Cytomegaloviruses in a Community of Wild Nonhuman Primates in Taï National Park, Côte D’Ivoire

Abstract
:1. Introduction
2. Materials and Methods
2.1. Field sites and Sample Collection
2.2. DNA Extraction
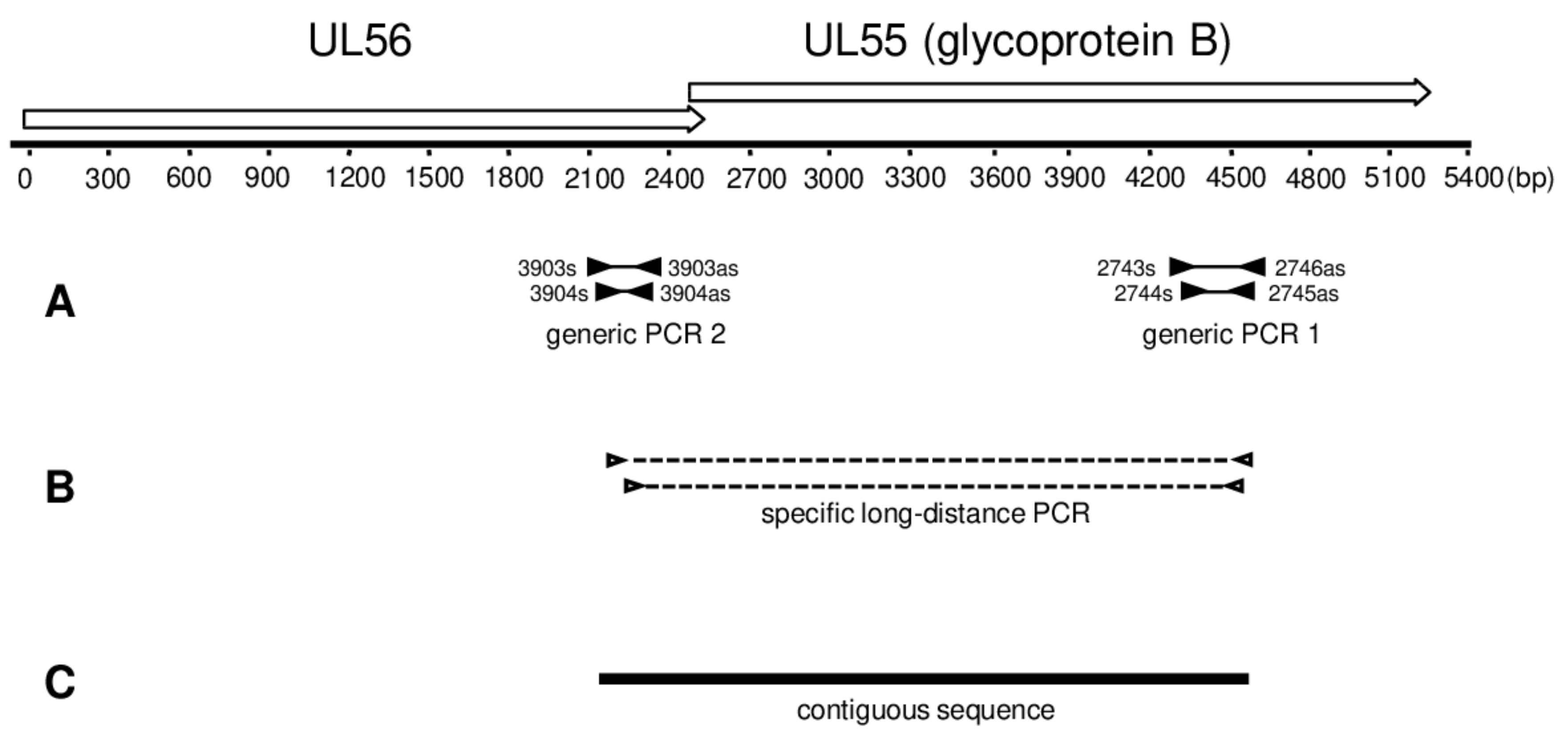
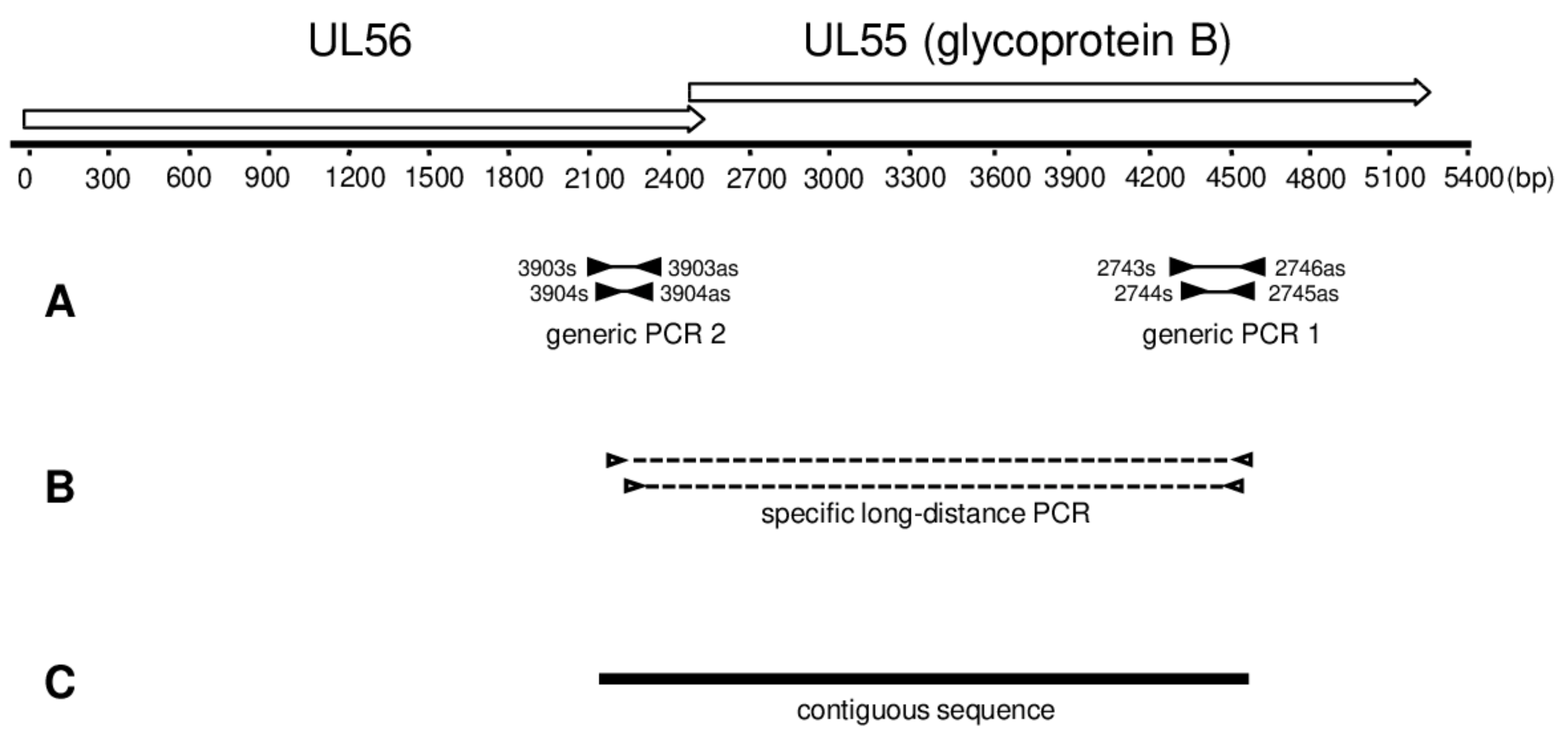
2.3. Generic PCR
2.4. Long-Distance Nested PCR
2.5. Cytochrome B PCR
2.6. Sequencing
2.7. BLAST Analysis
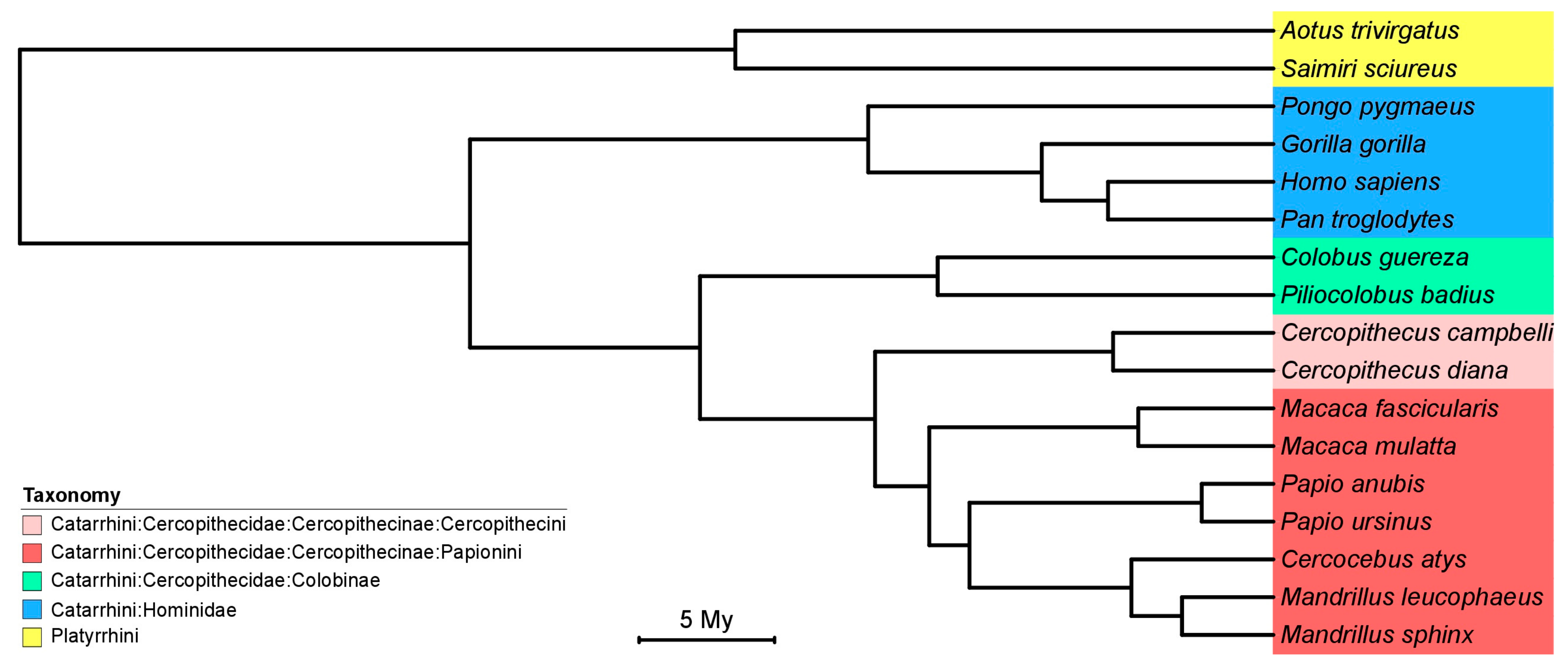
2.8. Phylogenetic and Co-Phylogenetic Analysis
3. Results
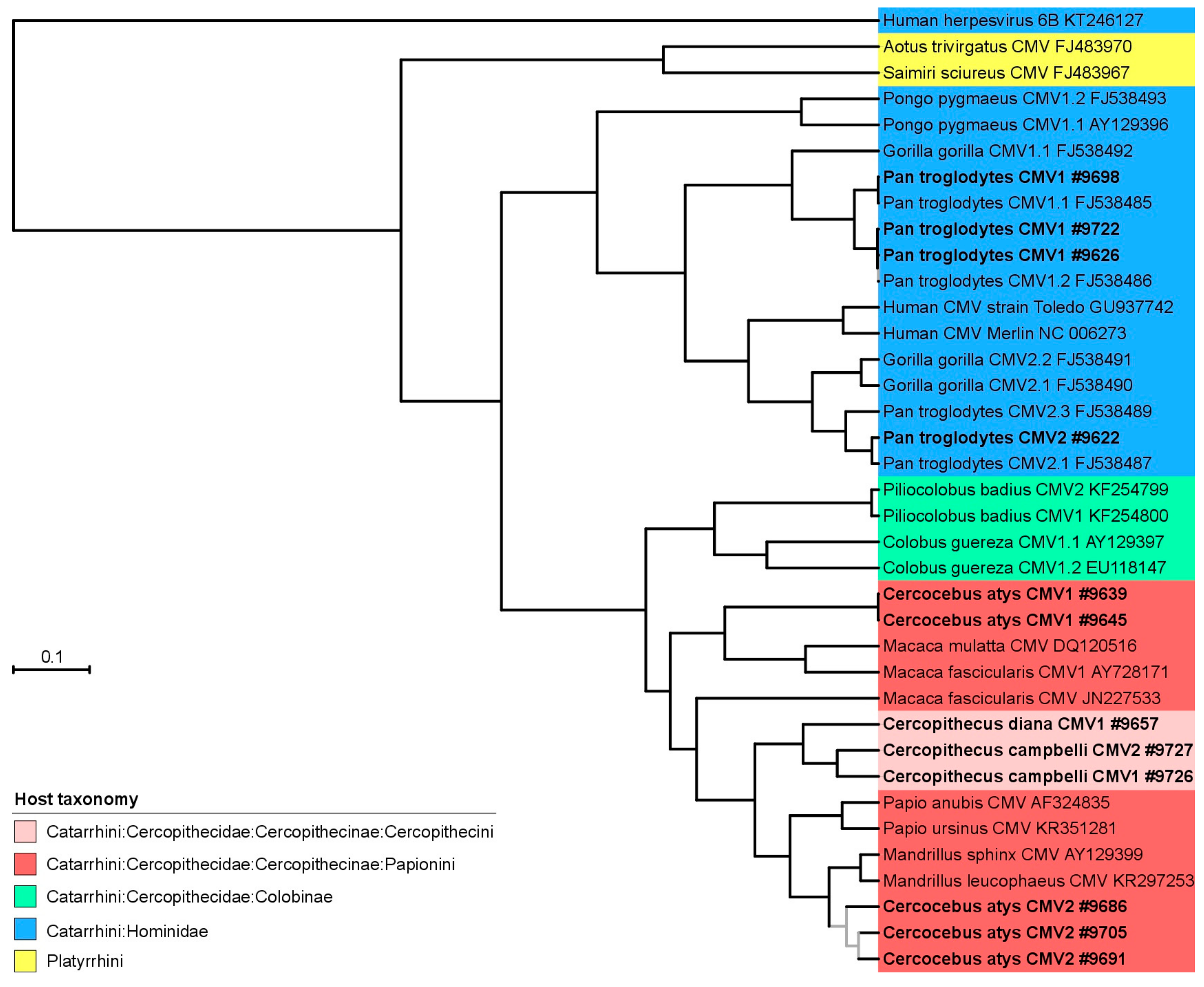
3.1. Detection of Cytomegaloviruses in Wild Nonhuman Primates from TNP
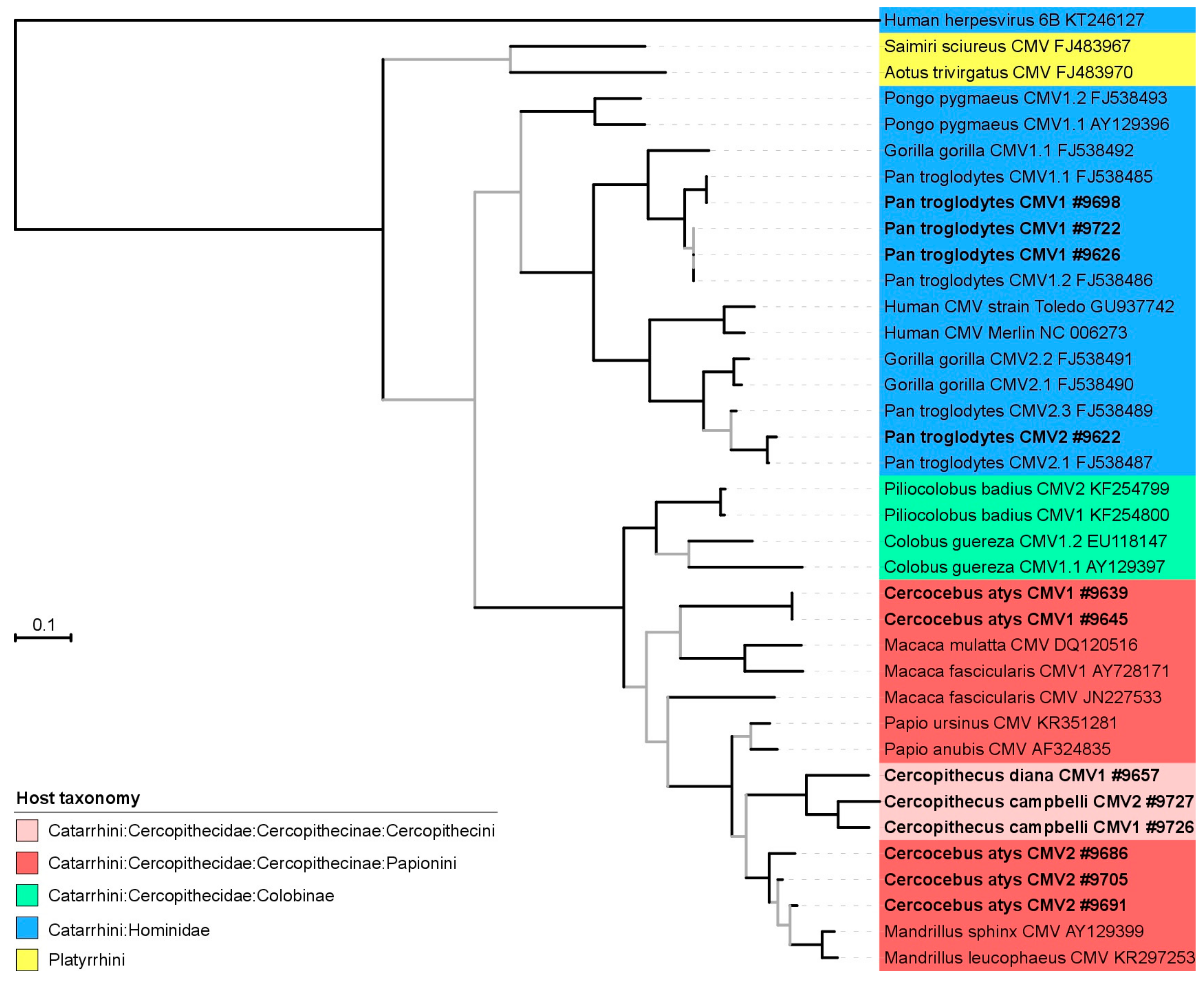
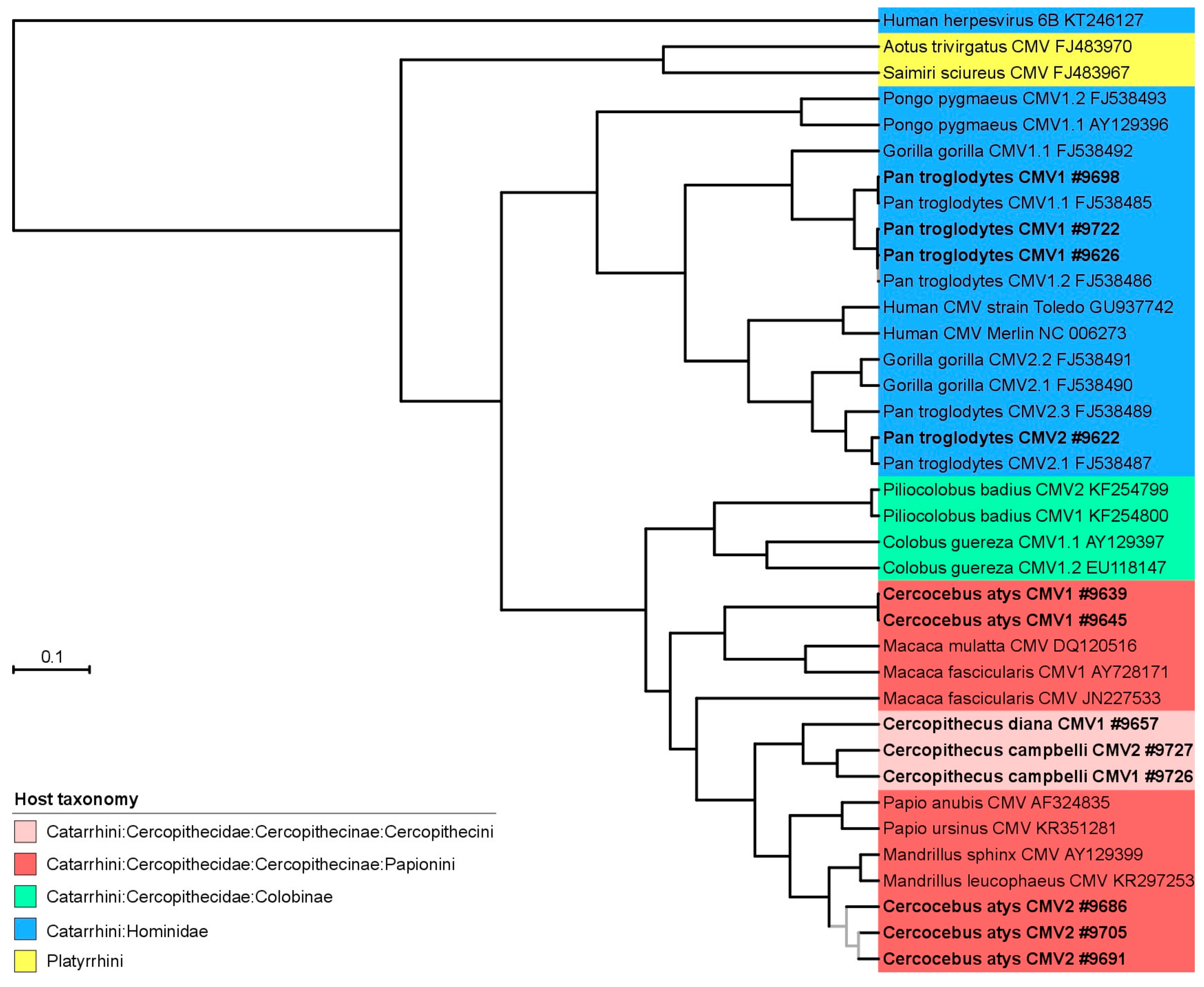
3.2. Phylogeny and Co-Phylogeny of Cytomegaloviruses and Their Hosts
4. Discussion
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Jabs, D.A. AIDS and ophthalmology, 2008. Arch. Ophthalmol. 2008, 126, 1143–1146. [Google Scholar] [CrossRef] [PubMed]
- Lischka, P.; Zimmermann, H. Antiviral strategies to combat cytomegalovirus infections in transplant recipients. Curr. Opin. Pharmacol. 2008, 8, 541–548. [Google Scholar] [CrossRef] [PubMed]
- Russell, M.Y.; Palmer, A.; Michaels, M.G. Cytomegalovirus infection in pediatric immunocompromised hosts. Infect. Disord. Drug Targets 2011, 11, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Vora, S.B.; Englund, J.A. Cytomegalovirus in immunocompromised children. Curr. Opin. Infect. Dis. 2015, 28, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Ho, M. The history of cytomegalovirus and its diseases. Med. Microbiol. Immunol. 2008, 197, 65–73. [Google Scholar] [CrossRef] [PubMed]
- Rafailidis, P.I.; Mourtzoukou, E.G.; Varbobitis, I.C.; Falagas, M.E. Severe cytomegalovirus infection in apparently immunocompetent patients: A systematic review. Virol. J. 2008, 5, 47. [Google Scholar] [CrossRef] [PubMed]
- Nassetta, L.; Kimberlin, D.; Whitley, R. Treatment of congenital cytomegalovirus infection: Implications for future therapeutic strategies. J. Antimicrob. Chemother. 2009, 63, 862–867. [Google Scholar] [CrossRef] [PubMed]
