
TiO2-Mediated Photocatalytic Mineralization of a Non-Ionic Detergent: Comparison and Combination with Other Advanced Oxidation Procedures

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
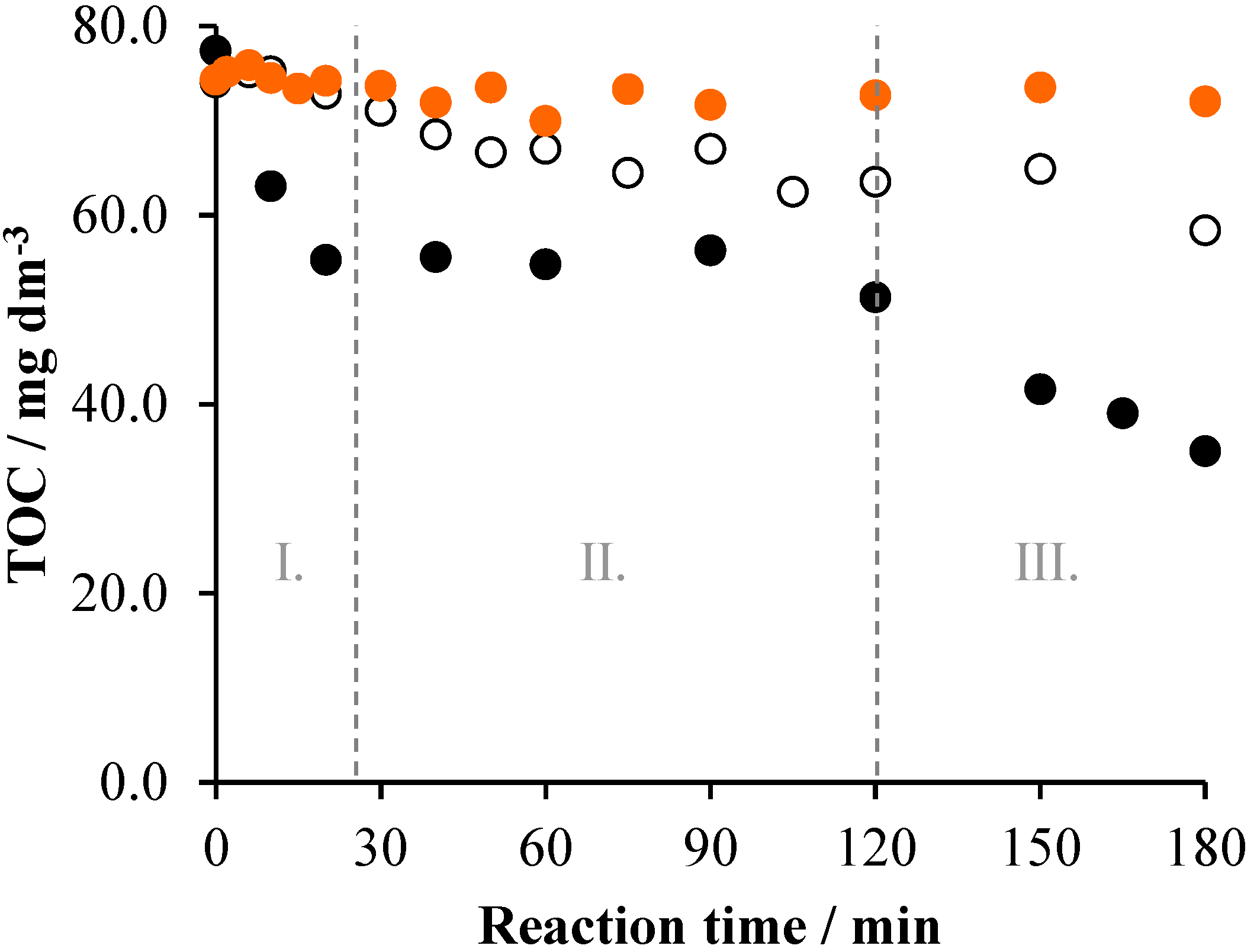
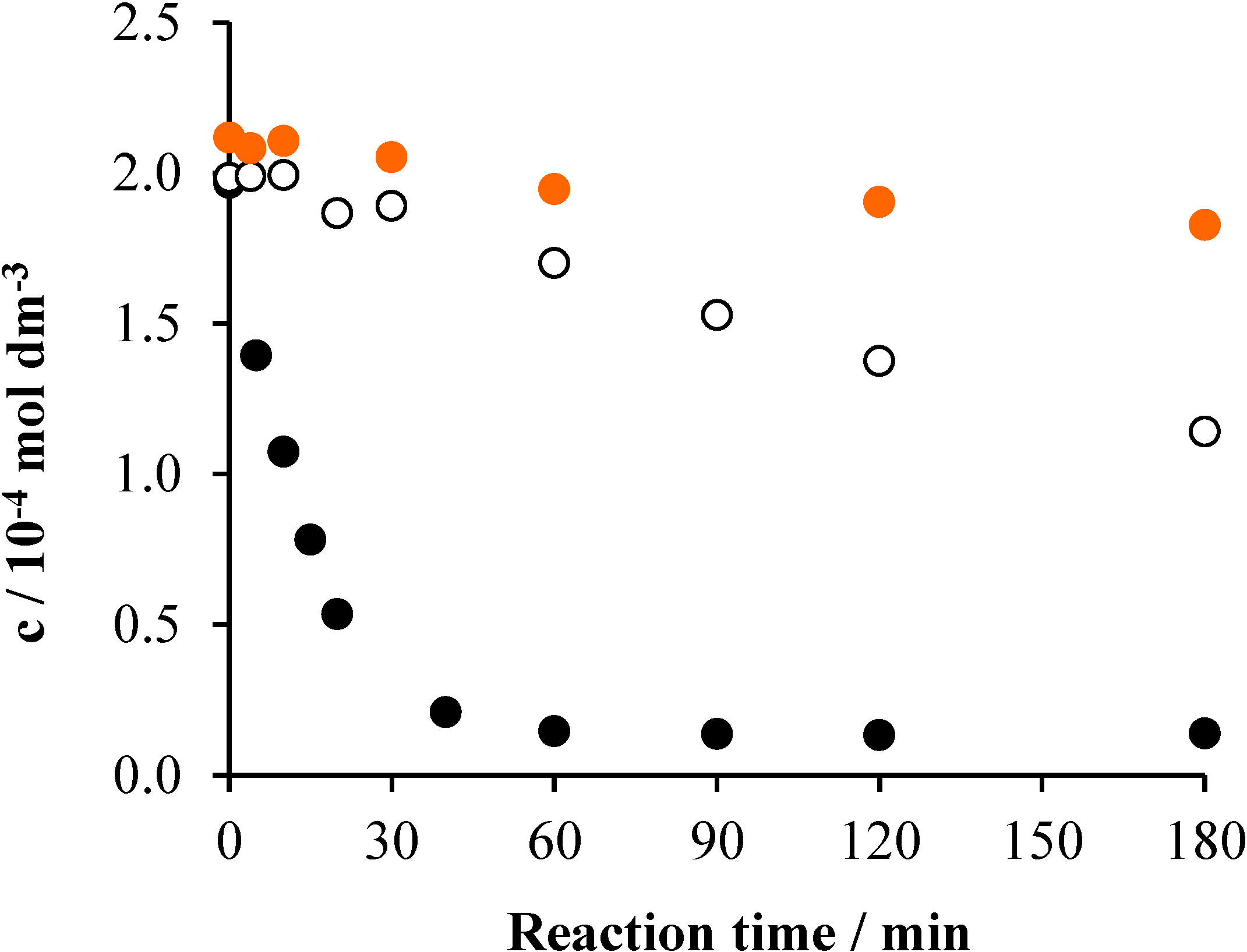
2. Results and Discussion
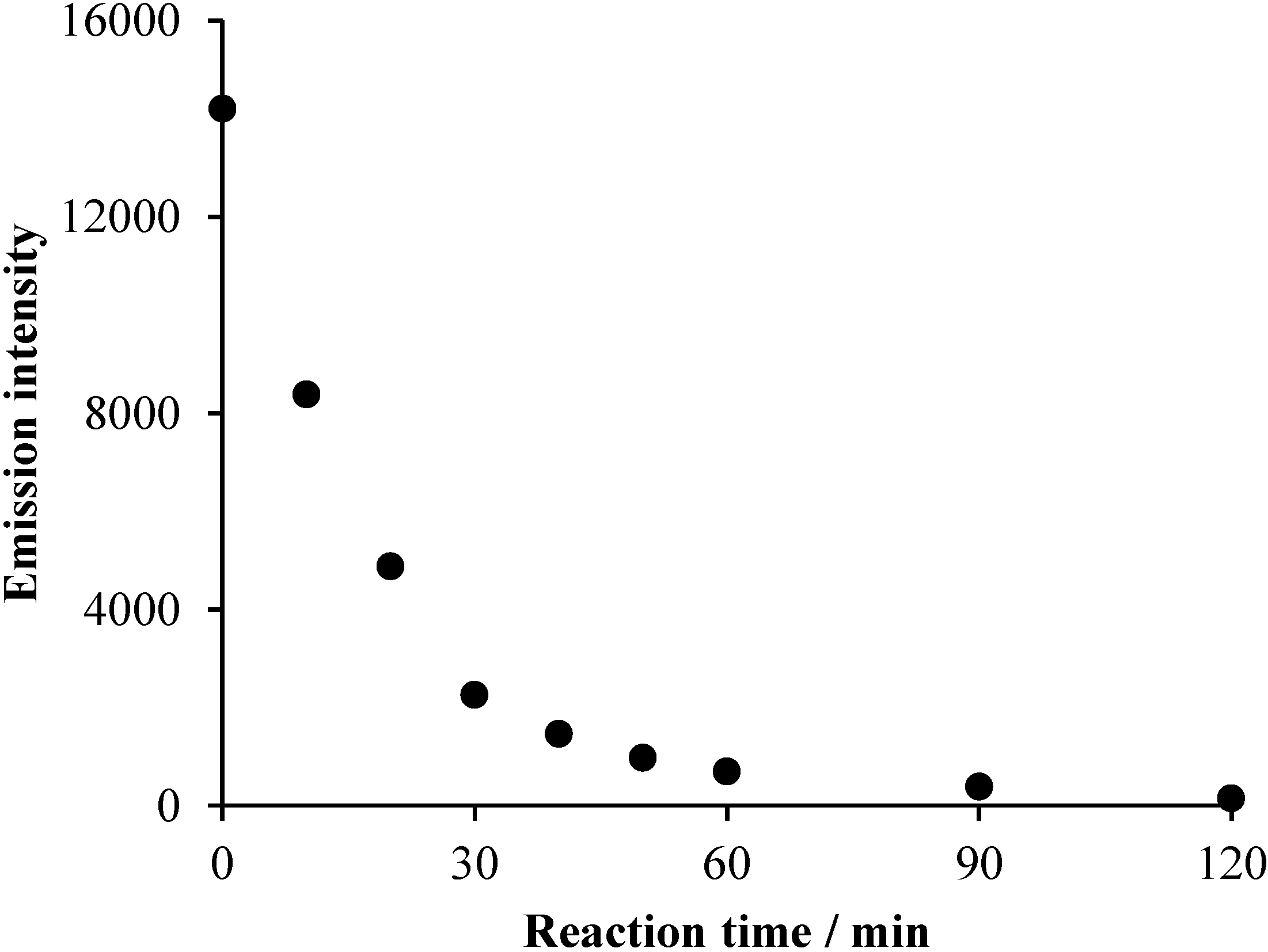
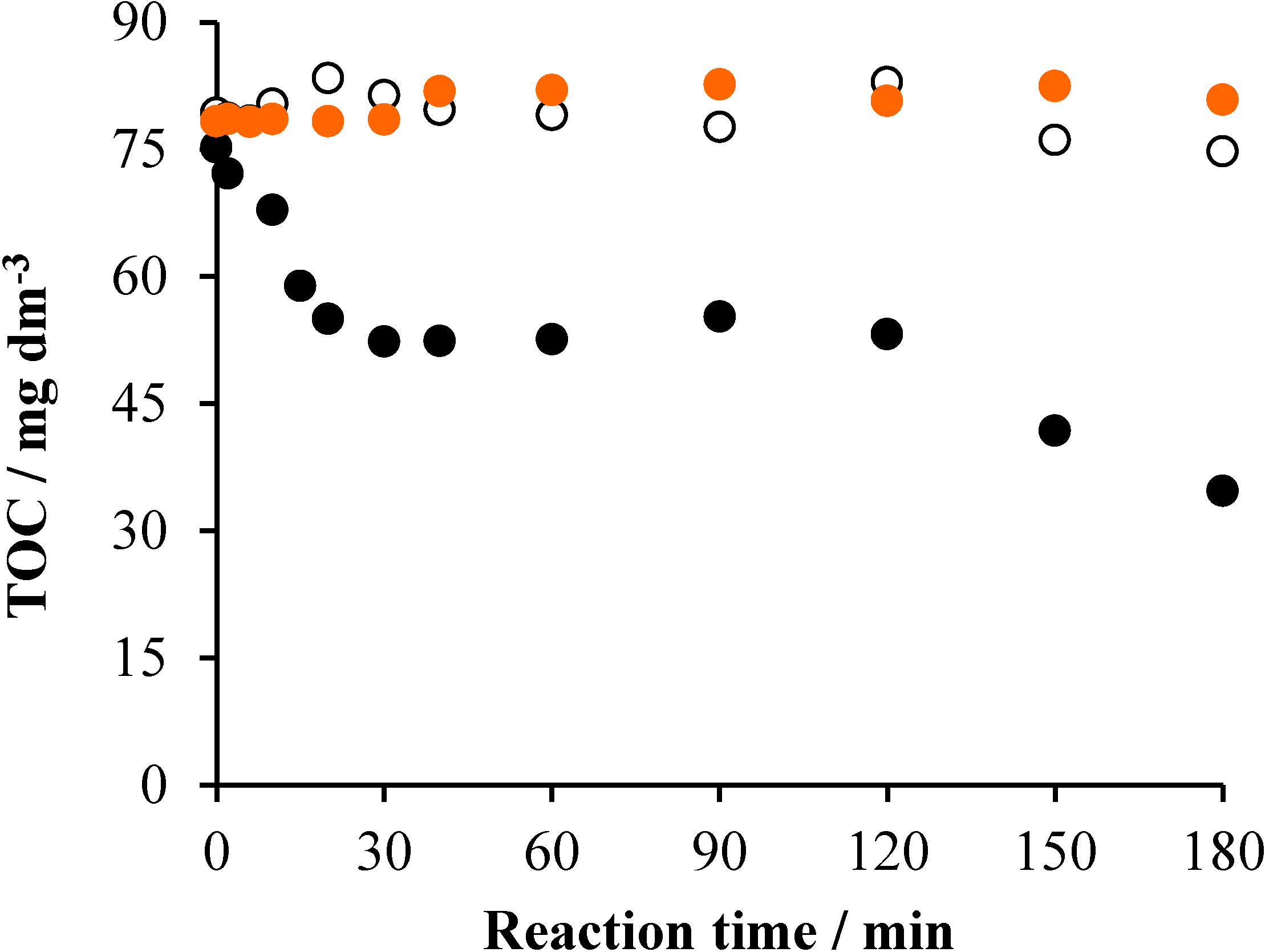
2.1. TiO2/UV/Air System
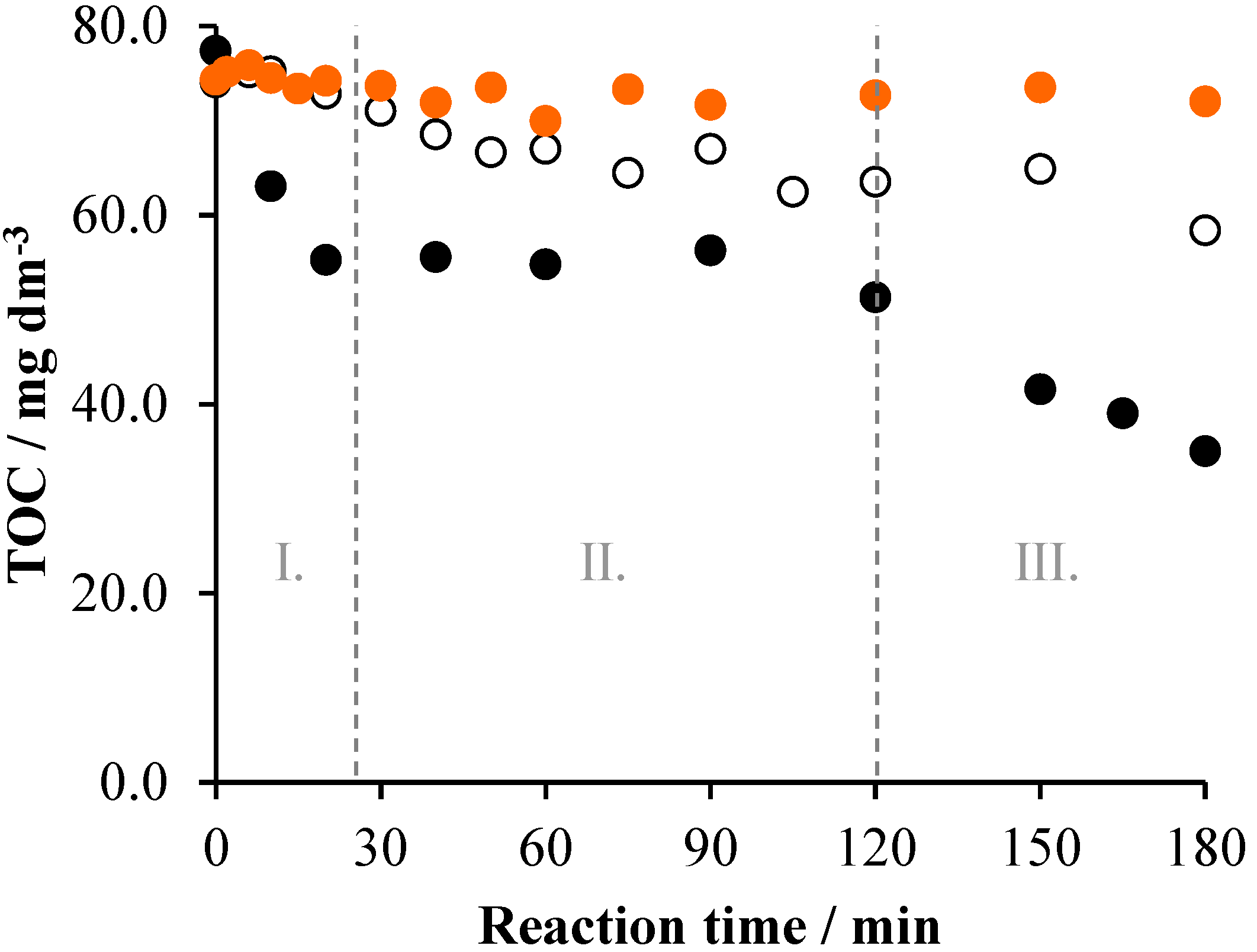

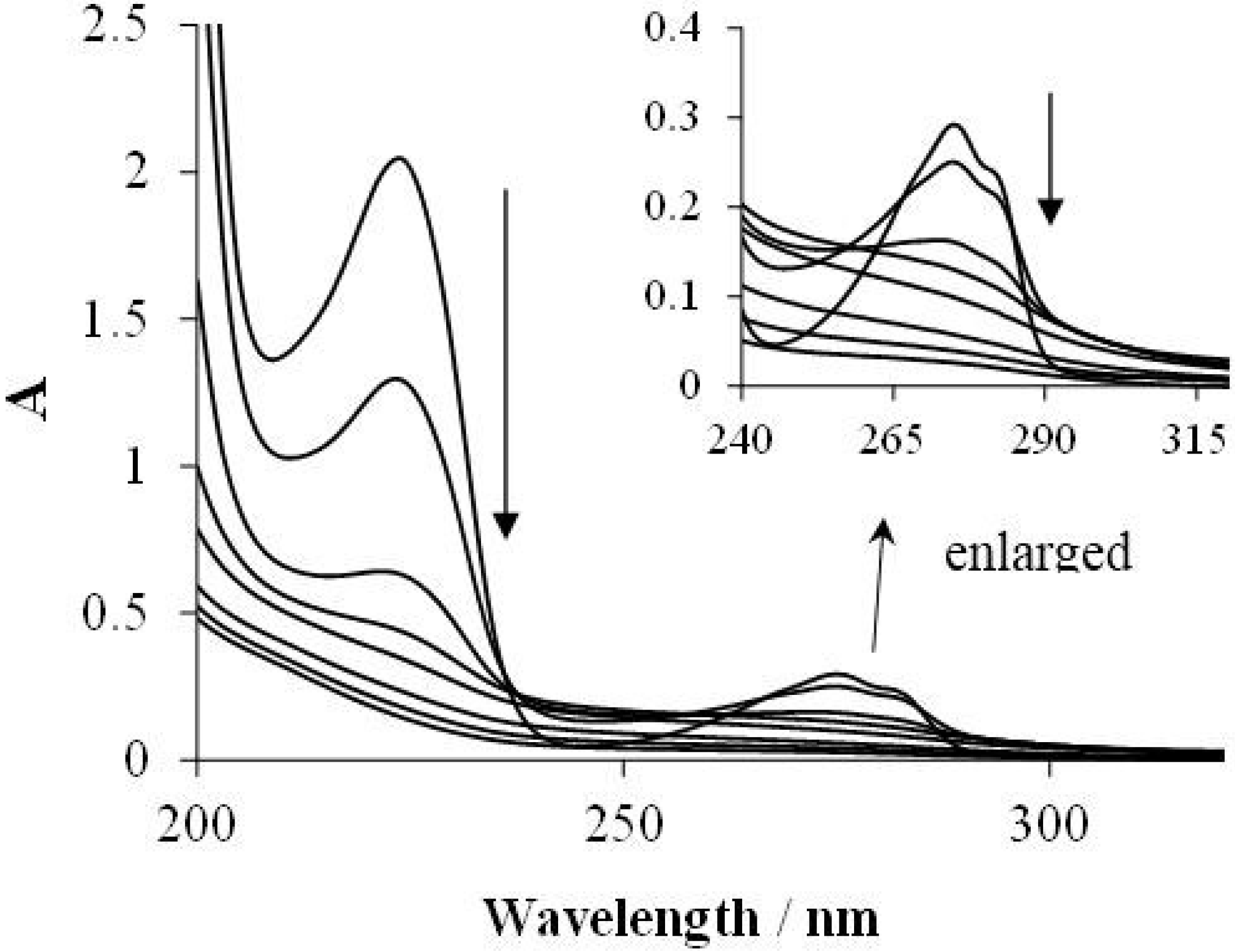

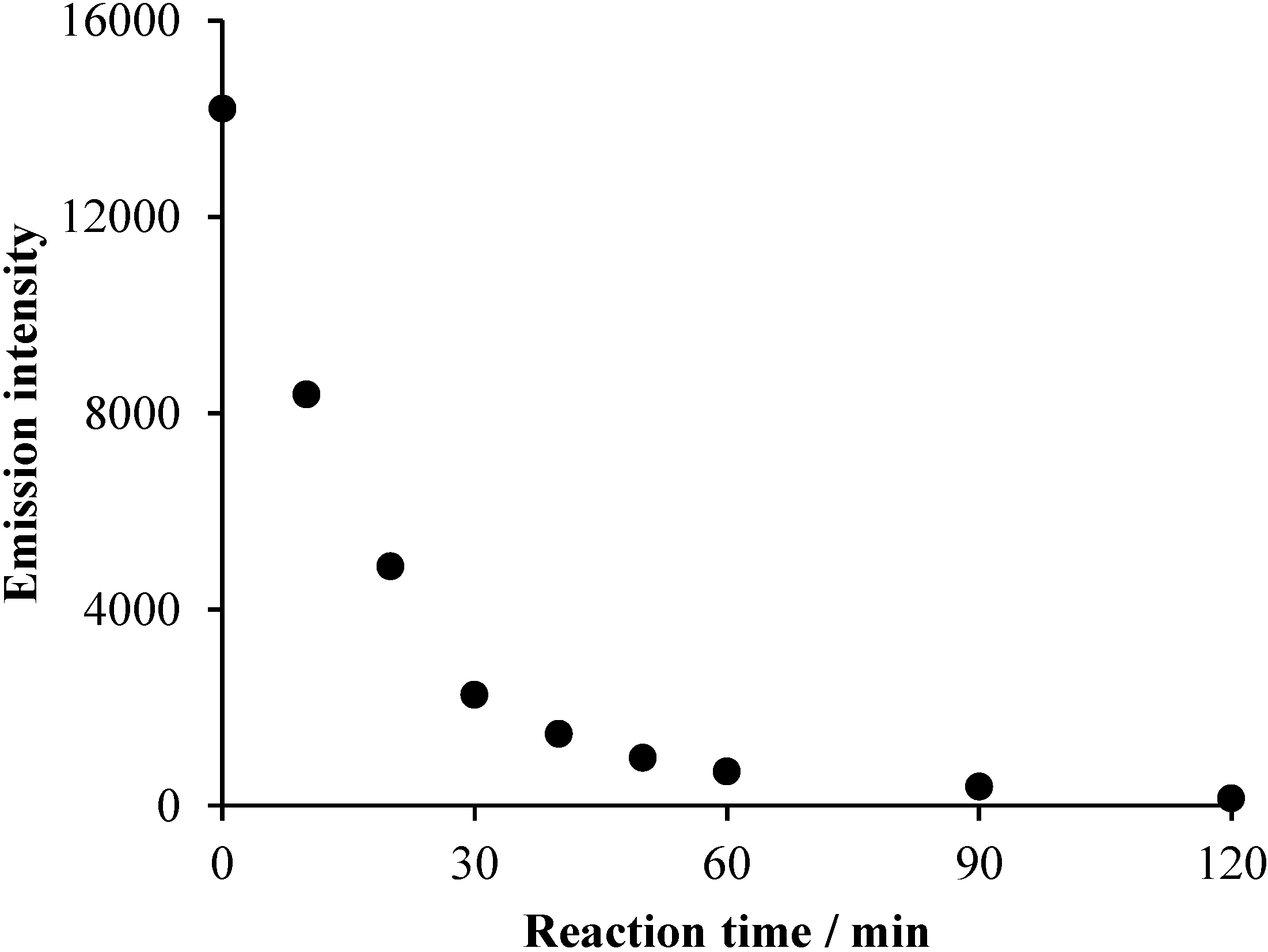
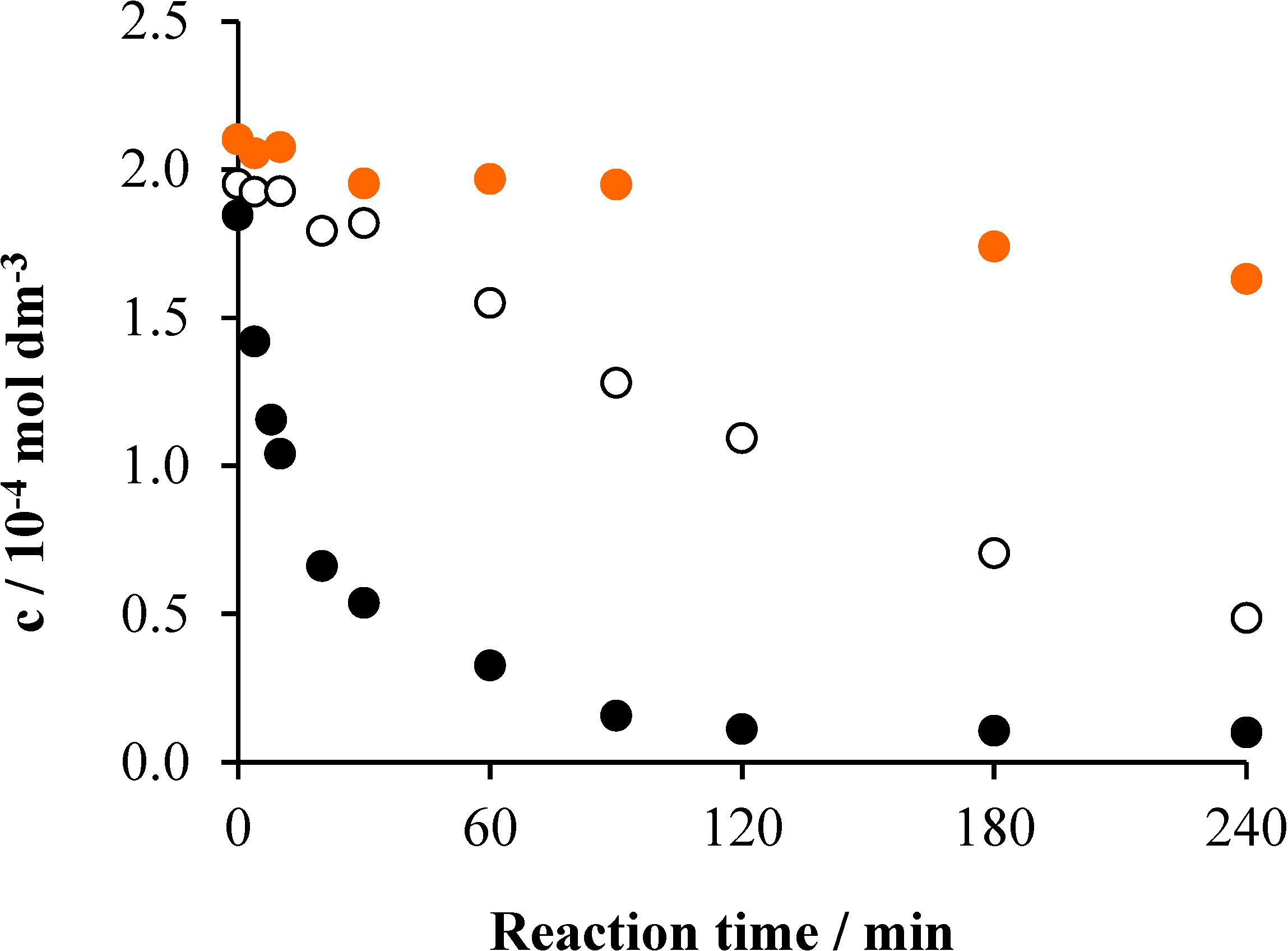
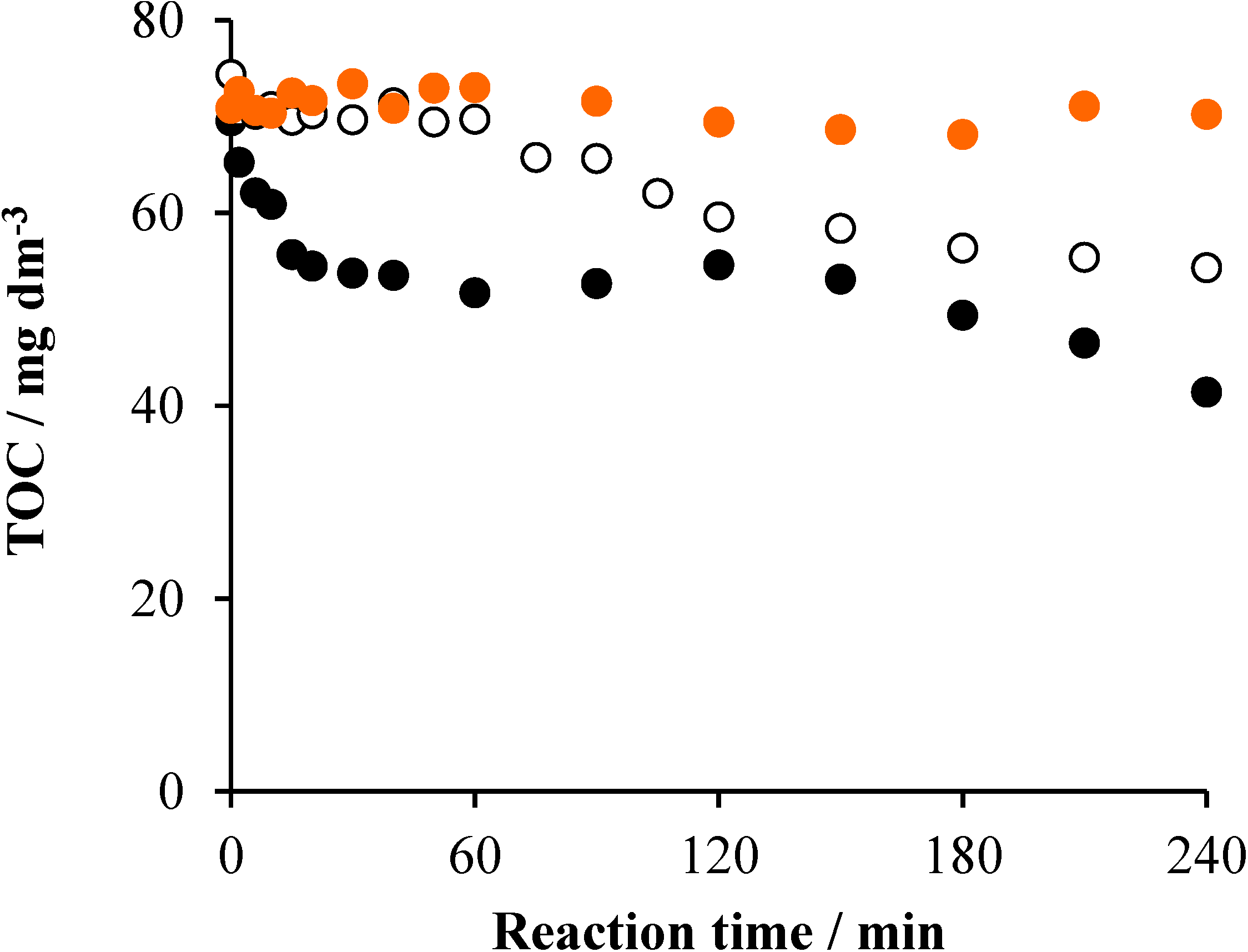
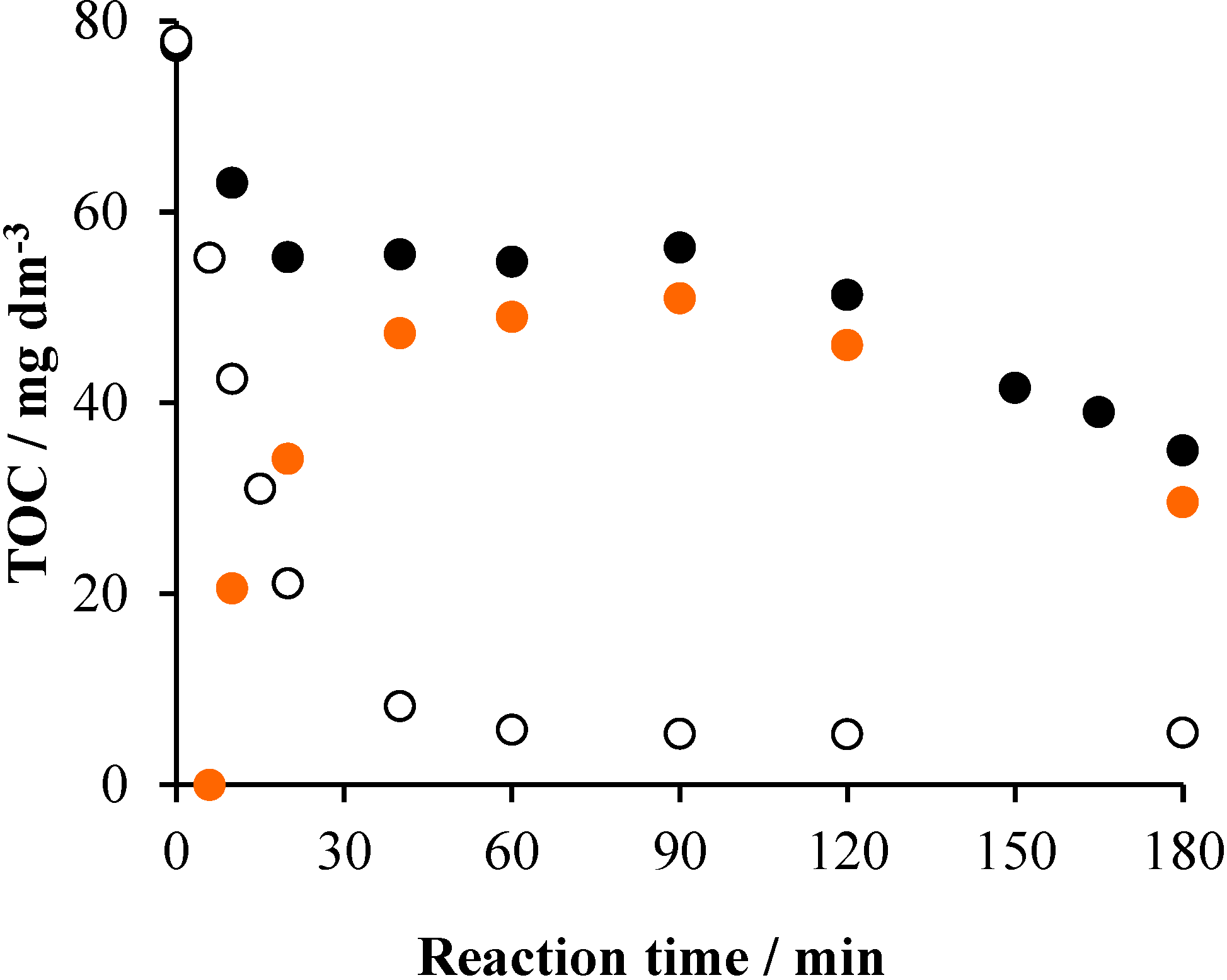
2.2. Effects of Na2S2O8
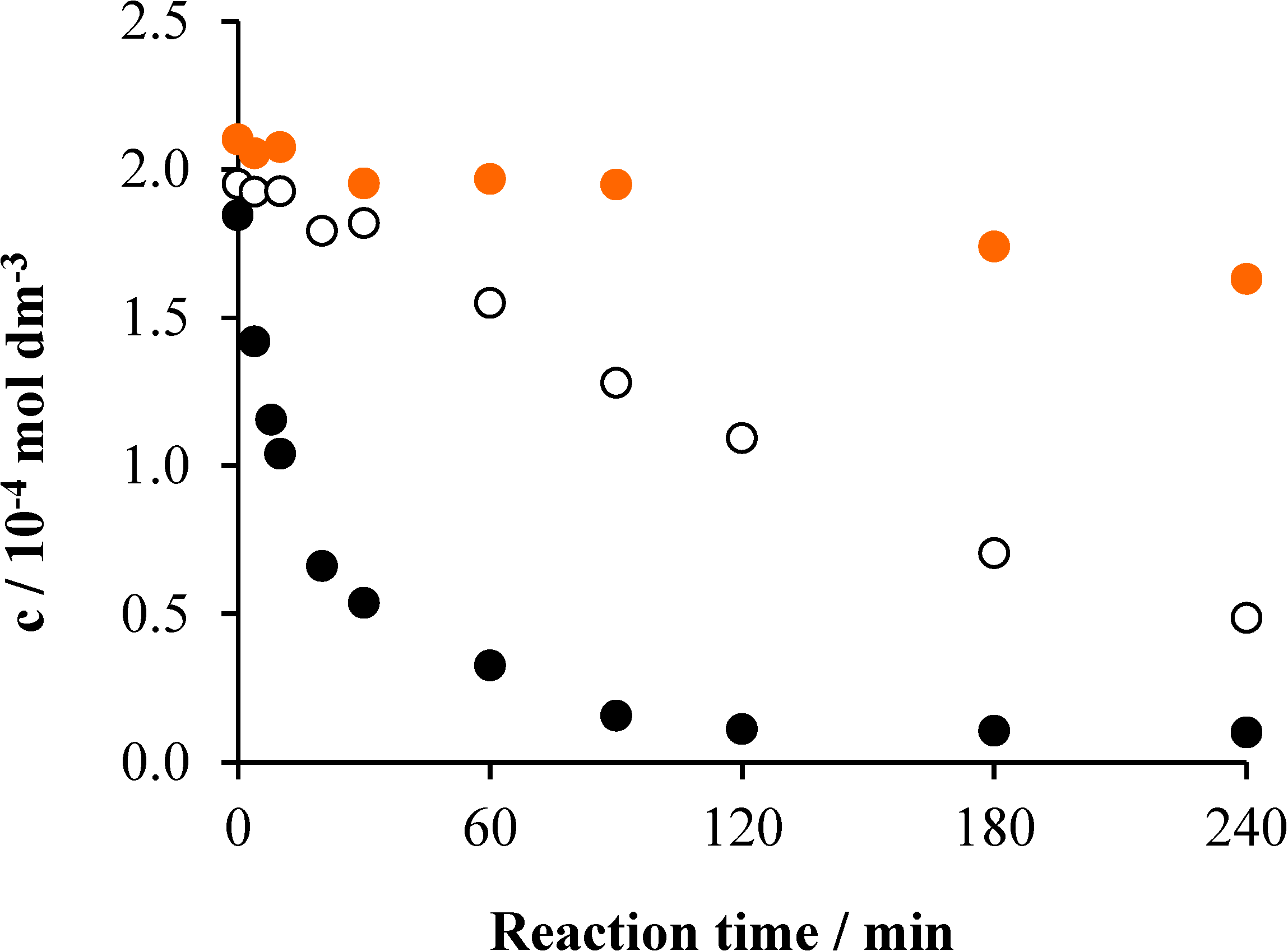

2.3. Effects of Ozonation
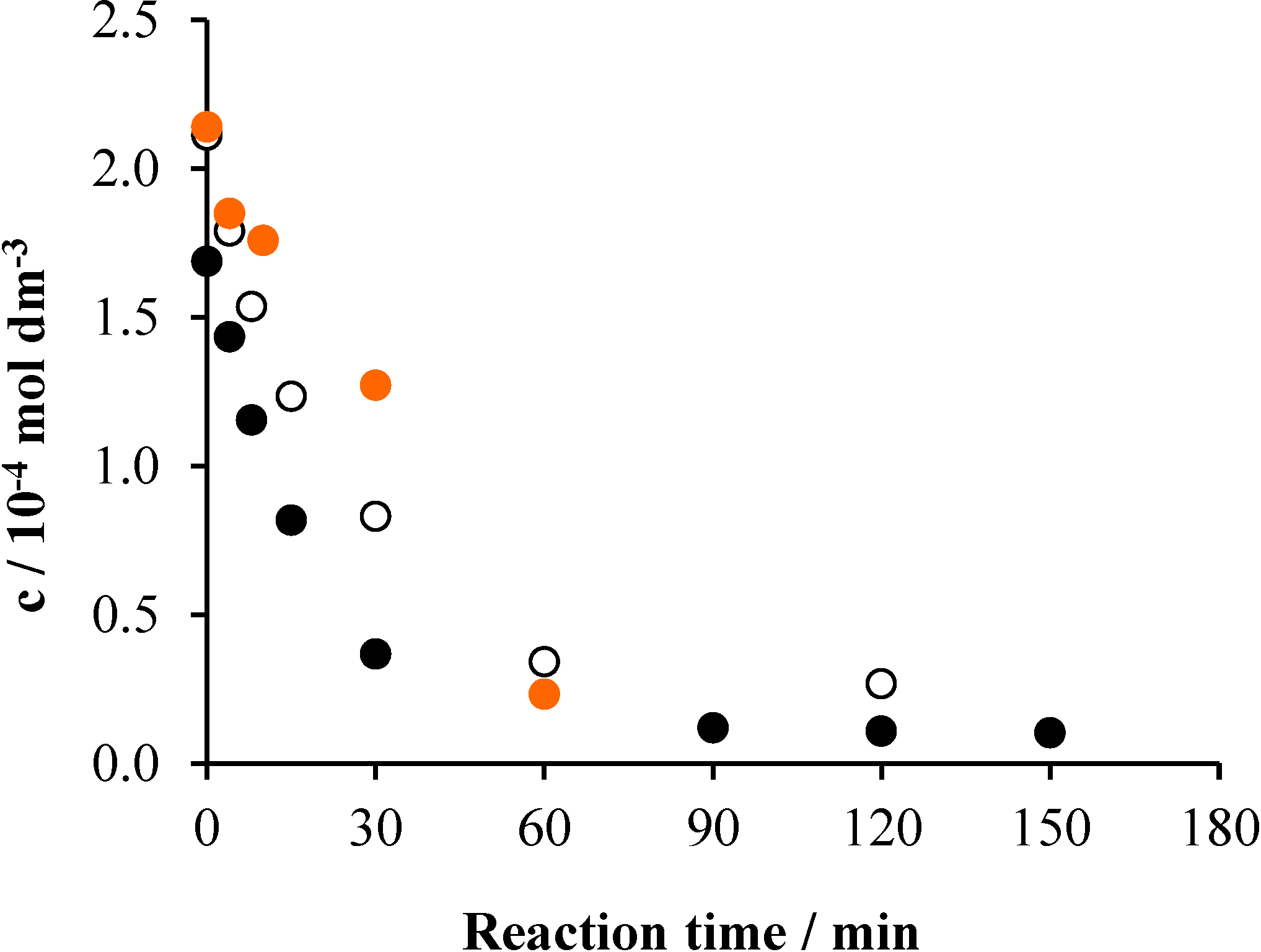
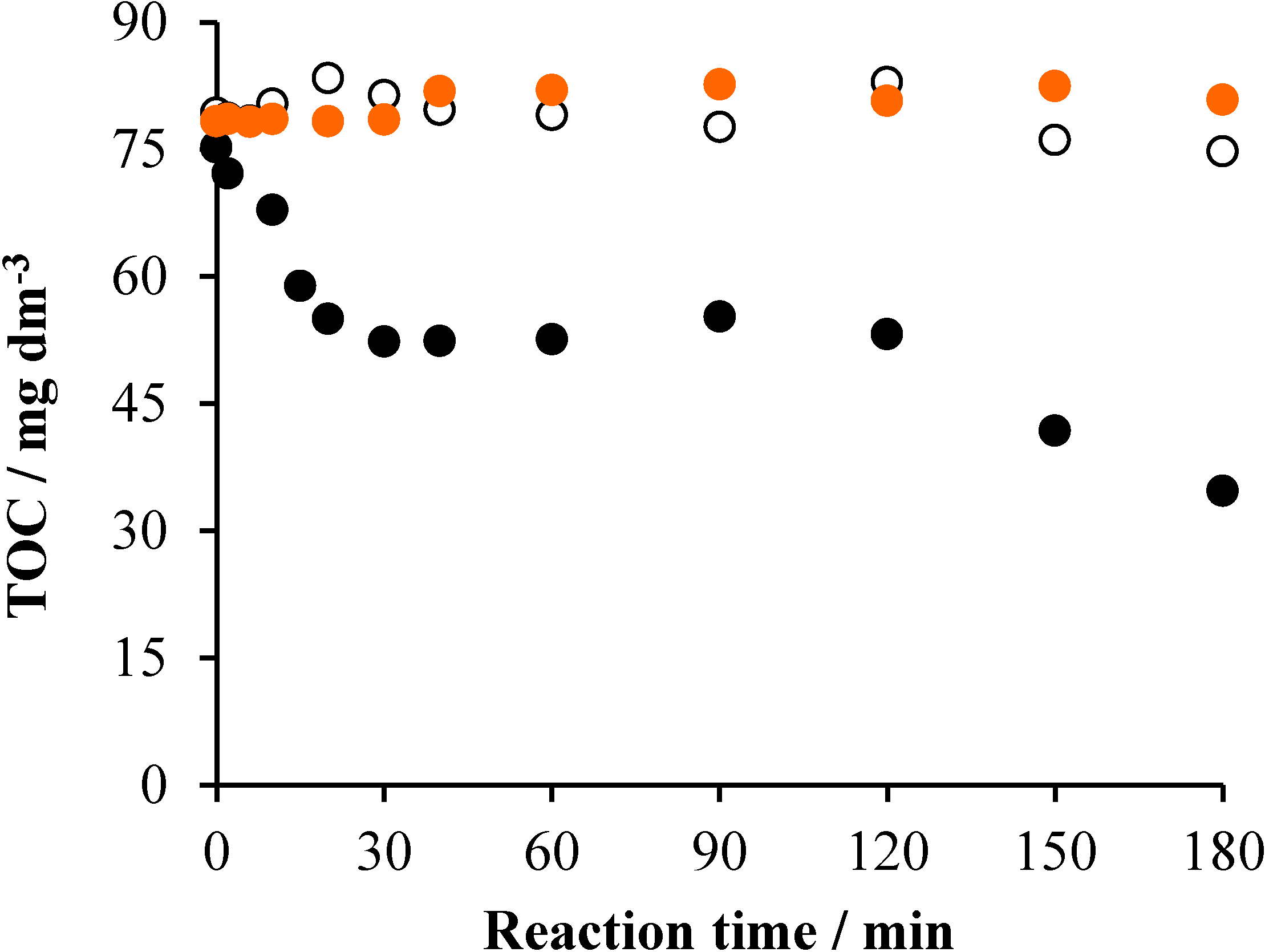
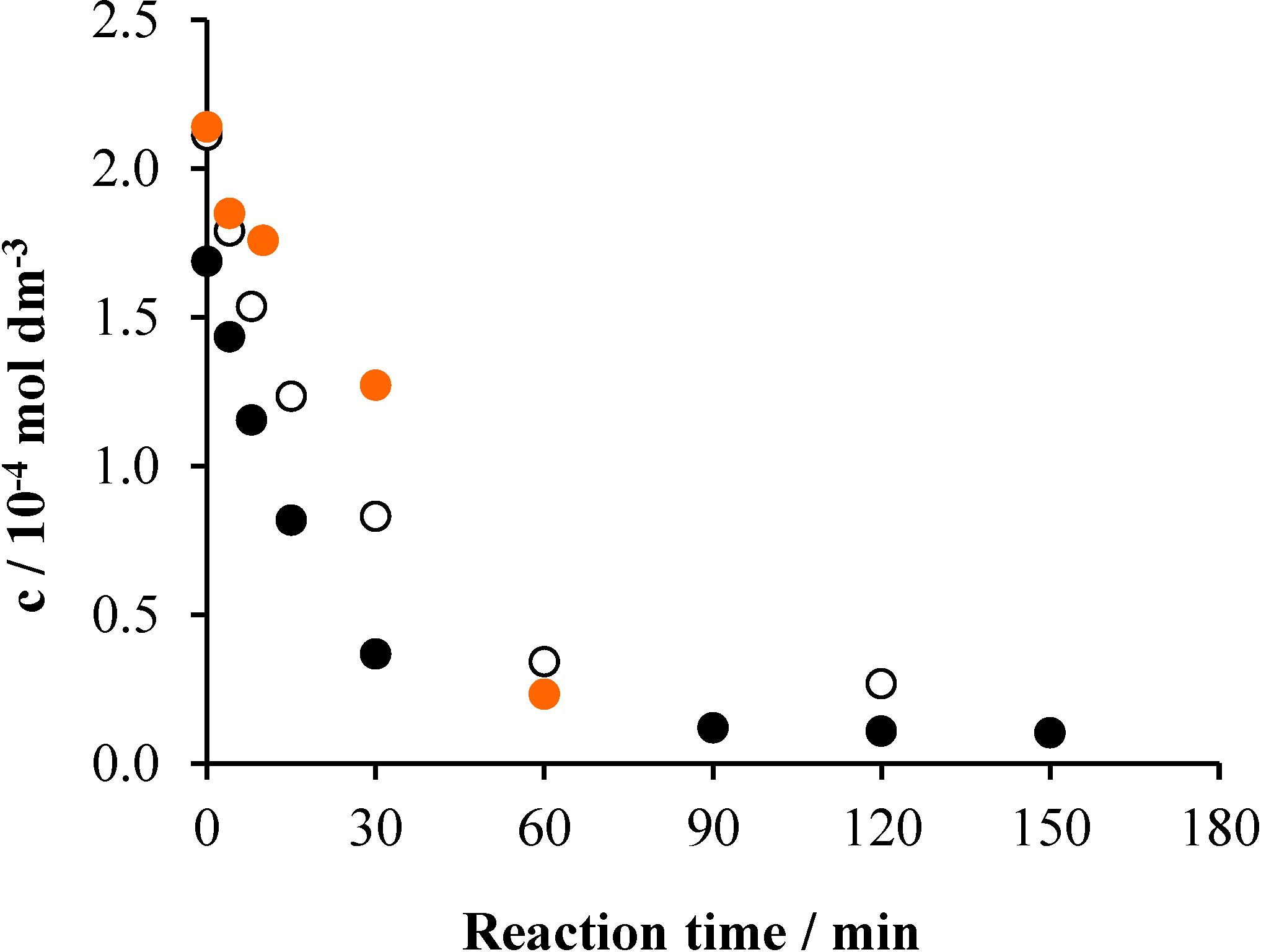
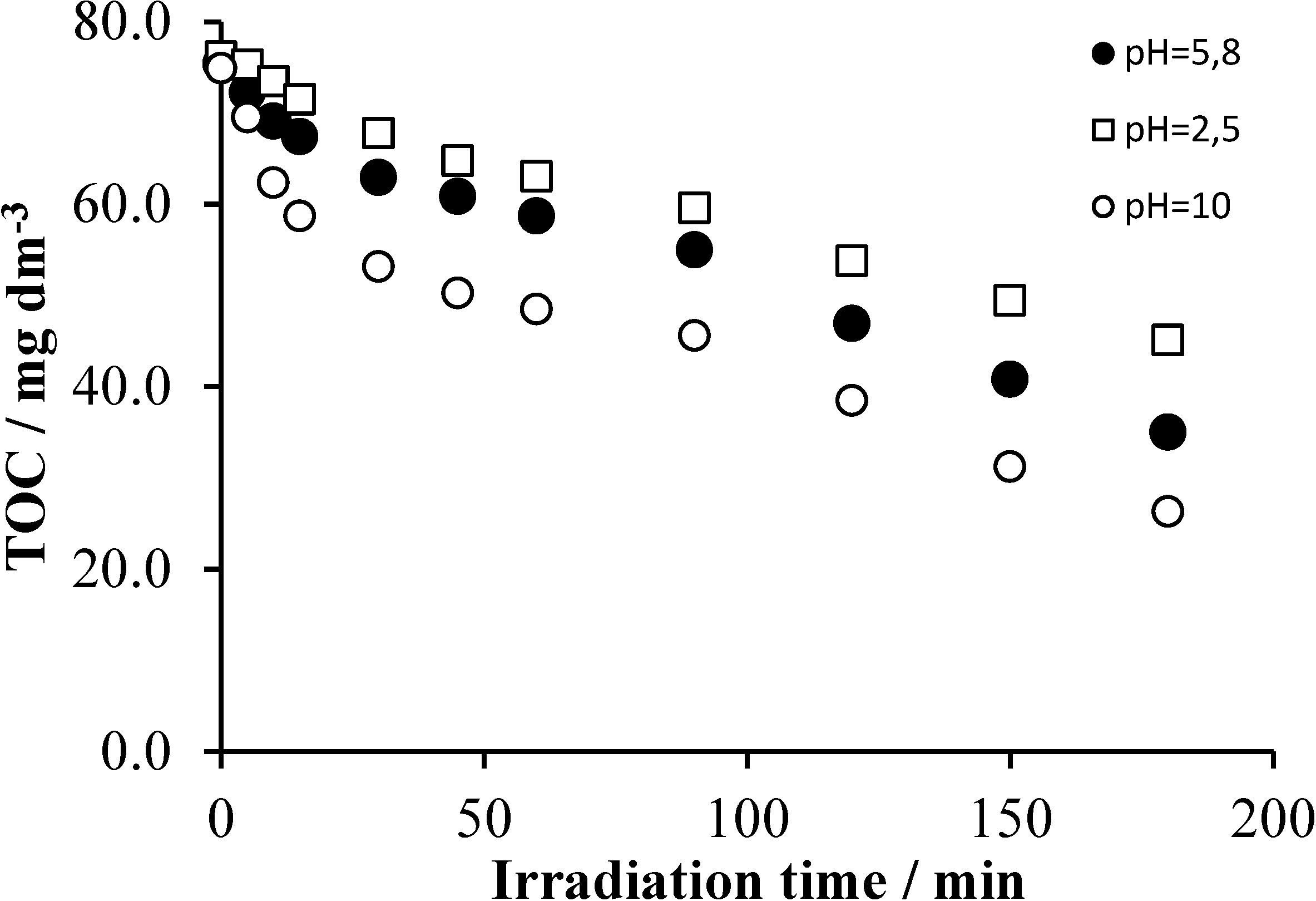
2.4. Effects of the Initial pH

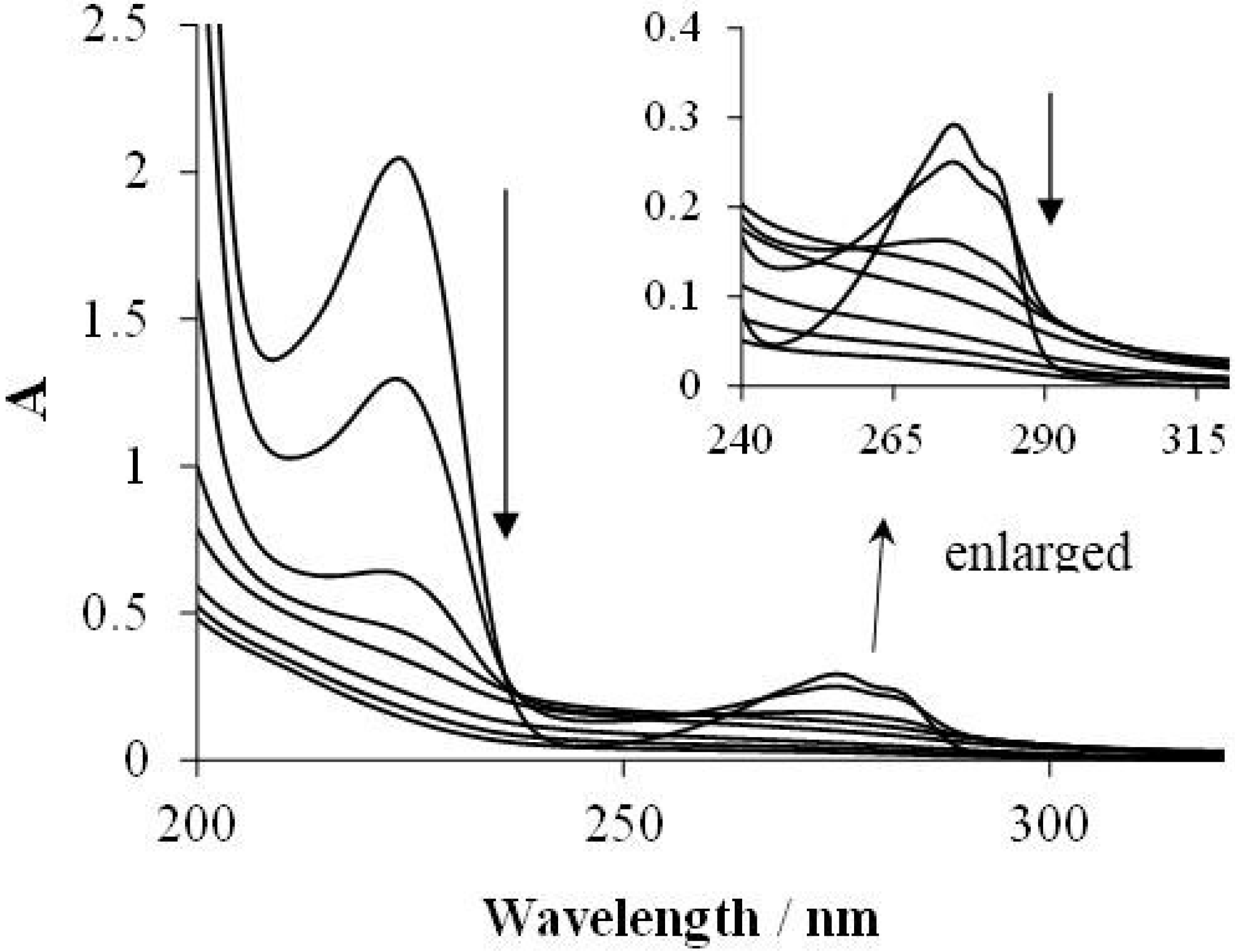
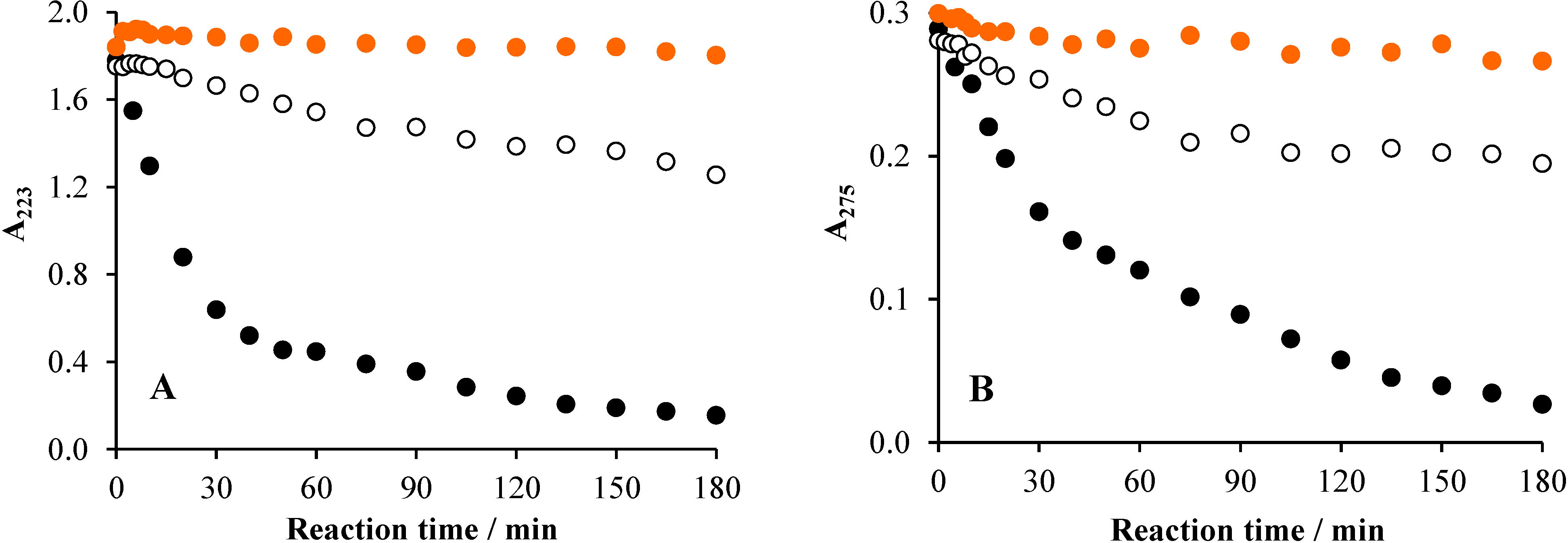
2.5. Mechanistic Considerations; Intermediates
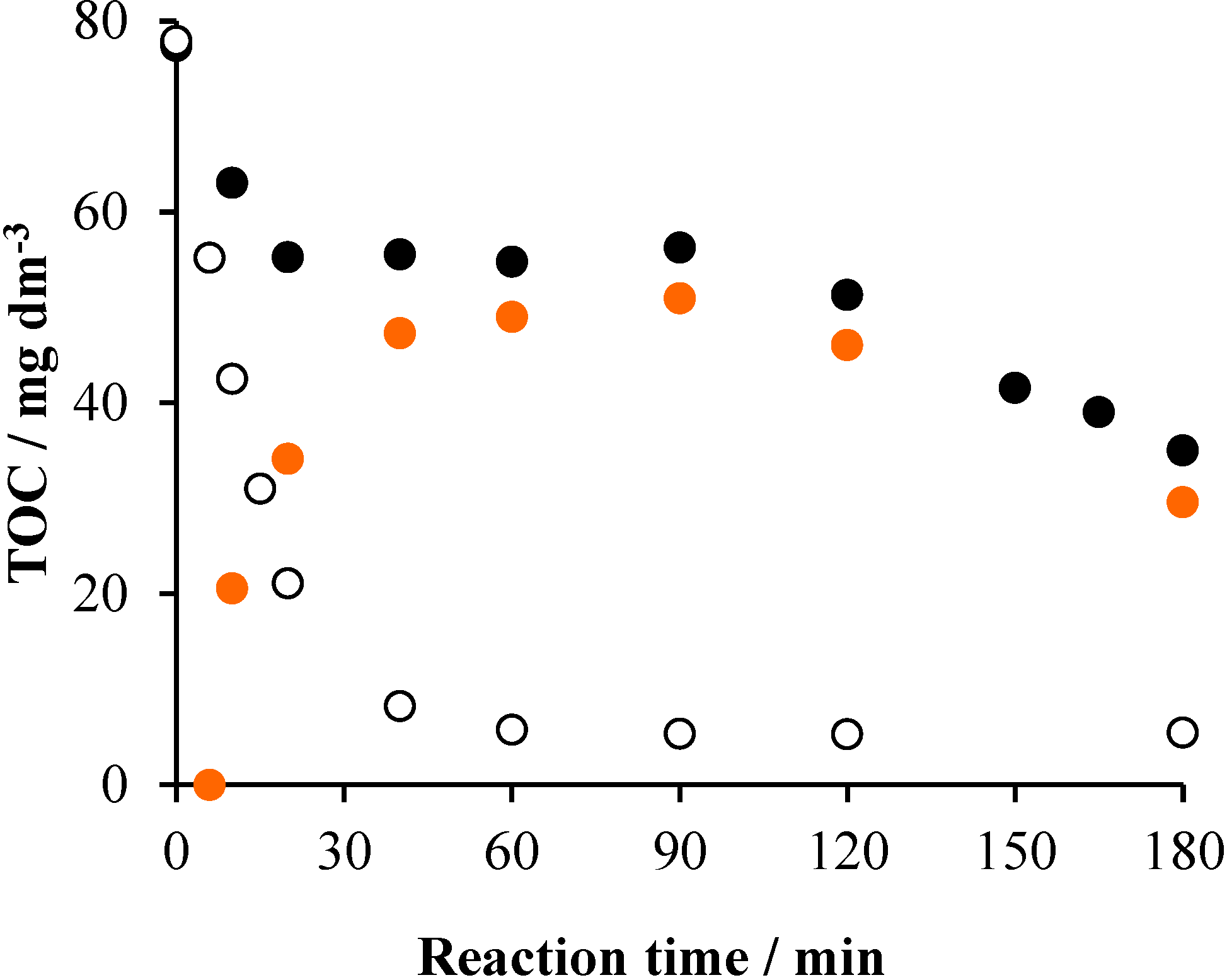
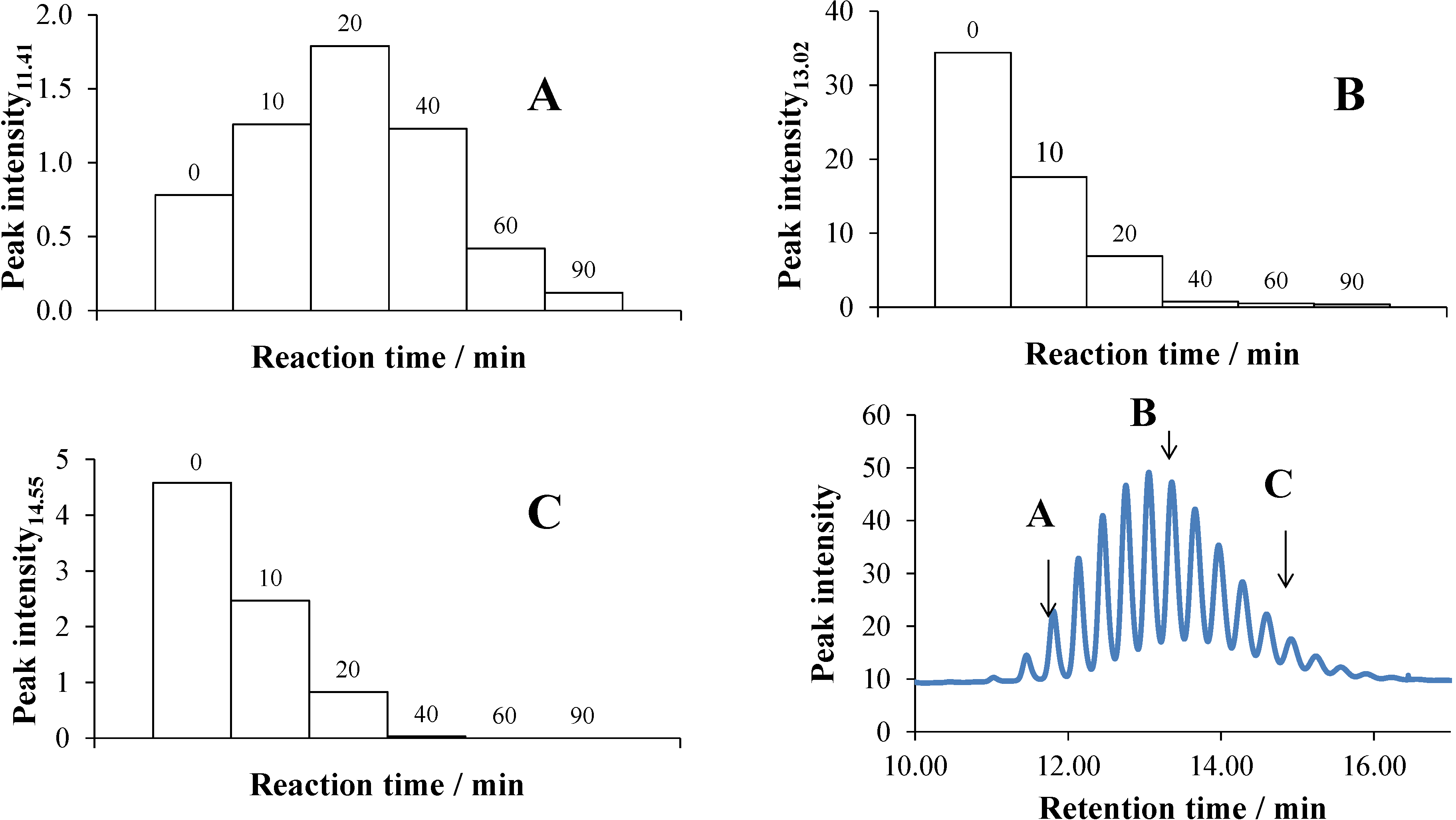
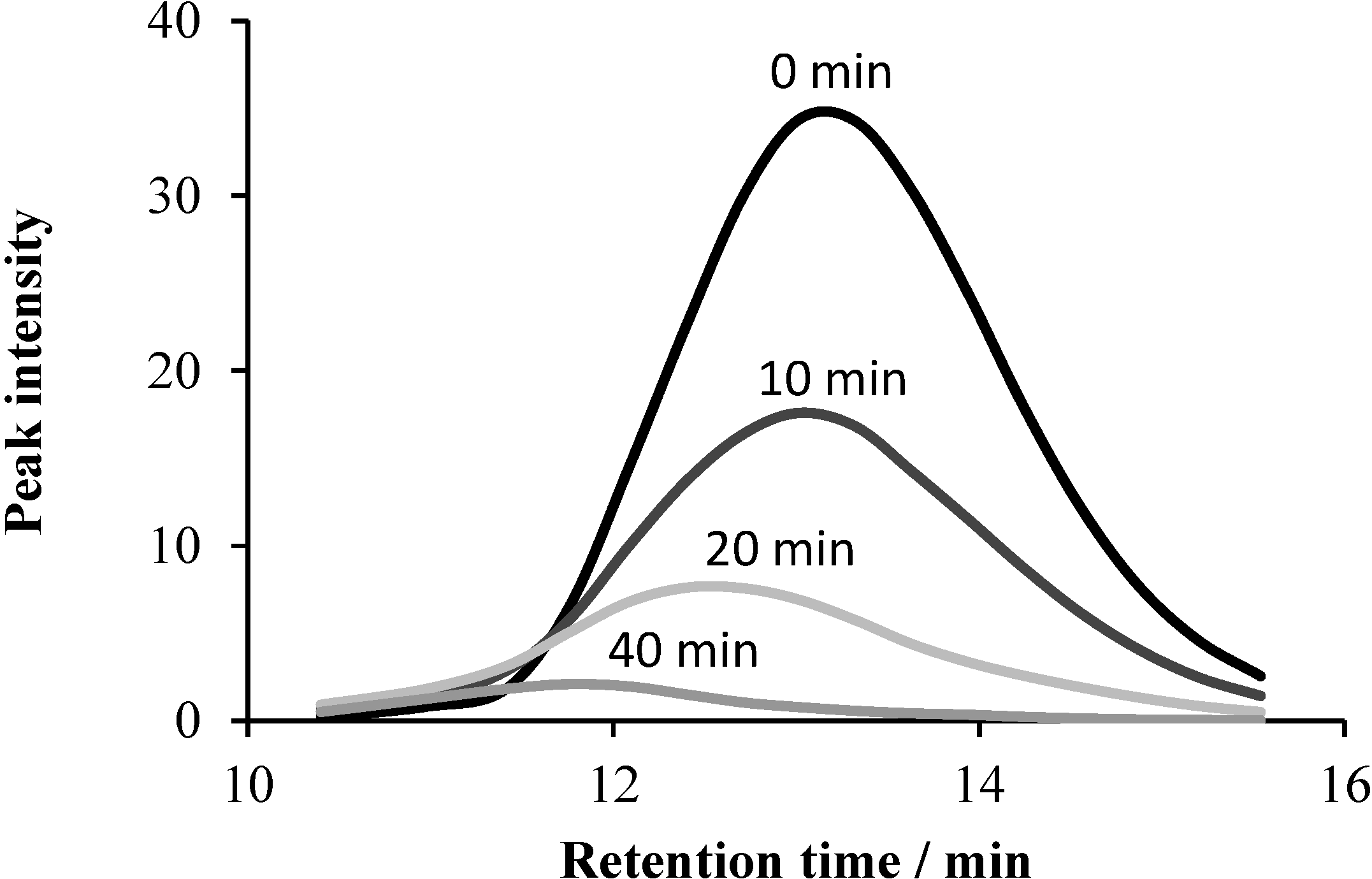
2.5.1. UHPLC Measurements

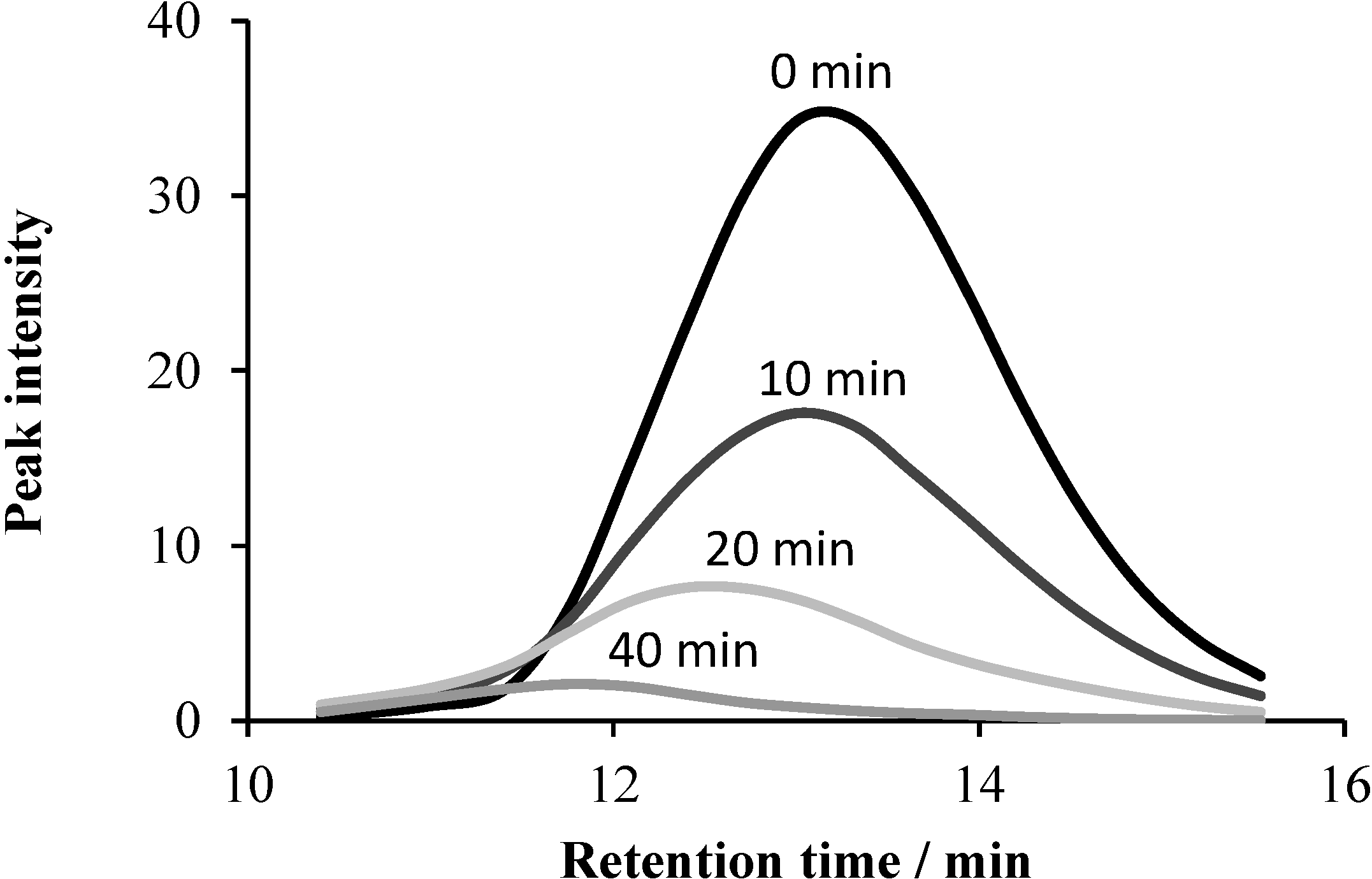
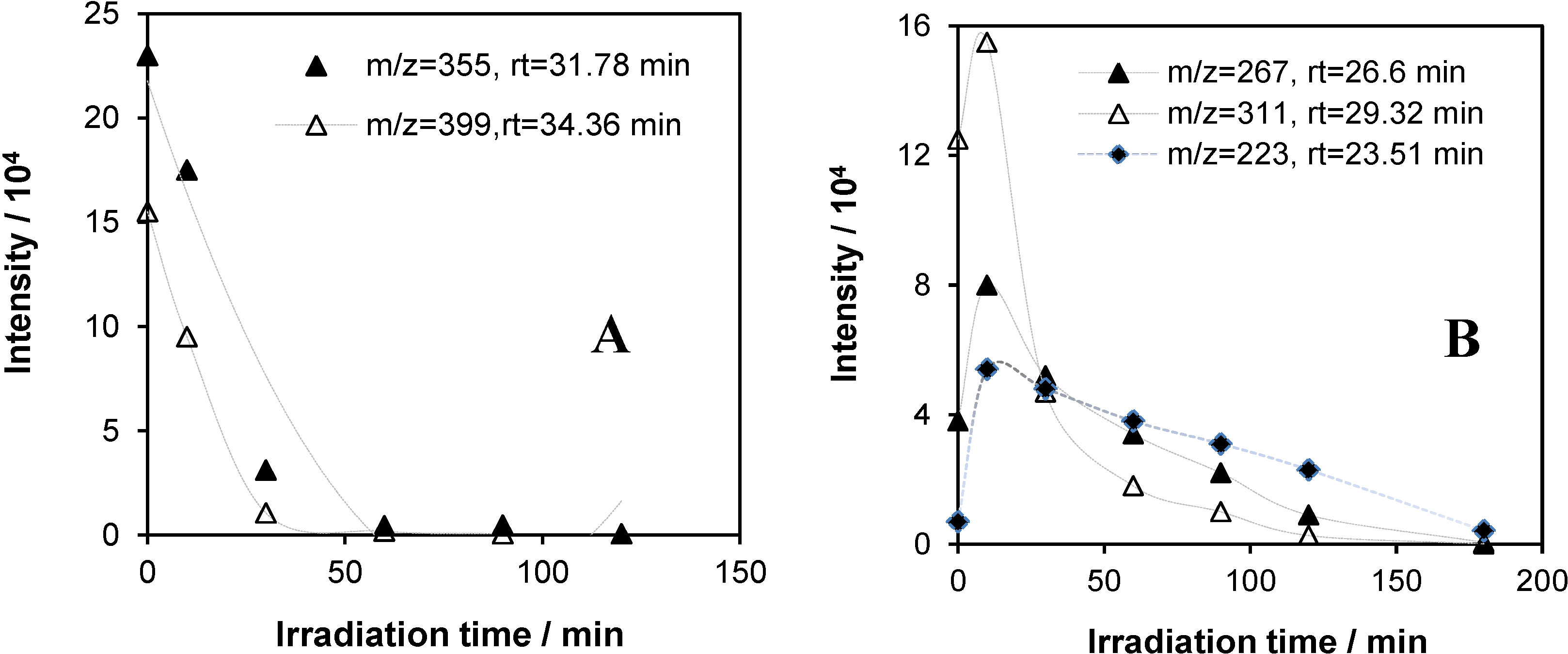
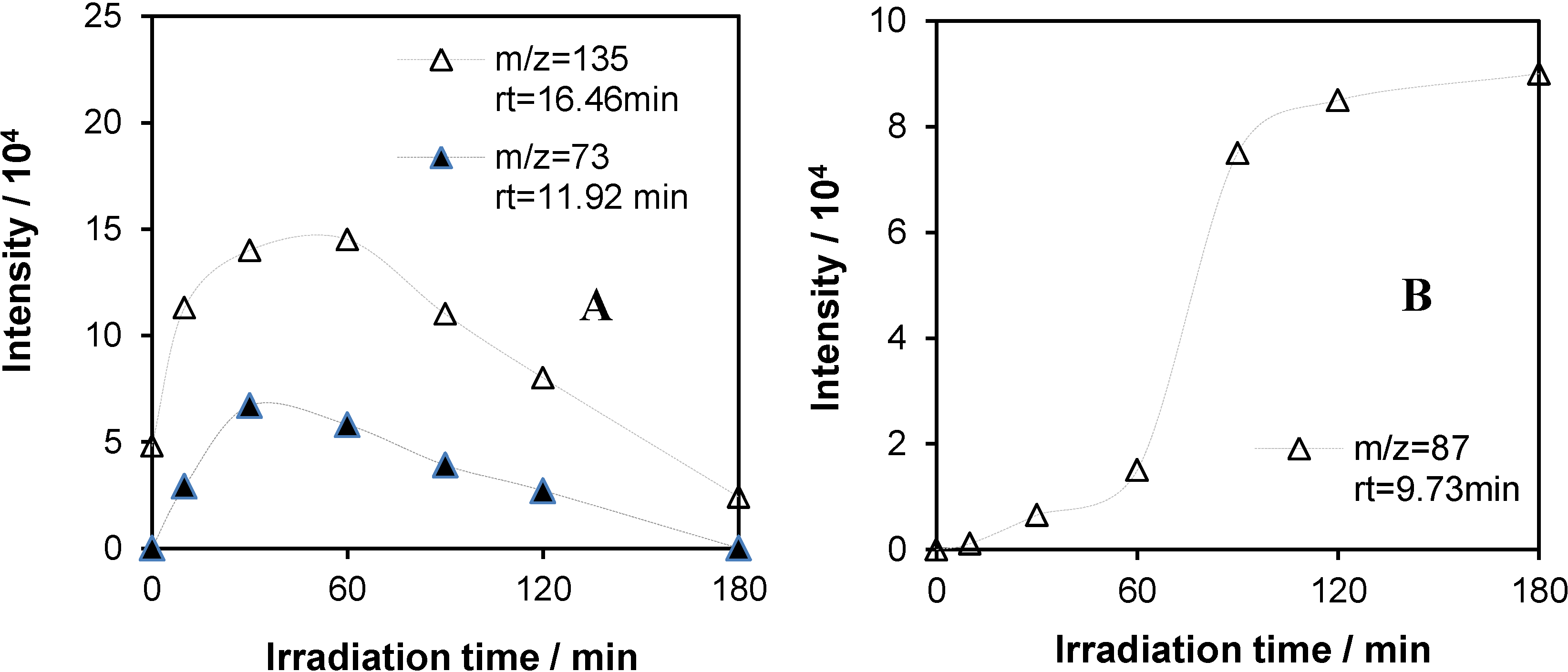
2.5.2. GC-MS Measurements


3. Experimental Section
