Toll-Like Receptor 3 Is Involved in Detection of Enterovirus A71 Infection and Targeted by Viral 2A Protease

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Ethics Statement
2.2. Cells, Viruses, and Reagents
2.3. Plasmids
2.4. RNA Interference
2.5. Luciferase Reporter Assay and ELISA
2.6. RT-PCR and Quantitative RT-PCR
2.7. Preparation and Enzyme Treatment of EV-A71-Infected RNA
2.8. Plaque Assay
2.9. In Vitro Cleavage Assay
2.10. Statistical Analysis
3. Results
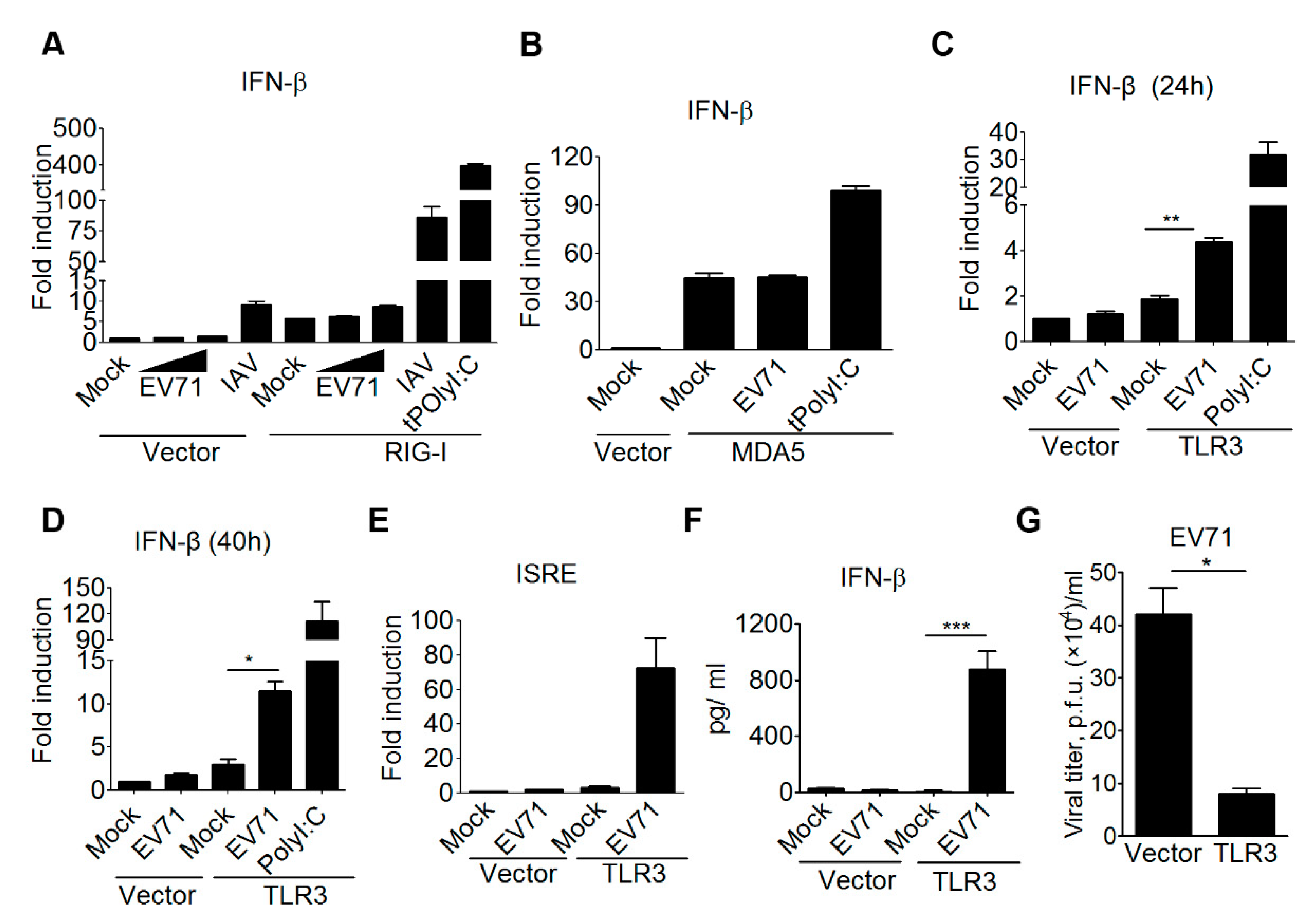
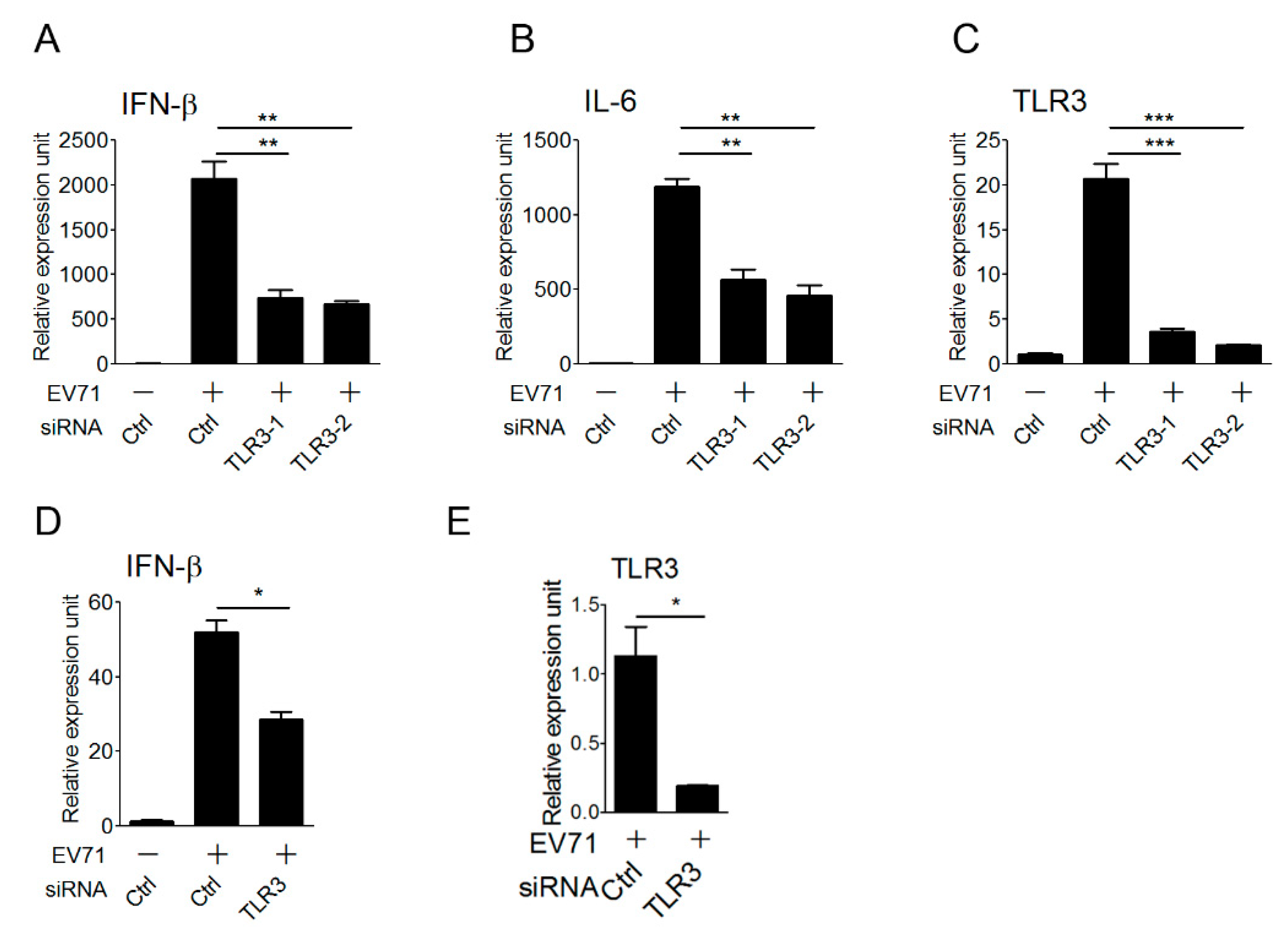
3.1. TLR3 is Involved in Detecting EV-A71 Infection to Induce Type I IFN Antiviral Responses
3.2. Double-Stranded Viral RNA Derived from EV-A71 Replication is Essential for TLR3 Detection
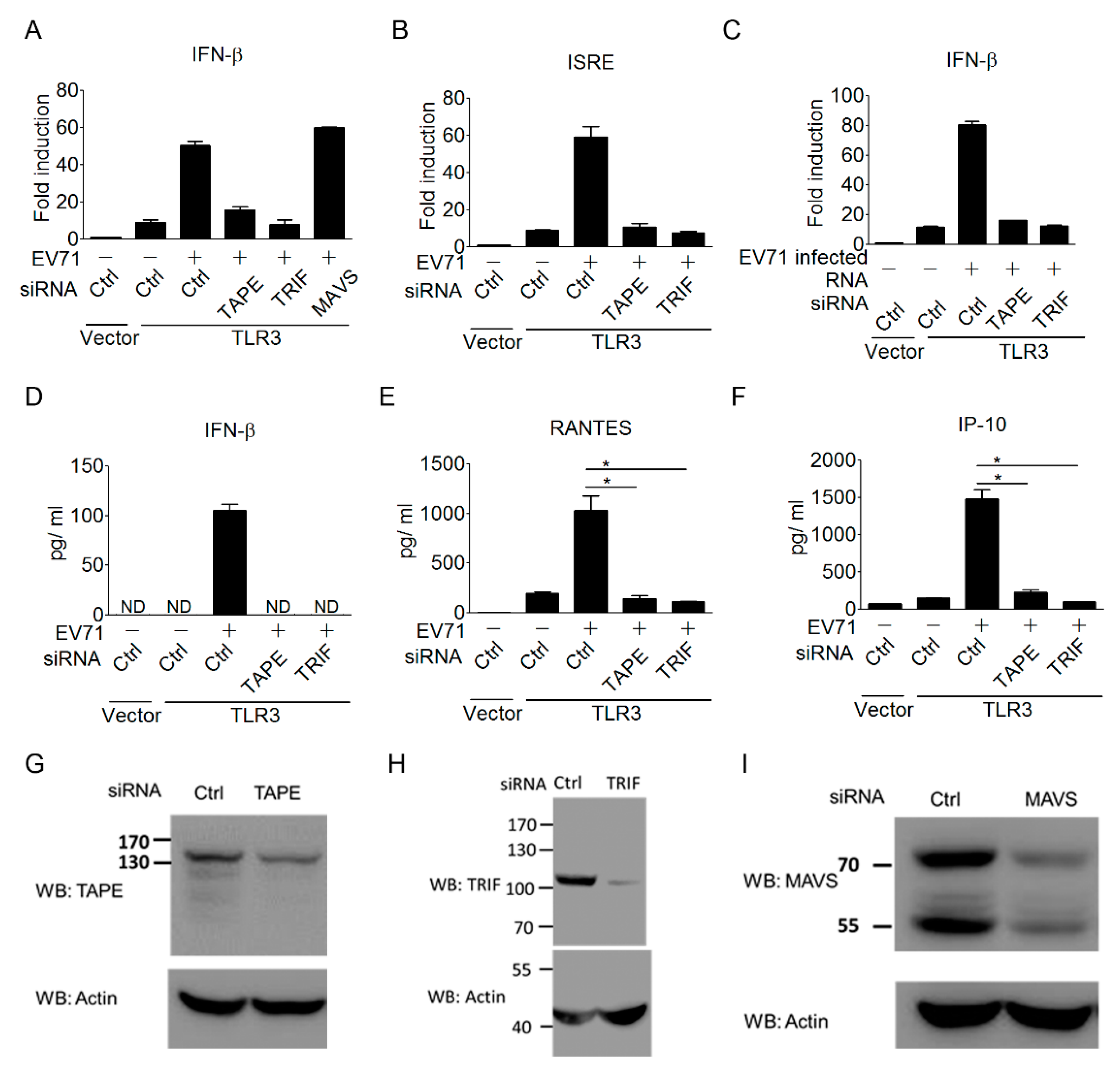
3.3. The TLR3 Signaling Pathway is Required for Triggering Type I IFN Response to EV-A71 Infection
3.4. TLR3 Detects EV-A71 Infection To Induce Type I IFN Response in Primary Myeloid Cells
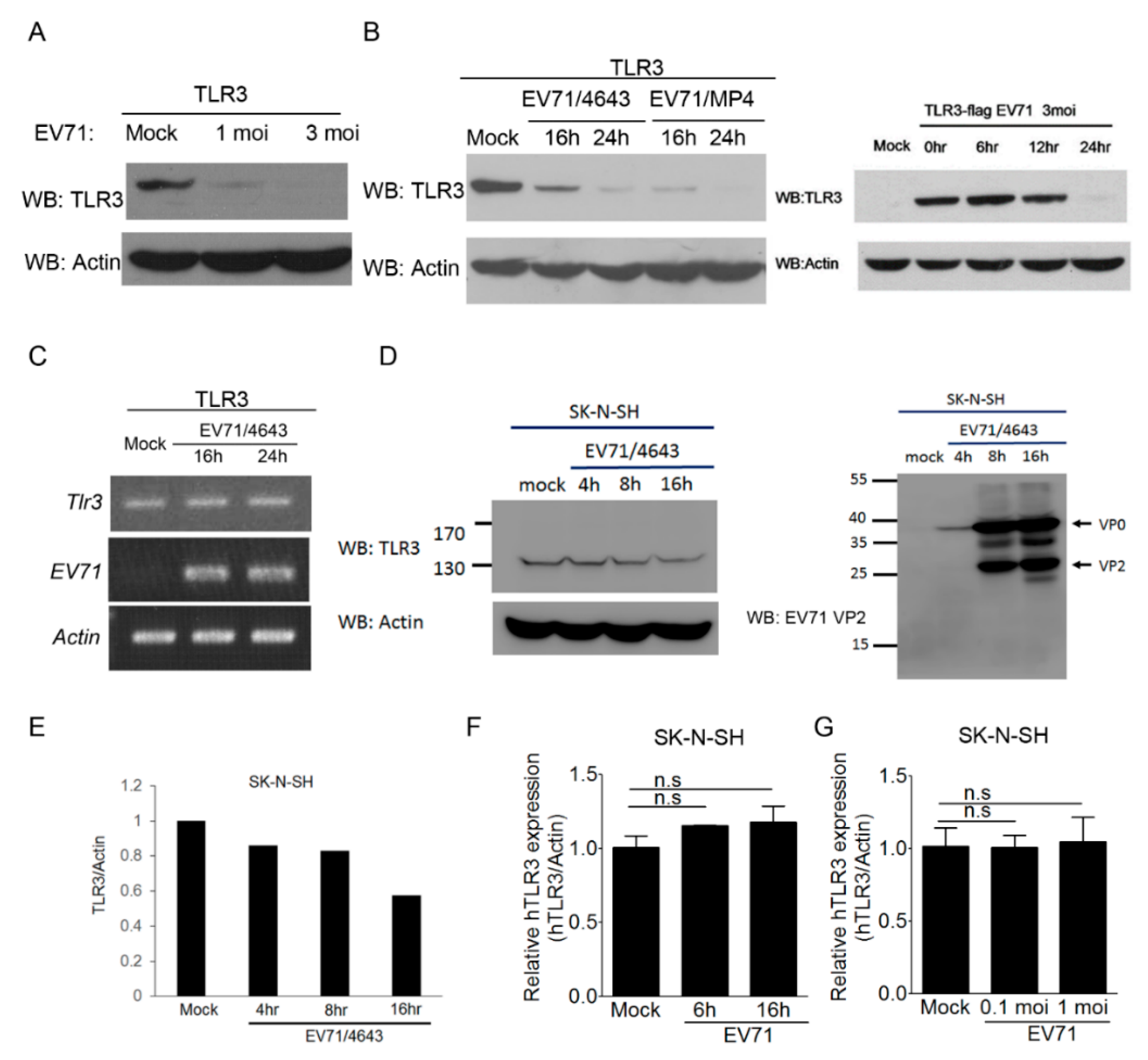
3.5. EV-A71 Infection Decreases the TLR3 Protein Level
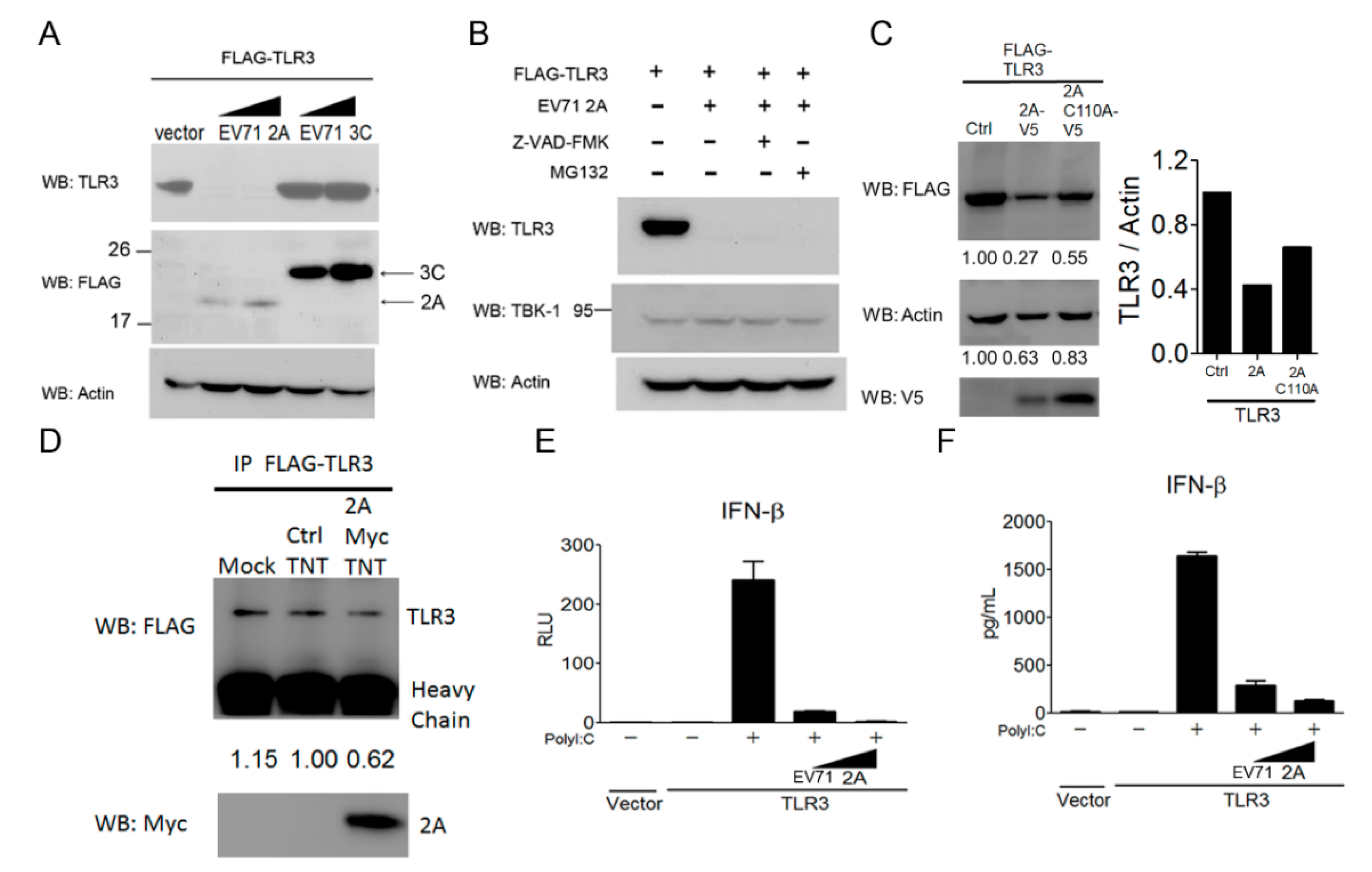
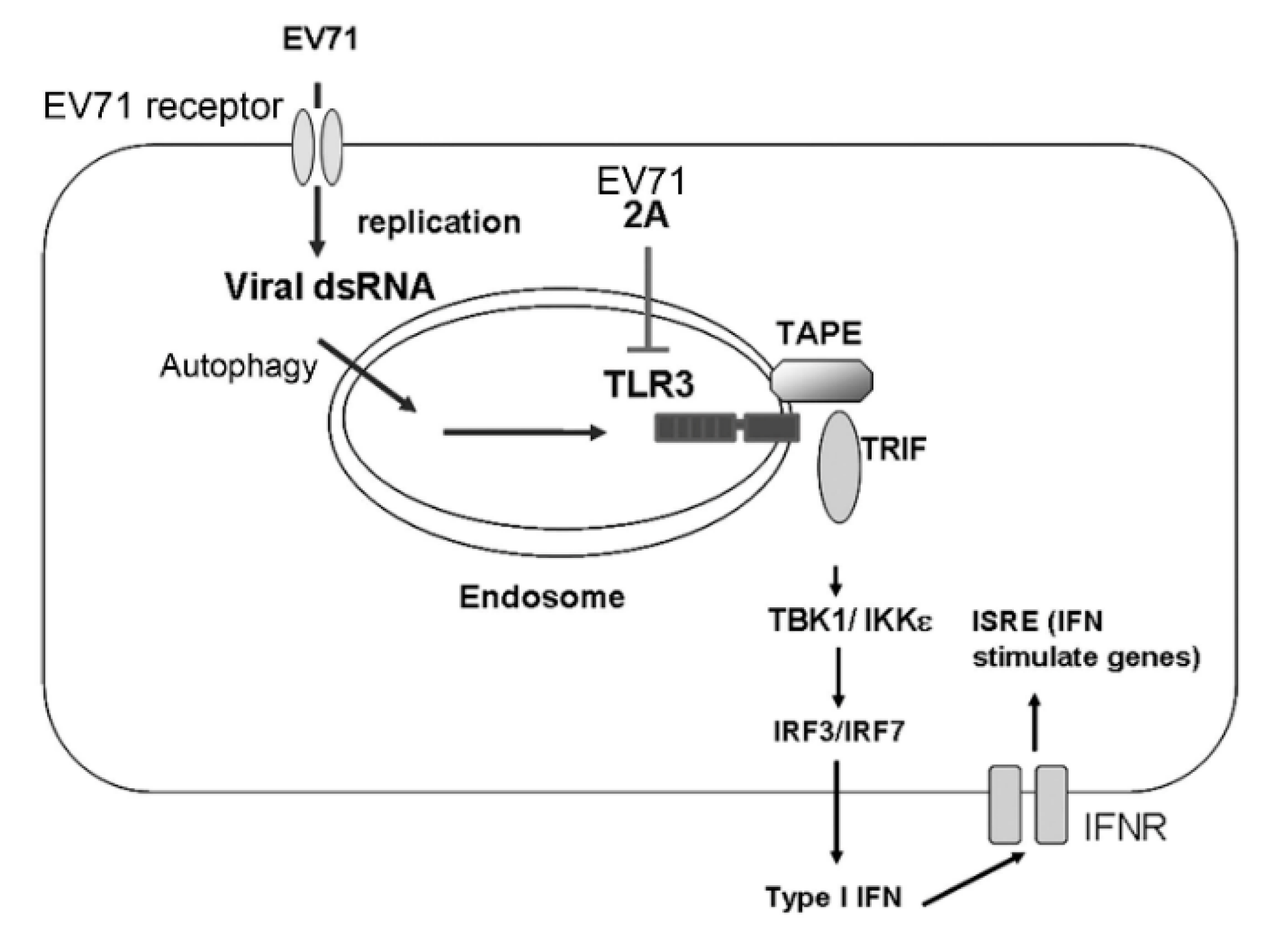
3.6. EV-A71 Protease 2A Targets TLR3 to Inhibit Type I IFN Induction
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Bek, E.J.; McMinn, P.C. The Pathogenesis and Prevention of Encephalitis due to Human Enterovirus 71. Curr. Infect. Dis. Rep. 2012, 14, 397–407. [Google Scholar] [CrossRef] [PubMed]
- Yip, C.C.; Lau, S.K.; Woo, P.C.; Yuen, K.Y. Human enterovirus 71 epidemics: What’s next? Emerg. Health Threats. 2013, 6, 19780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mao, Q.Y.; Wang, Y.; Bian, L.; Xu, M.; Liang, Z. EV71 vaccine, a new tool to control outbreaks of hand, foot and mouth disease (HFMD). Expert Rev. Vaccines 2016, 15, 599–606. [Google Scholar] [CrossRef] [PubMed]
- Kung, Y.A.; Hung, C.T.; Liu, Y.C.; Shih, S.R. Update on the development of enterovirus 71 vaccines. Exp. Opin. Biol. Ther. 2014, 14, 1455–1464. [Google Scholar] [CrossRef] [PubMed]
- Yamayoshi, S.; Yamashita, Y.; Li, J.; Hanagata, N.; Minowa, T.; Takemura, T.; Koike, S. Scavenger receptor B2 is a cellular receptor for enterovirus 71. Nat. Med. 2009, 15, 798–801. [Google Scholar] [CrossRef] [PubMed]
