
Oxygen Evolution at Manganite Perovskite Ruddlesden-Popper Type Particles: Trends of Activity on Structure, Valence and Covalence

 , ,
, ,
Abstract
:1. Introduction
2. Results and Discussion
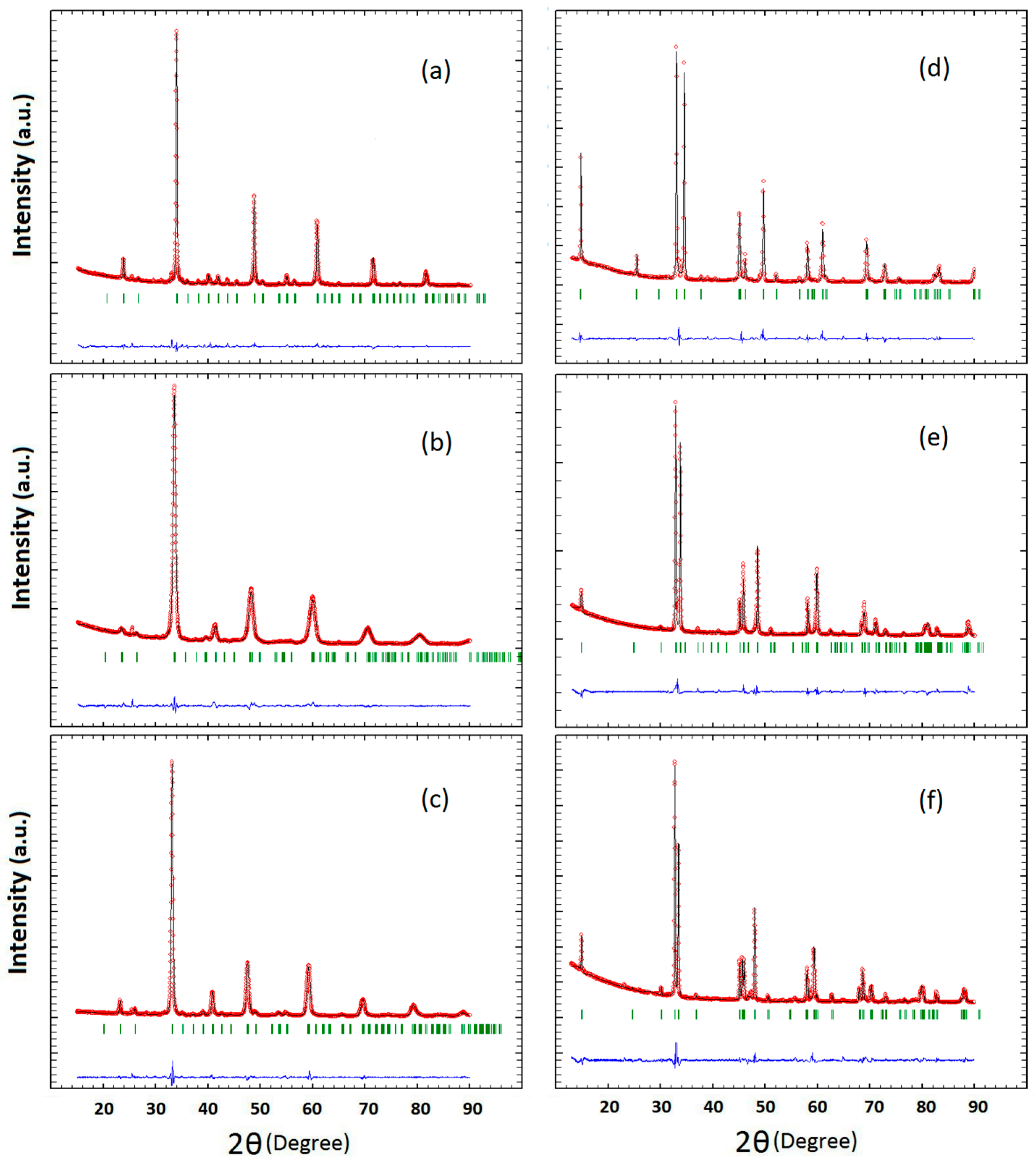
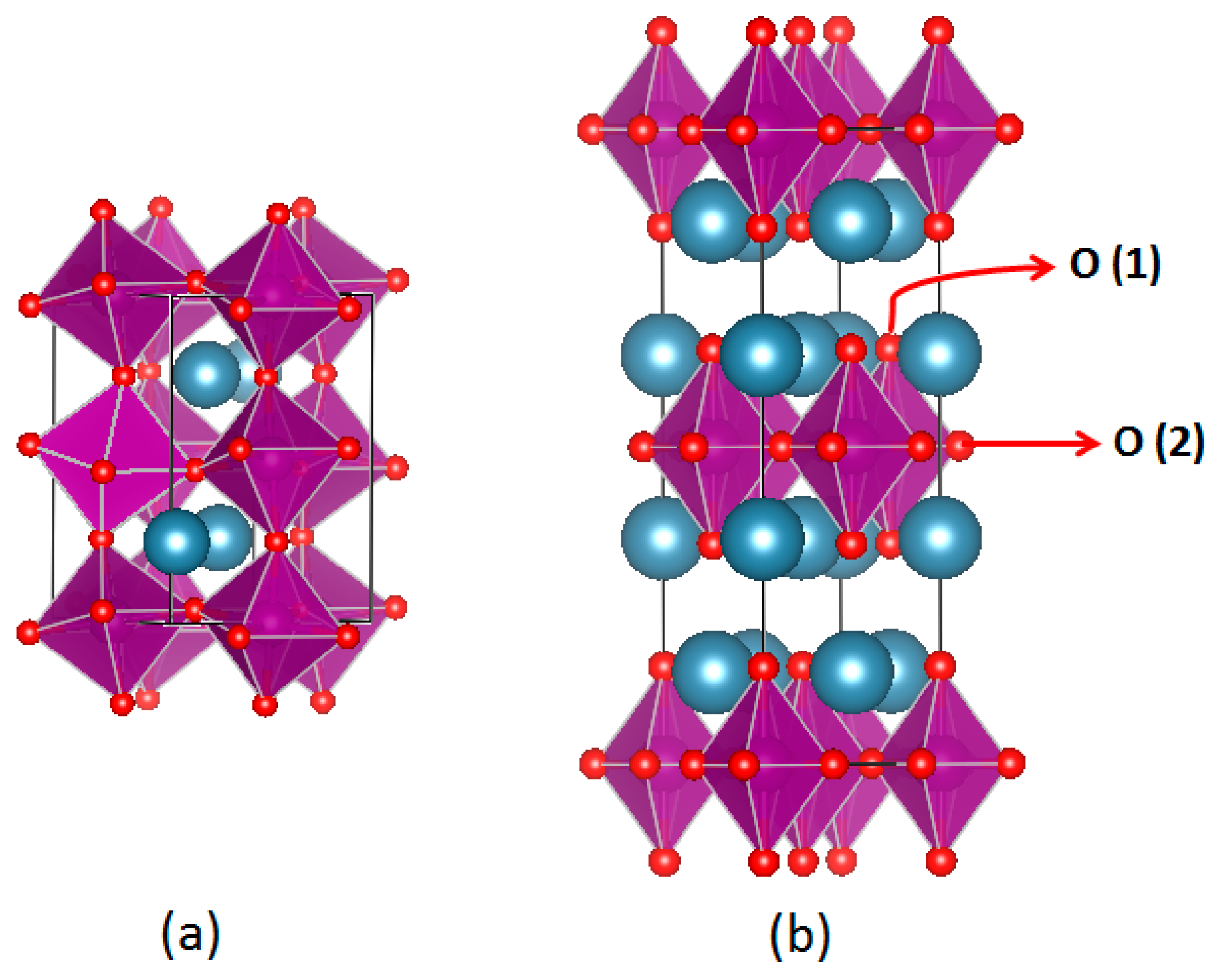
2.1. Crystal Structure
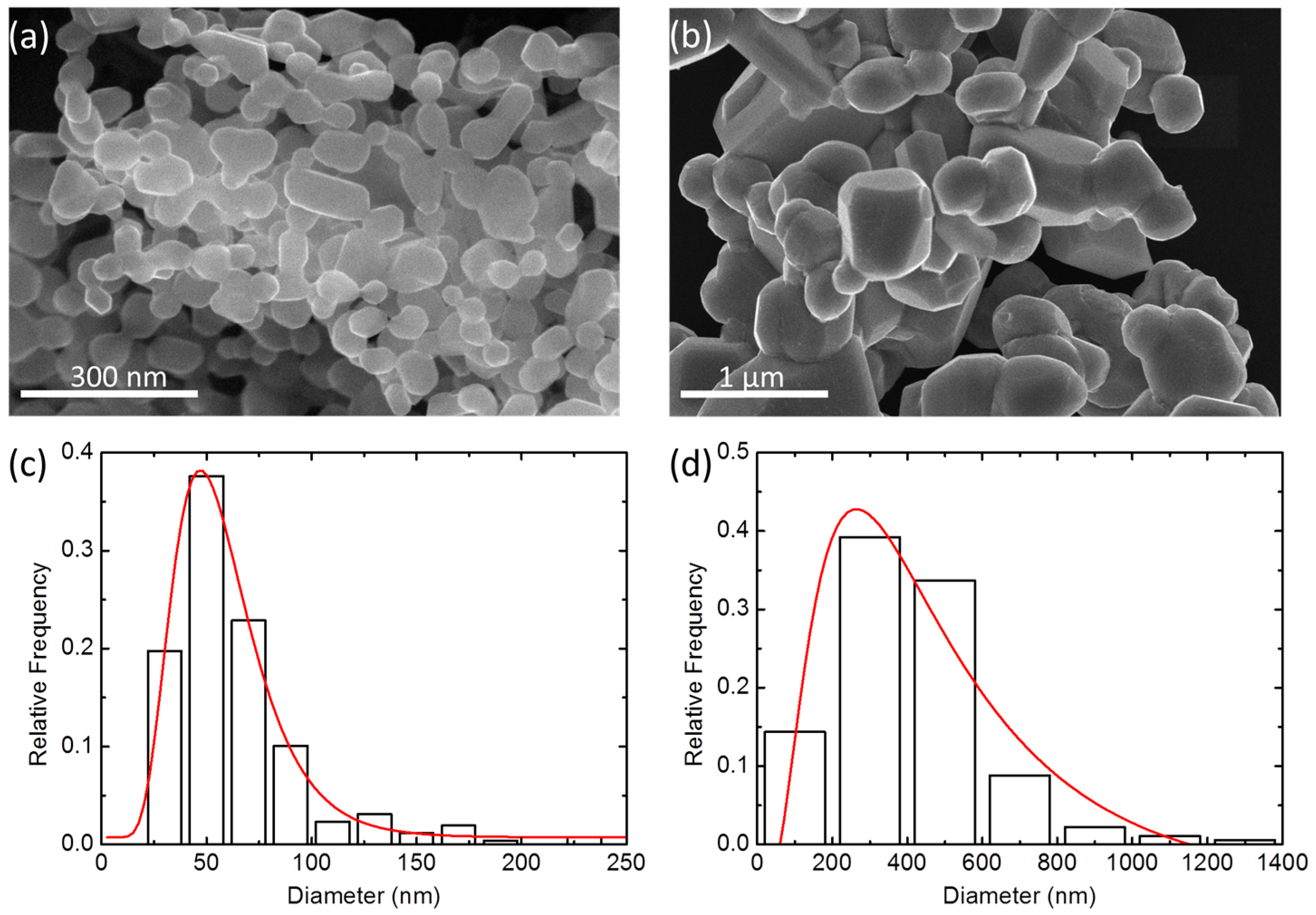
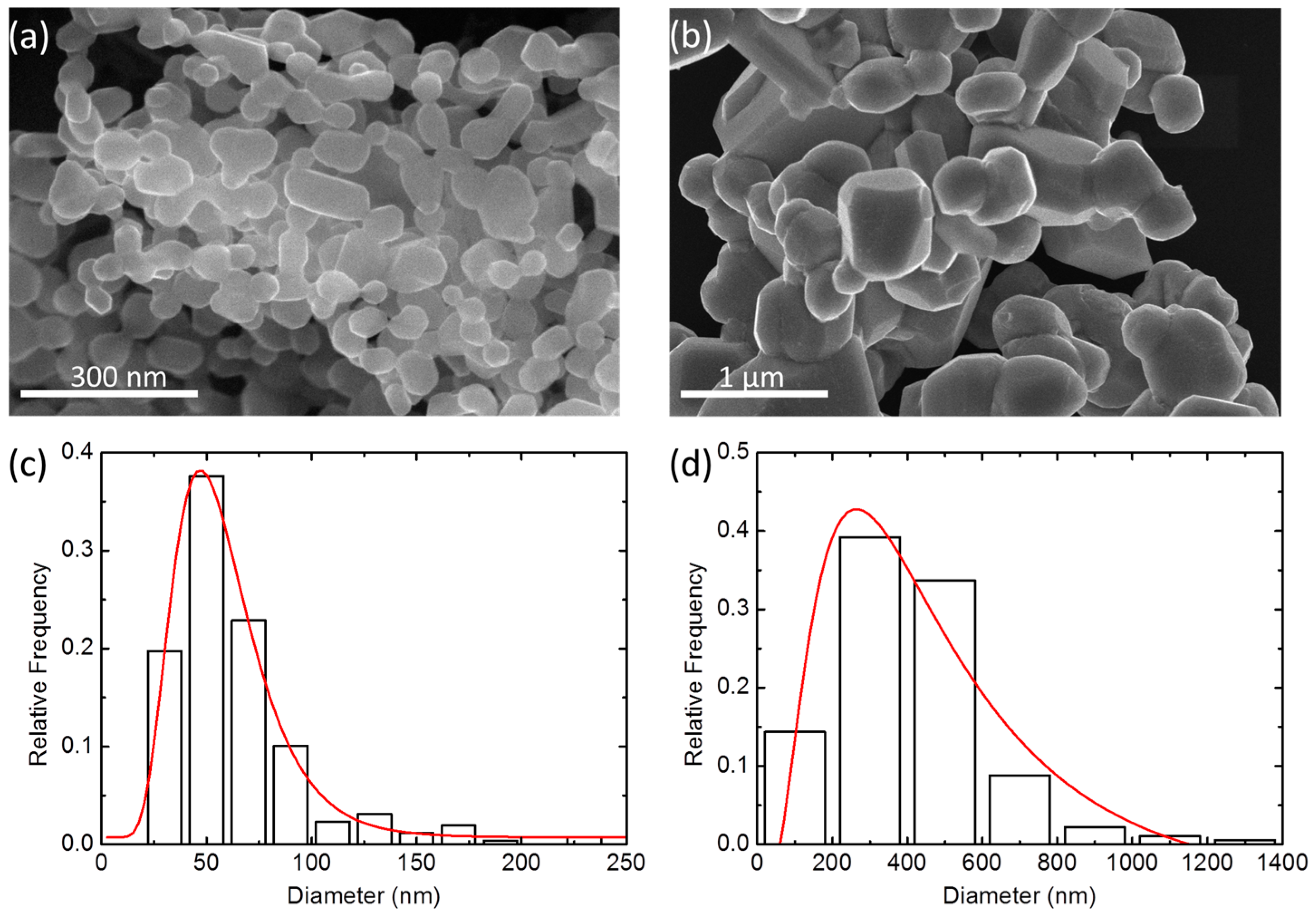
2.2. Particle Size and Microstructure
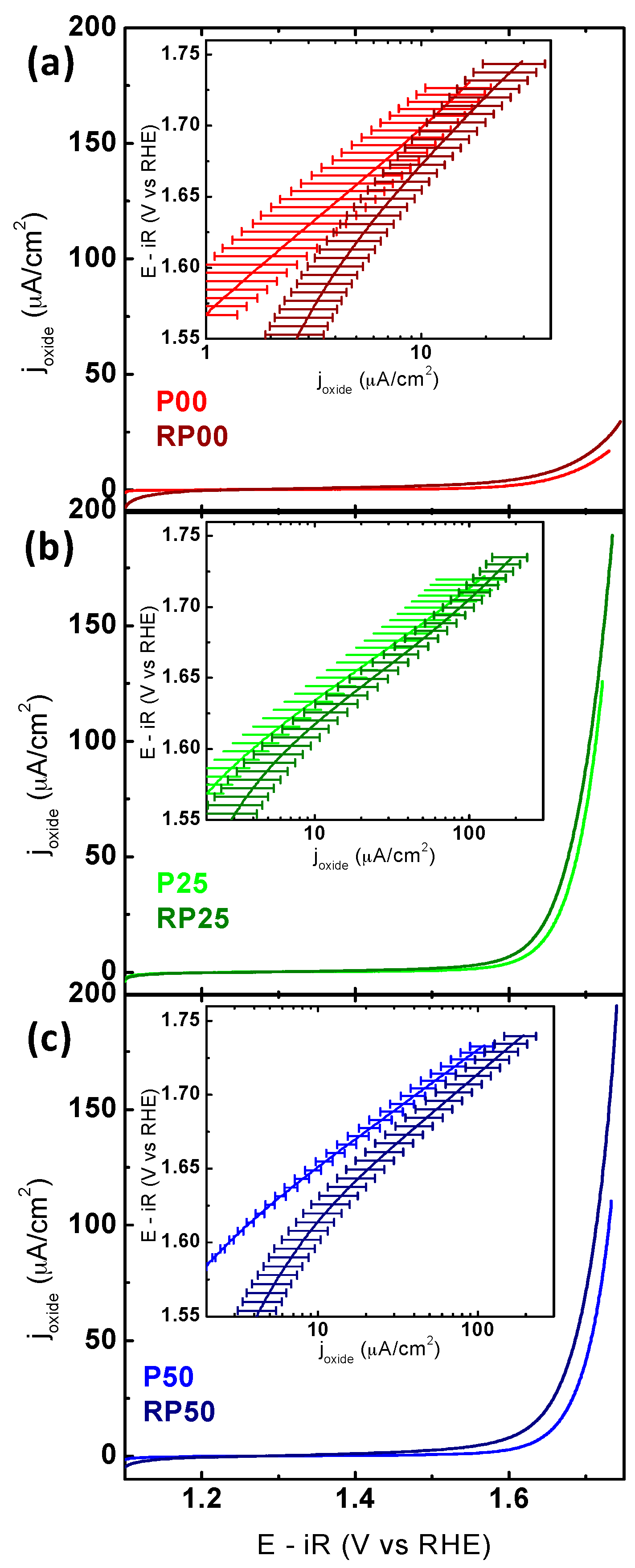
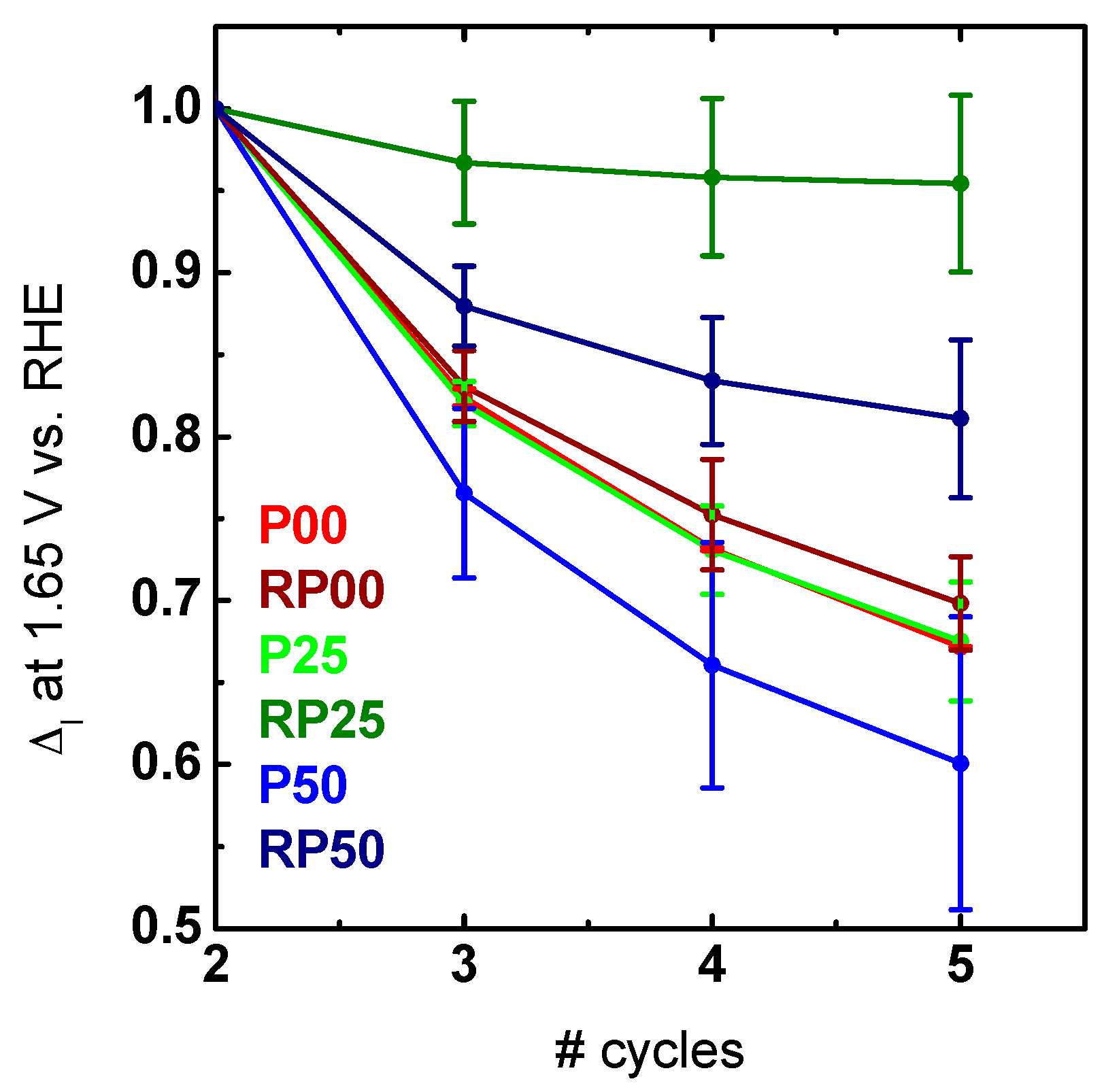
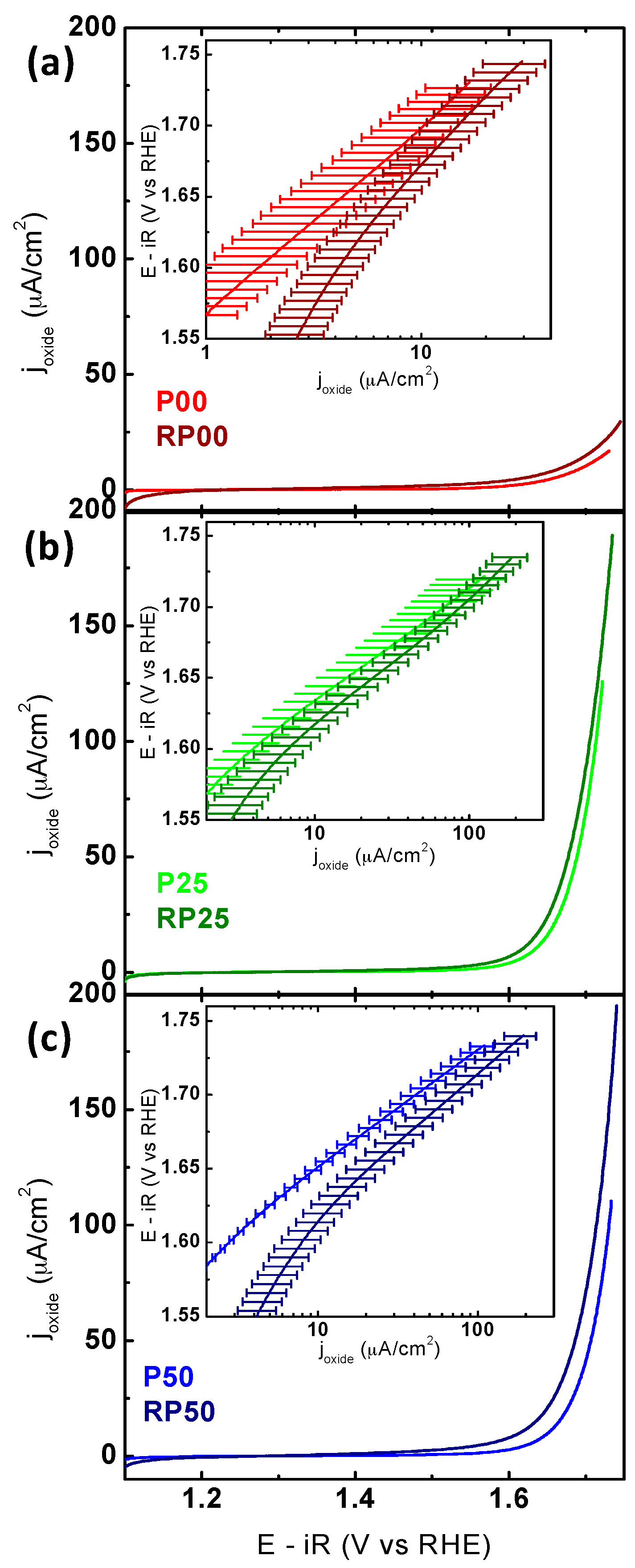
2.3. Electrochemical Activity and Stability Tests
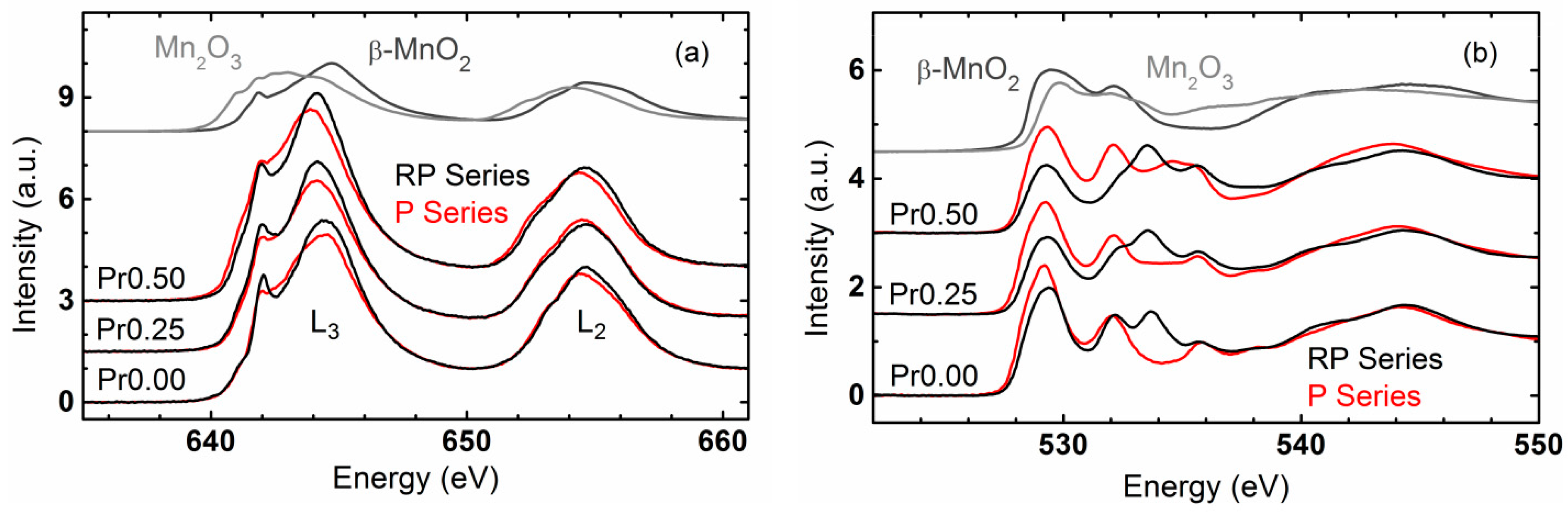
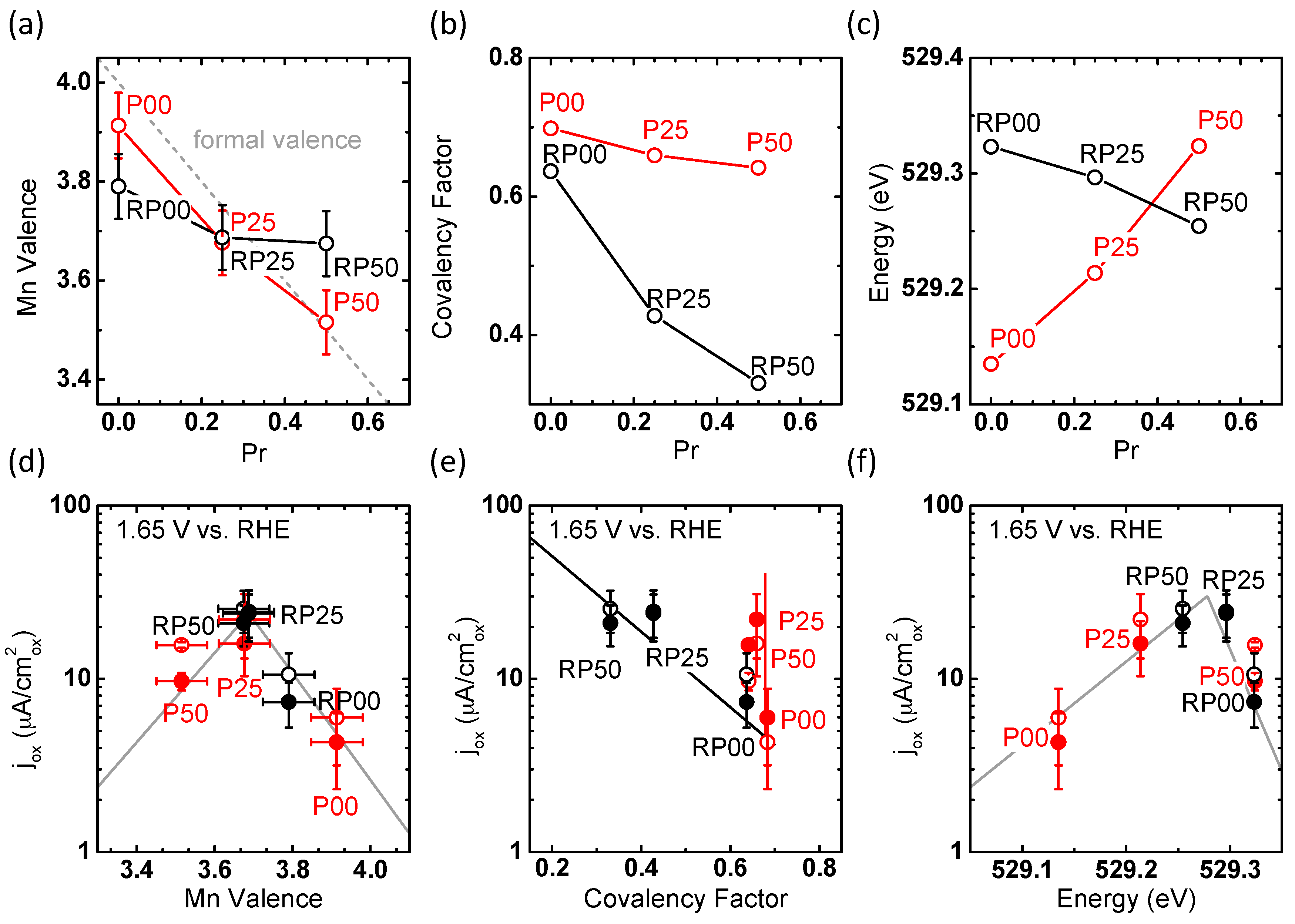
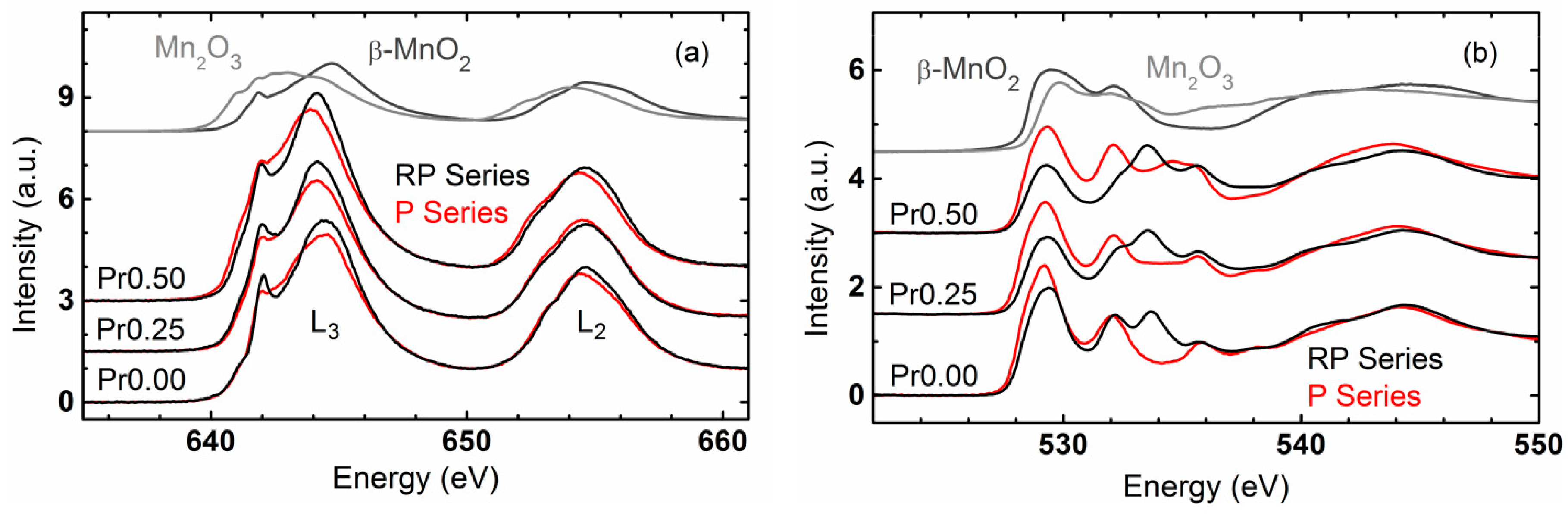
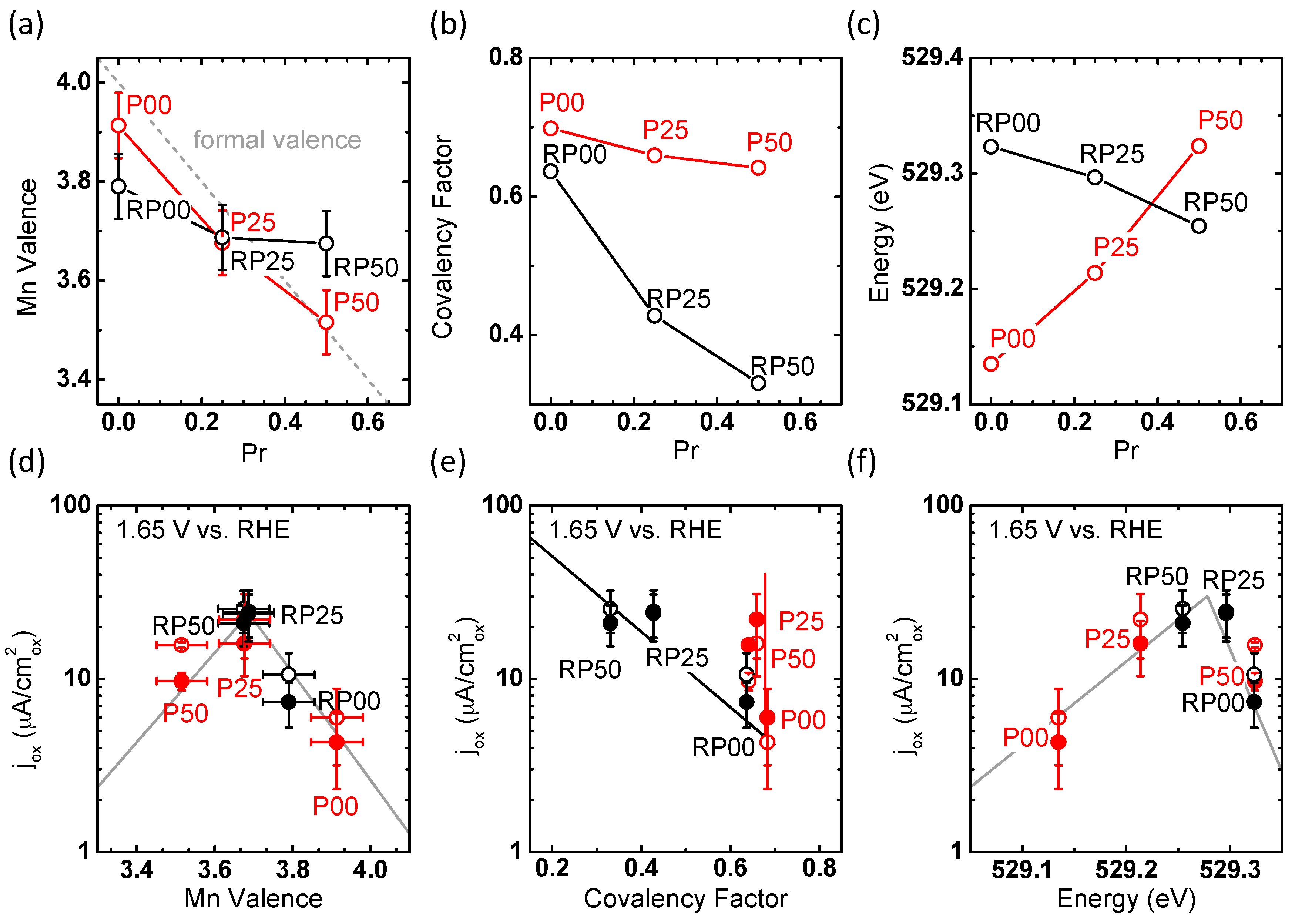
2.4. Trends of OER Activity with Valence State and Covalency
3. Materials and Methods
3.1. Synthesis Process
3.2. Characterizations
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Bockris, J.O.M.; Otagawa, T. The electrocatalysis of oxygen evolution on perovskites. J. Electrochem. Soc. 1984, 131, 290–302. [Google Scholar] [CrossRef]
- Bockris, J.O.; Otagawa, T. Mechanism of oxygen evolution on perovskites. J. Phys. Chem. 1983, 87, 2960–2971. [Google Scholar] [CrossRef]
- Matsumoto, Y.; Sato, E. Electrocatalytic properties of transition metal oxides for oxygen evolution reaction. Mater. Chem. Phys. 1986, 14, 397–426. [Google Scholar] [CrossRef]
- Bockris, J.O.M. Kinetics of Activation Controlled Consecutive Electrochemical Reactions: Anodic Evolution of Oxygen. J. Chem. Phys. 1956, 24, 817–827. [Google Scholar] [CrossRef]
- Hardin, W.G.; Mefford, J.T.; Slanac, D.A.; Patel, B.B.; Wang, X.; Dai, S.; Zhao, X.; Ruoff, R.S.; Johnston, K.P.; Stevenson, K.J. Tuning the Electrocatalytic Activity of Perovskites through Active Site Variation and Support Interactions. Chem. Mater. 2014, 26, 3368–3376. [Google Scholar] [CrossRef]
- Hardin, W.G.; Slanac, D.A.; Wang, X.; Dai, S.; Johnston, K.P.; Stevenson, K.J. Highly Active, Nonprecious Metal Perovskite Electrocatalysts for Bifunctional Metal–Air Battery Electrodes. J. Phys. Chem. Lett. 2013, 4, 1254–1259. [Google Scholar] [CrossRef] [PubMed]
