
The π-Electron Delocalization in 2-Oxazolines Revisited: Quantification and Comparison with Its Analogue in Esters †


Abstract
:
1. Introduction

2. Experimental Section
2.1. Materials
2.2. Instrumentation
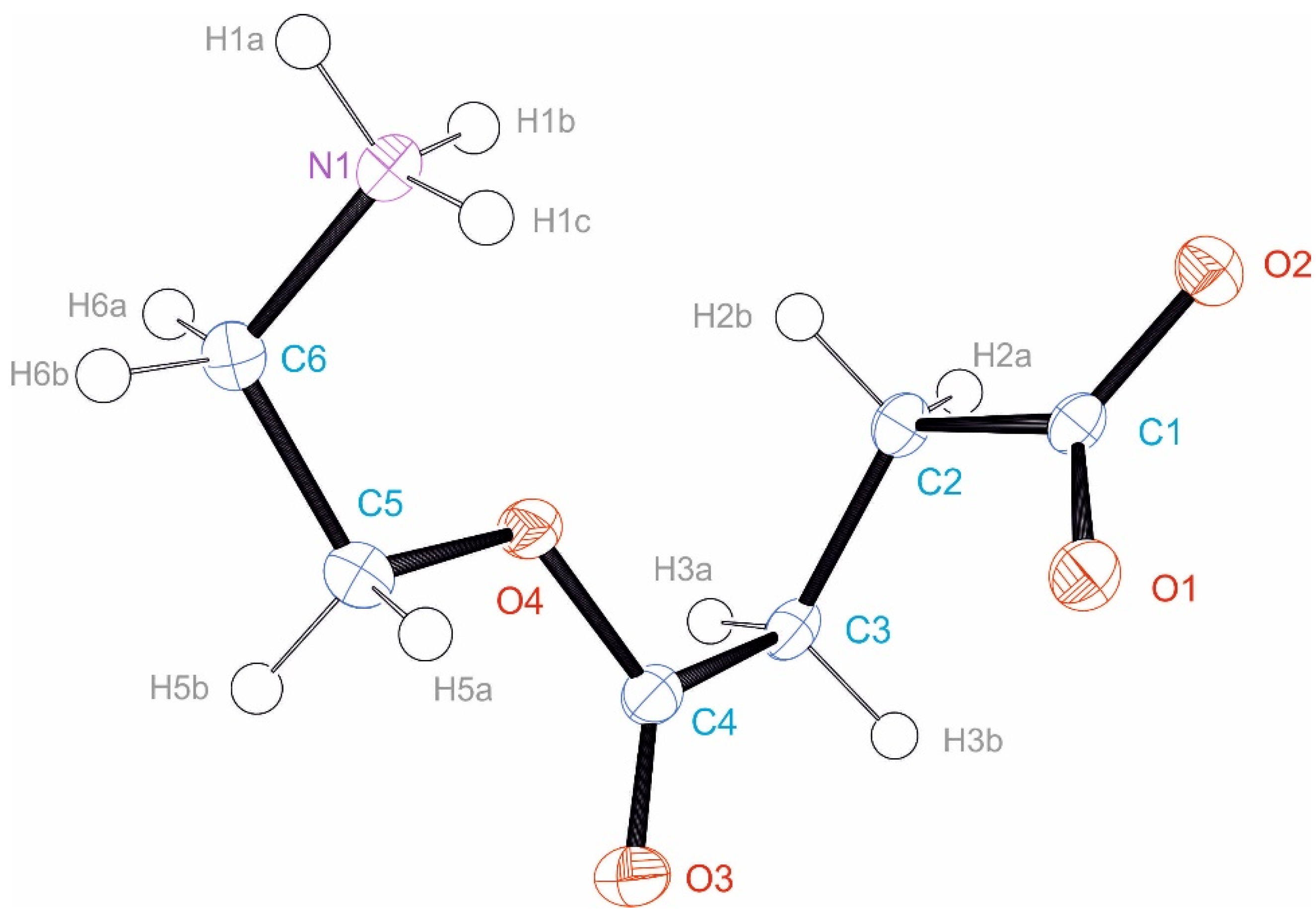
2.3. Single Crystal X-ray Diffraction Analyses

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Crystal Structure Analysis | Methyl 3-(4,5-dihydrooxazol-2-yl) Propanoate, EstOx | 4-(2-Aminoethoxy)-4-oxo-butanoic Acid, EstAA |
|---|---|---|
| Crystal data | ||
| CCDC No. | 1418758 | 1418759 |
| Crystal form | block | block |
| Crystal colour | colorless | colorless |
| Empirical formula | C7H11NO3 | C6H11NO4 |
| Formula weight | 157.17 | 161.16 |
| Crystal system | monoclinic | monoclinic |
| Space group | P21 | P21/c |
| a (Å) | 5.547 (2) | 10.3132 (8) |
| b (Å) | 6.765 (3) | 9.0304 (7) |
| c (Å) | 9.993 (4) | 8.0012 (7) |
| α (°) | 90 | 90 |
| β (°) | 91.583 (13) | 96.688 (5) |
| γ (°) | 90 | 90 |
| V (Å3) | 374.9 (3) | 740.10 (10) |
| ρcalc (g·cm−3) | 1.268 | 1.446 |
| Z | 2 | 4 |
| F(0 0 0) | 152 | 344 |
| µ (Mo-Kα) (cm−1) | 0.102 | 0.122 |
| Data Collection | ||
| Measured reflections | 4559 | 1786 |
| Unique reflections | 1278 | 1786 |