- Heberling, R. Viral diseases of nonhuman primates in the wild. Lab. Anim. Sci. 1971, 21, 1019–1022. [Google Scholar] [PubMed]
- Davison, A.J.; Dolan, A.; Akter, P.; Addison, C.; Dargan, D.J.; Alcendor, D.J.; McGeoch, D.J.; Hayward, G.S. The human cytomegalovirus genome revisited: Comparison with the chimpanzee cytomegalovirus genome FN1. J. Gen. Virol. 2003, 84, 17–28. [Google Scholar] [CrossRef] [PubMed]
- Leendertz, F.H.; Deckers, M.; Schempp, W.; Lankester, F.; Boesch, C.; Mugisha, L.; Dolan, A.; Gatherer, D.; McGeoch, D.I.; Ehlers, B. Novel cytomegaloviruses in free-ranging and captive great apes: Phylogenetic evidence for bidirectional horizontal transmission. J. Gen. Virol. 2009, 90, 2386–2394. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.G.; Strelow, L.I.; Franchi, D.C.; Anders, D.G.; Wong, S.W. Complete sequence and genomic analysis of rhesus cytomegalovirus. J. Virol. 2003, 77, 6620–6636. [Google Scholar] [CrossRef] [PubMed]
- Marsh, A.K.; Willer, D.O.; Ambagala, A.P.; Dzamba, M.; Chan, J.K.; Pilon, R.; Fournier, J.; Sandstrom, P.; Brudno, M.; MacDonald, K.S. Genomic sequencing and characterization of cynomolgus macaque cytomegalovirus. J. Virol. 2011, 85, 12995–13009. [Google Scholar] [CrossRef] [PubMed]
- Letvin, N.L.; Eaton, K.A.; Aldrich, W.R.; Sehgal, P.K.; Blake, B.J.; Schlossman, S.F.; King, N.W.; Hunt, R.D. Acquired immunodeficiency syndrome in a colony of macaque monkeys. Proc. Natl. Acad. Sci. USA 1983, 80, 2718–2722. [Google Scholar] [CrossRef] [PubMed]
- Black, P.H.; Hartley, J.W.; Rowe, W.P. Isolation of a cytomegalovirus from African green monkey. Proc. Soc. Exp. Biol. Med. 1963, 112, 601–605. [Google Scholar] [CrossRef] [PubMed]
- Blewett, E.; Lewis, J.; Gadsby, E.; Neubauer, S.; Eberle, R. Isolation of cytomegalovirus and foamy virus from the drill monkey (Mandrillus leucophaeus) and prevalence of antibodies to these viruses amongst wild-born and captive-bred individuals. Arch. Virol. 2003, 148, 423–433. [Google Scholar] [CrossRef] [PubMed]
