
Studying the Hydration of a Retarded Suspension of Ground Granulated Blast-Furnace Slag after Reactivation


Abstract
:1. Introduction
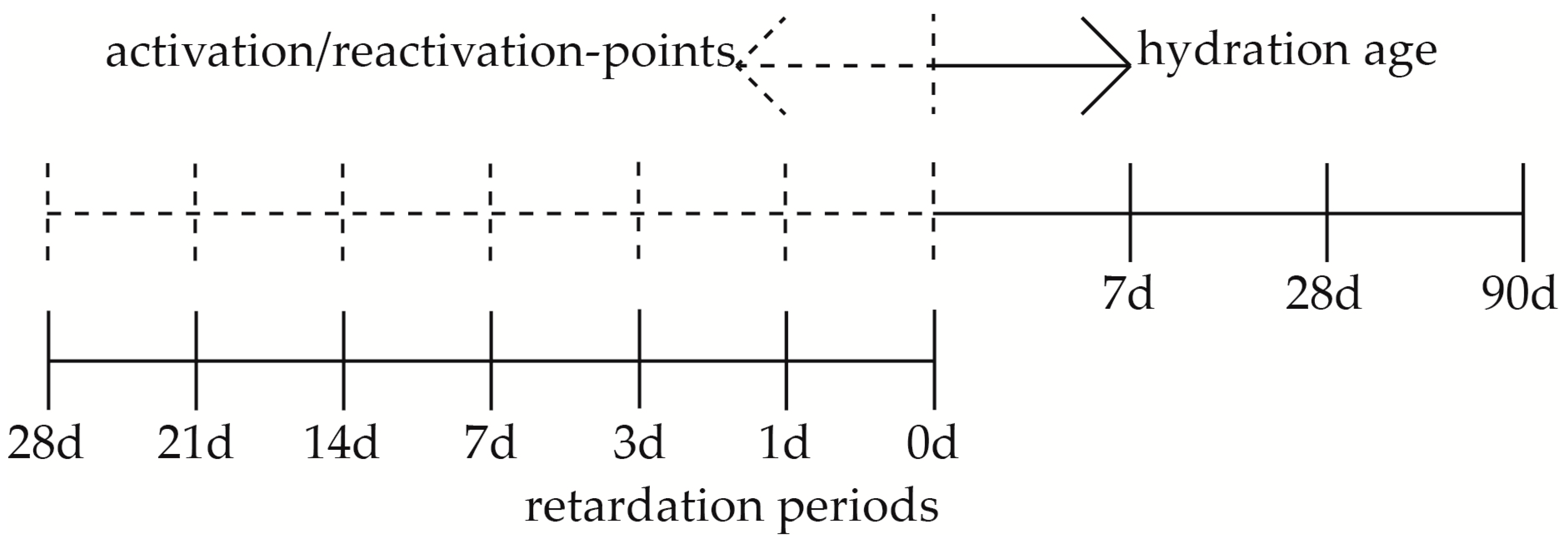
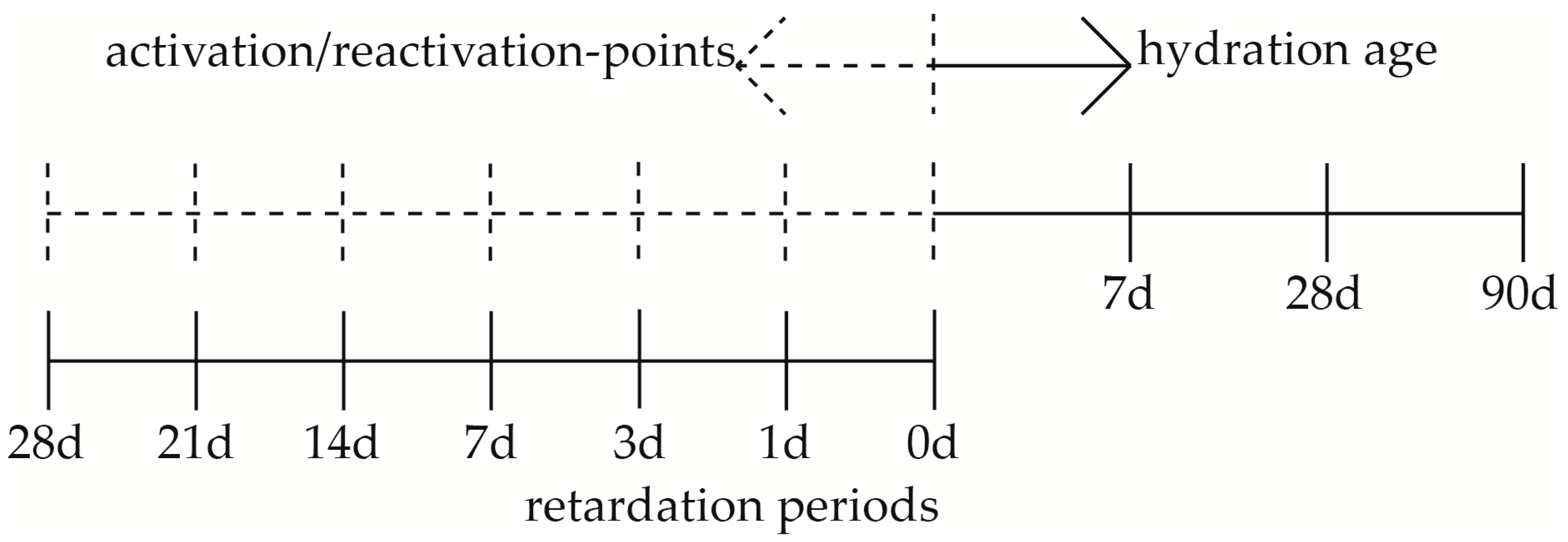
2. Materials and Methods
2.1. Compressive Strength
2.2. Thermogravimetry (TG), X-ray Diffraction (XRD), and Scanning Electron Microscopy (SEM)
3. Results and Discussion
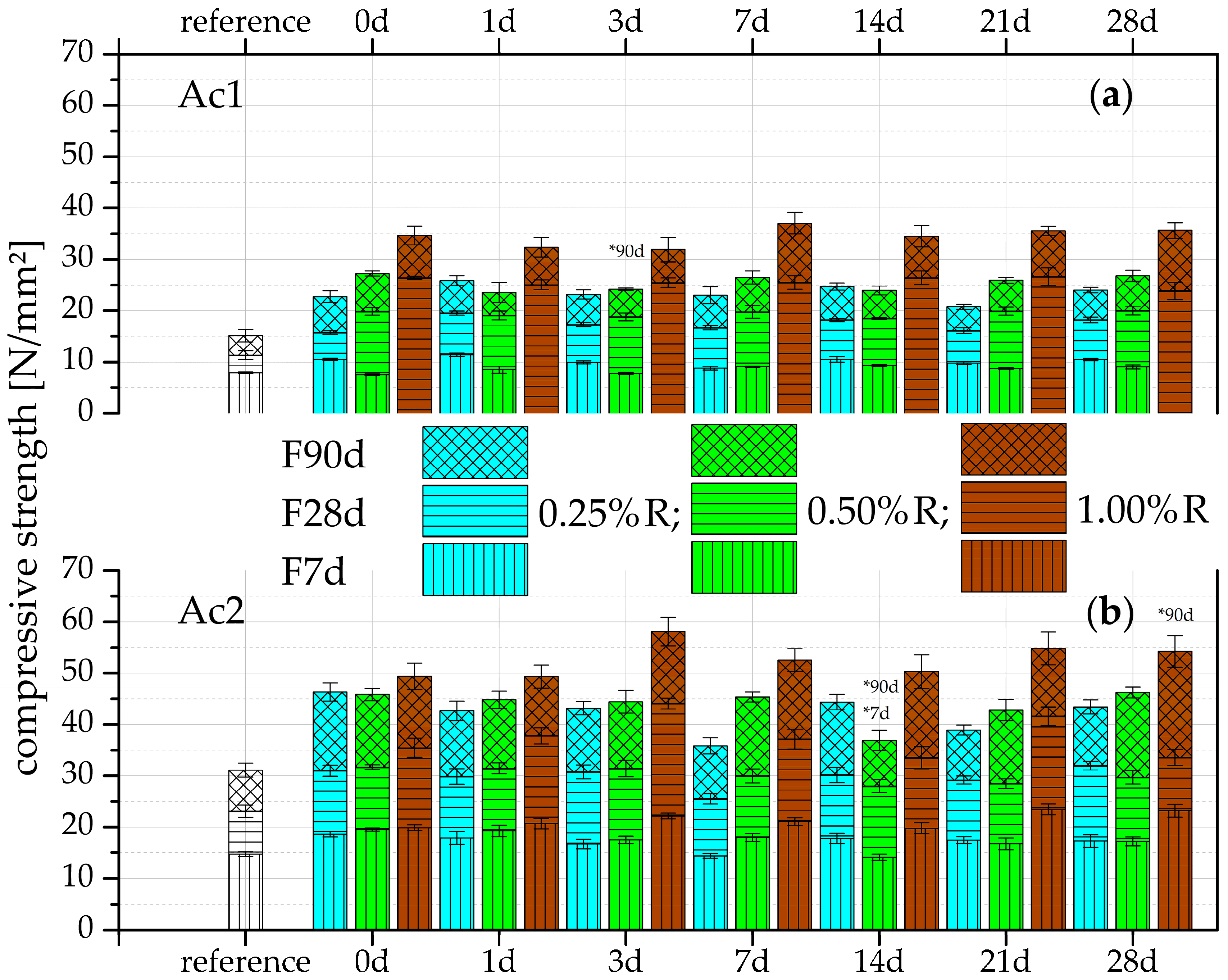
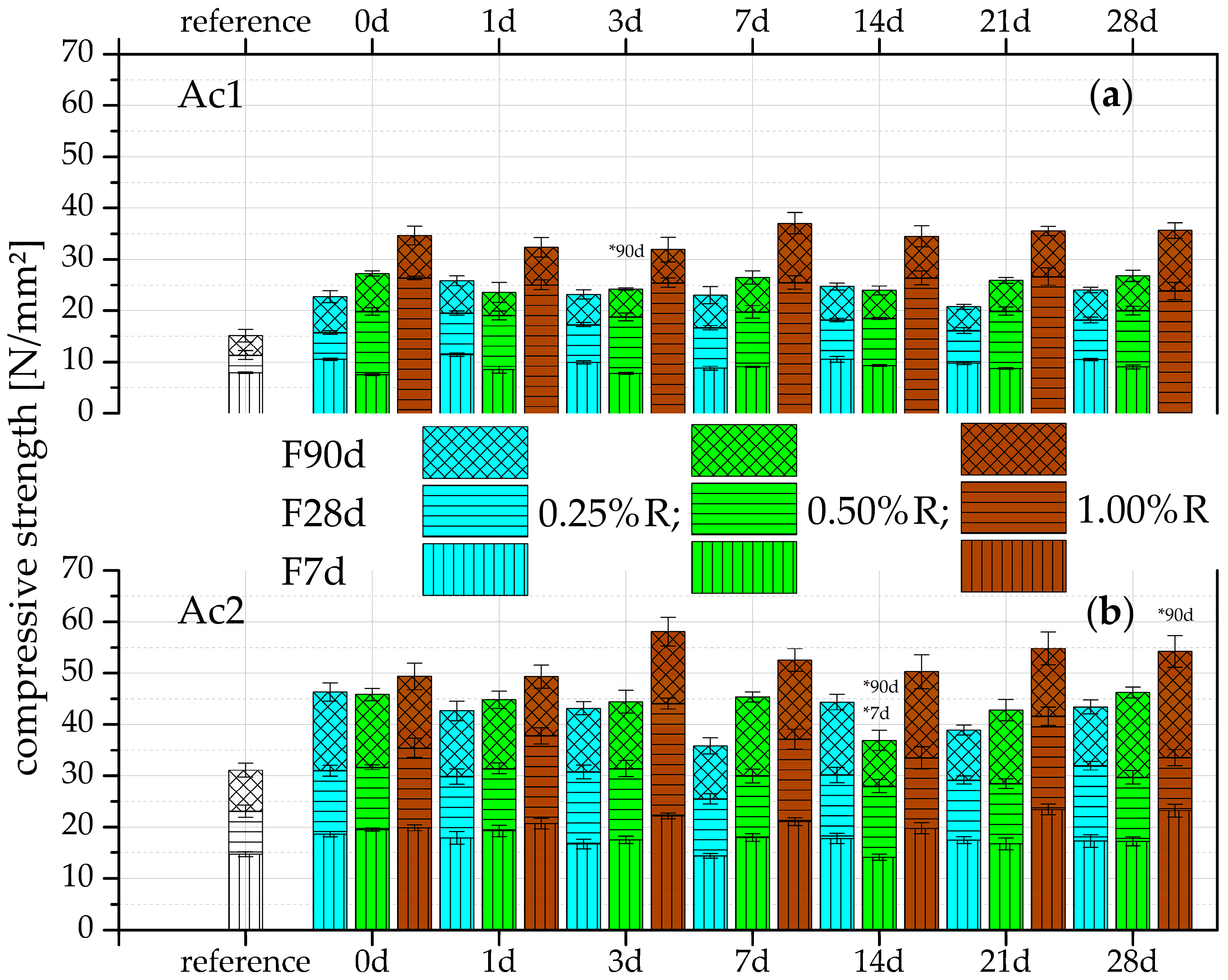
3.1. Compressive Strength
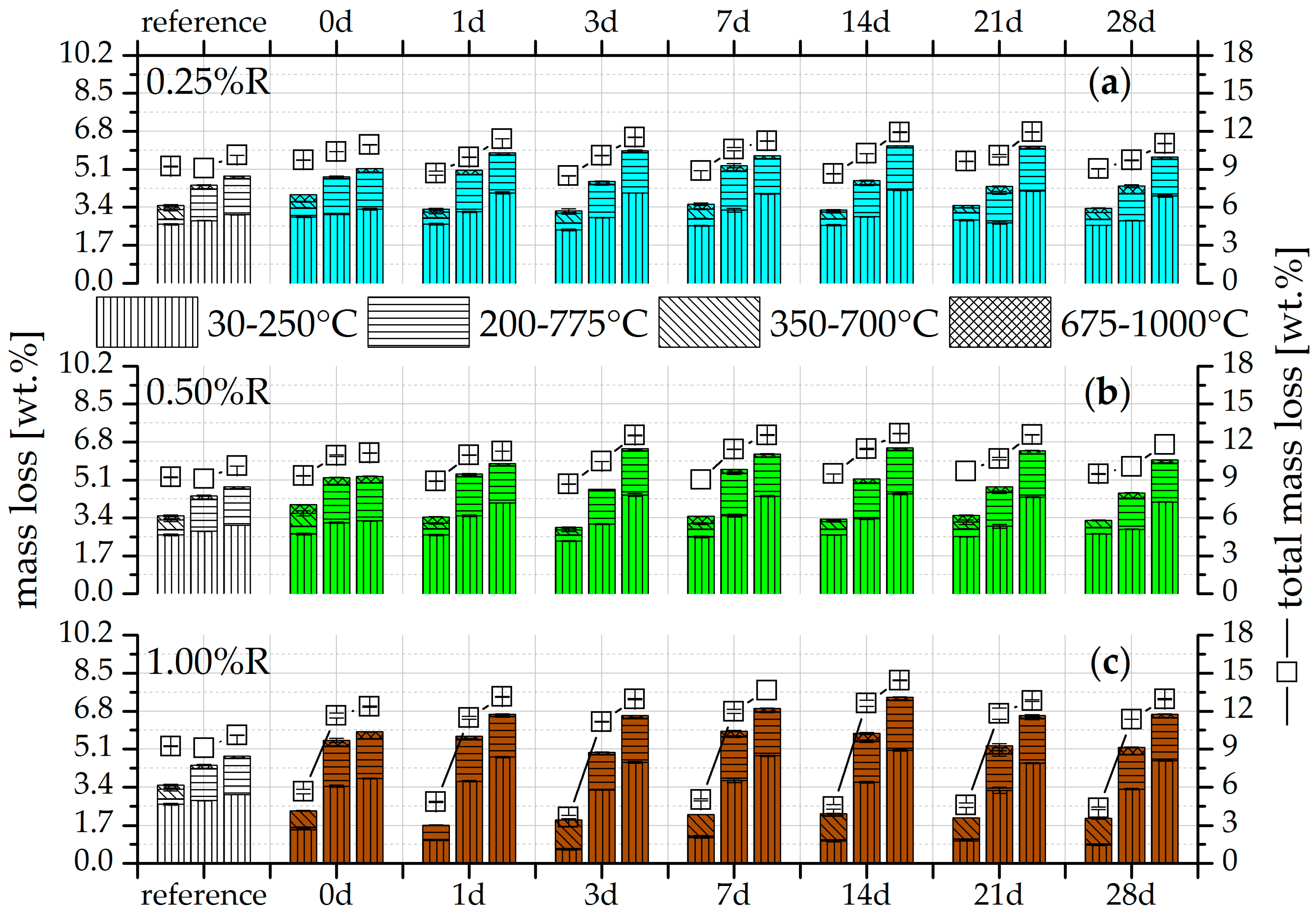
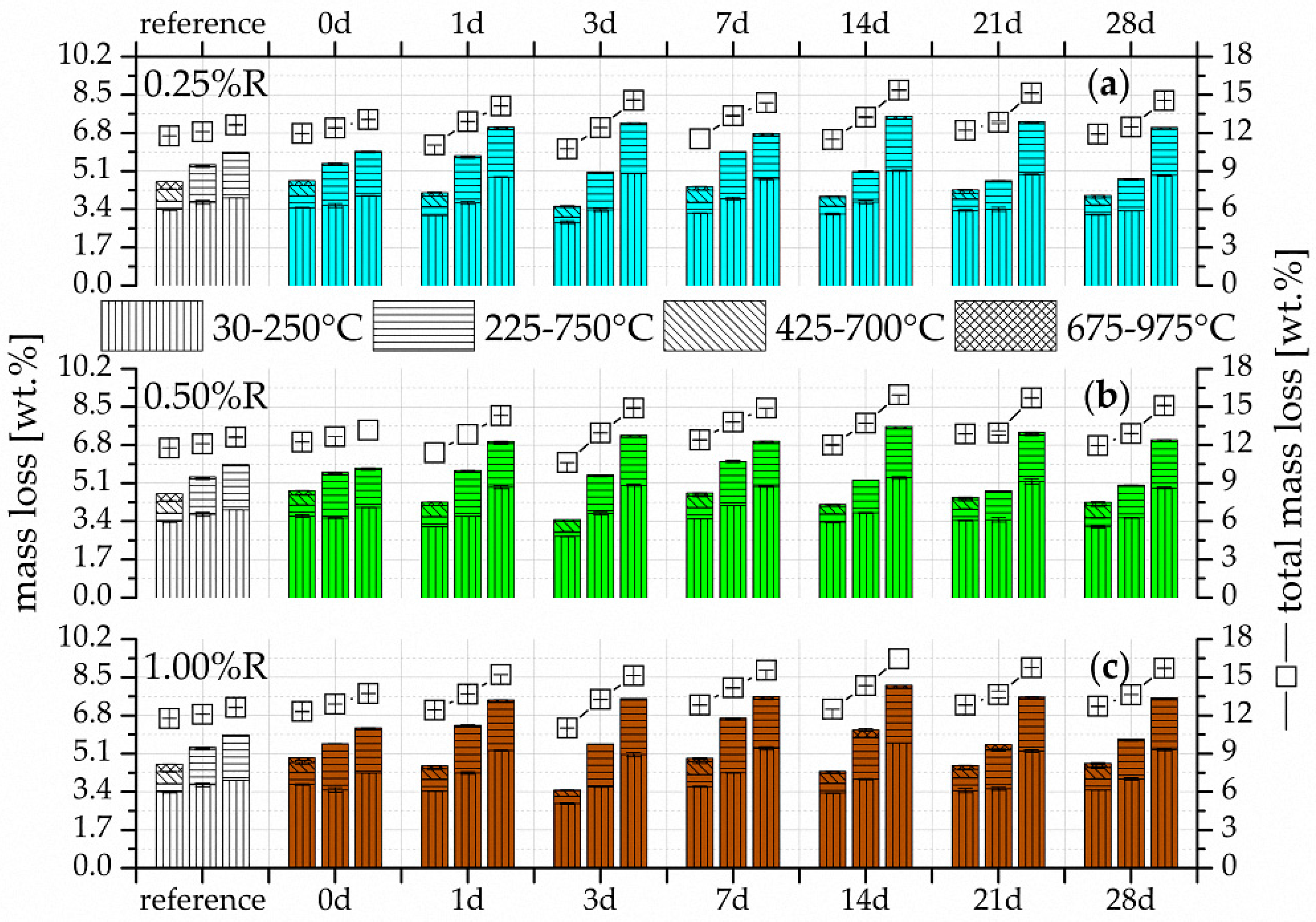
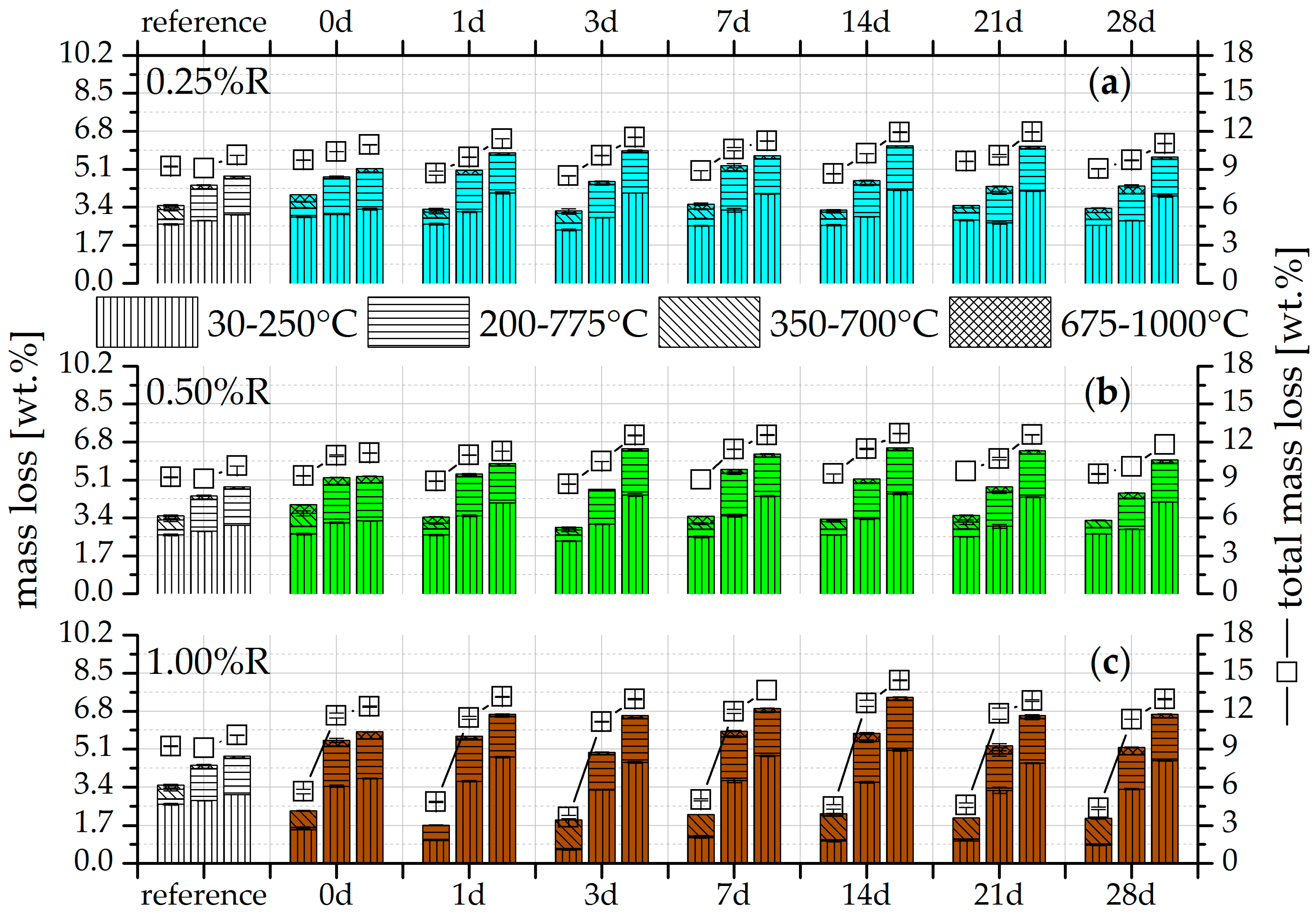
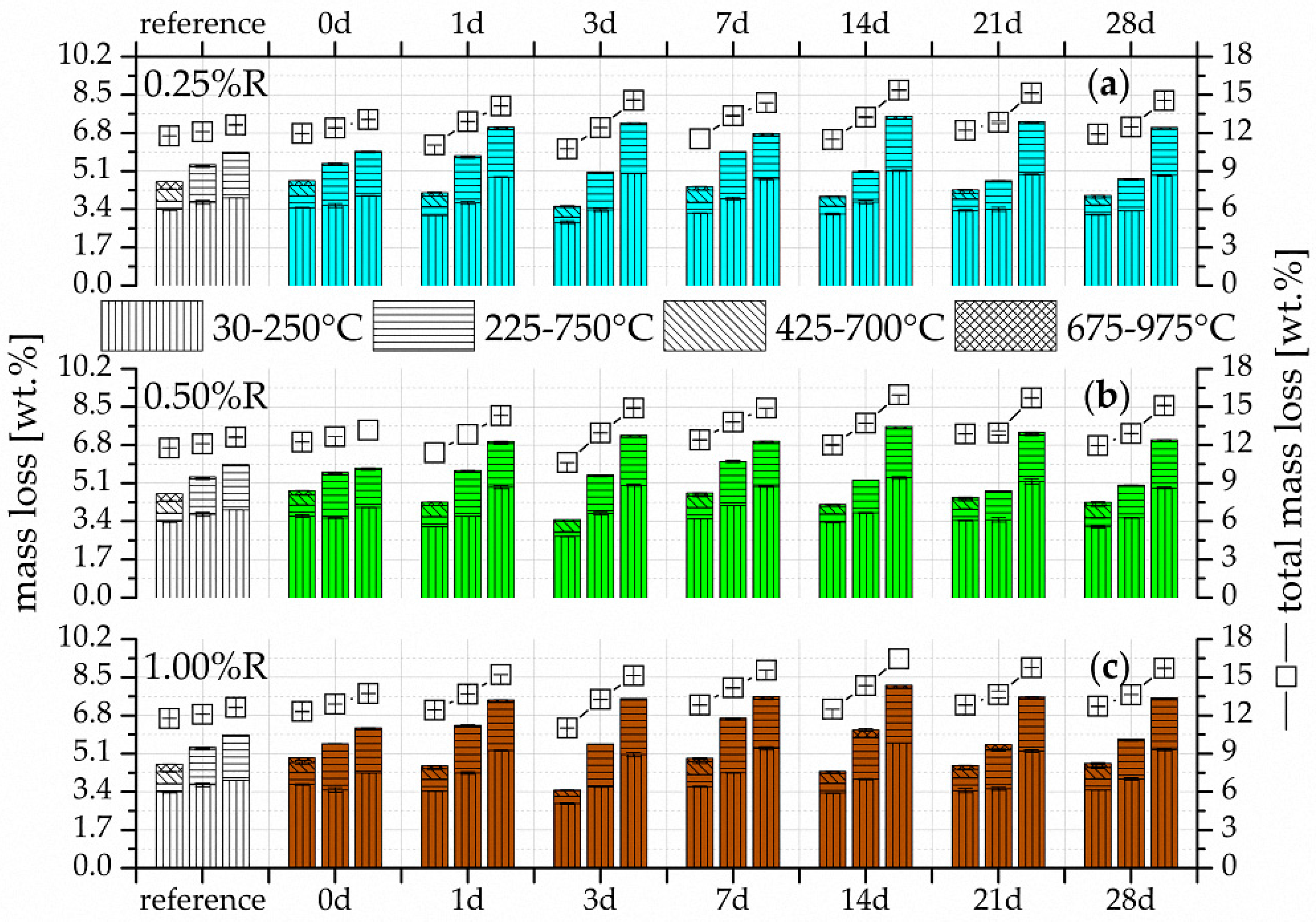
3.2. Thermogravimetry (TG)
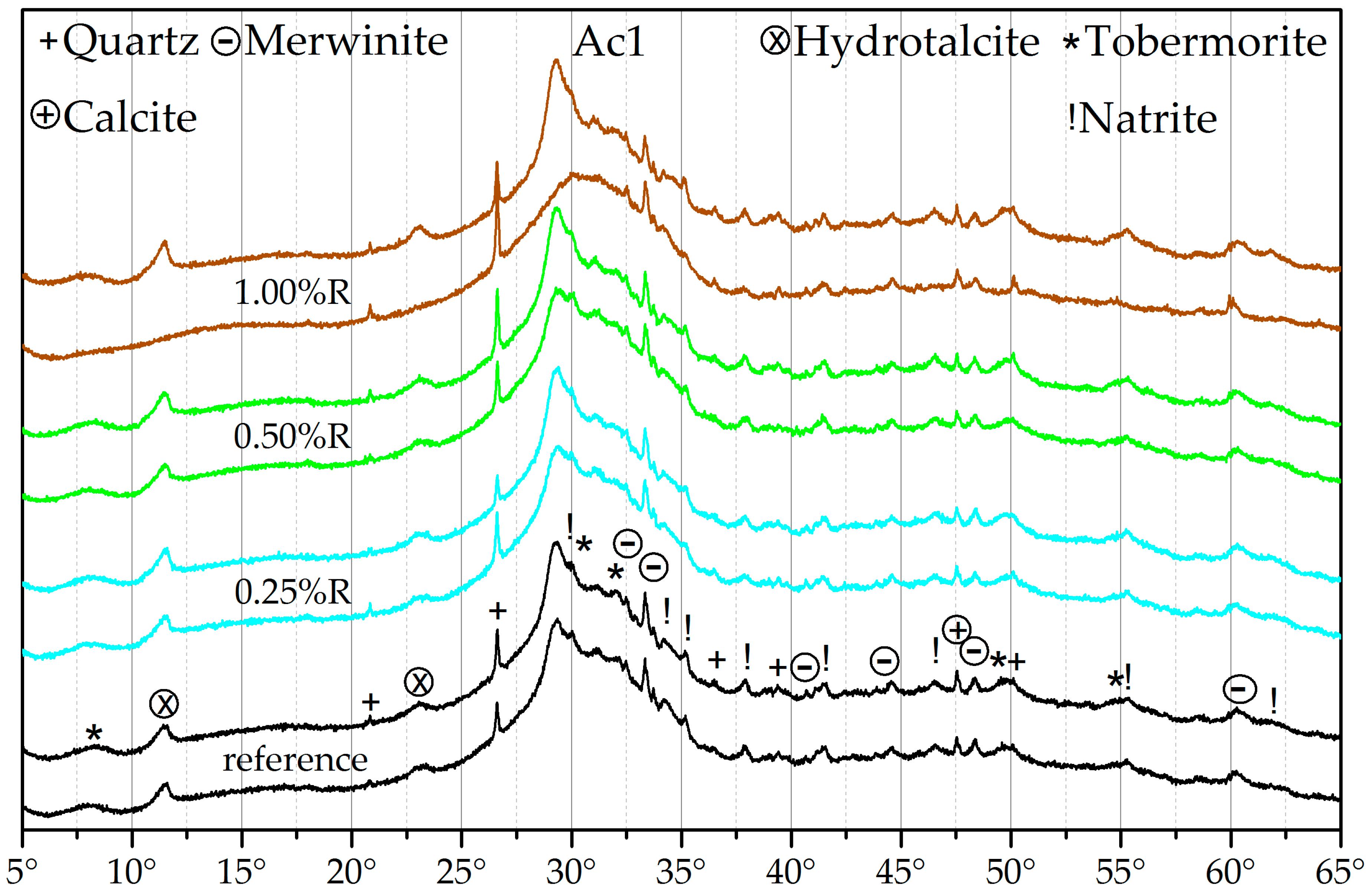
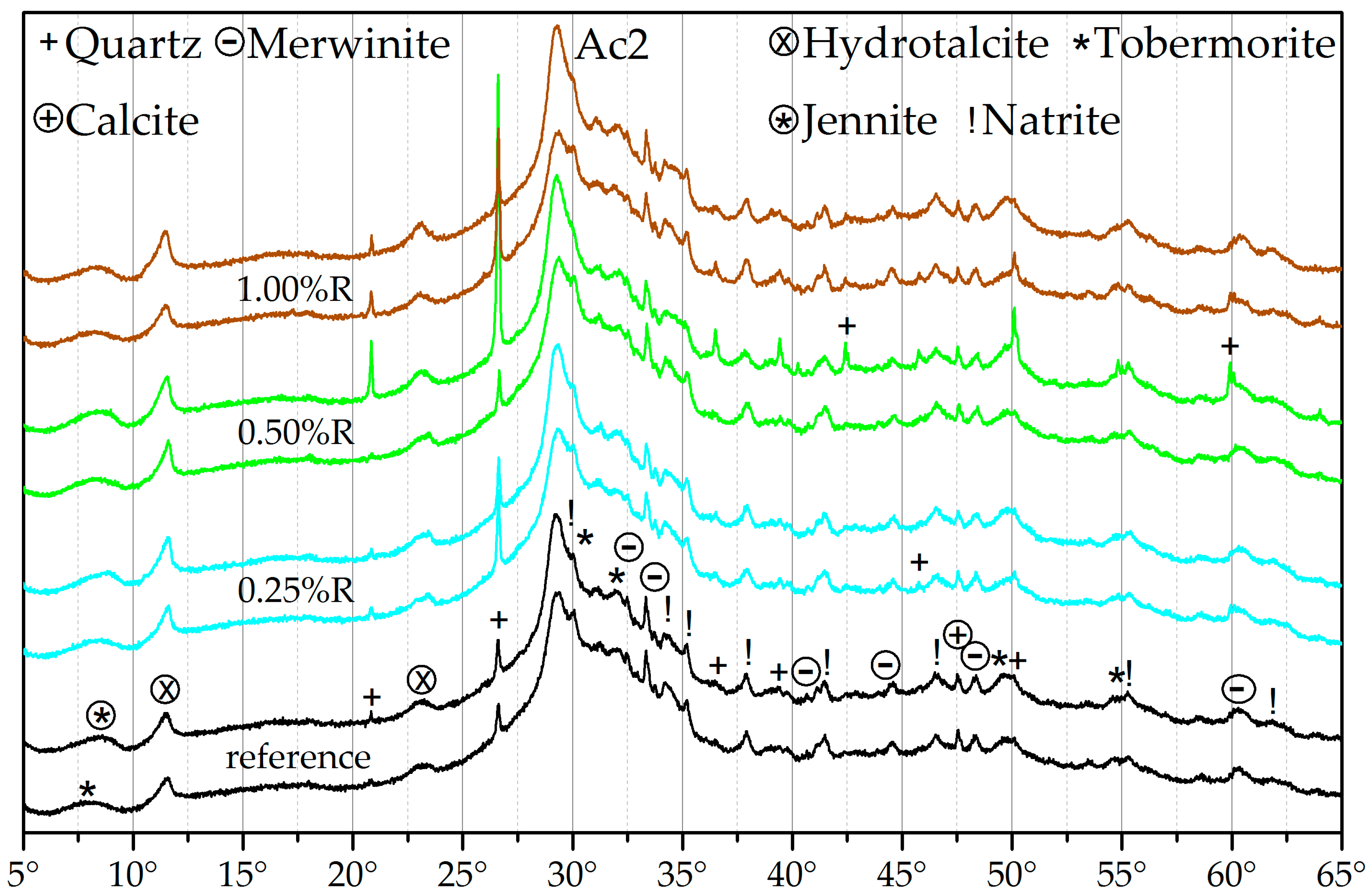
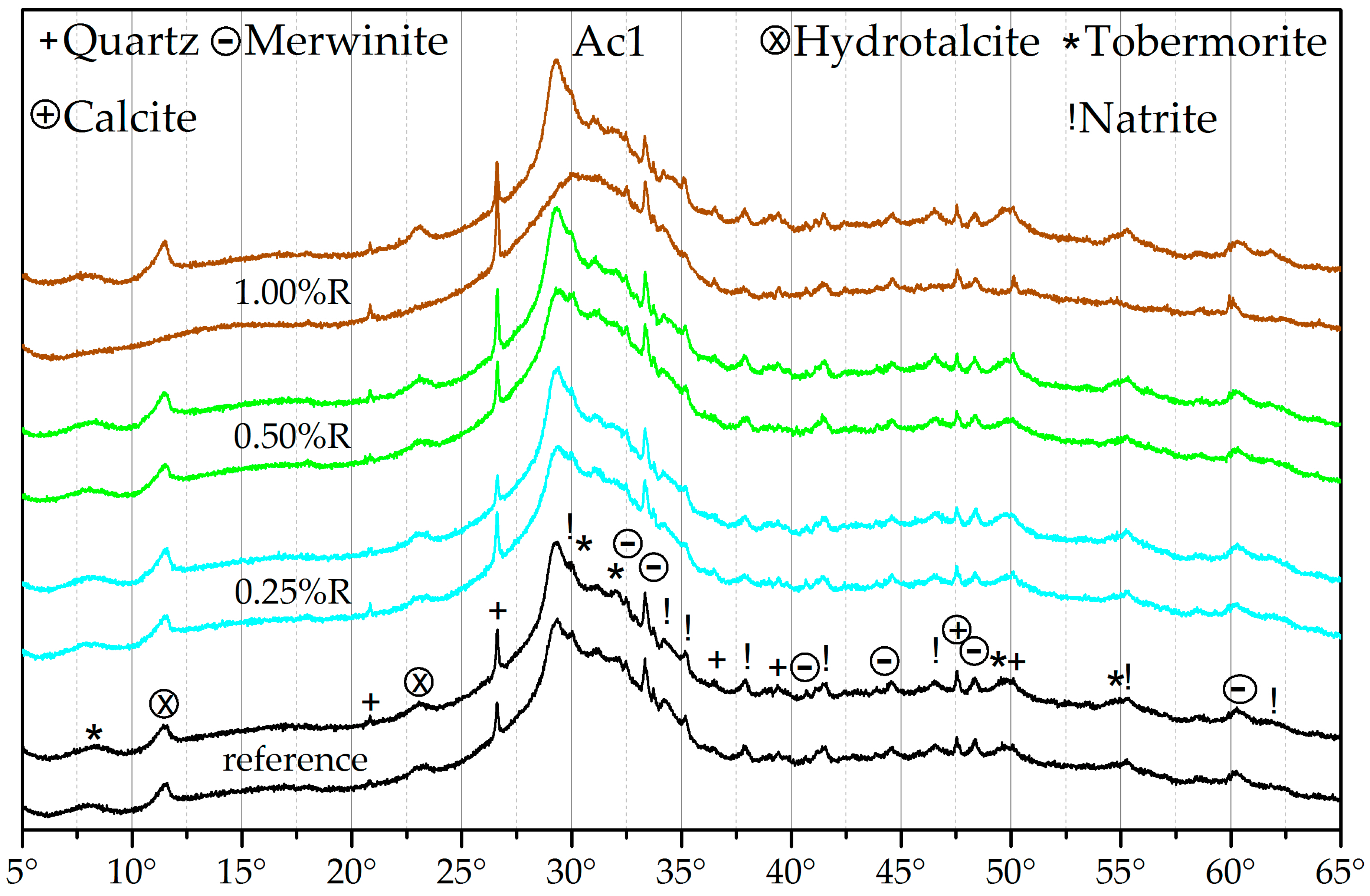
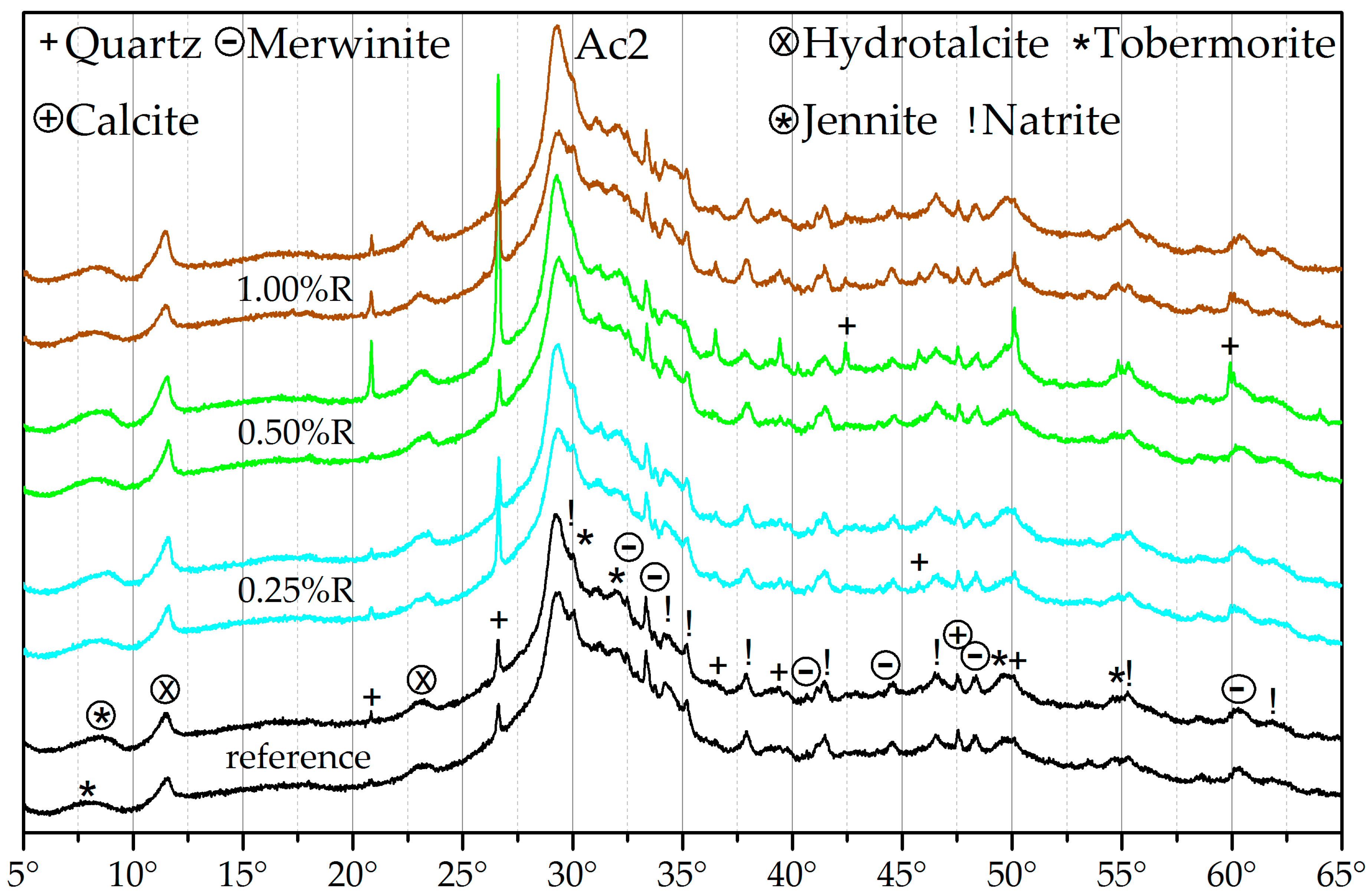
3.3. X-ray Diffraction (XRD)
3.4. Scanning Electron Microscopy (SEM)
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Burciaga-Díaz, O.; Escalante-García, J.I. Structure, mechanisms of reaction, and strength of an alkali-activated blast-furnace slag. J. Am. Ceram. Soc. 2013, 96, 3939–3948. [Google Scholar] [CrossRef]
