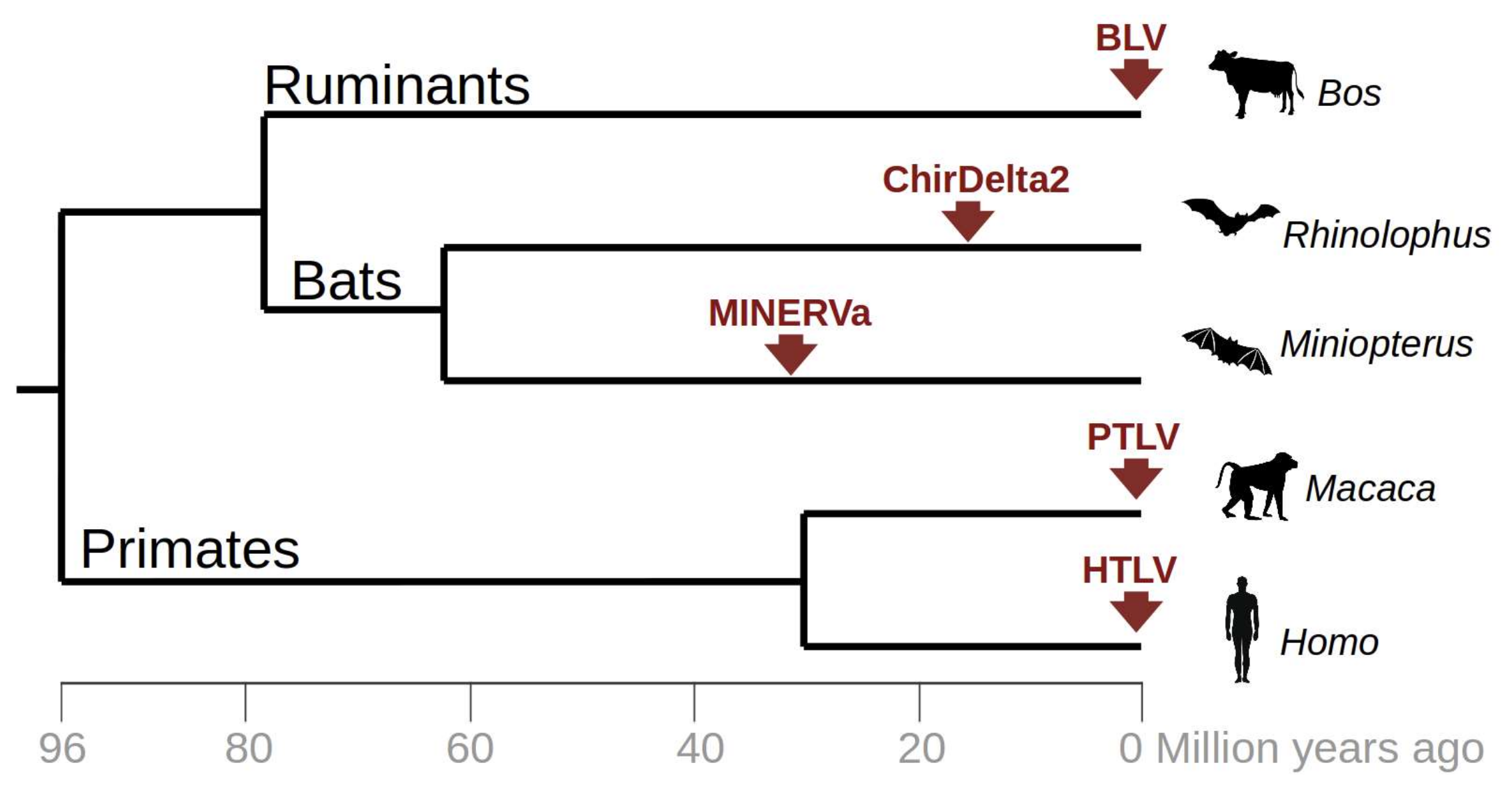
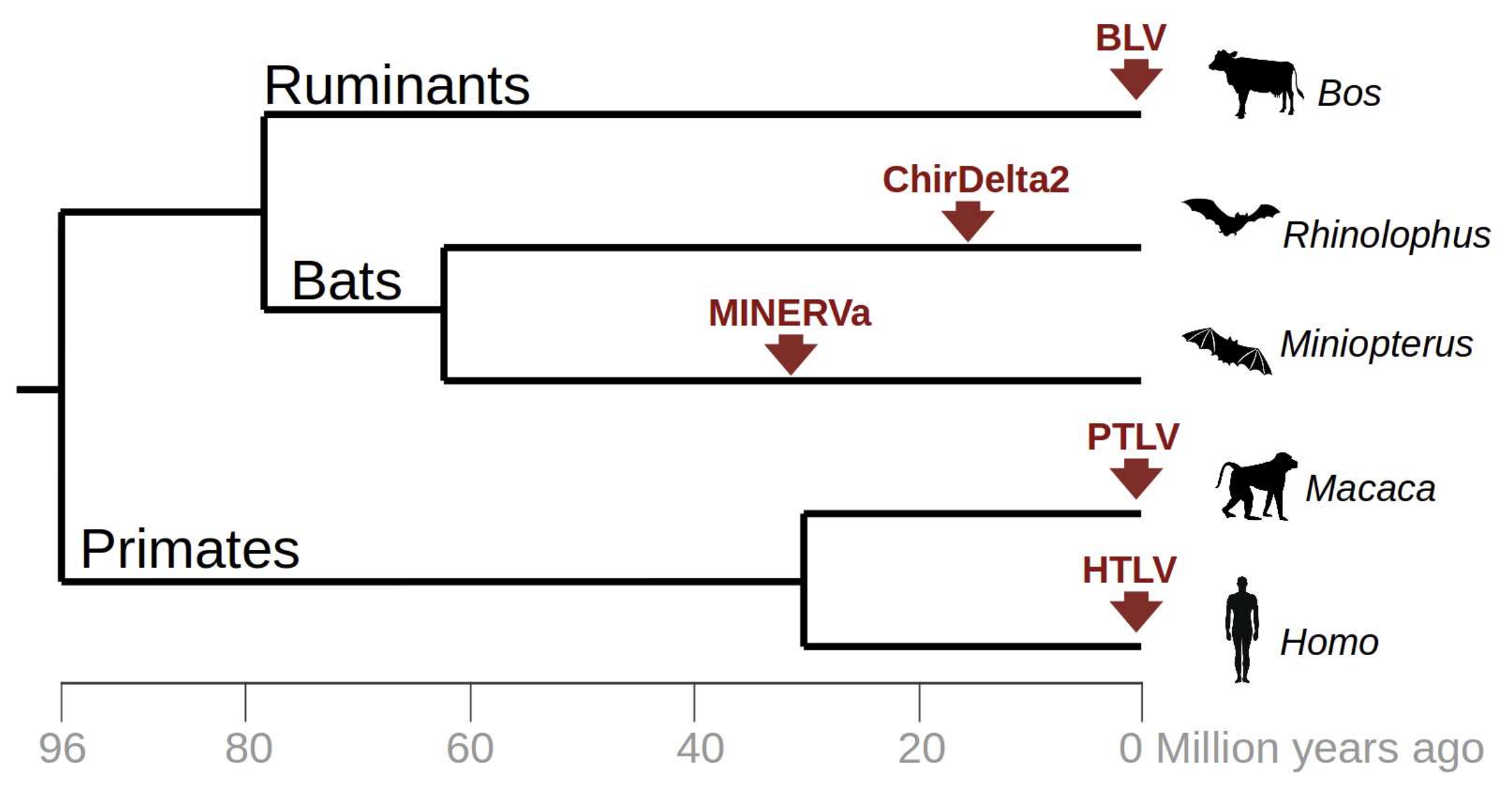
3. Results
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Johnson, W.E. Endogenous Retroviruses in the Genomics Era. Annu. Rev. Virol. 2015, 2, 135–159. [Google Scholar] [CrossRef] [PubMed]
- Greenwood, A.D.; Ishida, Y.; O’Brien, S.P.; Roca, A.L.; Eiden, M.V. Transmission, Evolution, and Endogenization: Lessons Learned from Recent Retroviral Invasions. Microbiol. Mol. Biol. Rev. 2018, 82. [Google Scholar] [CrossRef] [PubMed]
- Farkašová, H.; Hron, T.; Pačes, J.; Hulva, P.; Benda, P.; Gifford, R.J.; Elleder, D. Discovery of an endogenous Deltaretrovirus in the genome of long-fingered bats (Chiroptera: Miniopteridae). Proc. Natl. Acad. Sci. USA 2017, 114, 3145–3150. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.; Zaretskaya, I.; Raytselis, Y.; Merezhuk, Y.; McGinnis, S.; Madden, T.L. NCBI BLAST: A better web interface. Nucleic Acids Res. 2008, 36, W5–W9. [Google Scholar] [CrossRef] [PubMed]
- Serrao, E.; Ballandras-Colas, A.; Cherepanov, P.; Maertens, G.N.; Engelman, A.N. Key determinants of target DNA recognition by retroviral intasomes. Retrovirology 2015, 12, 39. [Google Scholar] [CrossRef] [PubMed]
- Bao, W.; Kojima, K.K.; Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA 2015, 6, 11. [Google Scholar] [CrossRef] [PubMed]
- Belshaw, R.; Watson, J.; Katzourakis, A.; Howe, A.; Woolven-Allen, J.; Burt, A.; Tristem, M. Rate of recombinational deletion among human endogenous retroviruses. J. Virol. 2007, 81, 9437–9442. [Google Scholar] [CrossRef] [PubMed]