- Blewett, E.; White, G.; Saliki, J.; Eberle, R. Isolation and characterization of an endogenous cytomegalovirus (BaCMV) from baboons. Arch. Virol. 2001, 146, 1723–1738. [Google Scholar] [CrossRef] [PubMed]
- Murthy, S.; Couacy-Hymann, E.; Metzger, S.; Nowak, K.; De Nys, H.; Boesch, C.; Wittig, R.; Jarvis, M.A.; Leendertz, F.H.; Ehlers, B. Absence of Frequent Herpesvirus Transmission in a Nonhuman Primate Predator-Prey System in the Wild. J. Virol. 2013, 87, 10651–10659. [Google Scholar] [CrossRef] [PubMed]
- Seimon, T.A.; Olson, S.H.; Lee, K.J.; Rosen, G.; Ondzie, A.; Cameron, K.; Reed, P.; Anthony, S.J.; Joly, D.O.; McAloose, D. Adenovirus and herpesvirus diversity in free-ranging great apes in the Sangha region of the Republic of Congo. PLoS ONE 2015, 10, e0118543. [Google Scholar] [CrossRef]
- Burwitz, B.J.; Malouli, D.; Bimber, B.N.; Reed, J.S.; Ventura, A.B.; Hancock, M.H.; Uebelhoer, L.S.; Bhusari, A.; Hammond, K.B.; Trethewy, R.G.E. Cross-Species Rhesus Cytomegalovirus Infection of Cynomolgus Macaques. PLoS Pathog. 2016, 12, e1006014. [Google Scholar] [CrossRef] [PubMed]
- Lilja, A.E.; Shenk, T. Efficient replication of rhesus cytomegalovirus variants in multiple rhesus and human cell types. Proc. Natl. Acad. Sci. USA 2008, 105, 19950–19955. [Google Scholar] [CrossRef] [PubMed]
- Swinkels, B.; Geelen, J.; Wertheim-van Dillen, P.; van Es, A.; van der Noordaa, J. Initial characterization of four cytomegalovirus strains isolated from chimpanzees. Arch. Virol. 1984, 82, 125–128. [Google Scholar] [CrossRef] [PubMed]
- Wroblewska, Z.; Gilden, D.; Devlin, M.; Huang, E.-S.; Rorke, L.B.; Hamada, T.; Furukawa, T.; Cummins, L.; Kalter, S.; Koprowski, H. Cytomegalovirus isolation from a chimpanzee with acute demyelinating disease after inoculation of multiple sclerosis brain cells. Infect. Immun. 1979, 25, 1008–1015. [Google Scholar] [PubMed]
