
Activation of Aspen Wood with Carbon Dioxide and Phosphoric Acid for Removal of Total Organic Carbon from Oil Sands Produced Water: Increasing the Yield with Bio-Oil Recycling


Abstract
:1. Introduction
2. Results and Discussion
2.1. TOC Removal by Activated Carbon from Wood

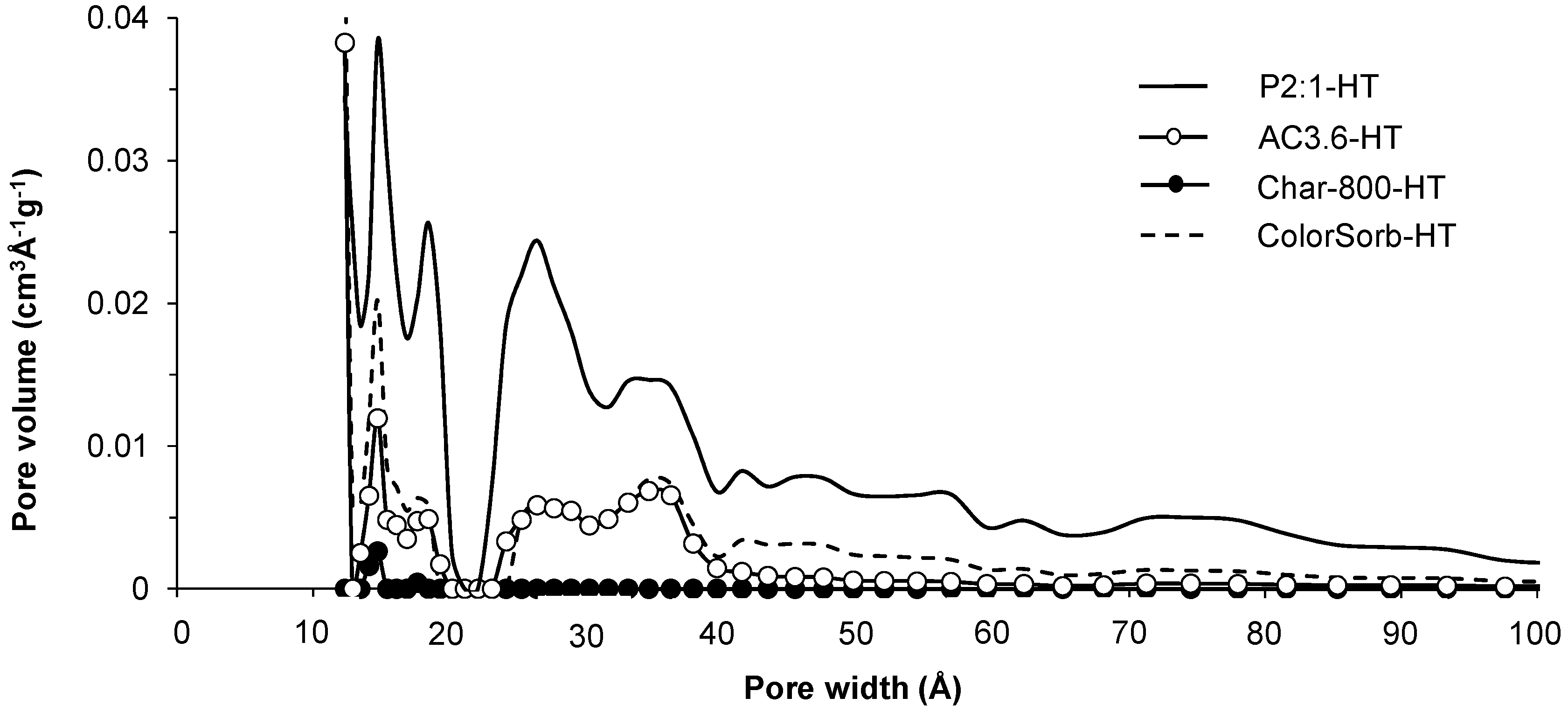{kind=link}
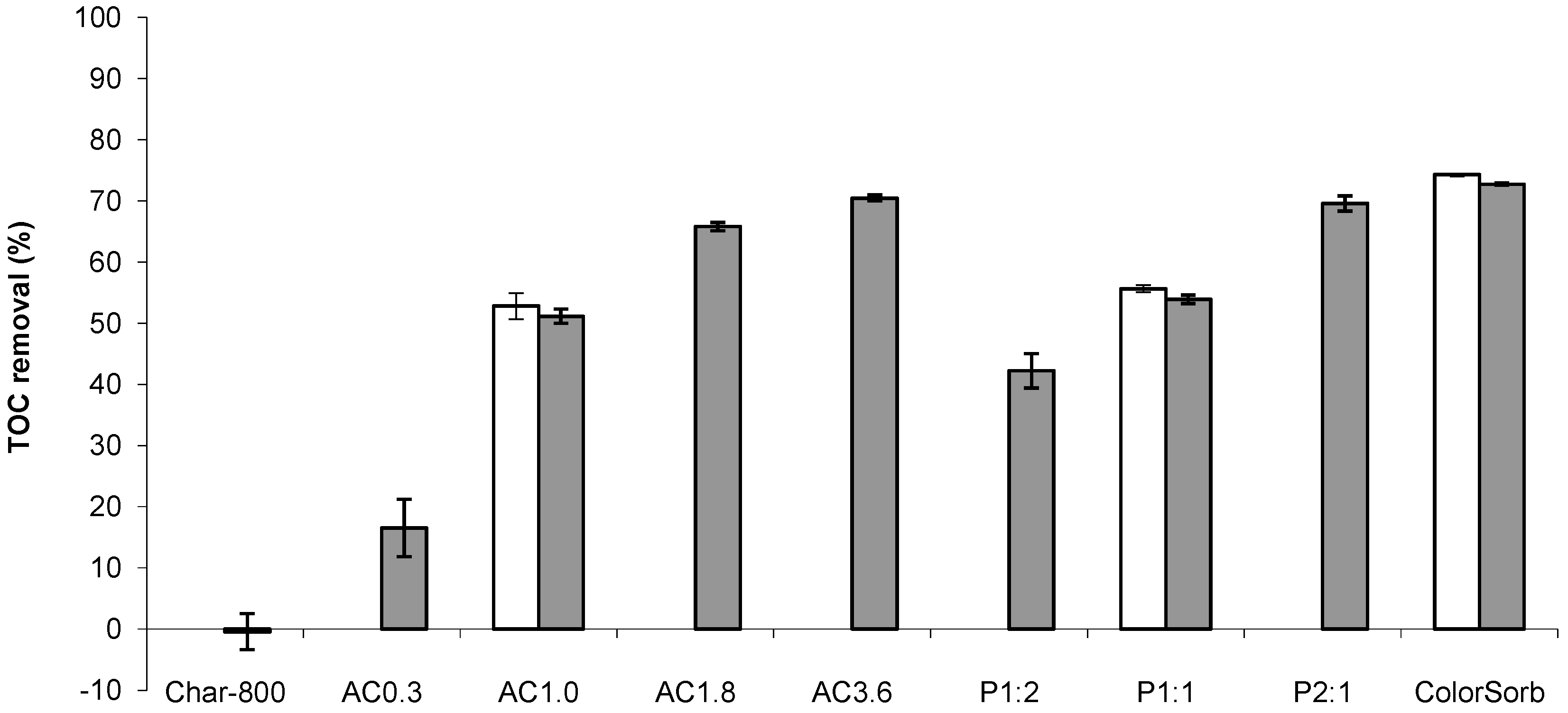{kind=link}
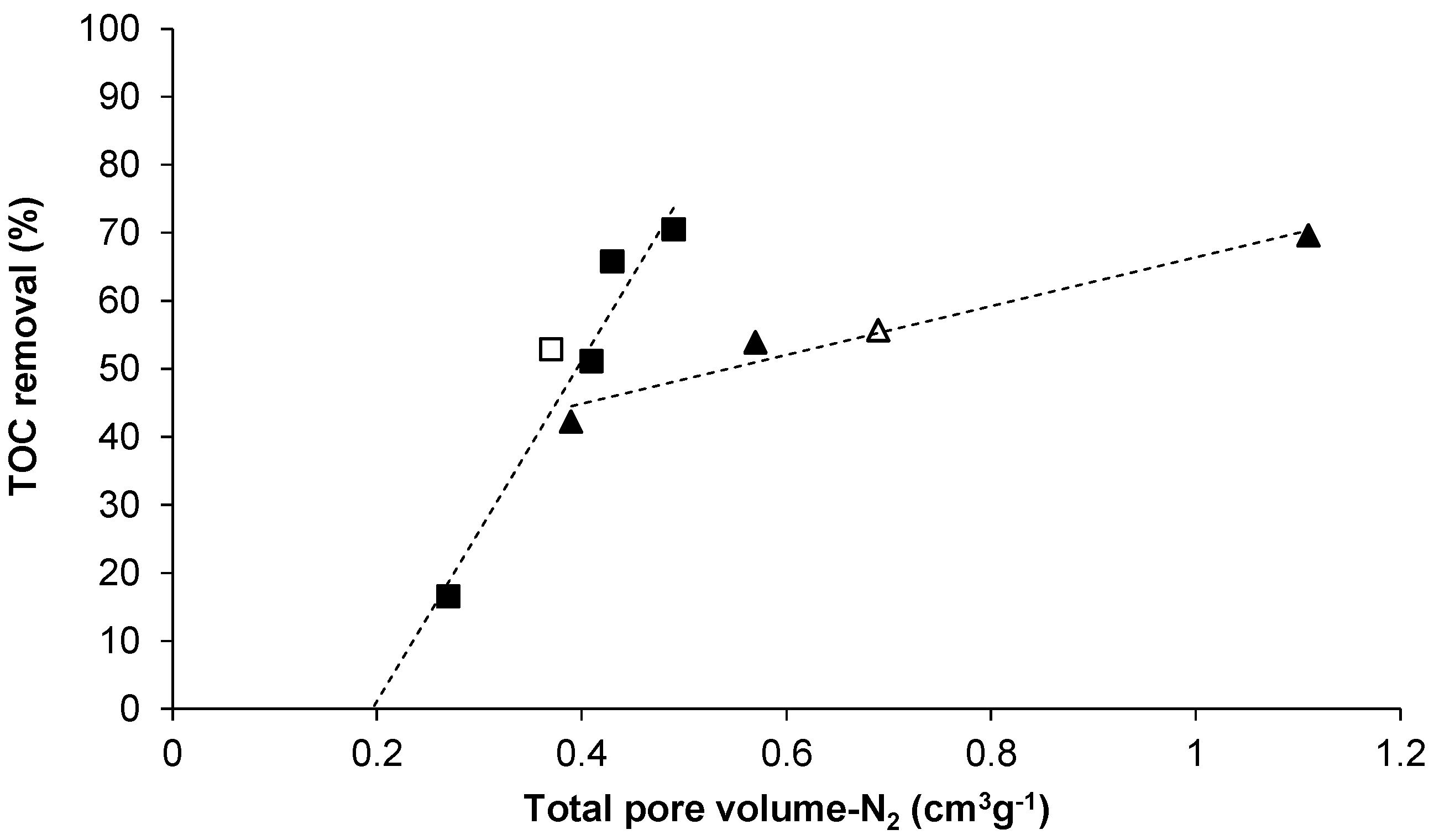{kind=link}
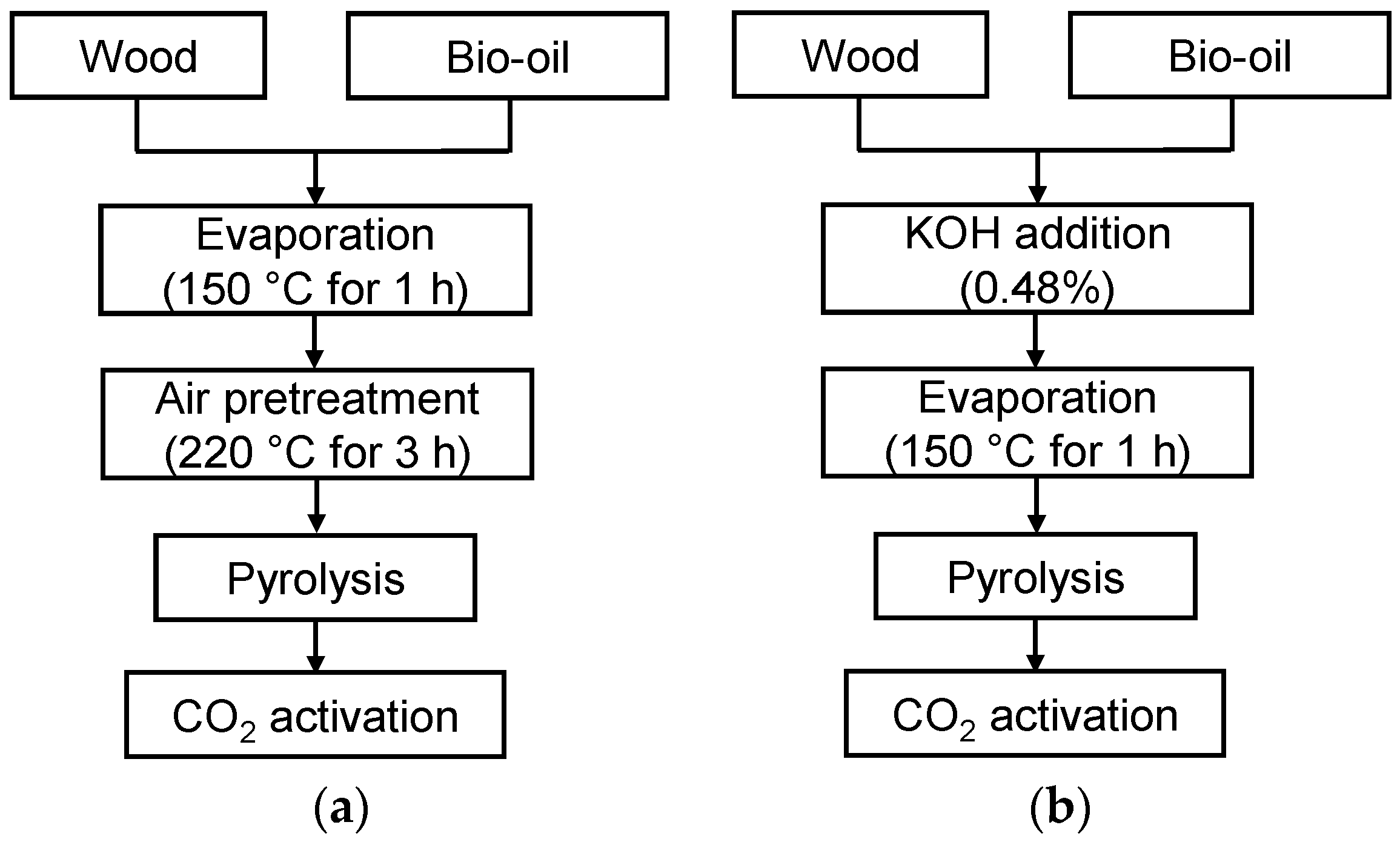{kind=link}
{kind=link}
| Sample | Yield (%) | Meso/Macropore Volume-N2 (cm3·g−1) | Micropore Volume-N2 (cm3·g−1) | Micropore Volume-CO2 (cm3·g−1) | BET-N2 Surface Area (m2·g−1) |
|---|---|---|---|---|---|
| Char | |||||
| Char-800-HT | 21 | 0.02 | 0.17 | 0.23 | 440 |
| CO2 Activated Carbon | |||||
| AC0.3-HT | 19 | 0.09 | 0.18 | 0.23 | 540 |
| AC1.0-HT | 15 ± 1 a | 0.22 | 0.19 | 0.28 | 690 |
| AC1.0 | 0.21 | 0.16 | 0.23 | 600 | |
| AC1.8-HT | 11 | 0.21 | 0.22 | 0.27 | 750 |
| AC3.6-HT | 6 | 0.19 | 0.30 | 0.32 | 910 |
| H3PO4 Activated Carbon | |||||
| P1:2-HT | 46 | 0.04 | 0.35 | 0.27 | 870 |
| P1:1-HT | 45 | 0.11 | 0.46 | 0.26 | 1110 |
| P1:1 | 0.09 | 0.60 | 0.24 | 1240 | |
| P2:1-HT | 41 | 0.71 | 0.40 | 0.26 | 1350 |
| Steam Activated Commercial Carbon | |||||
| ColorSorb-HT | - | 0.30 | 0.36 | 0.37 | 1140 |
| ColorSorb | 0.31 | 0.30 | 0.32 | 988 | |
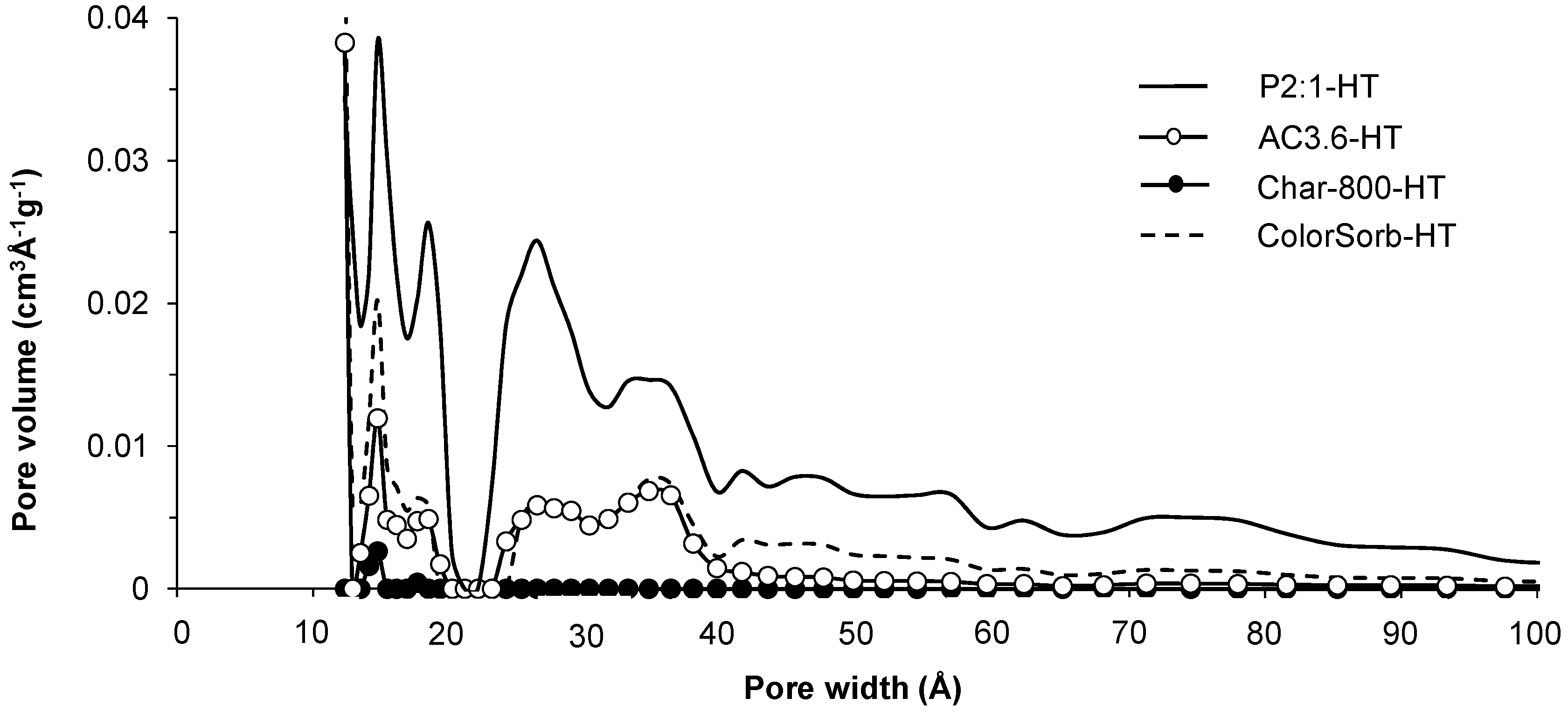
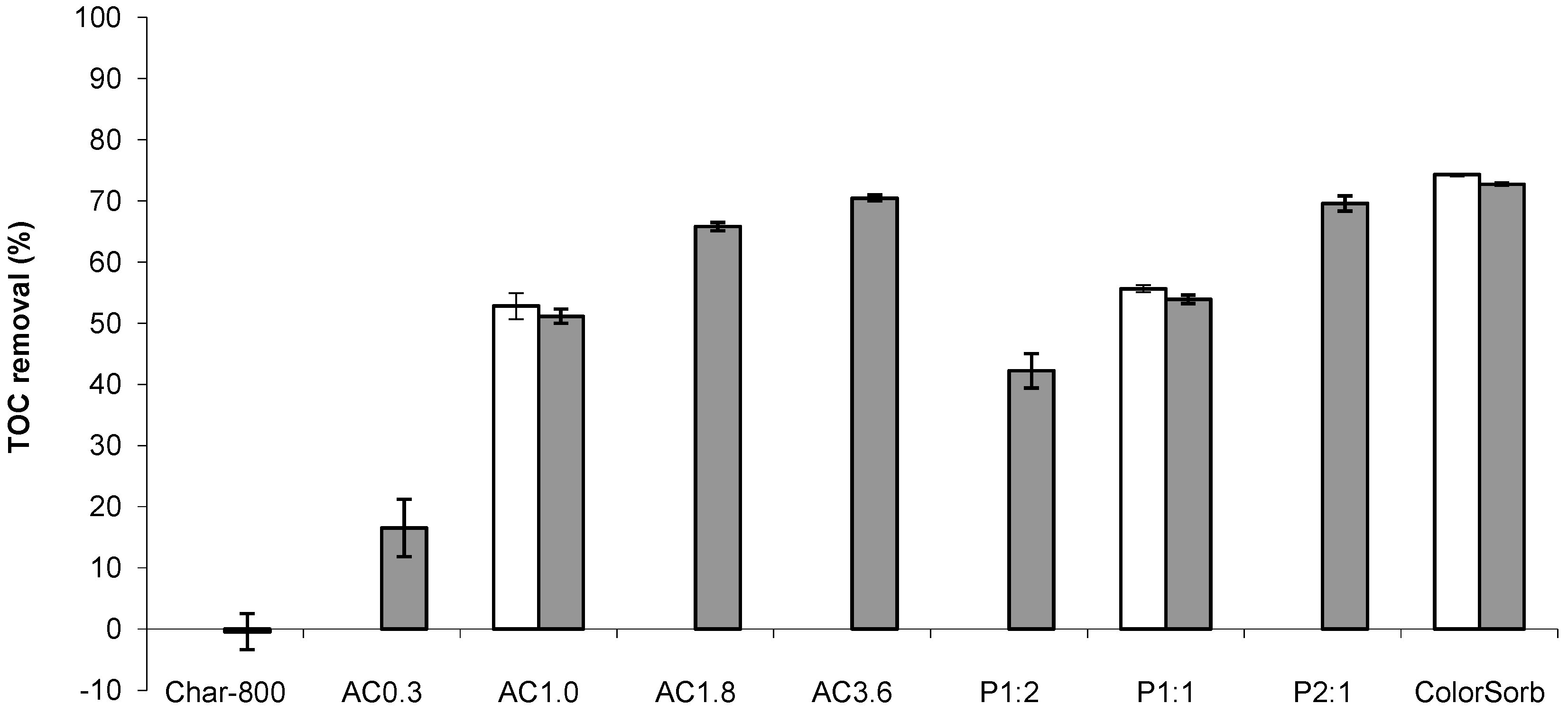