- Nishimura, Y.; Shimojima, M.; Tano, Y.; Miyamura, T.; Wakita, T.; Shimizu, H. Human P-selectin glycoprotein ligand-1 is a functional receptor for enterovirus 71. Nat. Med. 2009, 15, 794–797. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.W.; Yu, S.L.; Shao, H.Y.; Lin, H.Y.; Liu, C.C.; Hsiao, K.N.; Chitra, E.; Tsou, Y.L.; Chang, H.W.; Sia, C.; et al. Human SCARB2 transgenic mice as an infectious animal model for enterovirus 71. PLoS ONE 2013, 8, e57591. [Google Scholar] [CrossRef]
- Fujii, K.; Nagata, N.; Sato, Y.; Ong, K.C.; Wong, K.T.; Yamayoshi, S.; Shimanuki, M.; Shitara, H.; Taya, C.; Koike, S. Transgenic mouse model for the study of enterovirus 71 neuropathogenesis. Proc. Natl. Acad. Sci. USA 2013, 110, 14753–14758. [Google Scholar] [CrossRef] [Green Version]
- Tan, C.W.; Poh, C.L.; Sam, I.C.; Chan, Y.F. Enterovirus 71 uses cell surface heparan sulfate glycosaminoglycan as an attachment receptor. J. Virol. 2013, 87, 611–620. [Google Scholar] [CrossRef]
- Yang, B.; Chuang, H.; Yang, K.D. Sialylated glycans as receptor and inhibitor of enterovirus 71 infection to DLD-1 intestinal cells. Virol. J. 2009, 6, 141. [Google Scholar] [CrossRef] [Green Version]
- Su, P.Y.; Wang, Y.F.; Huang, S.W.; Lo, Y.C.; Wang, Y.H.; Wu, S.R.; Shieh, D.B.; Chen, S.H.; Wang, J.R.; Lai, M.D.; et al. Cell surface nucleolin facilitates enterovirus 71 binding and infection. J. Virol. 2015, 89, 4527–4538. [Google Scholar] [CrossRef]
- Yeung, M.L.; Jia, L.; Yip, C.C.Y.; Chan, J.F.W.; Teng, J.L.L.; Chan, K.H.; Cai, J.P.; Zhang, C.; Zhang, A.J.; Wong, W.M.; et al. Human tryptophanyl-tRNA synthetase is an IFN-gamma-inducible entry factor for Enterovirus. J. Clin. Investig. 2018. [Google Scholar] [CrossRef] [PubMed]
- Yamayoshi, S.; Fujii, K.; Koike, S. Receptors for enterovirus 71. Emerg. Microbes Infect. 2014, 3, e53. [Google Scholar] [CrossRef] [PubMed]
- Abzug, M.J. The enteroviruses: Problems in need of treatments. J. Infect. 2014, 68, 1081. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.H.; Shenoy, A.R.; Kumar, P.; Bradfield, C.J.; MacMicking, J.D. IFN-inducible GTPases in host cell defense. Cell. Host Microbe 2012, 12, 432–444. [Google Scholar] [CrossRef] [PubMed]
- Sadler, A.J.; Williams, B.R. Interferon-inducible antiviral effectors. Nat. Rev. Immunol. 2008, 8, 559–568. [Google Scholar] [CrossRef] [Green Version]
- Liu, M.L.; Lee, Y.P.; Wang, Y.F.; Lei, H.Y.; Liu, C.C.; Wang, S.M.; Su, I.J.; Wang, J.R.; Yeh, T.M.; Chen, S.H.; et al. Type I interferons protect mice against enterovirus 71 infection. J. Gen. Virol. 2005, 86, 3263–3269. [Google Scholar] [CrossRef] [Green Version]