| Rint | 0.0659 | 0.000 |
| Absorption | ||
| Tmin/Tmax | 0.9690/0.9808 | 0.9574/0.9855 |
| Refinement Results | ||
| Refined parameters | 101 | 101 |
| R1 a,b, wR2 a;c | 0.0530; 0.1352 | 0.0630; 0.1432 |
| a, b | 0.0887; 0.0457 | 0,000; 2.2816 |
| ρ (e·Å−3) | 0.306; −0.219 | 0.362; −0.325 |
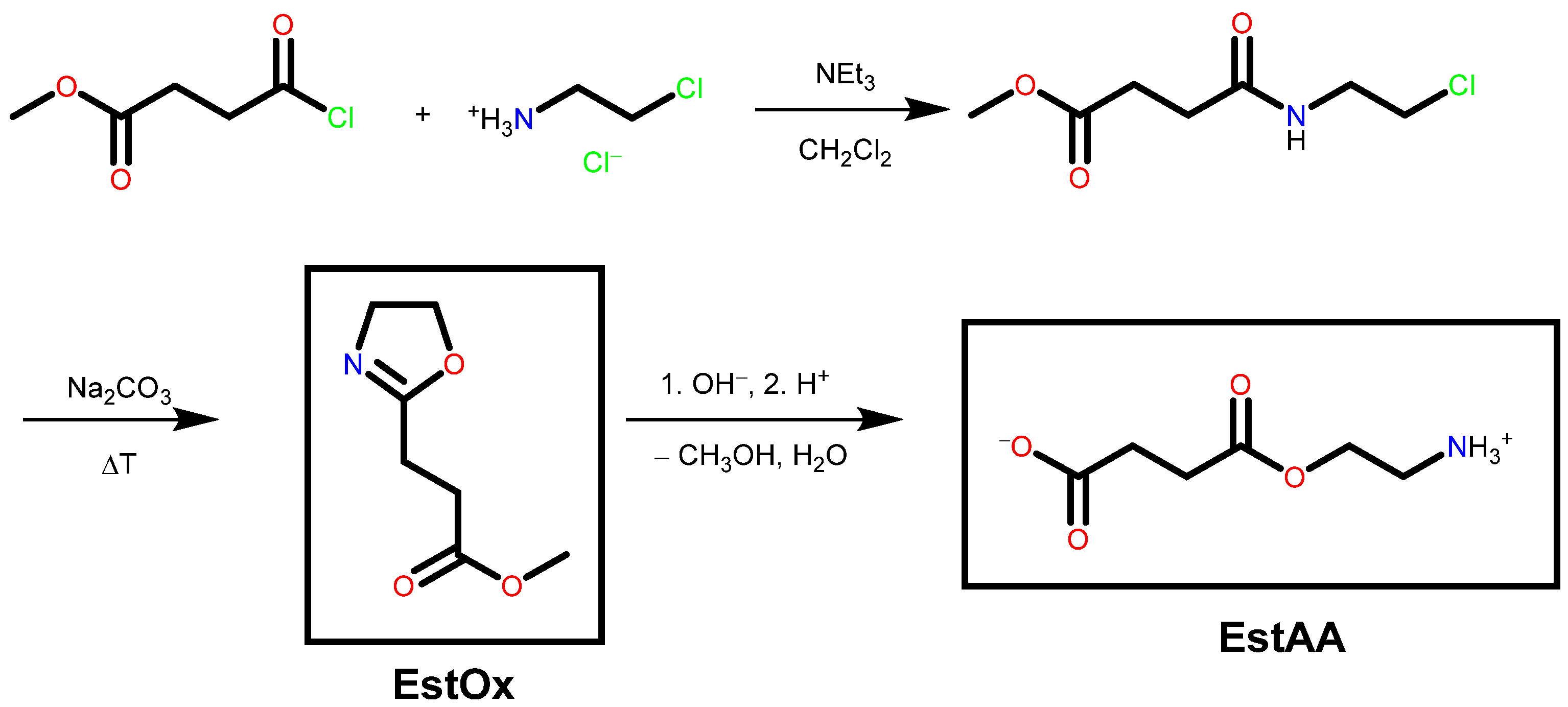
2.4. Preparation of Methyl 3-(4,5-Dihydrooxazol-2-yl)propanoate EstOx
2.5. Preparation of 4-(2-Aminoethoxy)-4-oxobutanoic Acid EstAA
3. Results and Discussion
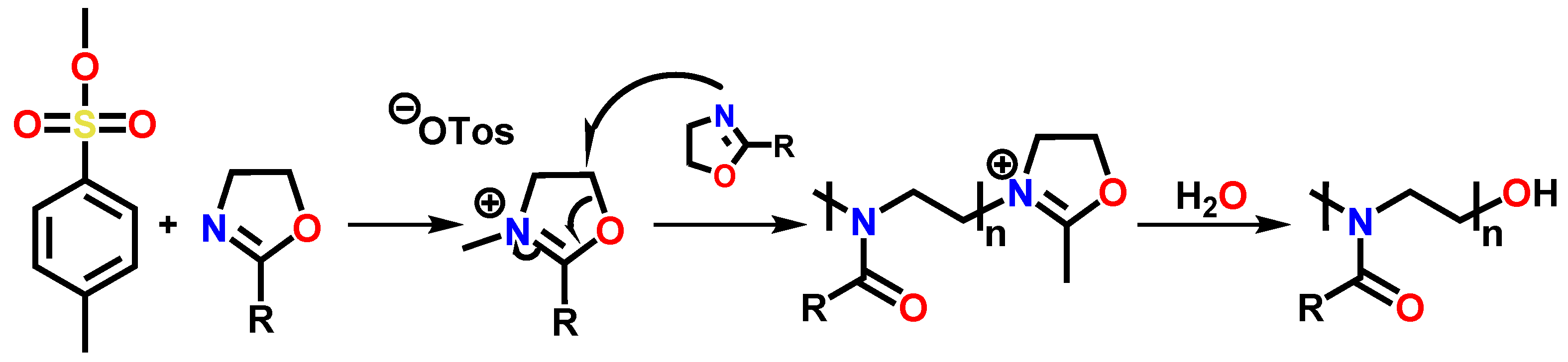
3.1. Synthesis of the Compounds EstOx and EstAA

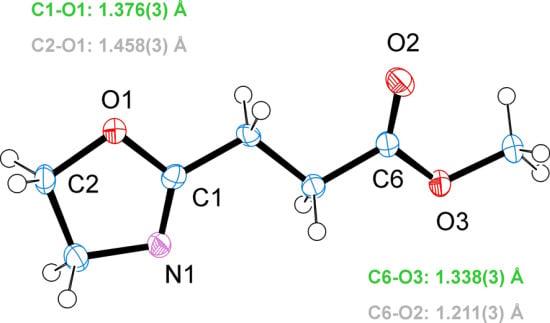
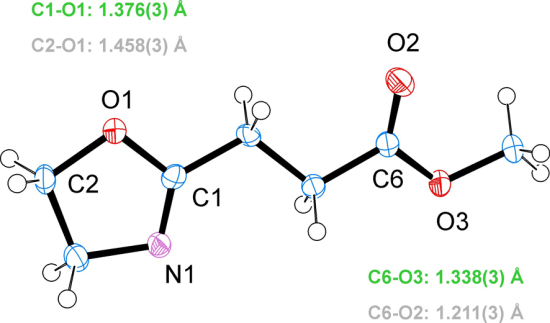
3.2. Crystal Structure of EstOx
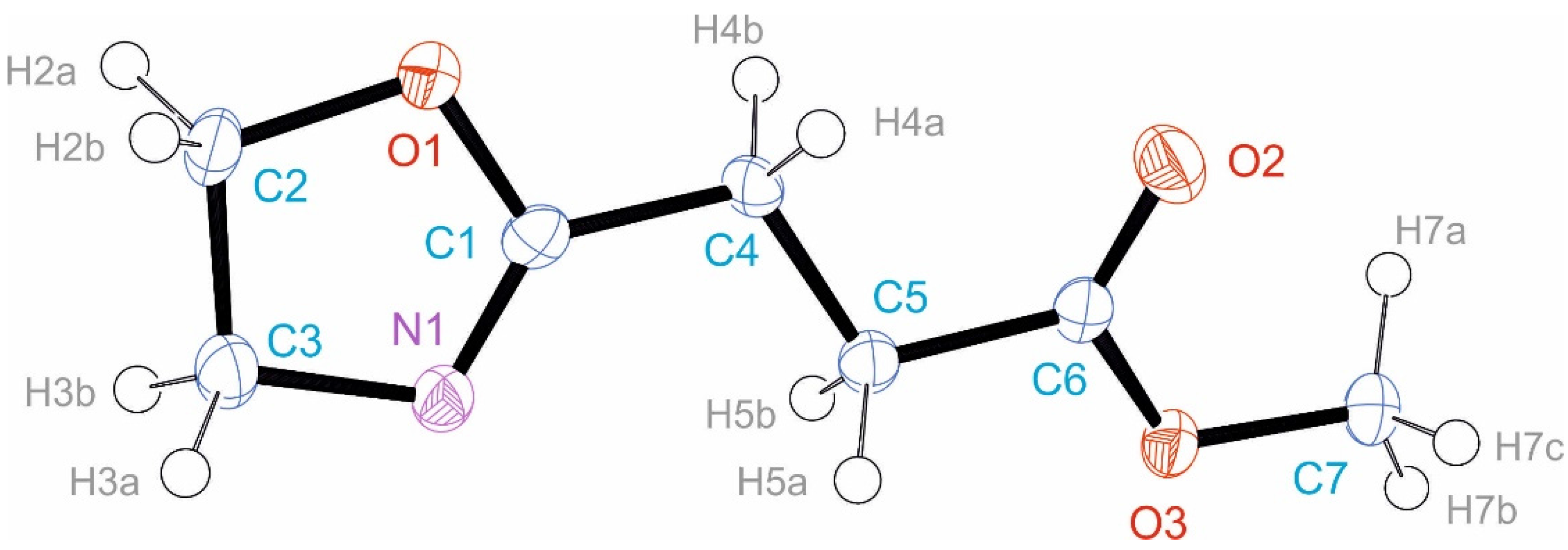
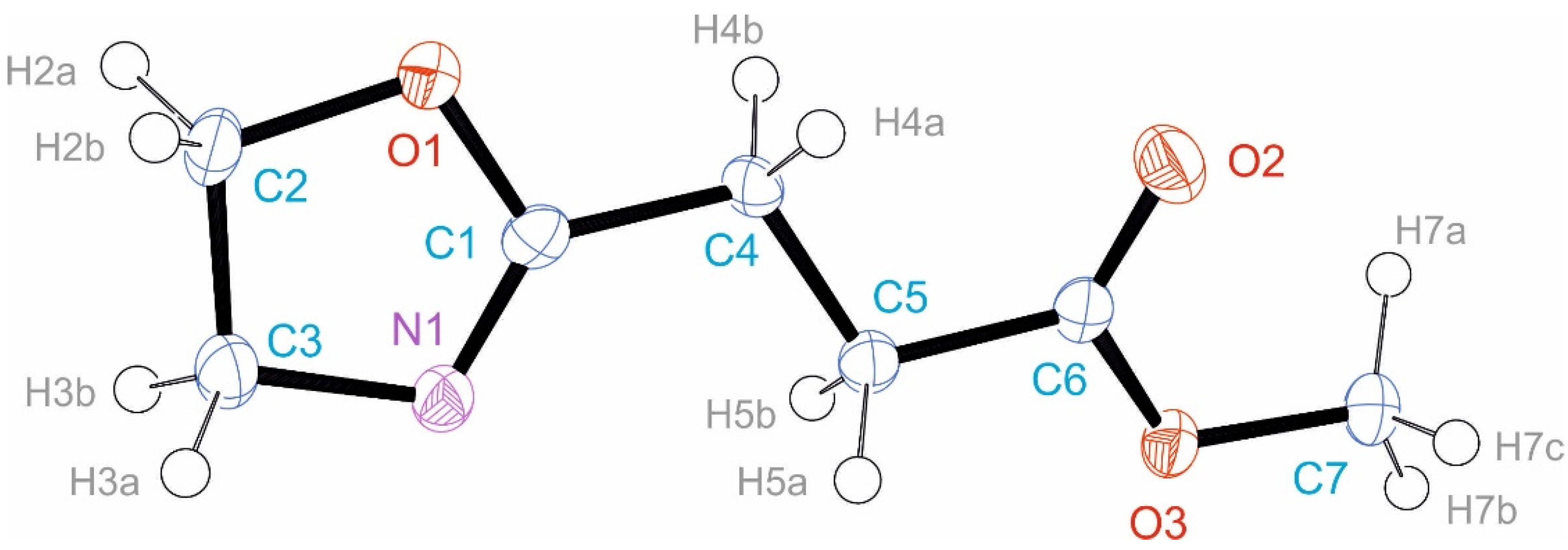
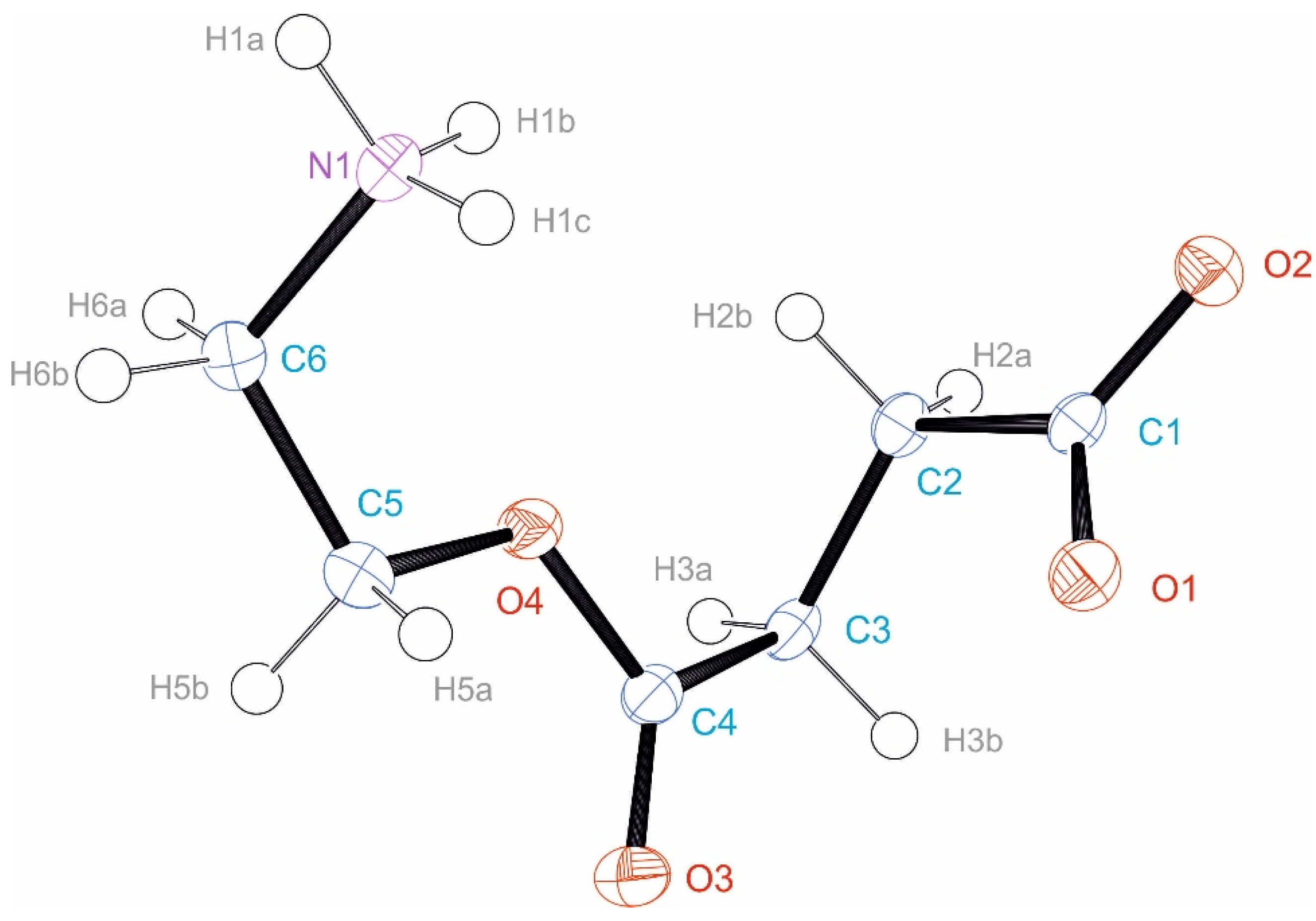
3.3. Crystal Structure of EstAA
| Atom Names | Bond Length N–H | Distance H···O | Distance N···O | Angle NHO |
|---|---|---|---|---|
| N1–H1A···O2 A | 0.91 Å | 1.84 Å | 2.739(3) Å | 171.1° |
| N1–H1B··O2 B | 0.91 Å | 1.86 Å | 2.741(3) Å | 162.7° |
| N1–H1C··O1 C | 0.91 Å | 1.84 Å | 2.747(3) Å | 172.2° |
3.4. Quantum Chemical Calculations of EstOx
