3.1. Phase Evolution and Microstructure Analysis
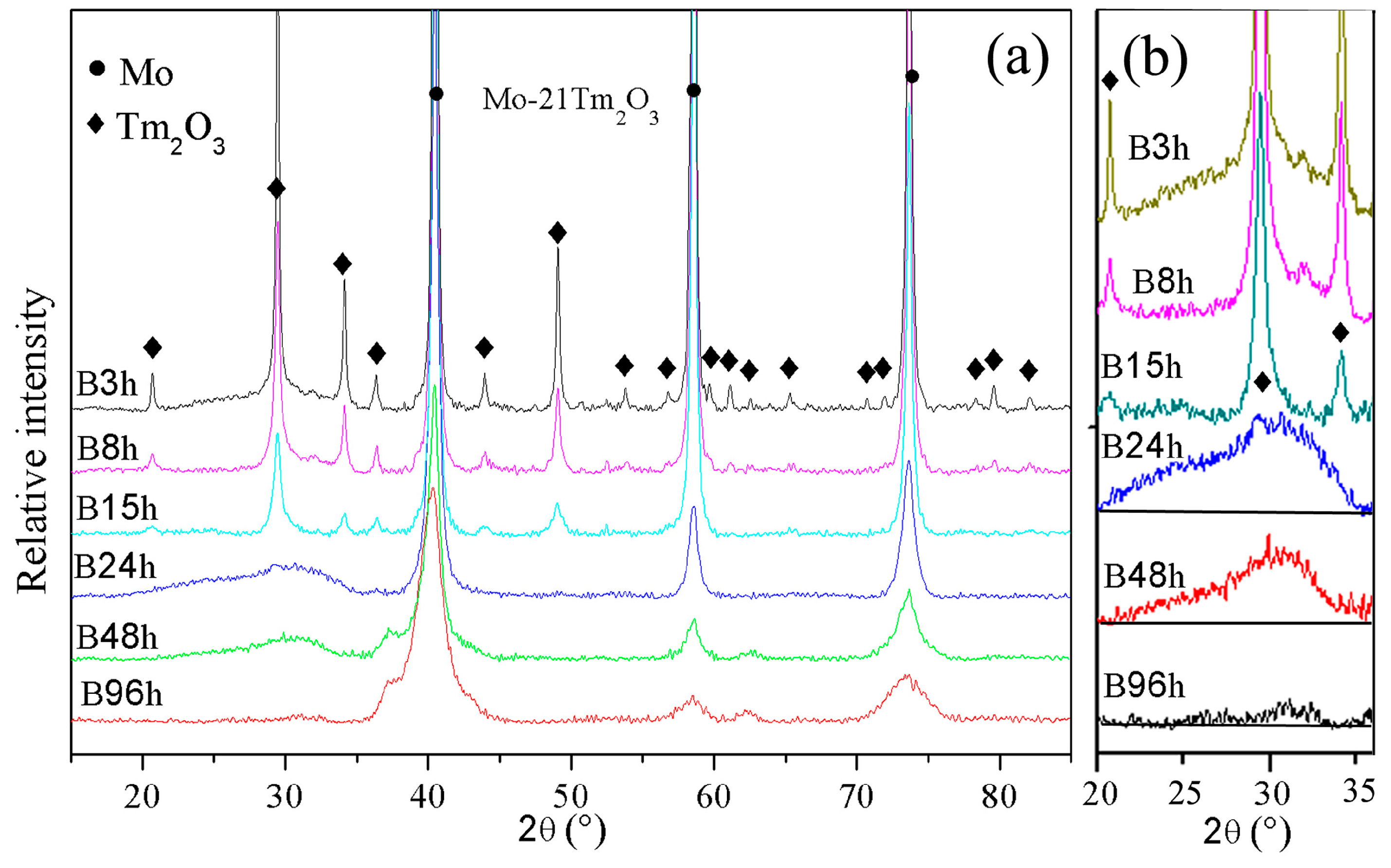
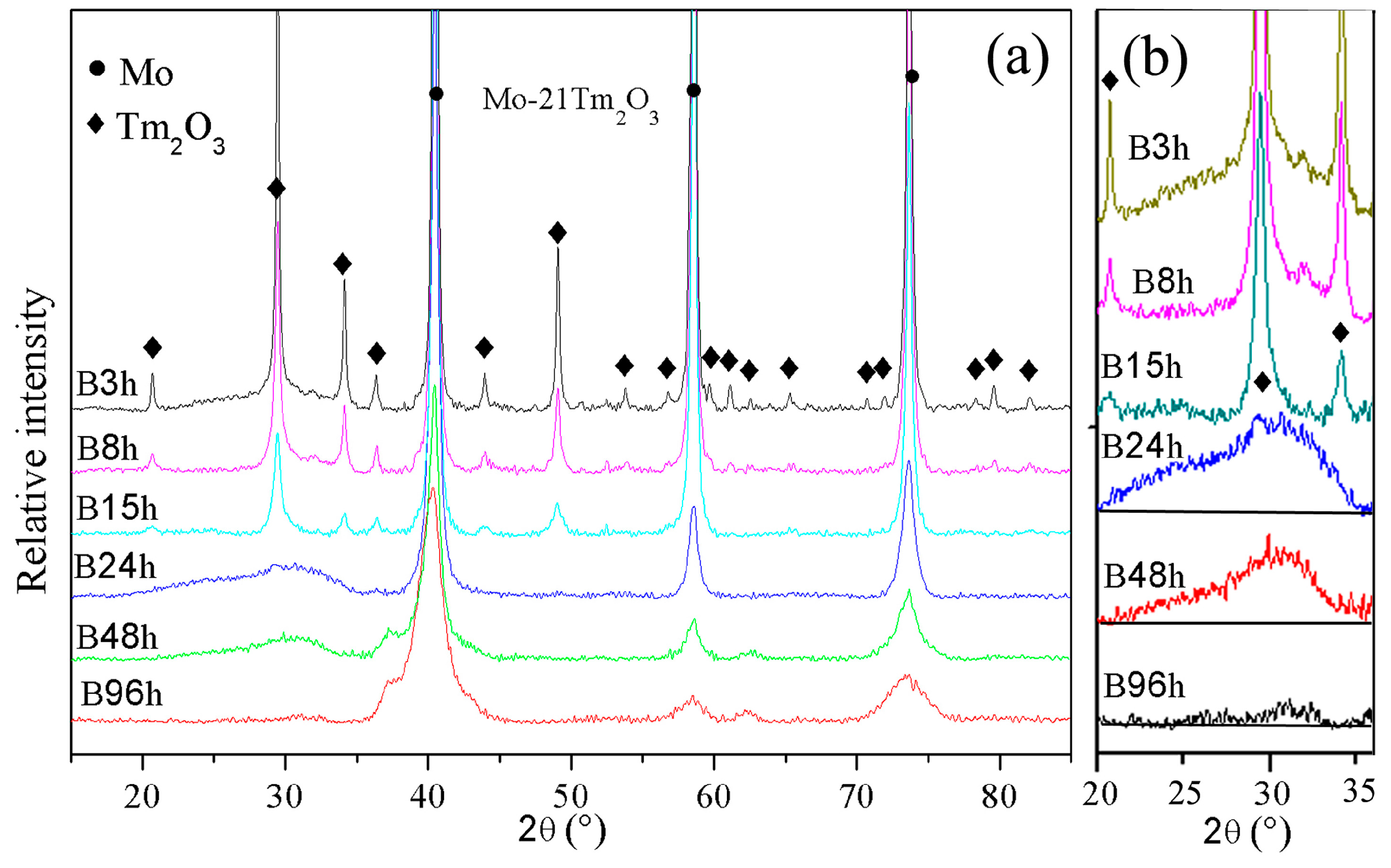
The XRD patterns of Mo (21 wt %) Tm
2O
3 powder mixtures milled for different times are shown in
Figure 1. The diffraction peaks of the Mo and Tm
2O
3 phases are broadened significantly and reduced in intensity with increasing milling time. Meanwhile, a small position shift of Mo peaks to a low diffraction angle is detected, which indicates the large-sized Tm atoms are dissolved into the Mo crystal structure.
The diffraction peaks indicate that the Tm
2O
3 crystal structure disappears after 24 h of ball milling, which is due to the destruction of the Tm
2O
3 crystal structure by ball milling. Only a diffraction hill indicating Tm
2O
3 amorphization can be observed in the XRD spectrum as shown in
Figure 1b. Meanwhile, the intensity of the diffraction hill decreases with increasing milling time. The diffraction hill disappears completely after 96 h of ball milling, which may be attributed to Tm and O atoms being dissolved into the Mo crystal structure. This result is similar with Zhang’s investigation that showed the intensity of Y
2O
3 diffraction peaks almost disappeared after 52 h of ball milling in a Co-based oxide dispersion strengthened alloy, and this was attributed to Y
2O
3 decomposition and dissolution into the Co crystal structure during ball milling [
18]. The effect of ball milling is more evident in the Tm
2O
3 phase than the Mo phase. In addition, no diffraction peaks of new phases among Mo, Tm, and O elements are observed, which indicates that no intermetallic compounds are formed during ball milling. Ball milling does not induce a chemical reaction between Mo and Tm
2O
3, and the amount of intermetallic compounds formed between Mo and Tm
2O
3 is also negligible even if they had a reaction.
The broadening of X-ray diffraction peaks is associated with the refinement of the grain size and lattice distortion during ball milling.