- Escalante-Garcia, J.I.; Fuentes, A.F.; Gorokhovsky, A.; Fraire-Luna, P.E.; Mendoza-Suarez, G. Hydration products and reactivity of blast-furnace slag activated by various alkalis. J. Am. Ceram. Soc. 2003, 86, 2148–2153. [Google Scholar] [CrossRef]
- Fernández-Jiménez, A.; Palomo, J.G.; Puertas, F. Alkali-activated slag mortars mechanical strength behaviour. Cem. Concr. Res. 1999, 29, 1313–1321. [Google Scholar] [CrossRef]
- Fernández-Jiménez, A.; Puertas, F. Effect of activator mix on the hydration and strength behaviour of alkali-activated slag cements. Adv. Cem. Res. 2003, 15, 129–136. [Google Scholar] [CrossRef]
- Roy, A.; Schilling, P.J.; Eaton, H.C.; Malone, P.G.; Brabston, W.N.; Wakeley, L.D. Activation of ground blast-furnace slag by alkali-metal and alkaline-earth hydroxides. J. Am. Ceram. Soc. 1992, 75, 3233–3240. [Google Scholar] [CrossRef]
- Song, S.; Sohn, D.; Jennings, H.M.; Mason, T.O. Hydration of alkali-activated ground granulated blast furnace slag. J. Mater. Sci. 2000, 35, 249–257. [Google Scholar] [CrossRef]
- Birchall, J.D.; Thomas, N.L. The mechanism of retardation of setting of OPC by sugars. Br. Ceram. Proc. 1984, 35, 305–315. [Google Scholar]