- Vojvodic, A.; Nørskov, J.K. Optimizing Perovskites for the Water-Splitting Reaction. Science 2011, 334, 1355–1356. [Google Scholar] [CrossRef] [PubMed]
- Seo, M.H.; Park, H.W.; Lee, D.U.; Park, M.G.; Chen, Z. Design of Highly Active Perovskite Oxides for Oxygen Evolution Reaction by Combining Experimental and ab Initio Studies. ACS Catal. 2015, 5, 4337–4344. [Google Scholar] [CrossRef]
- Trasatti, S. Electrocatalysis by oxides—Attempt at a unifying approach. J. Electroanal. Chem. Interfacial Electrochem. 1980, 111, 125–131. [Google Scholar] [CrossRef]
- Smith, R.D.L.; Prévot, M.S.; Fagan, R.D.; Zhang, Z.; Sedach, P.A.; Siu, M.K.J.; Trudel, S.; Berlinguette, C.P. Photochemical Route for Accessing Amorphous Metal Oxide Materials for Water Oxidation Catalysis. Science 2013, 340, 60–63. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.; Trudel, S.; Berlinguette, C.P. Water Oxidation Catalysis: Survey of Amorphous Binary Metal Oxide Films Containing Lanthanum and Late 3d Transition Metals. Eur. J. Inorgan. Chem. 2014, 2014, 660–664. [Google Scholar] [CrossRef]
- Zhang, Z.; Liu, J.; Gu, J.; Su, L.; Cheng, L. An overview of metal oxide materials as electrocatalysts and supports for polymer electrolyte fuel cells. Energy Environ. Sci. 2014, 7, 2535–2558. [Google Scholar] [CrossRef]
- Trotochaud, L.; Ranney, J.K.; Williams, K.N.; Boettcher, S.W. Solution-Cast Metal Oxide Thin Film Electrocatalysts for Oxygen Evolution. J. Am. Chem. Soc. 2012, 134, 17253–17261. [Google Scholar] [CrossRef] [PubMed]
- Doyle, R.L.; Godwin, I.J.; Brandon, M.P.; Lyons, M.E.G. Redox and electrochemical water splitting catalytic properties of hydrated metal oxide modified electrodes. Phys. Chem. Chem. Phys. 2013, 15, 13737–13783. [Google Scholar] [CrossRef] [PubMed]
- Hong, W.T.; Risch, M.; Stoerzinger, K.A.; Grimaud, A.; Suntivich, J.; Shao-Horn, Y. Toward the rational design of non-precious transition metal oxides for oxygen electrocatalysis. Energy Environ. Sci. 2015, 8, 1404–1427. [Google Scholar] [CrossRef]