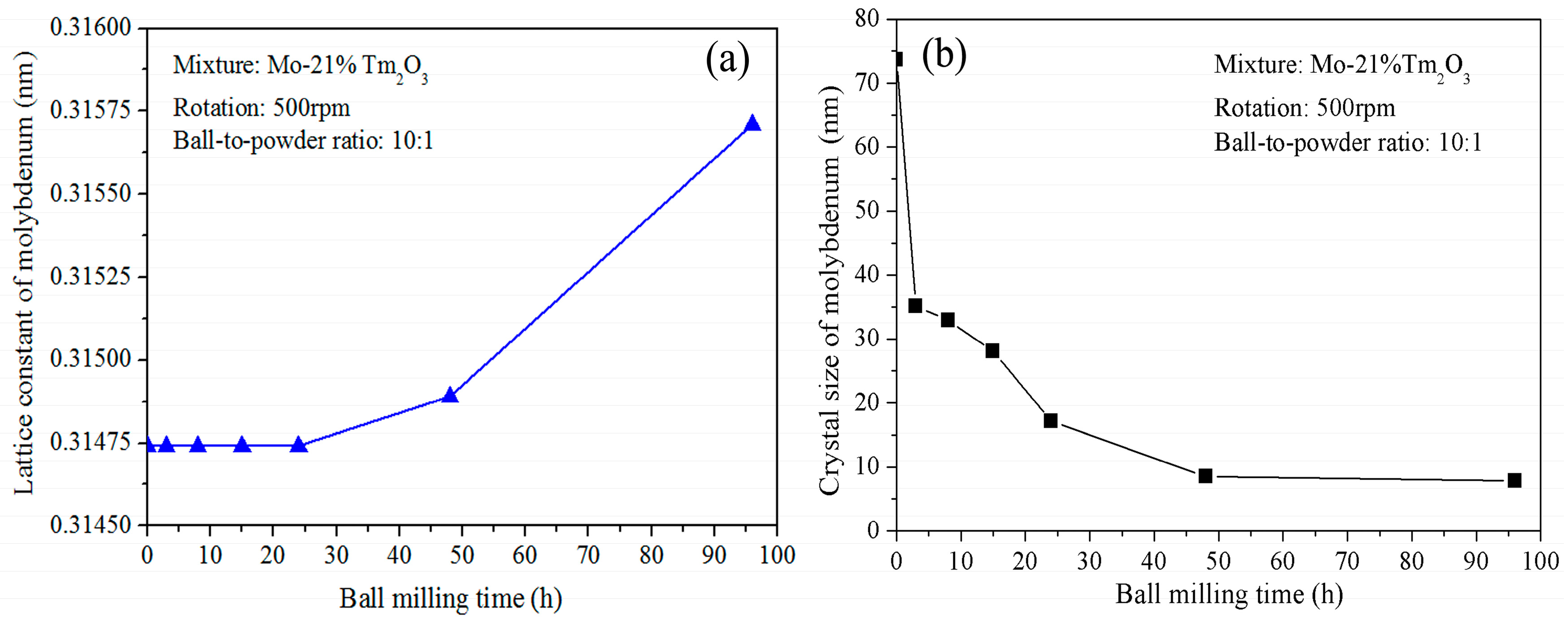
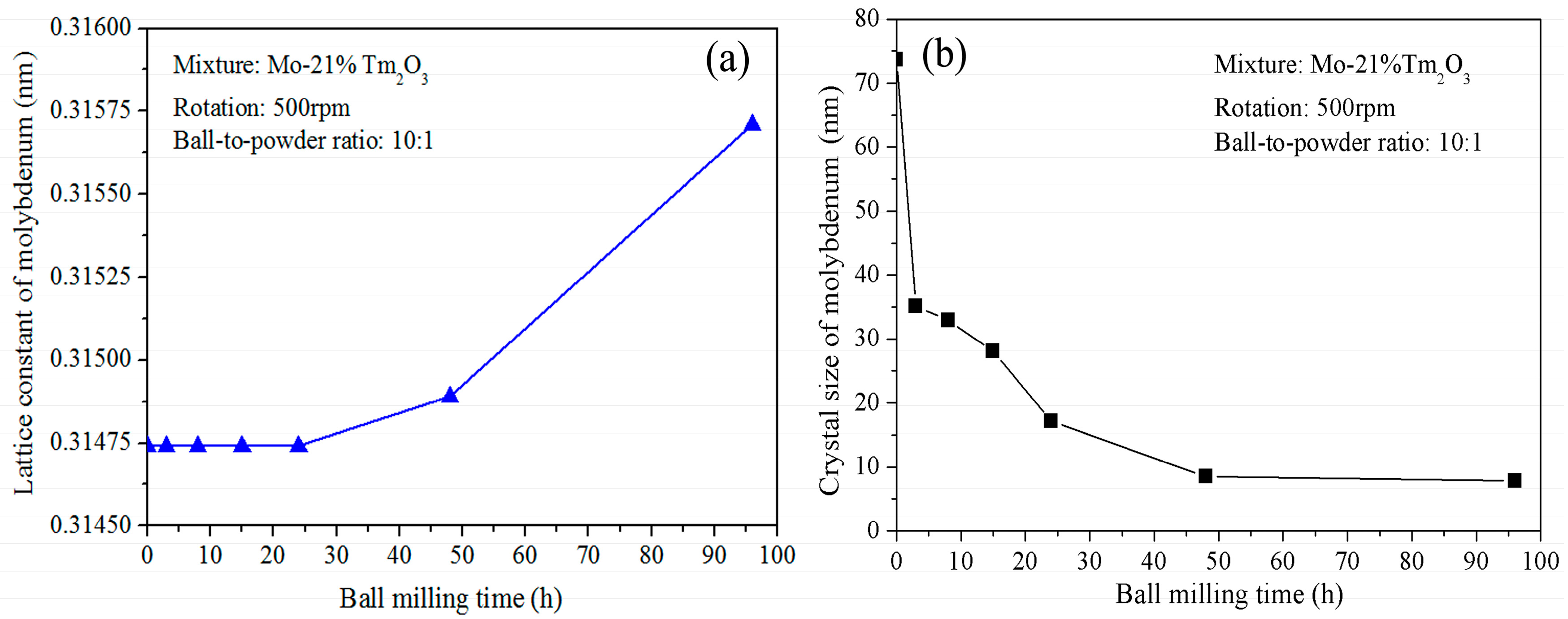
Figure 2a shows the variation of Mo lattice parameter with increasing ball-milling time. It is demonstrated that the lattice parameter increases from 0.31474 nm to 0.31564 nm as the ball-milling time increases from 0 to 96 h. In the initial stage, the Mo lattice parameter remains almost constant. After further ball milling, the Mo lattice parameter increases with increasing ball-milling time. The reason for the Mo lattice parameter increment can be attributed to the dissolution of Tm and O atoms. The supersaturated nanocrystalline solid solution of Mo (Tm, O) is formed after 96 h of ball milling in this system. This result differs from Raghavendra’s research, which showed that the diffraction peaks of the Fe and ZrO
2 phases could be still observed after 100 h of ball milling in a Fe (15 wt %) ZrO
2 system [
19]. However, Toualbi reported the diffraction peak disappearance of the Y
2O
3 phase in a Fe (9 wt %)-Cr (10 wt %) Y
2O
3 system during ball milling was due to the dissolution of a very small number of yttria particles into the matrix to form a solid solution and to the amorphization of a large amount of Y
2O
3 particles [
20].