- Perot, K.; Walker, C.M.; Spaete, R.R. Primary chimpanzee skin fibroblast cells are fully permissive for human cytomegalovirus replication. J. Gen. Virol. 1992, 73, 3281–3284. [Google Scholar] [CrossRef] [PubMed]
- Anoh, A.E.; Akoua-Koffi, C.; Couacy-Hymann, E.; Pauly, M.; Schubert, G.; Mossoun, A.; Weiss, S.; Leendertz, S.A.J.; Jarvis, M.A.; Leendertz, F.H.; et al. Genetic identification of cytomegaloviruses in a rural population of Côte d’Ivoire. Virol. J. 2015, 12, 155. [Google Scholar] [CrossRef] [PubMed]
- Mossoun, A.; Pauly, M.; Akoua-Koffi, C.; Couacy-Hymann, E.; Leendertz, S.A.; Anoh, A.E.; Gnoukpoho, A.H.; Leendertz, F.H.; Schubert, G. Contact to Non-human Primates and Risk Factors for Zoonotic Disease Emergence in the Tai Region, Cote d’Ivoire. Ecohealth 2015, 12, 580–591. [Google Scholar] [CrossRef] [PubMed]
- Ehlers, B.; Kuchler, J.; Yasmum, N.; Dural, G.; Voigt, S.; Schmidt-Chanasit, J.; Jakel, T.; Matuschka, F.R.; Richter, D.; Essbauer, S.; et al. Identification of novel rodent herpesviruses, including the first γherpesvirus of Mus musculus. J. Virol. 2007, 81, 8091–8100. [Google Scholar] [CrossRef] [PubMed]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic Local Alignment Search Tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gouy, M.; Guindon, S.; Gascuel, O. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol. Biol. Evol. 2010, 27, 221–224. [Google Scholar] [CrossRef] [PubMed]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [PubMed]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed]
- Guindon, S.; Lethiec, F.; Duroux, P.; Gascuel, O. PHYML Online—A web server for fast maximum likelihood-based phylogenetic inference. Nucleic Acids Res. 2005, 33, W557–559. [Google Scholar] [CrossRef] [PubMed]
- Anisimova, M.; Gil, M.; Dufayard, J.F.; Dessimoz, C.; Gascuel, O. Survey of branch support methods demonstrates accuracy, power, and robustness of fast likelihood-based approximation schemes. Syst. Biol. 2011, 60, 685–699. [Google Scholar] [CrossRef] [PubMed]