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kappe, C.O. Controlled microwave heating in modern organic synthesis. Angew. Chem. Int. Ed. 2004, 43, 6250–6284. [Google Scholar] [CrossRef] [PubMed]
- Ebner, C.; Bodner, T.; Stelzer, F.; Wiesbrock, F. One decade of microwave-assisted polymerizations: Quo vadis? Macromol. Rapid Commun. 2011, 32, 254–288. [Google Scholar] [CrossRef] [PubMed]
- Rossegger, E.; Schenk, V.; Wiesbrock, F. Design strategies for functionalized poly(2-oxazoline)s and derived materials. Polymers 2013, 5, 956–1011. [Google Scholar] [CrossRef]
- Kempe, K.; Becer, C.R.; Schubert, U.S. Microwave-assisted polymerizations: Recent status and future perspectives. Macromolecules 2011, 44, 5825–5842. [Google Scholar] [CrossRef]
- Tomalia, D.A.; Sheetz, D.P. Homopolymerization of 2-alkyl- and 2-aryl-2-oxazolines. J. Polym. Sci. A Polym. Chem. 1966, 4, 2253–2265. [Google Scholar] [CrossRef]
- Seeliger, W.; Aufderhaar, E.; Diepers, W.; Feinauer, R.; Nehring, R.; Thier, W.; Hellmann, H. Recent syntheses and reactions of cyclic imidic esters. Angew. Chem. Int. Ed. 1966, 5, 875–888. [Google Scholar] [CrossRef] [PubMed]