- Benachenhou, F.; Blikstad, V.; Blomberg, J. The phylogeny of orthoretroviral long terminal repeats (LTRs). Gene 2009, 448, 134–138. [Google Scholar] [CrossRef] [PubMed]
- Kypr, J.; Mrázek, J.; Reich, J. Nucleotide composition bias and CpG dinucleotide content in the genomes of HIV and HTLV 1/2. Biochim. Biophys. Acta 1989, 1009, 280–282. [Google Scholar] [CrossRef]
- Johnson, W.E.; Coffin, J.M. Constructing primate phylogenies from ancient retrovirus sequences. Proc. Natl. Acad. Sci. USA 1999, 96, 10254–10260. [Google Scholar] [CrossRef] [PubMed]
- Sagata, N.; Yasunaga, T.; Tsuzuku-Kawamura, J.; Ohishi, K.; Ogawa, Y.; Ikawa, Y. Complete nucleotide sequence of the genome of Bovine leukemia virus: Its evolutionary relationship to other retroviruses. Proc. Natl. Acad. Sci. USA 1985, 82, 677–681. [Google Scholar] [CrossRef] [PubMed]
- Dool, S.E.; Puechmaille, S.J.; Foley, N.M.; Allegrini, B.; Bastian, A.; Mutumi, G.L.; Maluleke, T.G.; Odendaal, L.J.; Teeling, E.C.; Jacobs, D.S. Nuclear introns outperform mitochondrial DNA in inter-specific phylogenetic reconstruction: Lessons from Horseshoe bats (Rhinolophidae: Chiroptera). Mol. Phylogenet. Evol. 2016, 97, 196–212. [Google Scholar] [CrossRef] [PubMed]
- Stoffberg, S.; Jacobs, D.S.; Mackie, I.J.; Matthee, C.A. Molecular phylogenetics and historical biogeography of Rhinolophus bats. Mol. Phylogenet. Evol. 2010, 54, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Soisook, P.; Struebig, M.J.; Noerfahmy, S.; Bernard, H.; Maryanto, I.; Chen, S.-F.; Rossiter, S.J.; Kuo, H.-C.; Deshpande, K.; Bates, P.J.J.; et al. Description of a New Species of the Rhinolophus trifoliatus-Group (Chiroptera: Rhinolophidae) from Southeast Asia. Acta Chiropterol. 2015, 17, 21–36. [Google Scholar] [CrossRef]