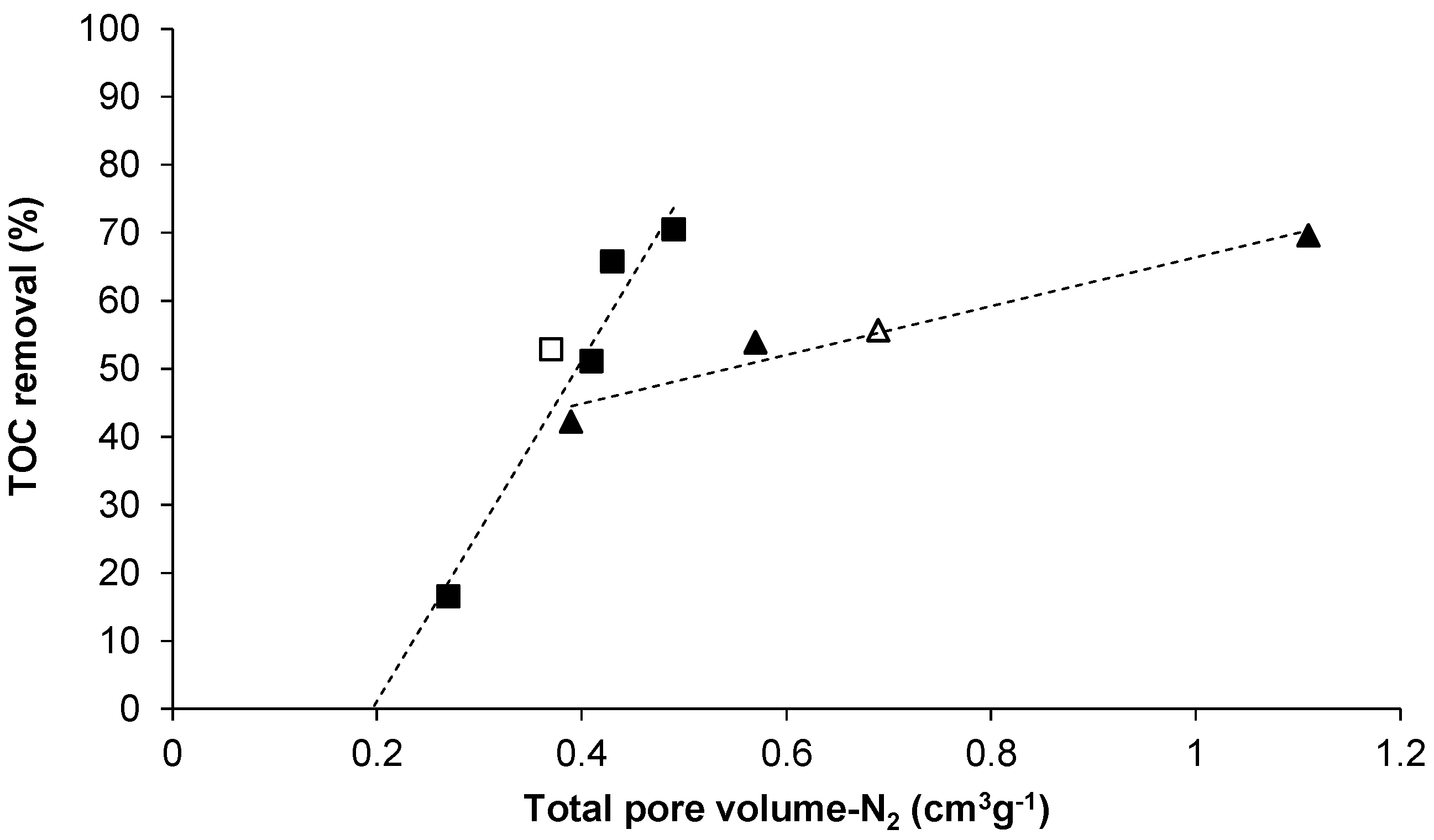
2.2. Increasing the Yield of CO2 Activated Carbon

| Sample | Yield (%) a | Meso/Macropore Volume-N2 (cm3·g−1) | Micropore Volume-N2 (cm3·g−1) | Micropore Volume-CO2 (cm3·g−1) | BET-N2 Surface Area (m2·g−1) |
|---|---|---|---|---|---|
| Char from Wood | |||||
| Char-600 | 23.7 ± 0.4 b | 0.02 b | 0.17 b | 0.18 b | 440 b |
| Char from Wood and Bio-Oil Mixture | |||||
| Char-600-Air | 30.4 ± 0.7 | 0.03 | 0.17 | 0.20 | 450 |
| Char-600-KOH | 30.9 ± 0.1 | 0.02 | 0.01 | 0.15 | 30 |
| Activated Carbon from Wood and Bio-Oil Mixture | |||||
| AC1.0-Air | 19.4 ± 0.1 | 0.15 | 0.19 | 0.25 | 600 |
| AC1.0-Air-HT | 0.19 | 0.32 | 0.33 | 930 | |
| AC1.0-KOH | 15.4 ± 0.1 | 0.20 | 0.18 | 0.24 | 640 |
| AC1.0-KOH-HT | 0.25 | 0.31 | 0.36 | 1020 | |
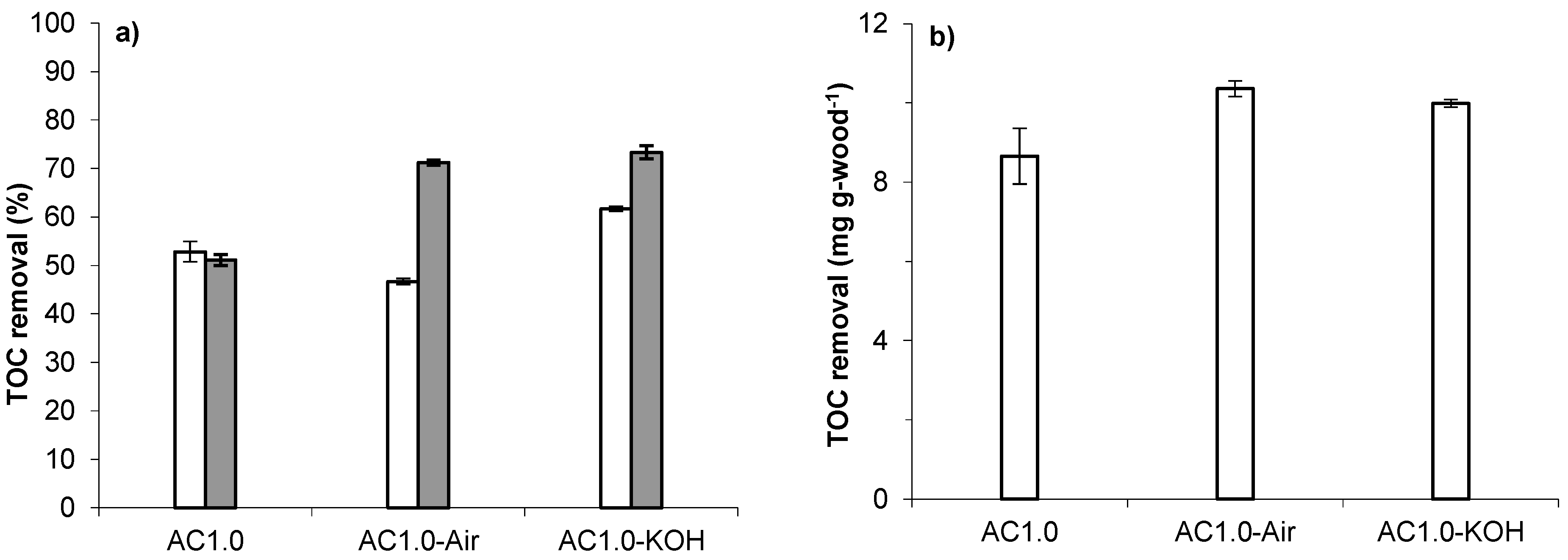
3. Materials and Methods
3.1. Preparation of Activated Carbon
3.2. Characterization of Char and Activated Carbon
3.3. Total Organic Carbon Removal from Water
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Moreno-Castilla, C. Adsorption of organic molecules from aqueous solutions on carbon materials. Carbon 2004, 42, 83–94. [Google Scholar] [CrossRef]
- Li, L.; Quinlivan, P.A.; Knappe, D.R.U. Effects of activated carbon surface chemistry and pore structure on the adsorption of organic contaminants from aqueous solution. Carbon 2002, 40, 2085–2100. [Google Scholar] [CrossRef]