Figure 2b shows the grain size of the ball-milled powders with increasing ball-milling time. It indicates that the refinement rate of crystallite size is roughly logarithmic with the ball-milling time. In fact, the grain size was calculated for the Mo phase, not for the Tm
2O
3 phase, because Tm
2O
3 peaks disappeared completely. It can be seen that ball milling results in a fast decrease of Mo grain size in the initial stage and a constant value in the later stage. The average grain size of the Mo phase is approximately 74 nm and 35 nm after ball milling of 0 and 3 h, respectively. The decrease rate is approximately 13 nm/h. After further ball milling from 3 to 24 h, the average grain size decreases from 35 to 17 nm. The decrease rate is approximately 0.85 nm/h. However, the average grain size only decreases from 17 to 8 nm after ball milling from 24 to 96 h. The decrease rate is only 0.125 nm/h, which indicates that further refinement of grain size occurs slowly after an extended milling time. In particular, Mo grain size remains almost constant at 8 nm from 48 to 96 h of ball milling. The lattice distortion calculated from the X-ray broadening exhibits an increasing tendency. The total lattice distortion is approximately 1.5% after 96 h of ball milling.
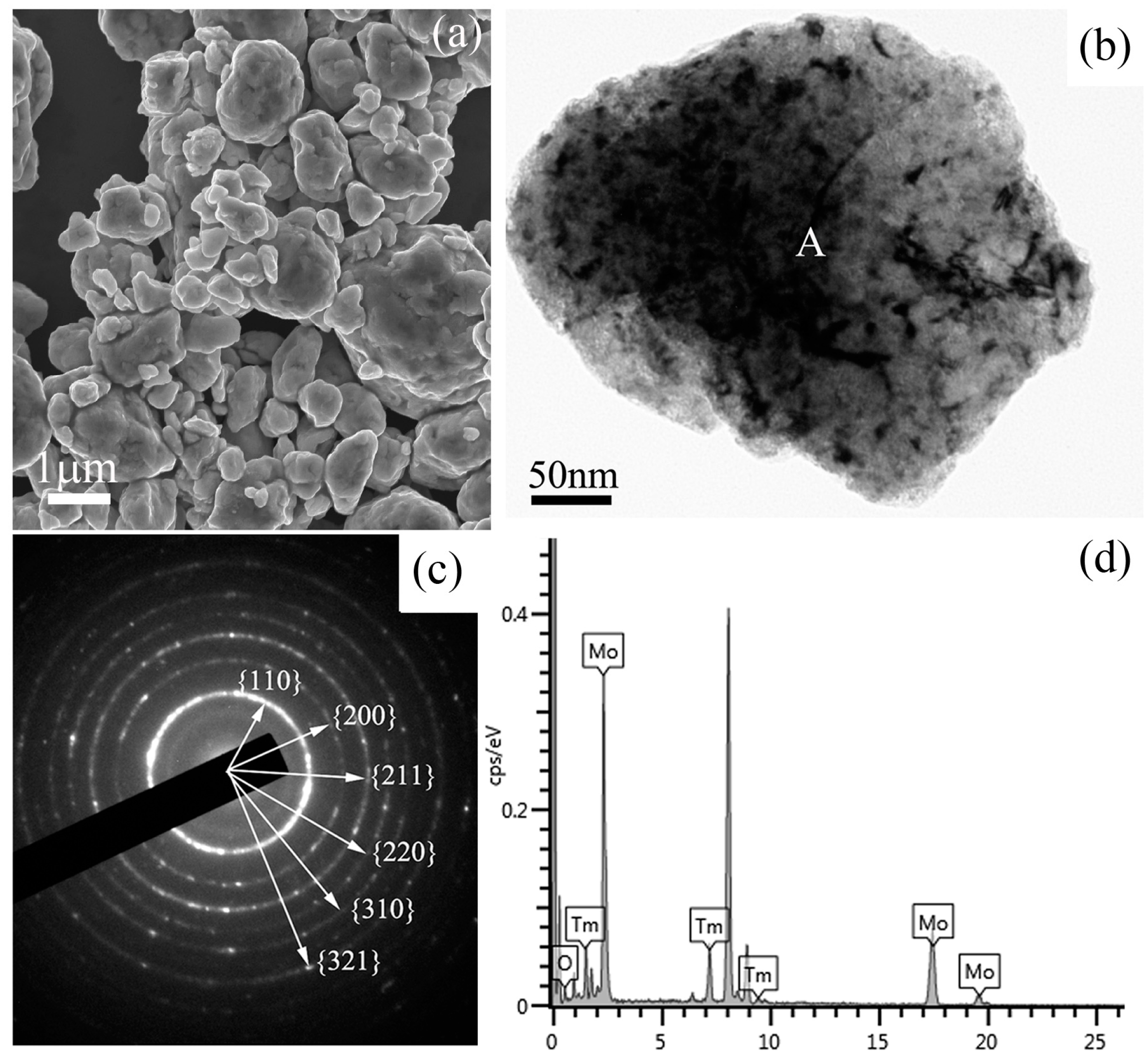
Figure 3 shows a SEM image, a bright field TEM image, the corresponding selected area electron diffraction (SAED) pattern, and the energy dispersive spectrometer (EDS) results of Mo (21 wt %) Tm
2O
3 powder mixtures milled for 48 h. The SEM image in
Figure 3a shows the morphology of the ball-milled powder mixtures, which indicates that many particles with small size aggregate to form a large-size particle. The size of the particle is over one micrometer. A spherical particle with a size of approximately 500 nm can be observed in the TEM image. Some defects, such as dislocations, voids, and crystal boundaries can also be observed in this particle, which were induced by heavy deformation of powder mixtures during ball milling. These defects enhance the diffusivity of solute atoms into the Mo matrix. The SAED pattern taken from the region marked ‘A’ in
Figure 3b is shown in
Figure 3c. After analyzing and indexing the ring-shaped SAED pattern, it is indicated that this SAED pattern belongs to the Mo phase, not the Tm
2O
3 phase. Meanwhile, it is also demonstrated that the Mo component has already been nano-crystallized, which is also confirmed by the calculation results of Mo crystallites derived from Bragg diffraction peaks in
Figure 1. Although the size of the particles is still at the micrometer scale, as shown in
Figure 3a, the grain size is up to the nanometer scale. The EDS spectrum taken from the ball-milled particle is shown in
Figure 3d, which includes Mo, O, and Tm element peaks. Similar EDS results are detected in other particles. Although the SAED results show that the particle belongs to a Mo crystal, Tm and O elements are also detected in this particle. Therefore, it indicates that Tm
2O
3 either uniformly distributes on the Mo particle surface in an amorphous form or dissolves into the Mo crystal structure in the form of Tm and O atoms. However, few diffraction halos related to the amorphous phase can be found in this SAED pattern, as show in