- Soisook, P.; Bumrungsri, S.; Satasook, C.; Thong, V.D.; Bu, S.S.H.; Harrison, D.L.; Bates, P.J.J. A taxonomic review of Rhinolophus stheno and R. malayanus (Chiroptera: Rhinolophidae) from continental Southeast Asia: An evaluation of echolocation call frequency in discriminating between cryptic species. Acta Chiropterol. 2008, 10, 221–242. [Google Scholar] [CrossRef]
- Volleth, M.; Loidl, J.; Mayer, F.; Yong, H.-S.; Müller, S.; Heller, K.-G. Surprising Genetic Diversity in Rhinolophus luctus (Chiroptera: Rhinolophidae) from Peninsular Malaysia: Description of a New Species Based on Genetic and Morphological Characters. Acta Chiropterol. 2015, 17, 1–20. [Google Scholar] [CrossRef]
- Camin, J.H.; Sokal, R.R. A Method for Deducing Branching Sequences in Phylogeny. Evolution 1965, 19, 311. [Google Scholar] [CrossRef]
- Madsen, O.; Scally, M.; Douady, C.J.; Kao, D.J.; DeBry, R.W.; Adkins, R.; Amrine, H.M.; Stanhope, M.J.; de Jong, W.W.; Springer, M.S. Parallel adaptive radiations in two major clades of placental mammals. Nature 2001, 409, 610–614. [Google Scholar] [CrossRef] [PubMed]
- Nishihara, H.; Hasegawa, M.; Okada, N. Pegasoferae, an unexpected mammalian clade revealed by tracking ancient retroposon insertions. Proc. Natl. Acad. Sci. USA 2006, 103, 9929–9934. [Google Scholar] [CrossRef] [PubMed]
- Simmons, N.B. Evolution. An Eocene big bang for bats. Science 2005, 307, 527–528. [Google Scholar] [CrossRef] [PubMed]
- Hayman, D.T.S. Bats as Viral Reservoirs. Ann. Rev. Virol. 2016, 3, 77–99. [Google Scholar] [CrossRef] [PubMed]