- Juenger, M.C.G.; Jennings, H.M. New insights into the effects of sugar on the hydration and microstructure of cement pastes. Cem. Concr. Res. 2002, 32, 393–399. [Google Scholar] [CrossRef]
- Puertas, F.; Palomo, A.; Fernández-Jiménez, A.; Izquierdo, J.D.; Granizo, M.L. Effect of superplasticisers on the behaviour and properties of alkaline cements. Adv. Cem. Res. 2003, 15, 23–28. [Google Scholar] [CrossRef]
- Langenfeld, M.; Stark, J. The Influence of Retarding Admixtures on the Early Hydration of Portland Cement Phases, Shown in ESEM-FEG; Wissenschaftliche Zeitschrift der Bauhaus-Universität Weimar: Weimar, Germany, 1998; pp. 82–90. [Google Scholar]
- Ludwig, U.; Urrutia Silva, C.R. The mechanism of the action of saccharose on the setting and hardening of cements. ZKG Int. 1989, 42, 431–436. [Google Scholar]
- Luke, K.; Luke, G.A. Effect of sucrose on retardation of Portland cement. Adv. Cem. Res. 2000, 12, 9–18. [Google Scholar] [CrossRef]
- Ma, B.G.; Xiao, J.; Tan, H.B. Effect of citric acid on cement hydration. Adv. Mater. Res. 2012, 393–395, 49–53. [Google Scholar] [CrossRef]