- Kagiya, T.; Narisawa, S.; Maeda, T.; Fukui, K. Ring-opening polymerisation of 2-substituted 2-oxazolines. J. Polym. Sci. B Polym. Lett. 1966, 4, 441–445. [Google Scholar] [CrossRef]
- Bassiri, T.G.; Levy, A.; Litt, M. Polymerization of cyclic imino ethers. I. Oxazolines. J. Polym. Sci. B Polym. Lett. 1967, 5, 871–879. [Google Scholar] [CrossRef]
- Hoogenboom, R. Poly(2-oxazoline)s: A polymer class with numerous potential applications. Angew. Chem. Int. Ed. 2009, 48, 7978–7994. [Google Scholar] [CrossRef] [PubMed]
- Hoogenboom, R.; Schlaad, H. Bioinspired poly(2-oxazoline)s. Polymers 2011, 3, 467–488. [Google Scholar] [CrossRef]
- Schlaad, H.; Diehl, C.; Gress, A.; Meyer, M.; Demirel, A.L.; Nur, Y.; Bertin, A. Poly(2-oxazoline)s as smart bioinspired polymers. Macromol. Rapid Commun. 2010, 31, 511–525. [Google Scholar] [CrossRef] [PubMed]
- Gaertner, F.C.; Luxenhofer, R.; Blechert, B.; Jordan, R.; Essler, M. Synthesis, biodistribution and excretion of radiolabeled poly(2-alkyl-2-oxazoline)s. J. Control. Release 2007, 119, 291–300. [Google Scholar] [CrossRef] [PubMed]
- Waschinski, C.J.; Tiller, J.C. Poly(oxazoline)s with telechelic antimicrobial functions. Biomacromolecules 2005, 6, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Waschinski, C.J.; Barnert, S.; Theobald, A.; Schubert, R.; Kleinschmidt, F.; Hoffmann, A.; Saalwächter, K.; Tiller, J.C. Insights in the antibacterial action of poly(methyloxazoline)s with a biocidal end group and varying satellite groups. Biomacromolecules 2008, 9, 1764–1771. [Google Scholar] [CrossRef] [PubMed]
- Kelly, A.; Kaltenhauser, V.; Mühlbacher, I.; Rametsteiner, K.; Kren, H.; Slugovc, C.; Stelzer, F.; Wiesbrock, F. Poly(2-oxazoline)-derived contact biocides: Contributions to the understanding of antimicrobial activity. Macromol. Biosci. 2013, 13, 116–125. [Google Scholar] [CrossRef] [PubMed]
- Kelly, A.M.; Wiesbrock, F. Strategies for the synthesis of poly(2-oxazoline)-based hydrogels. Macromol. Rapid Commun. 2012, 33, 1632–1647. [Google Scholar] [CrossRef] [PubMed]
- Schenk, V.; Ellmaier, L.; Rossegger, E.; Edler, M.; Griesser, T.; Weidinger, G.; Wiesbrock, F. Water-developable poly(2-oxazoline)-based negative photoresists. Macromol. Rapid Commun. 2012, 33, 396–400. [Google Scholar] [CrossRef] [PubMed]
- Fijten, M.W.M.; Haensch, C.; van Lankvelt, B.M.; Hoogenboom, R.; Schubert, U.S. Clickable poly(2-oxazoline)s as versatile building blocks. Macromol. Chem. Phys. 2008, 209, 1887–1895. [Google Scholar] [CrossRef]