- Drummond, A.J.; Suchard, M.A.; Xie, D.; Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 2012, 29, 1969–1973. [Google Scholar] [CrossRef] [PubMed]
- Arnold, C.; Matthews, L.J.; Nunn, C.L. The 10kTrees website: A new online resource for primate phylogeny. Evol. Anthropol. Issues News Rev. 2010, 19, 114–118. [Google Scholar] [CrossRef] [Green Version]
- Conow, C.; Fielder, D.; Ovadia, Y.; Libeskind-Hadas, R. Jane: A new tool for the cophylogeny reconstruction problem. Algorithms Mol. Biol. 2010, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Novak, Z.; Ross, S.A.; Patro, R.K.; Pati, S.K.; Kumbla, R.A.; Brice, S.; Boppana, S.B. Cytomegalovirus strain diversity in seropositive women. J. Clin. Microbiol. 2008, 46, 882–886. [Google Scholar] [CrossRef] [PubMed]
- Ross, S.A.; Arora, N.; Novak, Z.; Fowler, K.B.; Britt, W.J.; Boppana, S.B. Cytomegalovirus reinfections in healthy seroimmune women. J. Infect. Dis. 2010, 201, 386–389. [Google Scholar] [CrossRef] [PubMed]
- Meyer-König, U.; Ebert, K.; Schrage, B.; Pollak, S.; Hufert, F.T. Simultaneous infection of healthy people with multiple human cytomegalovirus strains. Lancet 1998, 352, 1280–1281. [Google Scholar] [CrossRef]
- Powers, C.; Frueh, K. Rhesus CMV: An emerging animal model for human CMV. Med. Microbiol. Immun. 2008, 197, 109–115. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.G.; Vieville, C.; Whizin, N.; Coyne-Johnson, L.; Siess, D.C.; Drummond, D.D.; Legasse, A.W.; Axthelm, M.K.; Oswald, K.; Trubey, C.M.; et al. Effector memory T cell responses are associated with protection of rhesus monkeys from mucosal simian immunodeficiency virus challenge. Nat. Med. 2009, 15, 293–299. [Google Scholar] [CrossRef] [PubMed]
- Hansen, S.G.; Powers, C.J.; Richards, R.; Ventura, A.B.; Ford, J.C.; Siess, D.; Axthelm, M.K.; Nelson, J.A.; Jarvis, M.A.; Picker, L.J.; et al. Evasion of CD8(+) T Cells Is Critical for Superinfection by Cytomegalovirus. Science 2010, 328, 102–106. [Google Scholar] [CrossRef] [PubMed]