- Ma, S.; Li, W.; Zhang, S.; Ge, D.; Yu, J.; Shen, X. Influence of sodium gluconate on the performance and hydration of Portland cement. Constr. Build. Mater. 2015, 91, 138–144. [Google Scholar] [CrossRef]
- Rickert, J. Influence of retarders on the hydration of clinker and cement (Part 1). Beton 2002, 52, 103–108. [Google Scholar]
- Rickert, J. Influence of retarders on the hydration of clinker and cement (Part 2). Beton 2002, 52, 159–164. [Google Scholar]
- Stephan, D.; Plank, J.P. Einfluss von Verzögerern auf Alit und Zemente mit unterschiedlichem Gehalt an Klinkerphasen. In Bauchemie von der Forschung bis zur Praxis: 5. Tagung Bauchemie in München [9.-10.10.2003]; Plank, J.P., Ed.; Gesellschaft Dt. Chemiker: Frankfurt am Main, Germany, 2003; Volume 27, pp. 31–38. [Google Scholar]
- Tenoutasse, N.; Singh, N.B. Effect of glucose and calcium gluconate on the hydration of Portland cement. Indian J. Technol. 1978, 16, 184–189. [Google Scholar]
- Thomas, N.L.; Birchall, J.D. The retarding action of sugars on cement hydration. Cem. Concr. Res. 1983, 13, 830–842. [Google Scholar] [CrossRef]