3.1. Materials
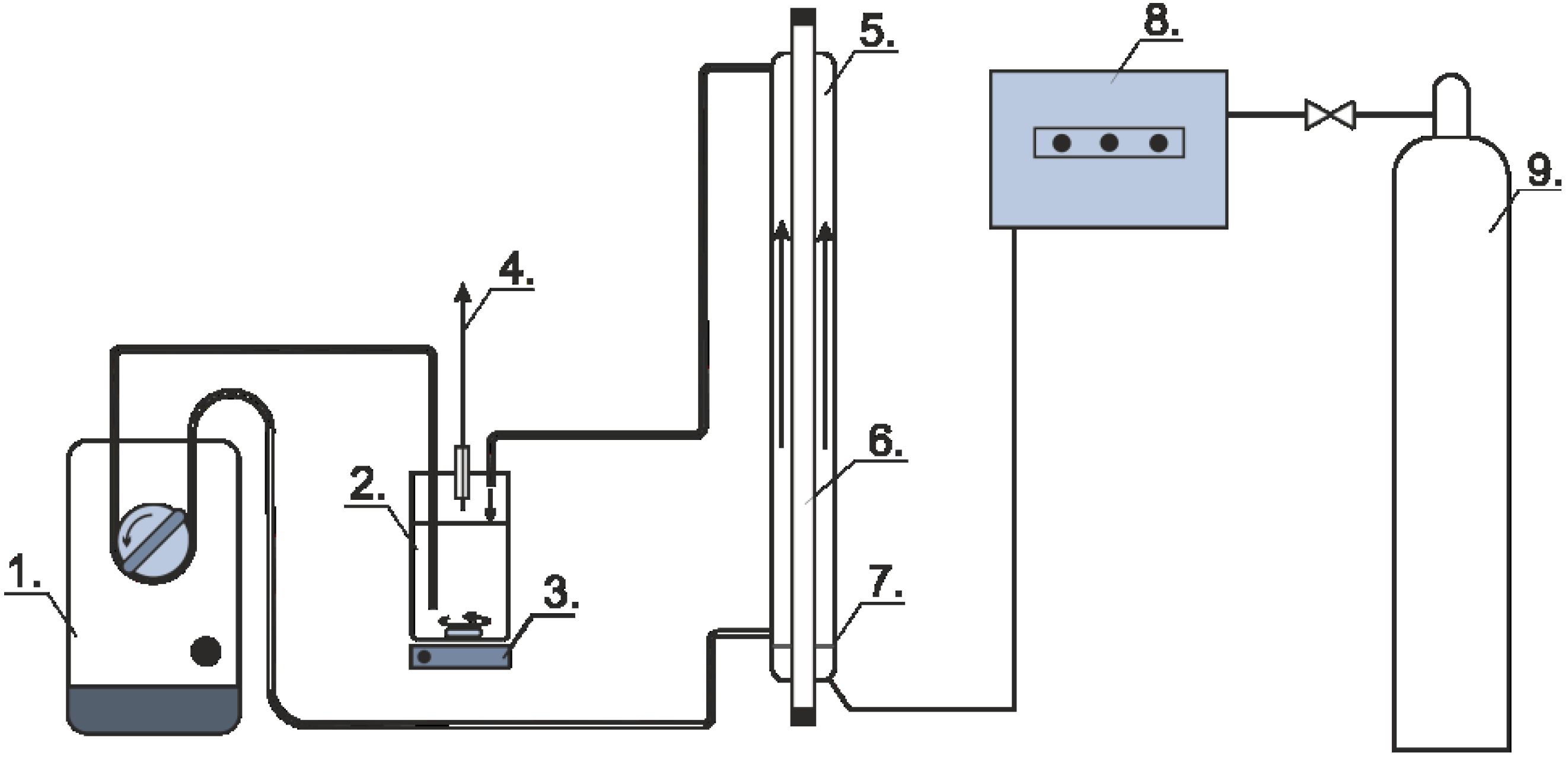
3.2. Photochemical Experiments

3.3. Analytical Procedures
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- White, M.A.; Clark, K.M.; Grayhack, E.J.; Dumont, M.E. Characteristics affecting expression and solubilization of yeast membrane proteins. J. Mol. Biol. 2007, 365, 621–636. [Google Scholar] [CrossRef] [PubMed]
- Kubak, B.M.; Yotis, W.W. Analysis of staphylococcus aureus cytoplasmic membrane proteins by isoelectric focusing. Biochim. Biophys. Acta 1981, 649, 642–650. [Google Scholar] [CrossRef] [PubMed]
- Anand, H.; Balasundarama, B.; Pandit, A.B.; Harrison, S.T.L. The effect of chemical pretreatment combined with mechanical disruption on the extent of disruption and release of intracellular protein from E. coli. Biochem. Eng. J. 2007, 35, 166–173. [Google Scholar] [CrossRef]
- Miller, D.M. Total solubilization of erythrocyte membranes by nonionic detergents. Biochem Biophys. Res. Commun. 1970, 40, 716–722. [Google Scholar] [CrossRef] [PubMed]
- Gomez, V.; Ferreres, L.; Pocurull, E.; Borrull, F. Determination of non-ionic and anionic surfactants in environmental water matrices. Talanta 2011, 84, 859–866. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.J.; Tseng, D.H.; Huang, S.L. Biodegradation of octylphenol polyethoxylate surfactant Triton X-100 by selected microorganisms. Bioresour. Technol. 2005, 96, 1483–1491. [Google Scholar] [CrossRef] [PubMed]