Figure 3c, which indicates that Tm
2O
3 does not exist mainly in the amorphous form in this particle.
Figure 4a is a high angle annual dark field STEM image of Mo (21 wt %) Tm
2O
3 powder mixtures milled for 96 h, which shows several small particles agglomerated to form a large-sized particle.
Figure 4b–d are Mo L, Tm L, and O K element mappings of the particles in
Figure 4a, respectively. It can be seen that Mo, Tm, and O atoms are uniformly distributed in the particles. Every location contains Mo, Tm, and O elements, which further demonstrates that Tm
2O
3 dissolves into the Mo crystal structure after 96 h of ball milling because the diffraction hill indicating Tm
2O
3 amorphization disappears completely after 96 h of ball milling, as shown in
Figure 1b.
In fact, the Tm
2O
3 component is a brittle powder, which is fragmented during ball milling. The particle size reduces continuously as a consequence of the energy provided by ball milling. However, the Mo component belongs to the ductile powder, which is repeatedly flattened, cold welded, fractured, and rewelded by the force of the impact. In the initial stage of ball milling, the microstructural evolution of this ductile-brittle Mo-Tm
2O
3 system is that the ductile Mo metal powder particles get flattened by the ball-powder-ball collisions, while the brittle Tm
2O
3 oxide get fragmented. These fragmented Tm
2O
3 particles tend to become occluded by the ductile Mo constituents and trapped in the ductile Mo particles. With further ball milling, Mo particles get work hardened, fractured, and refined. The newly created surface of the Mo component has a large number of dangling bonds that have high chemical activity, which makes the fined Mo particles aggregate together to form a large-sized particle, and also combined with dangling bonds of the fined Tm
2O
3 particles at an atomic level. Meanwhile, fragmented Tm
2O
3 particles will be continuously fined to smaller-sized particles with increasing ball-milling time and will be decreased to several nanometers. In particular, as the size of Tm
2O
3 is decreased to 1~2 nanometers, the ratio of atoms on the particle surface is very large. With increasing ball-milling time, Tm
2O
3 is transformed gradually from a crystal structure to an amorphous phase, as shown in
Figure 1b. The amorphous atom arrangement is disordered over a long distance, but ordered over a very short distance, which could induce Tm and O atoms to attach to a Mo particle surface in a state of dissociation. Tm and O atoms can easily diffuse into the Mo crystal structure. Meanwhile, heavy deformation is introduced continually into Mo particles. This is manifested by the presence of a variety of crystal defects, such as dislocations, vacancies, stacking faults, and an increased number of crystal defect boundaries [
21], which can enhance the diffusivity of Tm and O atoms into the Mo matrix. Moreover, the refined microstructural features decrease Tm and O atom diffusion distances. Additionally, the rise in temperature during ball milling further promotes diffusion. Consequently, Tm and O atoms dissolve into the Mo crystal structure. The longer the ball-milling time is, the greater the amount of dissolution of Tm atoms. After 96 h of ball milling, the supersaturated nanocrystalline solid solution of Mo (Tm, O) is formed.