- Meng, T.; Kwang, J. Attenuation of human enterovirus 71 high-replication-fidelity variants in AG129 mice. J. Virol. 2014, 88, 5803–5815. [Google Scholar] [CrossRef] [PubMed]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Goubau, D.; Deddouche, S.; Reis, E.S.C. Cytosolic sensing of viruses. Immunity 2013, 38, 855–869. [Google Scholar] [CrossRef]
- Kato, H.; Takeuchi, O.; Sato, S.; Yoneyama, M.; Yamamoto, M.; Matsui, K.; Uematsu, S.; Jung, A.; Kawai, T.; Ishii, K.J.; et al. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 2006, 441, 101–105. [Google Scholar] [CrossRef]
- Wang, J.P.; Cerny, A.; Asher, D.R.; Kurt-Jones, E.A.; Bronson, R.T.; Finberg, R.W. MDA5 and MAVS mediate type I interferon responses to coxsackie B virus. J. Virol. 2010, 84, 254–260. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; Hato, S.V.; Langereis, M.A.; Zoll, J.; Virgen-Slane, R.; Peisley, A.; Hur, S.; Semler, B.L.; van Rij, R.P.; van Kuppeveld, F.J. MDA5 detects the double-stranded RNA replicative form in picornavirus-infected cells. Cell. Rep. 2012, 2, 1187–1196. [Google Scholar] [CrossRef] [Green Version]
- Kuo, R.L.; Kao, L.T.; Lin, S.J.; Wang, R.Y.; Shih, S.R. MDA5 plays a crucial role in enterovirus 71 RNA-mediated IRF3 activation. PLoS ONE 2013, 8, e63431. [Google Scholar] [CrossRef] [PubMed]
- Oshiumi, H.; Okamoto, M.; Fujii, K.; Kawanishi, T.; Matsumoto, M.; Koike, S.; Seya, T. The TLR3/TICAM-1 pathway is mandatory for innate immune responses to poliovirus infection. J. Immunol. 2011, 187, 5320–5327. [Google Scholar] [CrossRef] [PubMed]
- Abe, Y.; Fujii, K.; Nagata, N.; Takeuchi, O.; Akira, S.; Oshiumi, H.; Matsumoto, M.; Seya, T.; Koike, S. The toll-like receptor 3-mediated antiviral response is important for protection against poliovirus infection in poliovirus receptor transgenic mice. J. Virol. 2012, 86, 185–194. [Google Scholar] [CrossRef]
- Feng, Q.; Langereis, M.A.; van Kuppeveld, F.J. Induction and suppression of innate antiviral responses by picornaviruses. Cytokine Growth Factor Rev. 2014. [Google Scholar] [CrossRef]
- Lei, X.; Liu, X.; Ma, Y.; Sun, Z.; Yang, Y.; Jin, Q.; He, B.; Wang, J. The 3C protein of enterovirus 71 inhibits retinoid acid-inducible gene I-mediated interferon regulatory factor 3 activation and type I interferon responses. J. Virol. 2010, 84, 8051–8061. [Google Scholar] [CrossRef]
- Lei, X.; Sun, Z.; Liu, X.; Jin, Q.; He, B.; Wang, J. Cleavage of the adaptor protein TRIF by enterovirus 71 3C inhibits antiviral responses mediated by Toll-like receptor 3. J. Virol. 2011, 85, 8811–8818. [Google Scholar] [CrossRef]