- Dastgheib, S.A.; Karanfil, T.; Cheng, W. Tailoring activated carbon for enhanced removal of natural organic matter from natural waters. Carbon 2004, 42, 547–557. [Google Scholar] [CrossRef]
- Dias, J.M.; Alvim-Ferraz, M.C.M.; Almeida, M.F.; Rivera-Utrilla, J.; Sanchez-Polo, M. Waste materials for activated carbon preparation and its use in aqueous-phase treatment: A review. J. Environ. Manag. 2007, 85, 833–846. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.; Yamamoto, N.; Kameshima, Y.; Yasumori, A. Adsorption properties of activated carbon from waste newspaper prepared by chemical and physical activation. J. Colloid Interface Sci. 2003, 262, 194–199. [Google Scholar] [CrossRef]
- Girgis, B.S.; Yunis, S.S.; Soliman, A.M. Characteristics of activated carbon from peanut hulls in relation to conditions of preparation. Mater. Lett. 2002, 57, 164–172. [Google Scholar] [CrossRef]
- Iranmanesh, S.; Harding, T.; Abedi, J.; Seyedeyn-Azad, F.; Layzell, D.B. Adsorption of naphthenic acids on high surface area activated carbon. J. Environ. Sci. Health A 2014, 49, 913–922. [Google Scholar] [CrossRef] [PubMed]
- Mestre, A.S.; Pires, R.A.; Aroso, I.; Fernandes, E.M.; Pinto, M.L.; Reis, R.L.; Andrade, M.A.; Pires, J.; Silva, S.P.; Carvalho, A.P. Activated carbon prepared from industrial pre-treated cork: Sustainable adsorbents for pharmaceutical compounds removal. Chem. Eng. J. 2014, 253, 408–417. [Google Scholar] [CrossRef]