- Atarashi, D.; Aikawa, Y.; Yoda, Y.; Miyauchi, M.; Sakai, E. Cement recycling system using sodium gluconate. In Proceedings of the 11th International Conference on Superplasticizers and Other Chemical Admixtures in Concrete, Ottawa, ON, Canada, 12–15 July 2015; pp. 437–447.
- Liao, T.-S.; Hwang, C.-L.; Ye, Y.-S.; Hsu, K.-C. Effects of a carboxylic acid/sulfonic acid copolymer on the material properties of cementitious materials. Cem. Concr. Res. 2006, 36, 650–655. [Google Scholar] [CrossRef]
- Lieber, W.; Richartz, W. Effect of triethanolamine, sugar and boric acids on the setting and hardening of cements. ZKG Int. 1972, 25, 403–409. [Google Scholar]
- Schneider, N.; Stephan, D. The effect of d-gluconic acid as a retarder of ground granulated blast-furnace slag pastes. Constr. Build. Mater. 2016, 123, 99–105. [Google Scholar] [CrossRef]
- Shi, C.; Day, R.L. A calorimetric study of early hydration of alkali-slag cements. Cem. Concr. Res. 1995, 25, 1333–1346. [Google Scholar] [CrossRef]
- Shi, C.; Day, R.L. Some factors affecting early hydration of alkali-slag cements. Cem. Concr. Res. 1996, 26, 439–447. [Google Scholar] [CrossRef]
- Chen, W.; Brouwers, H.J.H. The hydration of slag, part 1: Reaction models for alkali-activated slag. J. Mater. Sci. 2007, 42, 428–443. [Google Scholar] [CrossRef]