- Lei, X.; Xiao, X.; Xue, Q.; Jin, Q.; He, B.; Wang, J. Cleavage of interferon regulatory factor 7 by enterovirus 71 3C suppresses cellular responses. J. Virol. 2013, 87, 1690–1698. [Google Scholar] [CrossRef]
- Weng, K.F.; Li, M.L.; Hung, C.T.; Shih, S.R. Enterovirus 71 3C protease cleaves a novel target CstF-64 and inhibits cellular polyadenylation. PLoS Pathog. 2009, 5, e1000593. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Lei, X.; Xiao, X.; Yang, C.; Lu, W.; Huang, Z.; Leng, Q.; Jin, Q.; He, B.; Meng, G.; et al. Reciprocal Regulation between Enterovirus 71 and the NLRP3 Inflammasome. Cell. Rep. 2015, 12, 42–48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, Q.; Langereis, M.A.; Lork, M.; Nguyen, M.; Hato, S.V.; Lanke, K.; Emdad, L.; Bhoopathi, P.; Fisher, P.B.; Lloyd, R.E.; et al. Enterovirus 2Apro targets MDA5 and MAVS in infected cells. J. Virol. 2014, 88, 3369–3378. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Xi, X.; Lei, X.; Zhang, X.; Cui, S.; Wang, J.; Jin, Q.; Zhao, Z. Enterovirus 71 protease 2Apro targets MAVS to inhibit anti-viral type I interferon responses. PLoS Pathog. 2013, 9, e1003231. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Yi, L.; Zhao, J.; Yu, J.; Chen, Y.; Lin, M.C.; Kung, H.F.; He, M.L. Enterovirus 71 disrupts interferon signaling by reducing the level of interferon receptor 1. J. Virol. 2012, 86, 3767–3776. [Google Scholar] [CrossRef]
- Wang, L.C.; Chen, S.O.; Chang, S.P.; Lee, Y.P.; Yu, C.K.; Chen, C.L.; Tseng, P.C.; Hsieh, C.Y.; Chen, S.H.; Lin, C.F. Enterovirus 71 Proteins 2A and 3D Antagonize the Antiviral Activity of Gamma Interferon via Signaling Attenuation. J. Virol. 2015, 89, 7028–7037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathinayake, P.S.; Hsu, A.C.; Wark, P.A. Innate Immunity and Immune Evasion by Enterovirus 71. Viruses 2015, 7, 6613–6630. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.H.; Lai, L.C.; Cheng, H.C.; Chen, K.R.; Syue, Y.Z.; Lu, H.C.; Lin, W.Y.; Chen, S.H.; Huang, H.S.; Shiau, A.L.; et al. TBK1-associated protein in endolysosomes (TAPE) is an innate immune regulator modulating the TLR3 and TLR4 signaling pathways. J. Biol. Chem. 2011, 286, 7043–7051. [Google Scholar] [CrossRef]
- Wang, Y.F.; Chou, C.T.; Lei, H.Y.; Liu, C.C.; Wang, S.M.; Yan, J.J.; Su, I.J.; Wang, J.R.; Yeh, T.M.; Chen, S.H.; et al. A mouse-adapted enterovirus 71 strain causes neurological disease in mice after oral infection. J. Virol. 2004, 78, 7916–7924. [Google Scholar] [CrossRef]