- Zschoche, S.; Rueda, J.; Boyko, V.; Krahl, F.; Arndt, K.-F.; Voit, B. Thermo-responsive nanogels based on poly[NIPAAm-graft-(2-alkyl-2-oxazoline)]s crosslinked in the micellar state. Macromol. Chem. Phys. 2011, 211, 1035–1042. [Google Scholar] [CrossRef]
- Kempe, K.; Hoogenboom, R.; Jaeger, M.; Schubert, U.S. Three-fold metal-free efficient “click” reactions onto a multifunctional poly(2-oxazoline) designer scaffold. Macromolecules 2011, 44, 6424–6432. [Google Scholar] [CrossRef]
- Kelly, A.M.; Hecke, A.; Wirnsberger, B.; Wiesbrock, F. Synthesis of poly(2-oxazoline)-based hydrogels with tailor-made swelling degrees capable of stimuli-triggered compound release. Macromol. Rapid Commun. 2011, 32, 1815–1819. [Google Scholar] [CrossRef] [PubMed]
- Ten Brummelhuis, N.; Schlaad, H. Stimuli-responsive star polymers through thiol-yne core functionalization/crosslinking of block copolymer micelles. Polym. Chem. 2011, 2, 1180–1184. [Google Scholar] [CrossRef]
- Wiesbrock, F.; Hoogenboom, R.; Leenen, M.A.M.; Meier, M.A.R.; Schubert, U.S. Investigation of the living cationic ring-opening polymerization of 2-methyl-, 2-ethyl-, 2-nonyl-, and 2-phenyl-2-oxazoline in a single-mode microwave reactor. Macromolecules 2005, 38, 5025–5034. [Google Scholar] [CrossRef]