- Schneider, N.; Stephan, D. Langzeitverzögerung von Hüttensandmehl und dessen Wiederanregung. In 19. Internationale Baustofftagung, 700th ed.; F.A. Finger-Institut für Baustoffkunde: Weimar, Germany, 2015; pp. 875–882. [Google Scholar]
- Schneider, N.; Stephan, D. Reactivation of a retarded suspension of ground granulated blast-furnace slag. Materials 2016, 9, 174. [Google Scholar] [CrossRef]
- Pang, X.; Boontheung, P.; Boul, P.J. Dynamic retarder exchange as a trigger for Portland cement hydration. Cem. Concr. Res. 2014, 63, 20–28. [Google Scholar] [CrossRef]
- Brothers, L.E.; Pisklak, T.J. Set-Delayed Cement Compositions Comprising Pumice and Associated Methods. U.S. Patent 20140374098 A1, 9 March 2012. [Google Scholar]
- Bullard, J.W.; Jennings, H.M.; Livingston, R.A.; Nonat, A.; Scherer, G.W.; Schweitzer, J.S.; Scrivener, K.L.; Thomas, J.J. Mechanisms of cement hydration. Cem. Concr. Res. 2011, 41, 1208–1223. [Google Scholar] [CrossRef]
- Reiter, L.; Käßmann, R.; Shahab, A.R.; Wangler, T.; Flatt, R.J. Strategies to wake up sleeping concrete. In Proceedings of the 14th ICCC, Beijing, China, 13–16 October 2015.