3.2. Thermodynamics Analysis
As mentioned above, long time ball milling induces the Tm2O3 component to be decomposed and then dissolved into the Mo crystal structure. Therefore, thermodynamics will be used to analyze the dissolution behavior of Tm into the Mo crystal structure.
Based on Miedema semi-experimental theory [
22], the Gibbs free energy of a Mo-Tm solid solution is estimated as:
where
and
are mixing enthalpy and mixing entropy, respectively. With the assumption of an ideal solution,
can be expressed as:
where
and
are the molar fractions of element A and B,
+
= 1;
R is the gas constant; and
T is the reaction temperature.
The Gibbs free energy,
, can be rewritten as:
The enthalpy,
, of a solid solution formation can be written as [
23]:
where
,
, and
are the chemical, elastic, and structural contributions due to the mixing of the two different atoms, the atom size mismatch, and taking into account the difference in valence and crystal structure of the solute and solvent, respectively. Compared with the first two items, the structural contributions have only a minor effect [
24], which will be ignored here. Thus
will be rewritten as:
can be expressed by:
where
is the solution enthalpy of
i in
j given by:
is the parameter of adjacent atom in solution solid and can be expressed as:
where
,
, and
are the work function, molar volume, and electron density of the constituents, respectively.
P and
Q are empirical constants having the same value for widely different metal combinations.
is taken to be five for the short-range order and eight for the long-rang order, respectively.
can be expressed as:
where
is the size mismatch contribution to the enthalpy of a solution of