- Hoogenboom, R.; Fijten, M.W.M.; Kickelbick, G.; Schubert, U.S. Synthesis and crystal structures of multifunctional tosylates as basis for star-shaped poly(2-ethyl-2-oxazoline)s. Beilstein J. Org. Chem. 2010, 6, 773–783. [Google Scholar] [CrossRef] [PubMed]
- Luxenhofer, R.; Bezen, M.; Jordan, R. Kinetic investigations on the polymerization of 2-oxazolines using pluritriflate initiators. Macromol. Rapid Commun. 2008, 29, 1509–1513. [Google Scholar] [CrossRef]
- Wiesbrock, F.; Hoogenboom, R.; Abeln, C.H.; Schubert, U.S. Single-mode microwave ovens as new reaction devices: Accelerating the living polymerization of 2-ethyl-2-oxazoline. Macromol. Rapid Commun. 2004, 25, 1895–1899. [Google Scholar] [CrossRef]
- Bodner, T.; Ellmaier, L.; Schenk, V.; Albering, J.; Wiesbrock, F. Delocalized π-electrons in 2-oxazoline rings resulting in negatively charged nitrogen atoms: Revealing the selectivity during the initiation of cationic ring-opening polymerizations. Polym. Int. 2011, 60, 1173–1179. [Google Scholar] [CrossRef]
- SHELXS-97, Program for structure solution; University of Göttingen: Göttingen, Germany, 1997.