- Brough, A.R.; Holloway, M.S.; Sykes, J.M.; Atkinson, A. Sodium silicate-based alkali-activated slag mortars Part II. The retarding effect of additions of sodium chloride or malic acid. Cem. Concr. Res. 2000, 30, 1375–1379. [Google Scholar] [CrossRef]
- Pollard, S.J.T.; Montgomery, D.M.; Sollars, C.J.; Perry, R. Organic compounds in the cement-based stabilisation/solidification of hazardous mixed wastes—Mechanistic and process considerations. J. Hazard. Mater. 1991, 28, 313–327. [Google Scholar] [CrossRef]
- Milestone, N.B. Hydration of tricalcium silicate in the presence of lignosulfonates, glucose, and sodium gluconate. J. Am. Ceram. Soc. 1979, 62, 321–324. [Google Scholar] [CrossRef]
- Richartz, W. Einfluß von Zusätzen auf das Erstarrungsverhalten von Zement. Beton 1983, 33, 425–429. [Google Scholar]
- Taylor, H.F.W. Cement Chemistry, 2nd ed.; T. Telford: London, UK, 1997. [Google Scholar]
- Abd El Wahed, M.G.; Helmy, I.M.; El Didamony, H.; Ebied, E. Effect of admixtures on the electrical behaviour of Portland cement. J. Mater. Sci. Lett. 1993, 12, 40–42. [Google Scholar]
- Tsuyuki, N.; Oka, R.; Miyakawa, T.; Kasai, J. Effects of sodium gluconate on the formation of ettringite. J. Ceram. Soc. Jpn. 1990, 98, 332–338. [Google Scholar] [CrossRef]
- Smith, B.J.; Roberts, L.R.; Funkhouser, G.P.; Gupta, V.; Chmelka, B.F. Reactions and surface interactions of saccharides in cement slurries. Langmuir 2012, 28, 14202–14217. [Google Scholar] [CrossRef] [PubMed]
- Kühl, H. Zement-Chemie: Die Erhärtung und die Verarbeitung der Hydraulischen Bindemittel; Verlag Technik: Berlin, Germany, 1961. [Google Scholar]
- Ehrenberg, A.; Israel, D.; Kühn, A.; Ludwig, H.-M.; Tigges, V.; Wassing, W. Granulated blastfurnace slag: reaction potential and production of optimized cements, part 1. Cem. Int. 2008, 6, 90–96. [Google Scholar]
- Stark, J.; Wicht, B. Dauerhaftigkeit von Beton, 2nd ed.; Springer Vieweg: Berlin/Heidelberg, Germany, 2013. [Google Scholar]
- Richartz, W. Über die Gefüge- und Festigkeitsentwicklung des Zementsteins. Beton 1969, 19, 503–506. [Google Scholar]
- Walz, K.; Mathieu, H. Einfluß der Zusatzmenge von Betonverflüssigern auf die Festigkeitsentwicklung. Beton 1961, 11, 691–696. [Google Scholar]
- Han, M.-C.; Han, C.-G. Use of maturity methods to estimate the setting time of concrete containing super retarding agents. Cem. Concr. Compos. 2010, 32, 164–172. [Google Scholar] [CrossRef]
- Ravikumar, D.; Peethamparan, S.; Neithalath, N. Structure and strength of NaOH activated concretes containing fly ash or GGBFS as the sole binder. Cem. Concr. Compos. 2010, 32, 399–410. [Google Scholar] [CrossRef]
- Stephan, D.; Tänzer, R.; Braun, T.; Schmidt, M. Alkali activation—An alternative to binders that contain clinker; part 1. Cem. Int. 2010, 8, 72–85. [Google Scholar]
- Stephan, D.; Tänzer, R.; Braun, T.; Schmidt, M. Alkali activation – an alternative to binders that contain clinker; part 2. Cem. Int. 2010, 8, 74–81. [Google Scholar]
- Živica, V. Effects of type and dosage of alkaline activator and temperature on the properties of alkali-activated slag mixtures. Constr. Build. Mater. 2007, 21, 1463–1469. [Google Scholar] [CrossRef]
- Lothenbach, B.; Ben Haha, M.; Gruskovnjak, A.; Winnefeld, F. Influence of slag composition on hydrate composition and compressive strength. In 19. Internationale Baustofftagung, 700th ed.; F.A. Finger-Institut für Baustoffkunde: Weimar, Germany, 2015; pp. 131–138. [Google Scholar]
- Pacheco-Torgal, F.; Castro-Gomes, J.; Jalali, S. Alkali-activated binders: A review. Part 1. Historical background, terminology, reaction mechanisms and hydration products. Constr. Build. Mater. 2008, 22, 1305–1314. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.; Day, R.L. Selectivity of alkaline activators for the activation of slags. Cem. Concr. Aggreg. 1996, 18, 8–14. [Google Scholar]
- Wang, S.-D.; Scrivener, K.L.; Pratt, P.L. Factors affecting the strength of alkali-activated slag. Cem. Concr. Res. 1994, 24, 1033–1043. [Google Scholar] [CrossRef]
- Fernández-Jiménez, A.; Puertas, F.; Arteaga, Á. Determination of kinetic equations of alkaline activation of blast furnace slag by means of calorimetric data. J. Therm. Anal. Calorim. 1998, 52, 945–955. [Google Scholar] [CrossRef]
- Fernández-Jiménez, A.; Puertas, F. Setting of alkali-activated slag cement. Influence of activator nature. Adv. Cem. Res. 2001, 13, 115–121. [Google Scholar] [CrossRef]
- Tänzer, R.; Buchwald, A.; Stephan, D. Effect of slag chemistry on the hydration of alkali-activated blast-furnace slag. Mater. Struct. 2015, 48, 629–641. [Google Scholar] [CrossRef]
- Schwiete, H.-E.; Ludwig, U.; Würth, K.E.; Grieshammer, G. Neubildungen bei der Hydratation von Hochofenschlacken. ZKG Int. 1969, 21, 154–160. [Google Scholar]
- Pacheco-Torgal, F.; Castro-Gomes, J.; Jalali, S. Alkali-activated binders: A review. Part 1. Historical background, terminology, reaction mechanisms and hydration products. Constr. Build. Mater. 2008, 22, 1305–1314. [Google Scholar] [CrossRef] [Green Version]
- Puertas, F.; Fernández-Jiménez, A.; Blanco-Varela, M.T. Pore solution in alkali-activated slag cement pastes. Relation to the composition and structure of calcium silicate hydrate. Cem. Concr. Res. 2004, 34, 139–148. [Google Scholar] [CrossRef]
- Song, S.; Jennings, H.M. Pore solution chemistry of alkali-activated ground granulated blast-furnace slag. Cem. Concr. Res. 1999, 29, 159–170. [Google Scholar] [CrossRef]
- Ben Haha, M.; Lothenbach, B.; Le Saout, G.; Winnefeld, F. Influence of slag chemistry on the hydration of alkali-activated blast-furnace slag—Part I: Effect of MgO. Cem. Concr. Res. 2011, 41, 955–963. [Google Scholar] [CrossRef]
- Ben Haha, M.; Lothenbach, B.; Le Saout, G.; Winnefeld, F. Influence of slag chemistry on the hydration of alkali-activated blast-furnace slag—Part II: Effect of Al2O3. Cem. Concr. Res. 2012, 42, 74–83. [Google Scholar] [CrossRef]
- Fernández-Jiménez, A.; Puertas, F.; Sobrados, I.; Sanz, J. Structure of calcium silicate hydrates formed in alkaline-activated slag: Influence of the type of alkaline activator. J. Am. Ceram. Soc. 2003, 86, 1389–1394. [Google Scholar] [CrossRef]
- Wang, S.-D.; Scrivener, K.L. Hydration products of alkali activated slag cement. Cem. Concr. Res. 1995, 25, 561–571. [Google Scholar] [CrossRef]