- Toles, C.A.; Marshall, W.E.; Johns, M.M.; Wartelle, L.H.; McAloon, A. Acid-activated carbon from almond shells: Physical, chemical and adsorptive properties and estimated cost of production. Bioresour. Technol. 2000, 71, 87–92. [Google Scholar] [CrossRef]
- Toles, C.A.; Marshall, W.E.; Wartelle, L.H.; McAloon, A. Steam- or carbon dioxide-activated carbon from almond shells: Physical, chemical and adsorptive properties and estimated cost of production. Bioresour. Technol. 2000, 75, 197–203. [Google Scholar] [CrossRef]
- Ng, C.; Marshall, W.E.; Rao, R.M.; Bansode, R.R.; Losso, J.N. Activated carbon from pecan shell: Process description and economic analysis. Ind. Crops Prod. 2003, 17, 209–217. [Google Scholar] [CrossRef]
- Altenor, S.; Ncibi, M.C.; Brehm, N.; Emmanuel, E.; Gaspard, S. Pilot-scale synthesis of activated carbon from vetiver roots and sugar cane bagasse. Waste Biomass Valor. 2013, 4, 485–495. [Google Scholar] [CrossRef]
- Lee, Y.; Park, J.; Ryu, C.; Gang, K.S.; Yang, W.; Park, Y.K.; Jinho, J.; Hyun, S. Comparison of biochar properties from biomass residues produced by slow pyrolysis at 500 °C. Bioresour. Technol. 2013, 148, 196–201. [Google Scholar] [CrossRef] [PubMed]
- Antal, M.J., Jr.; Gronli, M. The art, science and technology of charcoal production. Ind. Eng. Chem. Res. 2003, 42, 1619–1640. [Google Scholar] [CrossRef]