- Mom, R.V.; Cheng, J.; Koper, M.T.M.; Sprik, M. Modeling the Oxygen Evolution Reaction on Metal Oxides: The Infuence of Unrestricted DFT Calculations. J. Phys. Chem. C 2014, 118, 4095–4102. [Google Scholar] [CrossRef]
- Su, H.-Y.; Gorlin, Y.; Man, I.C.; Calle-Vallejo, F.; Norskov, J.K.; Jaramillo, T.F.; Rossmeisl, J. Identifying active surface phases for metal oxide electrocatalysts: A study of manganese oxide bi-functional catalysts for oxygen reduction and water oxidation catalysis. Phys. Chem. Chem. Phys. 2012, 14, 14010–14022. [Google Scholar] [CrossRef] [PubMed]
- Arrigo, R.; Hävecker, M.; Schuster, M.E.; Ranjan, C.; Stotz, E.; Knop-Gericke, A.; Schlögl, R. In Situ Study of the Gas-Phase Electrolysis of Water on Platinum by NAP-XPS. Angew. Chem. Int. Ed. 2013, 52, 11660–11664. [Google Scholar] [CrossRef] [PubMed]
- Torrance, J.B.; Lacorre, P.; Asavaroengchai, C.; Metzger, R.M. Why are some oxides metallic, while most are insulating? Phys. C Supercond. 1991, 182, 351–364. [Google Scholar] [CrossRef]
- Suntivich, J.; Hong, W.T.; Lee, Y.-L.; Rondinelli, J.M.; Yang, W.; Goodenough, J.B.; Dabrowski, B.; Freeland, J.W.; Shao-Horn, Y. Estimating Hybridization of Transition Metal and Oxygen States in Perovskites from O K-edge X-ray Absorption Spectroscopy. J. Phys. Chem. C 2014, 118, 1856–1863. [Google Scholar] [CrossRef]
- Alghamdi, K.; Hargreaves, J.S.J.; Jackson, S.D. Base Catalysis with Metal Oxides. In Metal Oxide Catalysis; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2009; pp. 819–843. [Google Scholar]
- Rossmeisl, J.; Qu, Z.W.; Zhu, H.; Kroes, G.J.; Nørskov, J.K. Electrolysis of water on oxide surfaces. J. Electroanal. Chem. 2007, 607, 83–89. [Google Scholar] [CrossRef]
- Rossmeisl, J.; Logadottir, A.; Nørskov, J.K. Electrolysis of water on (oxidized) metal surfaces. Chem. Phys. 2005, 319, 178–184. [Google Scholar] [CrossRef]
- Raabe, S.; Mierwaldt, D.; Ciston, J.; Uijttewaal, M.; Stein, H.; Hoffmann, J.; Zhu, Y.; Blöchl, P.; Jooss, C. In Situ Electrochemical Electron Microscopy Study of Oxygen Evolution Activity of Doped Manganite Perovskites. Adv. Funct. Mater. 2012, 22, 3378–3388. [Google Scholar] [CrossRef]