- Chen, K.R.; Chang, C.H.; Huang, C.Y.; Lin, C.Y.; Lin, W.Y.; Lo, Y.C.; Yang, C.Y.; Hsing, E.W.; Chen, L.F.; Shih, S.R.; et al. TBK1-associated protein in endolysosomes (TAPE)/CC2D1A is a key regulator linking RIG-I-like receptors to antiviral immunity. J. Biol. Chem. 2012, 287, 32216–32221. [Google Scholar] [CrossRef]
- Hsu, Y.Y.; Liu, Y.N.; Wang, W.; Kao, F.J.; Kung, S.H. In vivo dynamics of enterovirus protease revealed by fluorescence resonance emission transfer (FRET) based on a novel FRET pair. Biochem. Biophys Res. Commun. 2007, 353, 939–945. [Google Scholar] [CrossRef] [PubMed]
- Tsai, M.T.; Cheng, Y.H.; Liu, Y.N.; Liao, N.C.; Lu, W.W.; Kung, S.H. Real-time monitoring of human enterovirus (HEV)-infected cells and anti-HEV 3C protease potency by fluorescence resonance energy transfer. Antimicrob. Agents Chemother. 2009, 53, 748–755. [Google Scholar] [CrossRef] [PubMed]
- Yang, C.H.; Li, H.C.; Jiang, J.G.; Hsu, C.F.; Wang, Y.J.; Lai, M.J.; Juang, Y.L.; Lo, S.Y. Enterovirus type 71 2A protease functions as a transcriptional activator in yeast. J. Biomed. Sci. 2010, 17, 65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Ji, L.; Yuan, X.; Jin, Y.; Cardona, C.J.; Xing, Z. Differential Regulation of TLR Signaling on the Induction of Antiviral Interferons in Human Intestinal Epithelial Cells Infected with Enterovirus 71. PLoS ONE 2016, 11, e0152177. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Rothenfusser, S.; Britsch, S.; Krug, A.; Jahrsdorfer, B.; Giese, T.; Endres, S.; Hartmann, G. Quantitative expression of toll-like receptor 1-10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J. Immunol. 2002, 168, 4531–4537. [Google Scholar] [CrossRef] [PubMed]
- Hornung, V.; Ellegast, J.; Kim, S.; Brzozka, K.; Jung, A.; Kato, H.; Poeck, H.; Akira, S.; Conzelmann, K.K.; Schlee, M.; et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science 2006, 314, 994–997. [Google Scholar] [CrossRef] [PubMed]
- Vercammen, E.; Staal, J.; Beyaert, R. Sensing of viral infection and activation of innate immunity by toll-like receptor 3. Clin. Microbiol. Rev. 2008, 21, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Bernard, J.J.; Cowing-Zitron, C.; Nakatsuji, T.; Muehleisen, B.; Muto, J.; Borkowski, A.W.; Martinez, L.; Greidinger, E.L.; Yu, B.D.; Gallo, R.L. Ultraviolet radiation damages self noncoding RNA and is detected by TLR3. Nat. Med. 2012, 18, 1286–1290. [Google Scholar] [CrossRef] [Green Version]