- Buck, C.B.; Van Doorslaer, K.; Peretti, A.; Geoghegan, E.M.; Tisza, M.J.; An, P.; Katz, J.P.; Pipas, J.M.; McBride, A.A.; Camus, A.C.; et al. The Ancient Evolutionary History of Polyomaviruses. PLoS Pathog. 2016, 12, e1005574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoppe, E.; Pauly, M.; Gillespie, T.R.; Akoua-Koffi, C.; Hohmann, G.; Fruth, B.; Karhemere, S.; Madinda, N.F.; Mugisha, L.; Muyembe, J.J.; et al. Multiple Cross-Species Transmission Events of Human Adenoviruses (HAdV) during Hominine Evolution. Mol. Biol. Evol. 2015, 32, 2072–2084. [Google Scholar] [CrossRef] [PubMed]
- Madinda, N.F.; Ehlers, B.; Wertheim, J.O.; Akoua-Koffi, C.; Bergl, R.A.; Boesch, C.; Akonkwa, D.B.; Eckardt, W.; Fruth, B.; Gillespie, T.R.; et al. Assessing Host-Virus Codivergence for Close Relatives of Merkel Cell Polyomavirus Infecting African Great Apes. J. Virol. 2016, 90, 8531–8541. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.A.; Redwood, A.J.; Jarvis, M.A. Self-disseminating vaccines for emerging infectious diseases. Expert Rev. Vaccines 2016, 15, 31–39. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Common Host Name | Host Species | Number of Individuals Tested | Number of Positives | % CMV Prevalence (95% CI §) | References |
|---|---|---|---|---|---|
| Western chimpanzee | Pan troglodytes verus | 38 (18 *) | 15 (9 *) | 0.39 (0.24–0.57) | [17] |
| Sooty mangabey | Cercocebus atys | 37 * | 7 * | 0.19 (0.09–0.36) | |
| Western red colobus | Piliocolobus badius | 31 (13 *) | 4 (0 *) | 0.13 (0.04–0.31) | [17] |
| Black-and-white colobus | Colobus polykomos | 12 (3 *) | 1 (0 *) | 0.08 (0.01–0.40) | [17] |
| Diana monkey | Cercopithecus diana | 8 * | 1 * | 0.12 (0.01–0.53) | |
| Campbell’s monkey | Cercopithecus campbelli | 4 * | 1 * | 0.25 (0.01–0.78) | |
| Potto | Perodicticus potto | 2 * | 0 * | 0 (0–0.90) | |
| Total | 132 | 29 | 22 |
| Host Family and Species | Sample Number | Full Virus Name | Abbreviated Virus Name | UL55 Sequence (kb) | References | Accession Number |