- Perkowski, J.; Mayer, J.; Kos, L. Reactions of non-ionic surfactants, Triton X-n type, with OH radicals. A review. Fibers Text. East. Eur. 2005, 13, 81–85. [Google Scholar]
- Manzano, M.A.; Derales, J.A.; Sales, D.; Quiroga, J.M. The effect of temperature on the biodegradation of a nonylphenol polyethoxylate in river water. Water Res. 1999, 33, 2593–2600. [Google Scholar] [CrossRef]
- Okpokwasili, G.C.; Olisa, A.O. River-water biodegradation of surfactants in liquid detergents and shampoos. Water Res. 1991, 25, 1425–1429. [Google Scholar] [CrossRef]
- Mohan, P.K.; Nakhla, G.; Yanful, E.K. Biokinetics of biodegradation of surfactants under aerobic, anoxic and anaerobic conditions. Water Res. 2006, 40, 533–540. [Google Scholar] [CrossRef] [PubMed]
- Ahel, M. Behaviour of Alkylphenol-Polyethoxylate Surfactants in the Aquatic Environment. Ph.D. Thesis, University of Zagreb, Zagreb, Croatia, 1987. [Google Scholar]
- Ahel, M.; Giger, W.; Koch, M. Behaviour of alkylphenol-polyethoxylate surfactants in the aquatic environment I. Occurrence and transformation in sewage treatment. Water Res. 1994, 28, 1131–1142. [Google Scholar] [CrossRef]
- Dias, N.; Mortara, R.A.; Lima, N. Morphological and physiological changes in Tetrahymena pyriformis for the in vitro cytotoxicity assessment of Triton X-100. Toxicol. In Vitro 2003, 17, 357–366. [Google Scholar] [CrossRef] [PubMed]