- Yasuda, K.; Yu, P.; Kirschning, C.J.; Schlatter, B.; Schmitz, F.; Heit, A.; Bauer, S.; Hochrein, H.; Wagner, H. Endosomal translocation of vertebrate DNA activates dendritic cells via TLR9-dependent and -independent pathways. J. Immunol. 2005, 174, 6129–6136. [Google Scholar] [CrossRef]
- Kato, H.; Takeuchi, O.; Mikamo-Satoh, E.; Hirai, R.; Kawai, T.; Matsushita, K.; Hiiragi, A.; Dermody, T.S.; Fujita, T.; Akira, S. Length-dependent recognition of double-stranded ribonucleic acids by retinoic acid-inducible gene-I and melanoma differentiation-associated gene 5. J. Exp. Med. 2008, 205, 1601–1610. [Google Scholar] [CrossRef]
- Zust, R.; Cervantes-Barragan, L.; Habjan, M.; Maier, R.; Neuman, B.W.; Ziebuhr, J.; Szretter, K.J.; Baker, S.C.; Barchet, W.; Diamond, M.S.; et al. Ribose 2′-O-methylation provides a molecular signature for the distinction of self and non-self mRNA dependent on the RNA sensor Mda5. Nat. Immunol. 2011, 12, 137–143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef]
- Pichlmair, A.; Schulz, O.; Tan, C.P.; Naslund, T.I.; Liljestrom, P.; Weber, F.; Reis e Sousa, C. RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 2006, 314, 997–1001. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, M.; Sato, S.; Mori, K.; Hoshino, K.; Takeuchi, O.; Takeda, K.; Akira, S. Cutting edge: A novel Toll/IL-1 receptor domain-containing adapter that preferentially activates the IFN-beta promoter in the Toll-like receptor signaling. J. Immunol. 2002, 169, 6668–6672. [Google Scholar] [CrossRef]
- Oshiumi, H.; Matsumoto, M.; Funami, K.; Akazawa, T.; Seya, T. TICAM-1, an adaptor molecule that participates in Toll-like receptor 3-mediated interferon-beta induction. Nat. Immunol. 2003, 4, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.C.; Yu, C.K.; Wang, Y.F.; Liu, C.C.; Su, I.J.; Lei, H.Y. A murine oral enterovirus 71 infection model with central nervous system involvement. J. Gen. Virol. 2004, 85, 69–77. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuo, R.L.; Kung, S.H.; Hsu, Y.Y.; Liu, W.T. Infection with enterovirus 71 or expression of its 2A protease induces apoptotic cell death. J. Gen. Virol. 2002, 83, 1367–1376. [Google Scholar] [CrossRef] [Green Version]
- Li, M.L.; Hsu, T.A.; Chen, T.C.; Chang, S.C.; Lee, J.C.; Chen, C.C.; Stollar, V.; Shih, S.R. The 3C protease activity of enterovirus 71 induces human neural cell apoptosis. Virology 2002, 293, 386–395. [Google Scholar] [CrossRef]