- Suntivich, J.; Gasteiger, H.A.; Yabuuchi, N.; Shao-Horn, Y. Electrocatalytic Measurement Methodology of Oxide Catalysts Using a Thin-Film Rotating Disk Electrode. J. Electrochem. Soc. 2010, 157, B1263–B1268. [Google Scholar] [CrossRef]
- May, K.J.; Carlton, C.E.; Stoerzinger, K.A.; Risch, M.; Suntivich, J.; Lee, Y.-L.; Grimaud, A.; Shao-Horn, Y. Influence of Oxygen Evolution during Water Oxidation on the Surface of Perovskite Oxide Catalysts. J. Phys. Chem. Lett. 2012, 3, 3264–3270. [Google Scholar] [CrossRef]
- Risch, M.; Grimaud, A.; May, K.J.; Stoerzinger, K.A.; Chen, T.J.; Mansour, A.N.; Shao-Horn, Y. Structural Changes of Cobalt-Based Perovskites upon Water Oxidation Investigated by EXAFS. J. Phys. Chem. C 2013, 117, 8628–8635. [Google Scholar] [CrossRef]
- Grimaud, A.; May, K.J.; Carlton, C.E.; Lee, Y.-L.; Risch, M.; Hong, W.T.; Zhou, J.; Shao-Horn, Y. Double perovskites as a family of highly active catalysts for oxygen evolution in alkaline solution. Nat. Commun. 2013, 4, 2439. [Google Scholar] [CrossRef] [PubMed]
- Bassat, J.-M.; Burriel, M.; Wahyudi, O.; Castaing, R.; Ceretti, M.; Veber, P.; Weill, I.; Villesuzanne, A.; Grenier, J.-C.; Paulus, W.; et al. Anisotropic Oxygen Diffusion Properties in Pr2NiO4+δ and Nd2NiO4+δ Single Crystals. J. Phys. Chem. C 2013, 117, 26466–26472. [Google Scholar] [CrossRef]
- Nakamura, T.; Yashiro, K.; Sato, K.; Mizusaki, J. Oxygen nonstoichiometry and defect equilibrium in La2−xSrxNiO4+δ. Solid State Ion. 2009, 180, 368–376. [Google Scholar] [CrossRef]
- Vashook, V.V.; Tolochko, S.P.; Yushkevich, I.I.; Makhnach, L.V.; Kononyuk, I.F.; Altenburg, H.; Hauck, J.; Ullmann, H. Oxygen nonstoichiometry and electrical conductivity of the solid solutions La2−xSrxNiOy (0 ≤ x ≤ 0.5). Solid State Ion. 1998, 110, 245–253. [Google Scholar] [CrossRef]
- Nakamura, T.; Ling, Y.; Amezawa, K. The effect of interstitial oxygen formation on the crystal lattice deformation in layered perovskite oxides for electrochemical devices. J. Mater. Chem. A 2015, 3, 10471–10479. [Google Scholar] [CrossRef]