- Koley, D.; Bard, A.J. Triton X-100 concentration effects on membrane permeability of a single HeLa cell by scanning electrochemical microscopy (SECM). Proc. Natl. Acad. Sci. USA 2010, 107, 16783–16787. [Google Scholar] [CrossRef] [PubMed]
- Riepl, R.G.; Vidaver, G.A. Effects of Triton X-100 treatments on the composition and activities of membrane vesicles from pigeon. Biochim. Biophys. Acta 1978, 507, 94–106. [Google Scholar] [CrossRef] [PubMed]
- Oz, M.; Spivak, C.E.; Lupica, C.R. The solubilizing detergents, Tween 80 and Triton X-100 non-competitively inhibit α7-nicotinic acetylcholine receptor function in Xenopus oocytes. J. Neurosci. Methods 2004, 137, 167–173. [Google Scholar] [CrossRef] [PubMed]
- Reemtsma, T. Methods of analysis of polar aromatic sulfonates from aquatic environments. J. Chromatogr. A 1996, 733, 473–489. [Google Scholar] [CrossRef]
- Manhas, M.S.; Khan, Z. Kinetics of oxidation of polyoxyethylene chain of polyoxyethylene-t-octyl phenol (Triton X-100) by diperiodatoargenate (III). Colloid. Surf. A 2006, 277, 207–213. [Google Scholar] [CrossRef]
- Perkowski, J.; Mayer, J. Gamma-radiolysis of Triton X-100 aqueous solution. J. Radioanal. Nucl. Chem. 1992, 157, 27–36. [Google Scholar] [CrossRef]
- Perkowski, J.; Bulska, A.; Józwiak, W.K. Titania-assisted photocatalytic decomposition of Triton X-100 detergent in aqueous solution. Environ. Prot. Eng. 2005, 31, 61–76. [Google Scholar]