- Dai, X.; Antal, M.J., Jr. Synthesis of a high-yield activated carbon by air gasification of macadamia nut shell charcoal. Ind. Eng. Chem. Res. 1999, 38, 3386–3395. [Google Scholar] [CrossRef]
- Tam, M.S.; Antal, M.J., Jr.; Jakab, E.; Varhegyi, G. Activated carbon from macadamia nut shell by air oxidation in boiling water. Ind. Eng. Chem. Res. 2001, 40, 578–588. [Google Scholar] [CrossRef]
- Py, X.; Guillot, A.; Cagnon, B. Activated carbon porosity tailoring by cyclic sorption/decomposition of molecular oxygen. Carbon 2003, 41, 1533–1543. [Google Scholar] [CrossRef]
- Kimetu, J.M.; Hill, J.M.; Husein, M.; Bergerson, J.; Layzell, D.B. Using activated biochar for greenhouse gas mitigation and industrial water treatment. Mitig. Adapt. Strateg. Glob. Change 2014. [Google Scholar] [CrossRef]
- Oasmaa, A.; Czernik, S. Fuel oil quality of biomass pyrolysis oils—State of the art for the end users. Energy Fuels 1999, 13, 914–921. [Google Scholar] [CrossRef]
- Huang, Y.; Kudo, S.; Norinaga, K.; Amaike, M.; Hayashi, J. Selective production of light oil by biomass pyrolysis with feedstock-mediated recycling of heavy oil. Energy Fuels 2012, 26, 256–264. [Google Scholar] [CrossRef]
- Huang, Y.; Kudo, S.; Masek, O.; Norinaga, K.; Hayashi, J. Simultaneous maximization of the char yield and volatility of oil from biomass pyrolysis. Energy Fuels 2013, 27, 247–254. [Google Scholar] [CrossRef]