- Grimaud, A.; Hong, W.T.; Shao-Horn, Y.; Tarascon, J.M. Anionic redox processes for electrochemical devices. Nat. Mater. 2016, 15, 121–126. [Google Scholar] [CrossRef] [PubMed]
- Rong, X.; Parolin, J.; Kolpak, A.M. A Fundamental Relationship between Reaction Mechanism and Stability in Metal Oxide Catalysts for Oxygen Evolution. ACS Catal. 2016, 6, 1153–1158. [Google Scholar] [CrossRef]
- Mefford, J.T.; Rong, X.; Abakumov, A.M.; Hardin, W.G.; Dai, S.; Kolpak, A.M.; Johnston, K.P.; Stevenson, K.J. Water electrolysis on La1−xSrxCoO3−δ perovskite electrocatalysts. Nat. Commun. 2016, 7, 11053. [Google Scholar] [CrossRef] [PubMed]
- Mildner, S.; Beleggia, M.; Mierwaldt, D.; Hansen, T.W.; Wagner, J.B.; Yazdi, S.; Kasama, T.; Ciston, J.; Zhu, Y.; Jooss, C. Environmental TEM Study of Electron Beam Induced Electrochemistry of Pr0.64Ca0.36MnO3 Catalysts for Oxygen Evolution. J. Phys. Chem. C 2015, 119, 5301–5310. [Google Scholar] [CrossRef]
- Mierwaldt, D.; Mildner, S.; Arrigo, R.; Knop-Gericke, A.; Franke, E.; Blumenstein, A.; Hoffmann, J.; Jooss, C. In Situ XANES/XPS Investigation of Doped Manganese Perovskite Catalysts. Catalysts 2014, 4, 129–145. [Google Scholar] [CrossRef]
- Umena, Y.; Kawakami, K.; Shen, J.-R.; Kamiya, N. Crystal structure of oxygen-evolving photosystem II at a resolution of 1.9 Å. Nature 2011, 473, 55–60. [Google Scholar] [CrossRef] [PubMed]
- Post, J.E. Manganese oxide minerals: Crystal structures and economic and environmental significance. Proc. Natl. Acad. Sci. USA 1999, 96, 3447–3454. [Google Scholar] [CrossRef] [PubMed]
- Siegbahn, P.E.M. Mechanisms for proton release during water oxidation in the S2 to S3 and S3 to S4 transitions in photosystem II. Phys. Chem. Chem. Phys. 2012, 14, 4849–4856. [Google Scholar] [CrossRef] [PubMed]
- Fawcett, I.D.; Sunstrom, J.E.; Greenblatt, M.; Croft, M.; Ramanujachary, K.V. Structure, Magnetism, and Properties of Ruddlesden−Popper Calcium Manganates Prepared from Citrate Gels. Chem. Mater. 1998, 10, 3643–3651. [Google Scholar] [CrossRef]
- Jirák, Z.; Krupička, S.; Šimša, Z.; Dlouhá, M.; Vratislav, S. Neutron diffraction study of Pr1−xCaxMnO3 perovskites. J. Magn. Magn. Mater. 1985, 53, 153–166. [Google Scholar] [CrossRef]