|---|---|---|---|---|---|---|
| Hominidae | ||||||
| Pan troglodytes verus | 9595 | Pan troglodytes cytomegalovirus 1 | PtroCMV1 | 2.34 | Current study | MG593784 |
| 9626 | 2.34 | MG593786 | ||||
| 9698 | 2.36 | MG593793 | ||||
| 9722 | 2.34 | MG593797 | ||||
| 3147 | 2.79 | [10] | FJ538485 | |||
| 9622 | Pan troglodytes cytomegalovirus 2 | PtroCMV2 | 2.39 | Current study | MG593785 | |
| 2296 | 1.83 | [10] | FJ538487 | |||
| Cercopithecidae | ||||||
| Cercocebus atys | 9639 | Cercocebus atys cytomegalovirus 1 | CatyCMV1 | 2.23 | Current study | MG593787 |
| 9645 | 2.23 | MG593788 | ||||
| 9646 | 2.23 | MG593789 | ||||
| 9686 | Cercocebus atys cytomegalovirus 2 | CatyCMV2 | 2.24 | Current study | MG593791 | |
| 9691 | 2.25 | MG593792 | ||||
| 9705 | 2.23 | MG593794 | ||||
| 9706 | Cercocebus atys cytomegalovirus 3 | CatyCMV3 | 0.20 | Current study | MG593795 | |
| 9720 | Cercocebus atys roseolovirus 1 | CatyROV1 | 0.22 | Current study | MG593796 | |
| Cercopithecus campbelli | 9726 | Cercopithecus campbelli cytomegalovirus 1 | CcamCMV1 | 2.24 | Current study | MG593798 |
| 9727 | Cercopithecus campbelli cytomegalovirus 2 | CcamCMV2 | 2.24 | Current study | MG593799 | |
| Cercopithecus diana | 9657 | Cercopithecus diana cytomegalovirus 1 | CdiaCMV1 | 2.24 | Current study | MG593790 |
| Piliocolobus badius | 6944 | Piliocolobus badius cytomegalovirus 1 | PbadCMV1 | 1.67 | [17] | KF254800 |
| 6940 | Piliocolobus badius cytomegalovirus 2 | PbadCMV2 | 1.67 | KF254799 | ||
| 4598 | Piliocolobus badius cytomegalovirus 3 | PbadCMV3 | 0.22 | KF318790 |
| Tissue | Cercopithecus diana | Cercopithecus campbelli | Cercocebus atys | Colobus polykomos | Pan troglodytes | Piliocolobus badius | Perodicticus poto |
|---|---|---|---|---|---|---|---|
| Kidney | 2 | 2 (1) | 5 (1) | - | 1 | 2 | - |
| Liver | 2 | 1 | 10 (2) | 1 | 9 (3) | 4 | 1 |
| Lung | 4 (1) a | - | 11 (3) | - | 7 (1) | 3 | 1 |