- Saien, J.; Ojaghloo, Z.; Soleymani, A.R.; Rasoulifard, M.H. Homogeneous and heterogeneous AOPs for rapid degradation of Triton X-100 in aqueous media via UV light, nano titania, hydrogen peroxide and potassium persulfate. Chem. Eng. J. 2011, 167, 172–182. [Google Scholar] [CrossRef]
- Perkowski, J.; Bulska, A.; Góralski, J.; Józwiak, W.K. Pt/TiO2-assisted photocatalytic decomposition of Triton X-100 detergent in aqueous solution. Environ. Prot. Eng. 2007, 33, 129–142. [Google Scholar]
- Zhang, Y.; Wan, Y. Heterogeneous photocatalytic degradation of Triton X-100 in aqueous TiO2 suspensions. Am. J. Environ. Prot. 2014, 3, 28–35. [Google Scholar] [CrossRef]
- Kohtani, S.; Koshiko, M.; Kudo, A.; Tokumura, K.; Ishigaki, Y.; Toriba, A.; Hayakawa, K.; Nakagaki, R. Photodegradation of 4-alkylphenols using BiVO4 photocatalyst under irradiation with visible light from a solar simulator. Appl. Catal. B 2003, 46, 573–586. [Google Scholar] [CrossRef]
- Naya, S.; Nikawa, T.; Kimura, K.; Tada, H. Rapid and complete removal of nonylphenol by gold nanoparticle/rutile titanium(IV) oxide plasmon photocatalyst. ACS Catal. 2013, 3, 903–907. [Google Scholar] [CrossRef]
- Szabó-Bárdos, E.; Zsilák, Z.; Lendvay, G.; Horváth, O.; Markovics, O.; Hoffer, A.; Törő, N. Photocatalytic degradation of 1,5-naphthalenedisulfonate on colloidal titanium dioxide. J. Phys. Chem. B 2008, 112, 14500–14508. [Google Scholar] [CrossRef] [PubMed]