- Shannon, R. Revised effective ionic radii and systematic studies of interatomic distances in halides and chalcogenides. Acta Crystallogr. Sect. A 1976, 32, 751–767. [Google Scholar] [CrossRef]
- Daoudi, A.; Le Flem, G. Sur une série de solutions solides de formule Ca2−xLnxMnO4 (Ln = Pr, Nd, Sm, Eu, Gd). J. Solid State Chem. 1972, 5, 57–61. [Google Scholar] [CrossRef]
- Ibarra, M.; Retoux, R.; Hervieu, M.; Autret, C.; Maignan, A.; Martin, C.; Raveau, B. Charge–orbital ordering above room temperature in the 2D Pr1−xCa1+xMnO4 manganites. J. Solid State Chem. 2003, 170, 361–367. [Google Scholar] [CrossRef]
- Chen, D.; Chen, C.; Baiyee, Z.M.; Shao, Z.; Ciucci, F. Nonstoichiometric oxides as low-cost and highly-efficient oxygen reduction/evolution catalysts for low-temperature electrochemical devices. Chem. Rev. 2015, 115, 9869–9921. [Google Scholar] [CrossRef] [PubMed]
- Aschauer, U.; Pfenninger, R.; Selbach, S.M.; Grande, T.; Spaldin, N.A. Strain-controlled oxygen vacancy formation and ordering in CaMnO3. Phys. Rev. B 2013, 88, 054111. [Google Scholar] [CrossRef]
- Suntivich, J.; May, K.J.; Gasteiger, H.A.; Goodenough, J.B.; Shao-Horn, Y. A Perovskite Oxide Optimized for Oxygen Evolution Catalysis from Molecular Orbital Principles. Science 2011, 334, 1383–1385. [Google Scholar] [CrossRef] [PubMed]
- Scholz, J.; Risch, M.; Stoerzinger, K.A.; Wartner, G.; Jooss, Ch. Rotating Ring Disk Study of Oxygen Evolution at a Perovskite Surface: Correlating Activity to Manganese Concentration. J. Phys. Chem. C 2016. [Google Scholar] [CrossRef]
- Binninger, T.; Mohamed, R.; Waltar, K.; Fabbri, E.; Levecque, P.; Kötz, R.; Schmidt, T.J. Thermodynamic explanation of the universal correlation between oxygen evolution activity and corrosion of oxide catalysts. Sci. Rep. 2015, 5, 12167. [Google Scholar] [CrossRef] [PubMed]
- Danilovic, N.; Subbaraman, R.; Chang, K.-C.; Chang, S.H.; Kang, Y.J.; Snyder, J.; Paulikas, A.P.; Strmcnik, D.; Kim, Y.-T.; Myers, D.; et al. Activity–Stability Trends for the Oxygen Evolution Reaction on Monometallic Oxides in Acidic Environments. J. Phys. Chem. Lett. 2014, 5, 2474–2478. [Google Scholar] [CrossRef] [PubMed]