| Spleen | 1 | 1 | 5 (1) | 1 | 11 (4) | 1 | 2 |
| Whole blood | 2 | - | 9 | 2 | - | - | - |
| Buffy coat | - b | - | 2 (1) | - | 1 | 7 | - |
| Heart | - | 1 | 3 (1) | - | 1 | - | - |
| Gastrointestinal tract | 2 | 1 (1) | 3 | - | 1 (1) | - | - |
| Lymph node | - | 2 | 2(1) | - | 4 (1) | 1 | - |
| Total | 13 (1) | 3 (0) | 50 (10) | 4 (0) | 37 (10) | 18 (0) | 4 (0) |
| Host Family and Species | Spleen | Blood/Buffy Coat | Lymph Node | Heart | Lung | Kidney | Liver | Oesophagus | Duodenum |
|---|---|---|---|---|---|---|---|---|---|
| Hominidae | |||||||||
| Pan troglodytes verus | PtroCMV1 a PtroCMV2 | PtroCMV2 | PtroCMV1 | PtroCMV1 | PtroCMV1 PtroCMV2 | PtroCMV2 | - | ||
| Cercopithecidae | |||||||||
| Cercocebus atys | CatyCMV1 | CatyCMV2 | CatyCMV3 | CatyCMV1, CatyCMV2 | CatyCMV1, CatyROV1 | CatyCMV1, CatyCMV2 | - | ||
| Cercopithecus campbelli | - | - | - | - | - | CcamCMV1 | - | - | CcamCMV2 |
| Cercopithecus diana | CdiaCMV1 | ||||||||
| Colobus polykomos | - | - | - | - | - | - | |||
| Perodicticus potto | - | - | - | - | - | - | |||
| Piliocolobus badius | - | - | - | - |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Anoh, A.E.; Murthy, S.; Akoua-Koffi, C.; Couacy-Hymann, E.; Leendertz, F.H.; Calvignac-Spencer, S.; Ehlers, B. Cytomegaloviruses in a Community of Wild Nonhuman Primates in Taï National Park, Côte D’Ivoire. Viruses 2018, 10, 11. https://doi.org/10.3390/v10010011
Anoh AE, Murthy S, Akoua-Koffi C, Couacy-Hymann E, Leendertz FH, Calvignac-Spencer S, Ehlers B. Cytomegaloviruses in a Community of Wild Nonhuman Primates in Taï National Park, Côte D’Ivoire. Viruses. 2018; 10(1):11. https://doi.org/10.3390/v10010011
Chicago/Turabian StyleAnoh, Augustin Etile, Sripriya Murthy, Chantal Akoua-Koffi, Emmanuel Couacy-Hymann, Fabian Hubertus Leendertz, Sébastien Calvignac-Spencer, and Bernhard Ehlers. 2018. "Cytomegaloviruses in a Community of Wild Nonhuman Primates in Taï National Park, Côte D’Ivoire" Viruses 10, no. 1: 11. https://doi.org/10.3390/v10010011