- Szabó-Bárdos, E.; Baja, B.; Horváth, E.; Horváth, A. Photocatalytic decomposition of L-serine and L-aspartic acid over bare and silver deposited TiO2. J. Photochem. Photobiol. A 2010, 213, 37–45. [Google Scholar] [CrossRef]
- Szabó-Bárdos, E.; Markovics, O.; Horváth, O.; Törő, N.; Kiss, G. Photocatalytic degradation of benzenesulfonate on colloidal titanium dioxide. Water Res. 2011, 45, 1617–1628. [Google Scholar] [CrossRef] [PubMed]
- Szabó-Bárdos, E.; Somogyi, K.; Törő, N.; Kiss, G.; Horváth, A. Photocatalytic decomposition of L-phenylalanine over TiO2: Identification of intermediates and the mechanism of photodegradation. Appl. Catal. B 2011, 101, 471–478. [Google Scholar] [CrossRef]
- Zsilák, Z.; Szabó-Bárdos, E.; Fónagy, O.; Horváth, O.; Horváth, K.; Hajós, P. Degradation of benzenesulfonate by heterogeneous photocatalysis combined with ozonation. Catal. Today 2014, 230, 55–60. [Google Scholar] [CrossRef]
- Zsilák, Z.; Fónagy, O.; Szabó-Bárdos, E.; Horváth, O.; Horváth, K.; Hajós, P. Degradation of industrial surfactants by photocatalysis combined with ozonation. Environ. Sci. Pollut. Res. 2014, 21, 11126–11134. [Google Scholar] [CrossRef]
- Yu, C.H.; Wu, C.H.; Ho, T.H.; Hong, P.K.A. Decolorization of C.I. Reactive Black 5 in UV/TiO2, UV/oxidant and UV/TiO2/oxidant systems: A comparative study. Chem. Eng. J. 2010, 158, 578–583. [Google Scholar] [CrossRef]