- De Groot, F.M.F.; Fuggle, J.C.; Thole, B.T.; Sawatzky, G.A. 2p X-ray absorption of 3d transition-metal compounds: An atomic multiplet description including the crystal field. Phys. Rev. B 1990, 42, 5459–5468. [Google Scholar] [CrossRef]
- Mildner, S.; Hoffmann, J.; Blöchl, P.E.; Techert, S.; Jooss, C. Temperature- and doping-dependent optical absorption in the small-polaron system Pr1−xCaxMnO3. Phys. Rev. B 2015, 92, 035145. [Google Scholar] [CrossRef]
- Hong, W.T.; Welsch, R.E.; Shao-Horn, Y. Descriptors of Oxygen-Evolution Activity for Oxides: A Statistical Evaluation. J. Phys. Chem. C 2016, 120, 78–86. [Google Scholar] [CrossRef]
- Trasatti, S.; Petrii, O.A. Real surface area measurements in electrochemistry. J. Electroanal. Chem. 1992, 327, 353–376. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA: A three-dimensional visualization system for electronic and structural analysis. J. Appl. Crystallogr. 2008, 41, 653–658. [Google Scholar] [CrossRef]
- Regier, T.; Krochak, J.; Sham, T.K.; Hu, Y.F.; Thompson, J.; Blyth, R.I.R. Performance and capabilities of the Canadian Dragon: The SGM beamline at the Canadian Light Source. Nucl. Instrum. Methods Phys. Res. Sect. A Accel. Spectrom. Detect. Assoc. Equip. 2007, 582, 93–95. [Google Scholar] [CrossRef]
- Hitchcock, A.P.; Brion, C.E. K-shell excitation spectra of CO, N2 and O2. J. Electron Spectrosc. Relat. Phenom. 1980, 18, 1–21. [Google Scholar] [CrossRef]
- Rheinländer, P.; Henning, S.; Herranz, J.; Gasteiger, H.A. Comparing hydrogen oxidation and evolution reaction kinetics on polycrystalline platinum in 0.1 M and 1 M KOH. ECS Trans. 2013, 50, 2163–2174. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| System | Composition | Space Group | a (Å) | b (Å) | c (Å) | V (Å3) | Density (g/cm3) |
|---|---|---|---|---|---|---|---|
| P00 | CaMnO3 | Pnma | 5.275 | 7.457 | 5.267 | 207.190 | 4.585 |
| P25 | Ca0.75Pr0.25MnO3 | Pnma | 5.328 | 7.589 | 5.325 | 215.302 | 5.190 |
| P50 | Ca0.50Pr0.50MnO3 | Pnma | 5.404 | 7.663 | 5.395 | 223.392 | 5.751 |