- SHELXL-97, Program for crystal structure analysis; University of Göttingen: Göttingen, Germany, 1997.
- Spek, A.L. Single-crystal structure validation with the program PLATON. J. Appl. Crystallogr. 2003, 36, 7–13. [Google Scholar] [CrossRef]
- Area-Detector Absorption Correction; Siemens Industrial Automation, Inc.: Madison, WI, USA, 1996.
- Zarka, M.T.; Nuyken, O.; Weberskirch, R. Amphiphilic polymer supports for the asymmetric hydrogenation of amino acid precursors in water. Chem. Eur. J. 2003, 9, 3228–3234. [Google Scholar] [CrossRef] [PubMed]
- Johnson, C.K. ORTEP; Report ORNL-5138; Oak Ridge National Laboratory: Oak Ridge, TN, USA, 1976. [Google Scholar]
- Cottrell, T.L. The Strengths of Chemical Bonds, 2nd ed.; Butterworths: London, UK, 1958. [Google Scholar]
- Benson, S.W. III - Bond energies. J. Chem. Educ. 1965, 42, 502–518. [Google Scholar] [CrossRef]
- Kállay, M.; Rolik, Z.; Csontos, J.; Ladjánszki, I.; Szegedy, L.; Ladóczki, B.; Samu, G.; MRCC. A Quantum Chemical Program Suite. Budapest University of Technology and Economics: Budapest, Hungary. Available online: http://www.mrcc.hu (accessed on 28 May 2015).
- Rolik, Z.; Szegedy, L.; Ladjánszki, I.; Ladóczki, B.; Kállay, M. An efficient linear-scaling CCSD(T) method based on local natural orbitals. J. Chem. Phys. 2013, 139, 094105. [Google Scholar] [CrossRef] [PubMed]
- Mulliken, R.S. Electronic population analysis on LCAO-MO molecular wave functions. J. Chem. Phys. 1955, 23, 1833–1840. [Google Scholar] [CrossRef]
- Mayer, I. Charge, bond order and valence in the ab initio SCF theory. Chem. Phys. Lett. 1983, 97, 270–274. [Google Scholar] [CrossRef]
- Dunning, T.H. Gaussian basis sets for use in correlated molecular calculations. I. The atoms boron through neon and hydrogen. J. Chem. Phys. 1989, 90, 1007–1023. [Google Scholar] [CrossRef]
- Weigend, F. Hartree-Fock exchange fitting basis sets for H to Rn. J. Comp. Chem. 2008, 29, 167–175. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Fimberger, M.; Luef, K.P.; Payerl, C.; Fischer, R.C.; Stelzer, F.; Kállay, M.; Wiesbrock, F. The π-Electron Delocalization in 2-Oxazolines Revisited: Quantification and Comparison with Its Analogue in Esters. Materials 2015, 8, 5385-5397. https://doi.org/10.3390/ma8085249
Fimberger M, Luef KP, Payerl C, Fischer RC, Stelzer F, Kállay M, Wiesbrock F. The π-Electron Delocalization in 2-Oxazolines Revisited: Quantification and Comparison with Its Analogue in Esters. Materials. 2015; 8(8):5385-5397. https://doi.org/10.3390/ma8085249
Chicago/Turabian StyleFimberger, Martin, Klaus P. Luef, Claudia Payerl, Roland C. Fischer, Franz Stelzer, Mihály Kállay, and Frank Wiesbrock. 2015. "The π-Electron Delocalization in 2-Oxazolines Revisited: Quantification and Comparison with Its Analogue in Esters" Materials 8, no. 8: 5385-5397. https://doi.org/10.3390/ma8085249