- Zhao, J.; Wu, K.; Hidaka, H.; Serpone, N. Photodegradation of dyes with poor solubility in an aqueous surfactant/TiO2 dispersion under visible light irradiation. J. Chem. Soc. Faraday Trans. 1998, 94, 673–676. [Google Scholar] [CrossRef]
- Olmez-Hanci, T.; Arslan-Alaton, I.; Genc, B. Degradation of the nonionic surfactant Triton™ X-45 with HO• and SO4•−—based advanced oxidation processes. Chem. Eng. J. 2014, 239, 332–340. [Google Scholar] [CrossRef]
- Nagarnaik, P.M.; Boulanger, B. Advanced oxidation of alkylphenol ethoxylates in aqueous systems. Chemosphere 2011, 85, 854–860. [Google Scholar] [CrossRef] [PubMed]
- Jahnke, A.; Gandrass, J.; Ruck, W. Simultaneous determination of alkylphenol ethoxylates and their biotransformation products by liquid chromatography/electrospray ionisation tandem mass spectrometry. J. Chromatogr. A 2004, 1035, 115–122. [Google Scholar] [CrossRef] [PubMed]
- Ying, G.G.; Williams, B.; Kookana, R. Environmental fate of alkylphenols and alkylphenolethoxylates—A review. Environ. Int. 2002, 28, 215–226. [Google Scholar] [CrossRef] [PubMed]
- Karcia, A.; Arslan-Alaton, I.; Bekbolet, M. Advanced oxidation of a commercially important nonionic surfactant: Investigation of degradation products and toxicity. J. Hazard. Mater. 2013, 263, 275–282. [Google Scholar] [CrossRef] [PubMed]