- Oh, J.E.; Monteiro, P.J.; Jun, S.S.; Choi, S.; Clark, S.M. The evolution of strength and crystalline phases for alkali-activated ground blast furnace slag and fly ash-based geopolymers. Cem. Concr. Res. 2010, 40, 189–196. [Google Scholar] [CrossRef]
- Gruskovnjak, A.; Lothenbach, B.; Holzer, L.; Figi, R.; Winnefeld, F. Hydration of alkali-activated slag: comparison with ordinary Portland cement. Adv. Cem. Res. 2006, 18, 119–128. [Google Scholar] [CrossRef]
- Richardson, I.G.; Brough, A.R.; Groves, G.W.; Dobson, C.M. The characterization of hardened alkali-activated blast-furnace slag pastes and the nature of the calcium silicate hydrate (C-S-H) phase. Cem. Concr. Res. 1994, 24, 813–829. [Google Scholar] [CrossRef]
- Brough, A.R.; Atkinson, A. Sodium silicate-based, alkali-activated slag mortars Part I. Strength, hydration and microstructure. Cem. Concr. Res. 2002, 32, 865–879. [Google Scholar] [CrossRef]
- Myers, R.J.; Bernal, S.A.; San Nicolas, R.; Provis, J.L. Generalized structural description of calcium–sodium aluminosilicate hydrate gels: The cross-linked substituted tobermorite model. Langmuir 2013, 29, 5294–5306. [Google Scholar] [CrossRef] [PubMed]
- Hong, S.-Y.; Glasser, F.P. Alkali binding in cement pastes: Part I. The C-S-H phase. Cem. Concr. Res. 1999, 29, 1893–1903. [Google Scholar] [CrossRef]
- Hong, S.-Y.; Glasser, F.P. Alkali sorption by C-S-H and C-A-S-H gels: Part II. Role of alumina. Cem. Concr. Res. 2002, 32, 1101–1111. [Google Scholar] [CrossRef]
- Nonat, A. The structure and stoichiometry of C-S-H. Cem. Concr. Res. 2004, 34, 1521–1528. [Google Scholar] [CrossRef]
- Richardson, I.G. Tobermorite/jennite- and tobermorite/calcium hydroxide-based models for the structure of C-S-H: Applicability to hardened pastes of tricalcium silicate, beta-dicalcium silicate, Portland cement, and blends of Portland cement with blast-furnace slag, metakaolin, or silica fume. Cem. Concr. Res. 2004, 34, 1733–1777. [Google Scholar]
- Richardson, I.G. The calcium silicate hydrates. Cem. Concr. Res. 2008, 38, 137–158. [Google Scholar] [CrossRef]
- Richartz, W.; Locher, F.W. Ein Beitrag zur Morphologie und Wasserbindung von Calciumsilicathydraten und zum Gefüge des Zementsteins. ZKG Int. 1965, 18, 449–459. [Google Scholar]
- Benedix, R. Bauchemie: Einführung in die Chemie für Bauingenieure und Architekten, 5th ed.; Vieweg + Teubner Verlag/Springer Fachmedien Wiesbaden GmbH, Wiesbaden: Wiesbaden, Germany, 2011. [Google Scholar]
- Ehrenberg, A.; Israel, D.; Kühn, A.; Ludwig, H.-M.; Tigges, V.; Wassing, W. Granulated blastfurnace slag: reaction potential and production of optimized cements, part 2. Cem. Int. 2008, 6, 82–92. [Google Scholar]
- DIN Deutsches Institut für Normung e.V. DIN 66145: Darstellung von Korn-(Teilchen-)größenverteilungen RRSB-Netz; Beuth: Berlin, Germany, 1976. [Google Scholar]
- DIN Deutsches Institut für Normung e.V. DIN EN 196-1: Methods of Testing Cement—Part 1: Determination of Strength; German Version EN 196-1; Beuth: Berlin, Germany, 2005. [Google Scholar]
- Ben Haha, M.; Le Saout, G.; Winnefeld, F.; Lothenbach, B. Influence of activator type on hydration kinetics, hydrate assemblage and microstructural development of alkali activated blast-furnace slags. Cem. Concr. Res. 2011, 41, 301–310. [Google Scholar] [CrossRef]
- Palmer, S.J.; Grand, L.M.; Frost, R.L. Thermal analysis of hydrotalcite synthesised from aluminate solutions. J. Therm. Anal. Calorim. 2011, 103, 473–478. [Google Scholar] [CrossRef] [Green Version]
- Cavani, F.; Trifirò, F.; Vaccari, A. Hydrotalcite-type anionic clays: Preparation, properties and applications. Catal. Today 1991, 11, 173–301. [Google Scholar] [CrossRef]
- Schneider, C.; Baetzner, S. X-ray diffractometric determination of the granulated blastfurnace slag content of cement. Cem. Int. 2005, 3, 96–105. [Google Scholar]
- Bernal, S.A.; Provis, J.L.; Myers, R.J.; San Nicolas, R.; van Deventer, J.S.J. Role of carbonates in the chemical evolution of sodium carbonate-activated slag binders. Mater. Struct. 2015, 48, 517–529. [Google Scholar] [CrossRef]
- Baston, G.M.N.; Clacher, A.P.; Heath, T.G.; Hunter, F.M.I.; Smith, V.; Swanton, S.W. Calcium silicate hydrate (C-S-H) gel dissolution and pH buffering in a cementitious near field. Mineral. Mag. 2012, 76, 3045–3053. [Google Scholar] [CrossRef]
- Zeng, H.-Y.; Wang, Y.-J.; Feng, Z.; You, K.-Y.; Zhao, C.; Sun, J.-W.; Liu, P.-L. Synthesis of propylene glycol monomethyl ether over Mg/Al hydrotalcite catalyst. Catal. Lett. 2010, 137, 94–103. [Google Scholar] [CrossRef]
- Rozov, K.; Berner, U.; Taviot-Gueho, C.; Leroux, F.; Renaudin, G.; Kulik, D.; Diamond, L.W. Synthesis and characterization of the LDH hydrotalcite–pyroaurite solid-solution series. Cem. Concr. Res. 2010, 40, 1248–1254. [Google Scholar] [CrossRef]
- Kanezaki, E. Thermal behavior of the hydrotalcite-like layered structure of Mg and Al-layered double hydroxides with interlayer carbonate by means of in situ powder HTXRD and DTA/TG. Solid State Ion. 1998, 106, 279–284. [Google Scholar] [CrossRef]
- Provis, J.L. Geopolymers and other alkali activated materials: Why, how, and what? Mater. Struct. 2014, 47, 11–25. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Components | Retarded Binder [wt.%] | Reactivated Binder [wt.%] | |
|---|---|---|---|
| GGBFS | 76.9 | 69.2 | |
| Retarder solution | 23.0 | 20.8 | |
| MC | 0.1 | 0.1 | |
| Activator solution | -/- | 9.9 | |
| Activator | -/- | Ac1 | Ac2 |
| w/b ratio | 0.30 | 0.40 | 0.37 |
| (a) | [wt.%] |
| SiO2 | 35.6 |
| Al2O3 | 10.6 |
| Fe2O3 | 0.7 |
| MnO | 0.2 |
| MgO | 7.4 |
| CaO | 43.2 |
| Na2O | 0.2 |
| K2O | 0.4 |
| TiO2 | 0.7 |
| (b) | |
| Density | 2.92 kg/dm3 |
| Specific surface area (Blaine) | 3218 cm²/g |
| Water demand (Puntke) | 19.1 wt.% |
| RRSB-Distribution x0/n (*) | 20.7/1.7 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Schneider, N.; Stephan, D. Studying the Hydration of a Retarded Suspension of Ground Granulated Blast-Furnace Slag after Reactivation. Materials 2016, 9, 933. https://doi.org/10.3390/ma9110933
Schneider N, Stephan D. Studying the Hydration of a Retarded Suspension of Ground Granulated Blast-Furnace Slag after Reactivation. Materials. 2016; 9(11):933. https://doi.org/10.3390/ma9110933
Chicago/Turabian StyleSchneider, Nick, and Dietmar Stephan. 2016. "Studying the Hydration of a Retarded Suspension of Ground Granulated Blast-Furnace Slag after Reactivation" Materials 9, no. 11: 933. https://doi.org/10.3390/ma9110933