i and
j per mol
i and can be estimated as [
25]:
where
,
, and
are the bulk modulus, shear modulus, and atomic volume, respectively.
can be expressed as:
where
c is constant.
The Gibbs free energy of formation of an amorphous phase can be estimated by [
26]:
where
and
are the enthalpy and entropy of mixing of the amorphous phase.
is the difference in Gibbs free energy between the amorphous and crystalline phases of the pure element at room temperature and can be calculated according to the formula proposed by Thompson:
where
and
Tm are the enthalpy of fusion and melting temperature, respectively.
contains only the chemical contribution due to the amorphous structure, which can be calculated by:
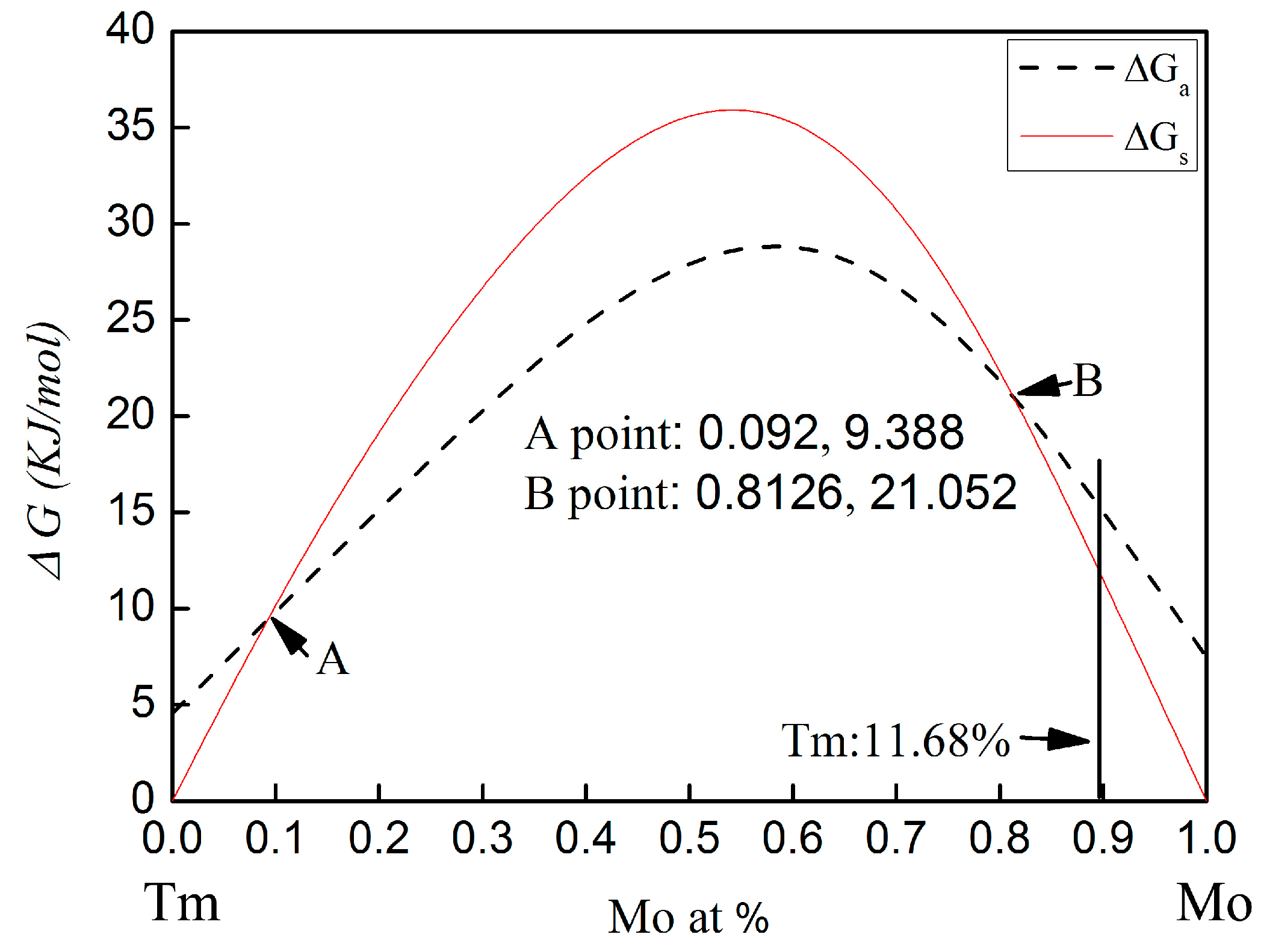
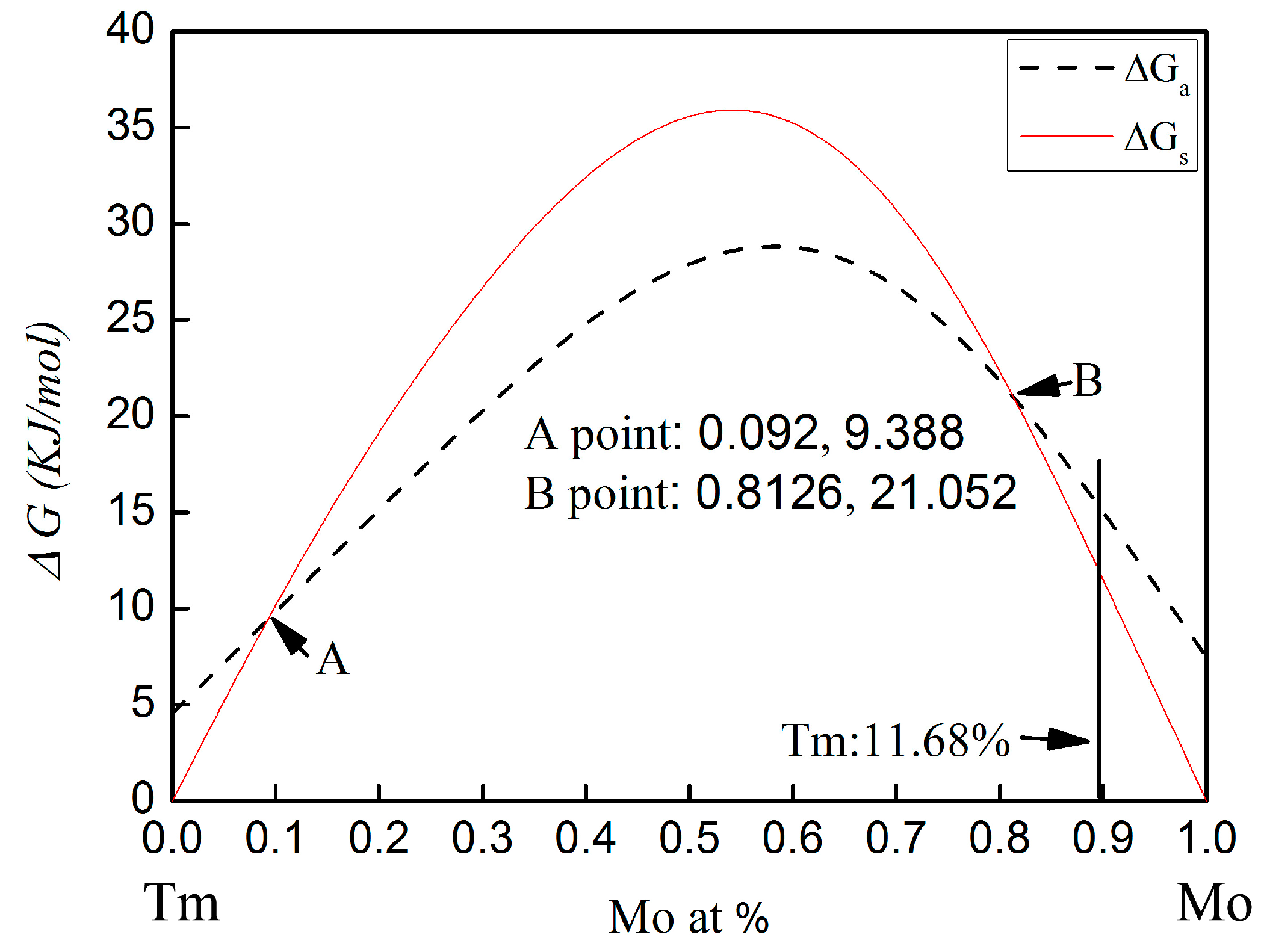
The calculated Gibbs free energy changes on forming an amorphous phase and crystalline solid solution in the Mo-Tm system is shown in
Figure 5. The temperature for calculating the Gibbs free energy is taken to be 373 K [
11]. The parameters used for calculation are listed in
Table 1 [
27,
28]. The calculated Gibbs free energy of both the crystalline solid solution and the amorphous phase are higher than zero for all compositions. Therefore, there is no chemical driving force to form a crystalline solid solution or an amorphous phase. From
Figure 5, it can be seen that, with molybdenum content in the range of 0~9.2 at % or 81.26~100 at %, the Gibbs free energy for the formation of an amorphous phase is higher than that of a crystalline solid solution. The Gibbs free energy curve of the crystalline solid solution intersects with that of the amorphous phase at the 9.2 at % and 81.26 at % Mo content.