- Rabek, J.F. Experimental Methods in Photochemistry and Photophysics; John Wiley & Sons Ltd.: New York, NY, USA, 1982; pp. 944–946. [Google Scholar]
- Kirk, A.D.; Namasivayam, C. Errors in ferrioxalate actinometry. Anal. Chem. 1983, 55, 2428–2429. [Google Scholar] [CrossRef]
- Tjahjanto, R.T.; Galuh, D.; Wardani, R.S. Ozone determination: A comparison of quantitative analysis methods. J. Pure Appl. Chem. Res. 2012, 1, 18–25. [Google Scholar]
© 2015 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hegedűs, P.; Szabó-Bárdos, E.; Horváth, O.; Horváth, K.; Hajós, P. TiO2-Mediated Photocatalytic Mineralization of a Non-Ionic Detergent: Comparison and Combination with Other Advanced Oxidation Procedures. Materials 2015, 8, 231-250. https://doi.org/10.3390/ma8010231
Hegedűs P, Szabó-Bárdos E, Horváth O, Horváth K, Hajós P. TiO2-Mediated Photocatalytic Mineralization of a Non-Ionic Detergent: Comparison and Combination with Other Advanced Oxidation Procedures. Materials. 2015; 8(1):231-250. https://doi.org/10.3390/ma8010231
Chicago/Turabian StyleHegedűs, Péter, Erzsébet Szabó-Bárdos, Ottó Horváth, Krisztián Horváth, and Péter Hajós. 2015. "TiO2-Mediated Photocatalytic Mineralization of a Non-Ionic Detergent: Comparison and Combination with Other Advanced Oxidation Procedures" Materials 8, no. 1: 231-250. https://doi.org/10.3390/ma8010231