- Veksha, A.; McLaughlin, H.; Layzell, D.B.; Hill, J.M. Pyrolysis of wood to biochar: Increasing yield while maintaining microporosity. Bioresour. Technol. 2014, 153, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Veksha, A.; Zaman, W.; Layzell, D.B.; Hill, J.M. Enhancing biochar yield by co-pyrolysis of bio-oil with biomass: Impacts of potassium hydroxide addition and air pretreatment prior to co-pyrolysis. Bioresour. Technol. 2014, 171, 88–94. [Google Scholar] [CrossRef] [PubMed]
- Kawaguchi, H.; Li, Z.; Masuda, Y.; Sato, K.; Nakagawa, H. Dissolved organic compounds in reused process water for steam-assisted gravity drainage oil sands extraction. Water Res. 2012, 46, 5566–5574. [Google Scholar] [CrossRef] [PubMed]
- Veksha, A.; Pandya, P.; Hill, J.M. The removal of methyl orange from aqueous solution by biochar and activated carbon under microwave irradiation and in the presence of hydrogen peroxide. J. Environ. Chem. Eng. 2015, 3, 1452–1458. [Google Scholar] [CrossRef]
- Petersen, M.A.; Grade, H. Analysis of steam assisted gravity drainage produced water using two-dimensional gas chromatography with time-of-flight mass spectrometry. Ind. Eng. Chem. Res. 2011, 50, 12217–12224. [Google Scholar] [CrossRef]
- Di Blasi, C. Modeling chemical and physical processes of wood and biomass pyrolysis. Prog. Energ. Combust. Sci. 2008, 34, 47–90. [Google Scholar] [CrossRef]