In the present work, the molar percentage of Tm atoms is 11.68 at %. The Gibbs free energy for the formation of an amorphous phase and a crystalline solid solution is 16.36 KJ/mol and 13.5 KJ/mol, respectively. The Gibbs free energy of amorphization is higher than that of the crystalline solid solution formation. Therefore, the formation of the crystalline solid solution is energetically preferred over the amorphous phase. However, the Gibbs free energy of the formation of the crystalline solid solution and the amorphization are both higher than zero for this chemical composition. There is no chemical driving force to form a crystalline solid solution and an amorphous phase. Meanwhile, there is also no chemical driving force for the transformation from crystalline to amorphous in this chemical composition. To more deeply understand the mechanism for microstructural evolution of Mo-Tm2O3 powder mixtures, especially in regard to the dissolution of Tm atoms into the Mo crystal structure, the reaction kinetics during ball milling must be considered.
In addition, although the calculated Gibbs free energy of both the crystalline solid solution and the amorphous phase are higher than zero for all compositions, as shown in
Figure 5, these calculated values are limitations and cannot completely represent actual situation. However, ball milling induced the decomposition of the Tm
2O
3 component, followed by solvation into the Mo crystal structure, the investigation of the Gibbs free energy using the Mo-Tm system to take the place of Mo-Tm
2O
3 system is also reasonable and feasible.
3.3. Dynamics Analysis
The atomic radii of the Mo atom and Tm atom is 0.19 nm and 0.222 nm, respectively [
29]. The difference in atomic radius is approximately 15%. At high pressure, the radii of the Mo atom and Tm atom will reach the same value. Under this condition, the Tm atom will easily dissolve into the Mo crystal structure. The required pressure is given by the following:
where
εMo =
q*/
EMo,
εTm =
q*/
ETm. Here,
R is the initial atom radius;
q* is the pressure;
E is the bulk elastic modulus; and
ε is the amount of strain. Therefore, Equation (19) can be rewritten as follows:
where
RMo = 0.19 nm,
RTm = 0.222 nm,
EMo = 230 GPa, and
ETm = 45 GPa. The
q* value can be obtained according to Equation (20) and is
q* = 7.79 GPa. Therefore, when the pressure is over 7.79 GPa, the radii of the Mo atom and Tm atom will become equivalent. In fact, during high-energy ball milling, a pressure of 7.79 GPa is not difficult to obtain and, in fact, the maximum pressure value
q0 will be up to 8 GPa according to the following theoretical calculations.
Assuming that two impacting balls have the same elastic properties, and taking
γ = 0.3, the formula is shown as follows [
30]:
where
α is the amount of elastic deformation between two colliding mill balls;
P is the compression force under the colliding process;
E is Young’s modulus;
R1 and
R2 are the radius of the colliding balls;
q0 is the pressure at the center of the surface of contact, which is the maximum pressure of the contacting surface; and
is the average pressure on the contact surface of two colliding mill balls.
The velocity of the ball in the present work is taken as
υ = 2 m/s. The weight of a mill ball is 0.51 g. The calculated impact energy is
Ek = 2 × 0.5 mυ
2 = 0.0024 J. Therefore, the pressure
q of two impacting balls can be obtained using the following integral:
Using Equations (21) and (23), Ek is: Ek = 1.923 × 109 α5/2. The compression force P can then be calculated and is P = 355.623 N. According to Equation (22), q0 = 8.369 GPa and = 5.58 GPa.
Therefore, the value of q0 = 8.369 GPa is larger than that of q* = 7.79 Gpa. The Tm atoms are easily dissolved into the Mo crystal structure when the Tm atom radius is less than or equal to the Mo atom radius. Certainly, in the above discussion, the calculation is an ideal condition. In actuality, the collision of two balls is random and is not always a frontal collision because of random motion of the grinding balls in the can. The values of collisional pressure are in a Gaussian distribution. The pressure of a part of collision in the actual can be over the q* value, which will induce the radius of Tm atom to reach that of Mo atoms. Therefore, a long time is needed to make all Tm atoms dissolve into the Mo crystal structure.
Simultaneously, the temperature at the colliding surface will instantaneously increase several hundred degrees Celsius, resulting from the impact of the balls, which promotes the diffusion and dissolution process of Tm atoms. Davis reported that the maximum temperature rise during ball milling was approximately 300 °C in a Fe (1.2 wt %)-C steel system [
31]. Tonejc’s research showed that the local rise in temperature was at least 570 °C in a Ga-Sb alloy system [
32]. Meanwhile, the large defect densities increase the free energies of atoms, which also promotes Tm atom dissolution.




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}