- Omatsu, T.; Watanabe, S.; Akashi, H.; Yoshikawa, Y. Biological characters of bats in relation to natural reservoir of emerging viruses. Comp. Immunol. Microbiol. Infect. Dis. 2007, 30, 357–374. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Tachedjian, M.; Wynne, J.W.; Boyd, V.; Cui, J.; Smith, I.; Cowled, C.; Ng, J.H.J.; Mok, L.; Michalski, W.P.; et al. Contraction of the type I IFN locus and unusual constitutive expression of IFN-α in bats. Proc. Natl. Acad. Sci. USA 2016, 113, 2696–2701. [Google Scholar] [CrossRef] [PubMed]
- Teeling, E.C.; Vernes, S.C.; Dávalos, L.M.; Ray, D.A.; Gilbert, M.T.P.; Myers, E. Bat1K Consortium Bat Biology, Genomes, and the Bat1K Project: To Generate Chromosome-Level Genomes for All Living Bat Species. Annu. Rev. Anim. Biosci. 2018, 6, 23–46. [Google Scholar] [CrossRef] [PubMed]



© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hron, T.; Farkašová, H.; Gifford, R.J.; Benda, P.; Hulva, P.; Görföl, T.; Pačes, J.; Elleder, D. Remnants of an Ancient Deltaretrovirus in the Genomes of Horseshoe Bats (Rhinolophidae). Viruses 2018, 10, 185. https://doi.org/10.3390/v10040185
Hron T, Farkašová H, Gifford RJ, Benda P, Hulva P, Görföl T, Pačes J, Elleder D. Remnants of an Ancient Deltaretrovirus in the Genomes of Horseshoe Bats (Rhinolophidae). Viruses. 2018; 10(4):185. https://doi.org/10.3390/v10040185
Chicago/Turabian StyleHron, Tomáš, Helena Farkašová, Robert J. Gifford, Petr Benda, Pavel Hulva, Tamás Görföl, Jan Pačes, and Daniel Elleder. 2018. "Remnants of an Ancient Deltaretrovirus in the Genomes of Horseshoe Bats (Rhinolophidae)" Viruses 10, no. 4: 185. https://doi.org/10.3390/v10040185