- Katyal, S.; Thambimuthu, K.; Valix, M. Carbonisation of bagasse in a fixed bed reactor: Influence of process variables on char yield and characteristics. Renew. Energy 2003, 28, 713–725. [Google Scholar] [CrossRef]
- Jagtoyen, M.; Derbyshire, F. Activated carbon from yellow poplar and white oak by H3PO4 activation. Carbon 1998, 36, 1085–1097. [Google Scholar] [CrossRef]
- Sams, D.A.; Shadman, F. Catalytic effect of potassium on the rate of char-CO2 gasification. Fuel 1983, 62, 880–882. [Google Scholar] [CrossRef]
- Wigmans, T.; Hoogland, A.; Tromp, P.; Moulijn, J.A. The influence of potassium carbonate on surface area development and reactivity during gasification of activated carbon by carbon dioxide. Carbon 1983, 21, 13–22. [Google Scholar] [CrossRef]
- Figueiredo, J.L.; Pereira, M.F.R.; Freitas, M.M.A.; Orfao, J.J.M. Modification of the surface chemistry of activated carbons. Carbon 1999, 37, 1379–1389. [Google Scholar] [CrossRef]
- Figueiredo, J.L.; Pereira, M.F.R.; Freitas, M.M.A.; Orfao, J.J.M. Characterization of active sites on carbon catalysts. Ind. Eng. Chem. Res. 2007, 46, 4110–4115. [Google Scholar] [CrossRef]
- Zielke, U.; Hüttinger, K.J.; Hoffman, W.P. Surface-oxidized carbon fibers: I. Surface structure and chemistry. Carbon 1996, 34, 983–998. [Google Scholar] [CrossRef]
- Brunauer, S.; Emmett, P.H.; Teller, E. Adsorption of gases in multimolecular layers. J. Am. Chem. Soc. 1938, 60, 309–319. [Google Scholar] [CrossRef]