- Trinchieri, G. Type I interferon: Friend or foe? J. Exp. Med. 2010, 207, 2053–2063. [Google Scholar] [CrossRef]
- Lee, H.K.; Lund, J.M.; Ramanathan, B.; Mizushima, N.; Iwasaki, A. Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 2007, 315, 1398–1401. [Google Scholar] [CrossRef]
- Gorbea, C.; Makar, K.A.; Pauschinger, M.; Pratt, G.; Bersola, J.L.; Varela, J.; David, R.M.; Banks, L.; Huang, C.H.; Li, H.; et al. A role for Toll-like receptor 3 variants in host susceptibility to enteroviral myocarditis and dilated cardiomyopathy. J. Biol. Chem. 2010, 285, 23208–23223. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.Y.; Jouanguy, E.; Ugolini, S.; Smahi, A.; Elain, G.; Romero, P.; Segal, D.; Sancho-Shimizu, V.; Lorenzo, L.; Puel, A.; et al. TLR3 deficiency in patients with herpes simplex encephalitis. Science 2007, 317, 1522–1527. [Google Scholar] [CrossRef] [PubMed]
- Lafaille, F.G.; Pessach, I.M.; Zhang, S.Y.; Ciancanelli, M.J.; Herman, M.; Abhyankar, A.; Ying, S.W.; Keros, S.; Goldstein, P.A.; Mostoslavsky, G.; et al. Impaired intrinsic immunity to HSV-1 in human iPSC-derived TLR3-deficient CNS cells. Nature 2012, 491, 769–773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hardarson, H.S.; Baker, J.S.; Yang, Z.; Purevjav, E.; Huang, C.H.; Alexopoulou, L.; Li, N.; Flavell, R.A.; Bowles, N.E.; Vallejo, J.G. Toll-like receptor 3 is an essential component of the innate stress response in virus-induced cardiac injury. Am. J. Physiol. Heart Circ. Physiol. 2007, 292, 251–258. [Google Scholar] [CrossRef] [PubMed]
- Negishi, H.; Osawa, T.; Ogami, K.; Ouyang, X.; Sakaguchi, S.; Koshiba, R.; Yanai, H.; Seko, Y.; Shitara, H.; Bishop, K.; et al. A critical link between Toll-like receptor 3 and type II interferon signaling pathways in antiviral innate immunity. Proc. Natl. Acad. Sci. USA 2008, 105, 20446–20451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, K.R. Biochemical and functional study of antiviral innate immunity against RNA virus infection. Ph.D. Thesis, National Cheng Kung University, Taiwan, 2016. [Google Scholar]
- Zhu, K.; Yang, J.; Luo, K.; Yang, C.; Zhang, N.; Xu, R.; Chen, J.; Jin, M.; Xu, B.; Guo, N.; et al. TLR3 signaling in macrophages is indispensable for the protective immunity of invariant natural killer T cells against enterovirus 71 infection. PLoS Pathog. 2015, 11, e1004613. [Google Scholar] [CrossRef] [PubMed]