| RP00 | Ca2MnO4 | I 41/acd | 5.191 | 5.191 | 24.082 | 648.858 | 4.074 |
| RP25 | Ca1.75Pr0.25MnO4 | I 4/mmm | 3.748 | 3.748 | 11.880 | 166.926 | 4.463 |
| RP50 | Ca1.50Pr0.50MnO4 | Fmm2 | 5.365 | 5.354 | 11.840 | 340.095 | 4.872 |
| System | Composition | Mn-O(1) | Mn-O(2) | Mn-O(1)-Mn | Mn-O(2)-Mn |
|---|---|---|---|---|---|
| P00 | CaMnO3 | 1.926 | 1.853, 1.929 | 151.0 | 160.5 |
| P25 | Ca0.75Pr0.25MnO3 | 1.919 | 1.842, 2.026 | 153.7 | 162.7 |
| P50 | Ca0.50Pr0.50MnO3 | 1.941 | 1.890, 2.015 | 155.6 | 161.4 |
| RP00 | Ca2MnO4 | 2.050 | 1.853 | – | 164.1 |
| RP25 | Ca1.75Pr0.25MnO4 | 2.035 | 1.874 | – | 180.0 |
| RP50 | Ca1.50Pr0.50MnO4 | 2.014, 1.920 * | 1.895 | – | 180.0 |
| System | Tafel Slope (CV) | V vs. RHE (10 μA/cm2) | V vs. RHE (50 µA/cm2) | joxide (1.65 V vs. RHE) (μA/cm2) |
|---|---|---|---|---|
| P00 | 130 ± 7 | 1.70 ± 0.02 | n/a | 4.3 ± 2.0 |
| P25 | 79 ± 4 | 1.63 ± 0.01 | 1.69 ± 0.02 | 16.0 ± 5.7 |
| P50 | 80 ± 3 | 1.65 ± 0.01 | 1.71 ± 0.01 | 9.7 ± 1.2 |
| RP00 | 165 ± 6 | 1.68 ± 0.01 | n/a | 7.4 ± 2.1 |
| RP25 | 86 ± 8 | 1.62 ± 0.01 | 1.68 ± 0.01 | 24.0 ± 6.7 |
| RP50 | 93 ± 4 | 1.61 ± 0.02 | 1.69 ± 0.01 | 21.0 ± 5.6 |
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ebrahimizadeh Abrishami, M.; Risch, M.; Scholz, J.; Roddatis, V.; Osterthun, N.; Jooss, C. Oxygen Evolution at Manganite Perovskite Ruddlesden-Popper Type Particles: Trends of Activity on Structure, Valence and Covalence. Materials 2016, 9, 921. https://doi.org/10.3390/ma9110921
Ebrahimizadeh Abrishami M, Risch M, Scholz J, Roddatis V, Osterthun N, Jooss C. Oxygen Evolution at Manganite Perovskite Ruddlesden-Popper Type Particles: Trends of Activity on Structure, Valence and Covalence. Materials. 2016; 9(11):921. https://doi.org/10.3390/ma9110921
Chicago/Turabian StyleEbrahimizadeh Abrishami, Majid, Marcel Risch, Julius Scholz, Vladimir Roddatis, Norbert Osterthun, and Christian Jooss. 2016. "Oxygen Evolution at Manganite Perovskite Ruddlesden-Popper Type Particles: Trends of Activity on Structure, Valence and Covalence" Materials 9, no. 11: 921. https://doi.org/10.3390/ma9110921