- Lippens, B.C.; de Boer, J.H. Studies on pore systems in catalysts: V. the t method. J. Catal. 1968, 4, 319–323. [Google Scholar] [CrossRef]
- Dubinin, M.M. Physical adsorption of gases and vapors in micropores. Prog. Surf. Membr. Sci. 1975, 9, 1–70. [Google Scholar]
- Lastoskie, C.; Gubbins, K.E.; Quirkeft, N. Pore size distribution analysis of microporous carbons: A density functional theory approach. J. Phys. Chem. 1993, 97, 4786–4796. [Google Scholar] [CrossRef]
© 2016 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Veksha, A.; Bhuiyan, T.I.; Hill, J.M. Activation of Aspen Wood with Carbon Dioxide and Phosphoric Acid for Removal of Total Organic Carbon from Oil Sands Produced Water: Increasing the Yield with Bio-Oil Recycling. Materials 2016, 9, 20. https://doi.org/10.3390/ma9010020
Veksha A, Bhuiyan TI, Hill JM. Activation of Aspen Wood with Carbon Dioxide and Phosphoric Acid for Removal of Total Organic Carbon from Oil Sands Produced Water: Increasing the Yield with Bio-Oil Recycling. Materials. 2016; 9(1):20. https://doi.org/10.3390/ma9010020
Chicago/Turabian StyleVeksha, Andrei, Tazul I. Bhuiyan, and Josephine M. Hill. 2016. "Activation of Aspen Wood with Carbon Dioxide and Phosphoric Acid for Removal of Total Organic Carbon from Oil Sands Produced Water: Increasing the Yield with Bio-Oil Recycling" Materials 9, no. 1: 20. https://doi.org/10.3390/ma9010020