- Lin, Y.L.; Hu, Y.C.; Liang, C.C.; Lin, S.Y.; Liang, Y.C.; Yuan, H.P.; Chiang, B.L. Enterovirus-71 virus-like particles induce the activation and maturation of human monocyte-derived dendritic cells through TLR4 signaling. PLoS ONE 2014, 9, e111496. [Google Scholar] [CrossRef] [PubMed]
- Heil, F.; Hemmi, H.; Hochrein, H.; Ampenberger, F.; Kirschning, C.; Akira, S.; Lipford, G.; Wagner, H.; Bauer, S. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 2004, 303, 1526–1529. [Google Scholar] [CrossRef]
- Diebold, S.S.; Kaisho, T.; Hemmi, H.; Akira, S.; Reis e Sousa, C. Innate antiviral responses by means of TLR7-mediated recognition of single-stranded RNA. Science 2004, 303, 1529–1531. [Google Scholar] [CrossRef]
- Schoenemeyer, A.; Barnes, B.J.; Mancl, M.E.; Latz, E.; Goutagny, N.; Pitha, P.M.; Fitzgerald, K.A.; Golenbock, D.T. The interferon regulatory factor, IRF5, is a central mediator of toll-like receptor 7 signaling. J. Biol Chem. 2005, 280, 17005–17012. [Google Scholar] [CrossRef]
- Shih, S.R.; Stollar, V.; Li, M.L. Host factors in enterovirus 71 replication. J. Virol. 2011, 85, 9658–9666. [Google Scholar] [CrossRef] [PubMed]
- Lin, J.Y.; Shih, S.R. Cell and tissue tropism of enterovirus 71 and other enteroviruses infections. J. Biomed. Sci. 2014, 21, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, J.; Zhang, J.; Yanagawa, B.; Luo, Z.; Yang, X.; Chang, J.; McManus, B.; Luo, H. Cleavage of serum response factor mediated by enteroviral protease 2A contributes to impaired cardiac function. Cell. Res. 2012, 22, 360–371. [Google Scholar] [CrossRef] [PubMed]







© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, K.-R.; Yu, C.-K.; Kung, S.-H.; Chen, S.-H.; Chang, C.-F.; Ho, T.-C.; Lee, Y.-P.; Chang, H.-C.; Huang, L.-Y.; Lo, S.-Y.; et al. Toll-Like Receptor 3 Is Involved in Detection of Enterovirus A71 Infection and Targeted by Viral 2A Protease. Viruses 2018, 10, 689. https://doi.org/10.3390/v10120689
Chen K-R, Yu C-K, Kung S-H, Chen S-H, Chang C-F, Ho T-C, Lee Y-P, Chang H-C, Huang L-Y, Lo S-Y, et al. Toll-Like Receptor 3 Is Involved in Detection of Enterovirus A71 Infection and Targeted by Viral 2A Protease. Viruses. 2018; 10(12):689. https://doi.org/10.3390/v10120689
Chicago/Turabian StyleChen, Kuan-Ru, Chun-Keung Yu, Szu-Hao Kung, Shun-Hua Chen, Chuan-Fa Chang, Tzu-Chuan Ho, Yi-Ping Lee, Hung-Chuan Chang, Lan-Yin Huang, Shih-Yen Lo, and et al. 2018. "Toll-Like Receptor 3 Is Involved in Detection of Enterovirus A71 Infection and Targeted by Viral 2A Protease" Viruses 10, no. 12: 689. https://doi.org/10.3390/v10120689